CHAPTER 4 DISORDERS OF MEMORY
DEFINITIONS AND TERMINOLOGY
Declarative and Nondeclarative Memory
In general, when patients speak of memory complaints, they are referring to the recollection of information for which there is conscious awareness. In neuropsychological terminology, this is called declarative, or explicit, memory. The corollary is nondeclarative, or implicit, memory, which refers to phenomena such as motor skill learning, priming and classical conditioning (Table 4-1). For instance, with appropriate training, a tennis player can be seen to “learn,” as evidenced by improved performance; however, in the course of a rally, the player is not consciously recollecting the motor sequence required to execute each shot. Priming refers to a situation in which prior exposure leads to altered performance and can be shown experimentally in language tasks such as naming and lexical decision making. For example, imagine being presented with words on a computer screen and being asked to read them as soon as they appear; the latency between exposure and response (reaction time) is shorter if a related, as opposed to a nonrelated, item is presented immediately before the test item (e.g., a subject will respond faster to the word tiger if it is preceded by lion rather than house). Highlighting the dissociation between declarative and nondeclarative memory is the fact that patients with Alzheimer’s disease have marked declarative deficits and yet may respond even faster with priming (hyperpriming) than control subjects. Although nondeclarative memory is crucial for many functions, it is not what patients usually mean when they report memory symptoms and is not discussed further.
Episodic and Semantic Memory
Declarative memory is also considered in terms of encoding, storage, and retrieval. In other words, a neurophysiological change takes place during learning (encoding); this change must leave some enduring trace (storage), and there must be a mechanism by which this trace can be reactivated, at will, to lead to the subjective experience of remembering (retrieval). Often, these processes cannot be disentangled in the clinic; for instance, a patient with no recall of new information on formal neuropsychological testing may have a deficit at any or all of these stages. Nevertheless, neuropsychological tests can, to some extent, tease these stages apart by varying task demands. For example, a patient who requires an excessive number of learning trials to reach a criterion and yet retains a lot of this information after a delay can be considered as having an encoding problem. This profile is often indicative of an attention disorder rather than a true amnesic syndrome. Storage problems may be suggested by an accelerated decay in recall performance between two time points (e.g., immediately and 30 minutes after encoding). However, it is important to realize that a degree of decay is seen under normal circumstances; therefore, interpretation requires comparison with demographically matched norms. Retrieval deficits can be investigated by comparing free recall (“What did I ask you to remember?”) with recognition memory (“Which of these did I show you earlier?”). Recognition is typically tested by either forced-choice questions—in which target answers and foils (incorrect alternatives) are presented simultaneously in pairs and the patient has to choose those that he or she has seen before—or by asking for yes/no responses as targets and foils are presented in a random sequence. Because the patient is presented the previously studied material, retrieval demands are minimized, and hence a retrieval deficit is suggested where there is disproportionate impairment of free recall in comparison with recognition memory. Recognition is, however, an easier task than is free recall, and so this dissociation also needs to be assessed by comparison with control norms. It is likely that the neural systems responsible for these processes partially overlap, in that some brain areas such as the hippocampus may be necessary for all aspects, whereas other areas may be differentially engaged at a specific stage. For example, functional activation studies of changes in cerebral blood flow suggest greater engagement of the left and right prefrontal cortices during encoding and retrieval, respectively.1
Amnesia
Amnesia is the generic term for loss of memory. An inability to establish new memories after the pathological event is called anterograde amnesia, whereas the inability to recall memories that had been established before the pathological event is called retrograde amnesia (Fig. 4-1). The fact that the distinction between anterograde and retrograde is referenced to the pathological insult is important. Students often think erroneously that retrograde amnesia refers to memory loss for past events, whereby “past” is loosely defined as any time before the clinical consultation. This leads to the absurd situation of testing for retrograde deficits by asking the patient what they did “yesterday,” which makes the distinction from anterograde deficits meaningless, as it becomes referenced to the ever-changing present. Patients with dense amnesia, especially in the acute phase, may confabulate: that is, fill in memory gaps with false statements.
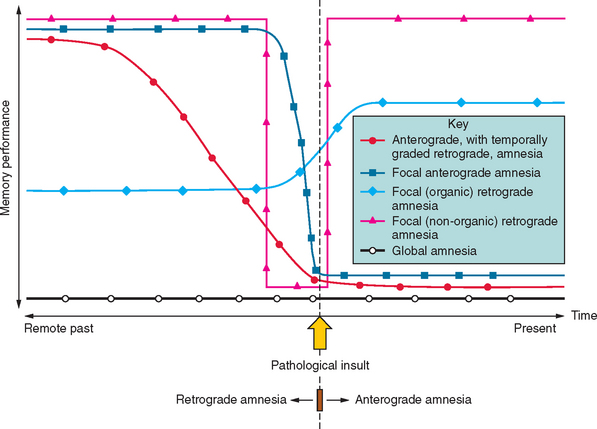
The situation becomes more ambiguous with regard to factual semantic knowledge (public events, famous people), including personal semantic facts (e.g., names of schools attended, name of employers). Variable degrees of impairment in factual semantic knowledge are usual in amnesia, and this information can be objectively dated in time (in contrast to an individual’s episodic recollections, which are more difficult to date or verify). As a consequence, evidence for temporal gradients in memory impairment is typically compiled from these datable facts. For instance, a patient with a new-onset pathological process who knows that the Berlin Wall was pulled down, that apartheid ended, and that Iraq invaded Kuwait but does not remember a tsunami killing hundreds of thousands, the World Trade Center being destroyed, or Princess Diana dying has evidence for a retrograde amnesia extending back to the early to mid-1990s. It should be noted that the distinction between episodic and semantic memory—the degree of “semanticization”—becomes blurred at this point, inasmuch as some facts may be recalled from the specific context in which they were encountered—as so-called “flashbulb” memories. In addition, the frequency of exposure to particular facts varies between individuals. For example, the average American has most likely had many more encounters with stories about the World Trade Center than with stories about the Berlin Wall, and this will bias successful recollection towards the more recent, semantically reinforced event.
Working Memory
An important concept to distinguish from both episodic and semantic memory is working memory, frequently referred to by experimental psychologists as short-term memory. This refers to information that, at any given moment, is held “on-line” by the brain—that is, in consciousness—and that is not recollected after a distraction period (unless incidental episodic encoding of the material has also taken place). This faculty is exemplified by the ability to keep a telephone number in the mind in the interval between looking it up the telephone directory and dialing the number. Models of working memory posit auditory-verbal and visuospatial components that are coordinated and manipulated by a central executive system.2 A highly distributed network of cortical and subcortical areas supports working memory, although the dorsolateral prefrontal cortex appears to be particularly important for the executive component. It is therefore unsurprising that working memory is most vulnerable to diffuse insults such as those found in metabolic encephalopathies and closed head injury. It can be dissociated from the long-term declarative memory systems described previously; patients with even the densest amnesia can still have intact working memory.
NEUROBIOLOGY OF LEARNING AND MEMORY
Cellular Mechanisms
These observations suggest that the transmission properties of existing synapses can be modified by usage, but it is notable that the “hard wiring” of the nervous system is also dynamic. The dendritic spines, the sites of synaptic connections in the neuronal dendritic arbor, also display plasticity over time, which suggests that the neural networks themselves may be adaptive.3 Technical developments, enabling dendritic spines to be labeled and studied in real time, have shown that changes in structure, including the generation of new dendritic spine protrusions, can be triggered by LTP-inducing activity. Conversely, some LTD-inducing stimuli have been shown to cause loss of dendritic spines. It must be acknowledged, however, that although these morphological and physiological findings suggest mechanisms by which learning and memory may be supported, the precise relevance of these specific processes to memory in vivo is far from established in the mature brain.
For further reading on these topics and other current issues in the neuroscience of memory, the excellent essays by Dudai (2002) are recommended.
The Topography of Memory
Between synaptic plasticity and the topographical anatomy of regions implicated in amnesia, the precise nature of the memory trace remains difficult to delineate. As already discussed, the information that constitutes episodic memory comprises multiple sensory modalities. The prevailing hypothesis is that structures such as the hippocampus do not “store” this information, but rather act to bind the disparate components that constitute a memory trace. To illustrate this model, consider an episodic memory of a specific conversation comprising, for simplicity, an auditory component (the dialogue) and a visual component (the scene). At the time of encoding, visual and auditory inputs contemporaneously activate a neural network in the isocortex, including auditory and visual association cortex, as well as the mesial temporal lobe. The temporospatial firing pattern that represents the memory trace can then be subsequently reactivated via the hippocampus. The traditional view (Fig. 4-2) is that with time, this neural representation gradually becomes independent of the hippocampus: so-called consolidation. This is the explanation for the Ribot effect, a temporal gradient in retrograde amnesia in which amnesia is most dense for the time immediately before the pathological insult and is progressively less dense for earlier periods. This view is almost certainly an oversimplification; many amnesic patients do not exhibit a Ribot effect but, rather, a flat profile of memory loss extending back over their whole life. Furthermore, when it is found, the temporal gradient often extends back over several decades, which suggests that consolidation is an implausible entity in evolutionary terms, inasmuch as it implies that in old age, the brain is still consolidating memories from early adult life.4
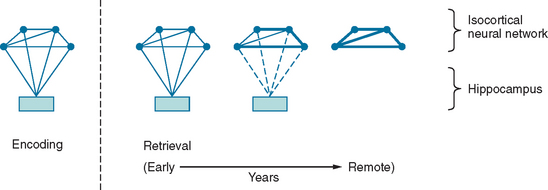
Another hotly debated topic is whether the distinction between semantic and episodic memory is meaningful in biological terms. Evidence for two separable memory systems comes from the observation that individuals with episodic memory amnesia have preservation of general semantic knowledge. Furthermore, neuropsychological studies have shown that patients with hippocampal amnesia, including those with damage sustained in infancy,5 can acquire new semantic knowledge in spite of the amnesia. This suggests that semantic memory, including its acquisition, can be supported independently of the hippocampus. The opposing view is that the acquisition of both semantic and episodic memory is dependent on a unitary system. Evidence for this comes from the observation that although acquisition of semantic knowledge may occur in patients with hippocampal amnesia, it is not completely normal.6 Whereas each episodic memory is unique in time and space, semantic facts are experienced in multiple contexts. This means that there is an enormous encoding advantage for the latter, which may explain the dissociation.
In addition to the interaction of the hippocampus and isocortical sensory association areas in sustaining declarative memory, the amygdala and prefrontal cortex merit mention. The amygdala is involved in emotional processes, such as recognition of fear in other’s faces, but with regard to memory, it is thought to reinforce encoding of emotionally salient events. Clearly, people do not recollect their entire life experience as a bland, continuous narrative. The fact that some experiences are forgotten but others are remembered is related in part to their differing emotional significance, and it is thought that encoding of emotionally significant events is facilitated by an interaction of the amygdala with other mesial temporal structures.7 The prefrontal cortex is thought to have a “meta” role in mnemonic processing, being involved in focusing attention for encoding, forming retrieval strategies, and monitoring output.8 For instance, when a person is asked to produce a list of animals, performance is enhanced if the list is clustered into semantic categories (“category clustering”) such as zoo animals, farm animals, and domestic animals. The prefrontal cortex is specifically engaged in this type of processing, but not in storing the memories per se. Consequently, frontal lesions can cause deficits in free recall with relative preservation of recognition, because the latter does not require an active retrieval strategy.
ASSESSMENT OF MEMORY
Clinical Assessment
Wherever possible, a history should be obtained from a close relative of the patient to cross-reference to the patient’s account. After the clinician explains the procedure to the patient and obtains his or her consent, the informant should be seen alone, to allow frank discussion without fear of embarrassment. A common mistake in assessing patients with memory symptoms is failure to examine memory adequately. There is often insufficient time in a general clinic to test delayed recall over a long interval; however, new learning can be informally assessed by giving the patient some material to encode and then testing recall and recognition after a 5-minute distraction period filled with other elements of the examination. A seven-item name and address recall can be assessed without any special equipment; this test yields indices of encoding, recall, and recognition (Fig. 4-3). Asking the patient to copy abstract designs, followed by testing of spontaneous recall and recognition, can help assess nonverbal memory.

Neuropsychological Assessment
The neuropsychologist also documents the patient’s history, including the demographic information necessary for test interpretation (age, education, handedness). Memory performance also needs to be interpreted in the context of general cognitive abilities; thus, the assessment is never restricted to memory alone. A vast array of tests are available that probe various aspects of memory: encoding, retrieval, and recognition; verbal and nonverbal memory; remote memory; and so on. Some of the memory tests in common usage are listed in Table 4-2. Reliable interpretation of neuropsychological performance requires that the tests be administered under standardized conditions by an appropriately trained examiner. Ad hoc incorporation of neuropsychological tests into the bedside examination is therefore best avoided.
| Test | Description | Reference or Source |
|---|---|---|
| New Learning | ||
| Rey Auditory Verbal Learning Test (RAVLT) | Word list encoding, free recall, and recognition | Rey A: L’Examen Clinique en Psychologie. Paris: Presses Universitaires de France, 1964 |
| California Verbal Learning Test (CVLT) | Word list semantic clustering, encoding, free recall and recognition | Delis DC, Kaplan E, Kramer J, et al: California Verbal Learning Test (CVLT-II) Second Edition—Adult Version. San Antonio, TX: The Psychological Corporation, 2000 |
| Story Recall (Logical Memory) | Story free recall ± recognition | Wechsler D: Wechsler Memory Scale—Revised. San Antonio, TX: The Psychological Corporation, 1987. |
| Recognition Memory Test (RMT) | Face and word recognition | Warrington, EK: Recognition Memory Test. Windsor, UK: NFER-Nelson, 1984 |
| Doors and People Test | Verbal and nonverbal recall and recognition | Baddeley A, Emslie H, Nimmo-Smith I: Doors and people. Oxford, UK: Thames Valley Test Company, 1994 |
| Rey-Osterrieth Complex Figure* | Nonverbal recall ± recognition | Osterrieth PA: Le Test de Copie d’Une Figure Complex [Complex Figure Copy Test]. Arch Psychol 1944; 30:206-356. |
| Paired Associates Learning (PAL) | Object/spatial location recall | Cambridge Neuropsychological Test Automated Battery. Cambridge, UK: Cambridge Cognition, Ltd. |
| Rivermead Behavioural Memory Test (RBMT) | Various tests including some “real-life” memory tasks | Wilson BA, Cockburn J, Baddeley A: Rivermead Behavioural Memory Test (RBMT-II). Oxford, UK: Thames Valley Test Company, 2003 |
| Remote Memory | ||
| Autobiographical Memory Interview (AMI) | Personal semantics and episodes from childhood, early adulthood, and recent past | Kopelman M, Wilson B, Baddeley A: The Autobiographical Memory Interview (AMI). Oxford, UK: Thames Valley Test Company, 1990 |
| Galton-Crovitz cue-word test | Specific autobiographical episodes generated to word prompts | Crovitz HF, Schiffman H: Frequency of episodic memories as a function of their age. Bull Psychonom Soc 1974; 4:517-518 |
| General Semantic Memory | ||
| Category fluency | Generation of exemplars to a target category | |
| Boston Naming Test | Picture naming | Kaplan E, Goodglass H, Weintraub S: Boston Naming Test. Philadelphia: Lea & Febiger (Also published by Psychcorp and LWW.), 1983 |
| Graded Naming Test | Picture naming | McKenna P, Warrington EK: The Graded Naming Test. Windsor, UK: NFER-Nelson, 1993 |
| Pyramids and Palm Trees Test | Word and picture forced-choice associative knowledge | Howard D, Patterson K: Pyramids and Palm Trees Test. Oxford, UK: Thames Valley Test Company, 1992 |
| Factual Semantic Memory | ||
| Variations on tests of famous people, news events, etc.† | ||
† These tests are typically used to assess the temporal extent of amnesia; however, because knowledge of such events varies greatly between communities, these tests are typically devised by individual neuropsychology laboratories.
SYNDROMES OF MEMORY DISTURBANCE
Transient Memory Disorders
Transient Global Amnesia
Transient global amnesia (TGA) is a syndrome of sudden onset, occurring in late middle or old age. An affected patient is characteristically densely amnesic and appears confused, repeatedly asking questions such as “Where am I?” or “What are we doing?” The event typically lasts for several hours, and during this time there is both anterograde and retrograde amnesia, although knowledge of personal identity is maintained. After the acute attack resolves, the patient is left with a period of amnesia for the time of the episode. The cause is uncertain, with suggested possibilities including a vascular transient ischemic attack, a seizure, or migraine. In single photon emission computed tomography images, transient perfusion defects in the mesial temporal lobe are evident during the episode,9 which highlights the presence of hippocampal dysfunction even if not identifying the underlying cause. Although TGA has an acute onset, it is seldom confused with more dangerous causes of acute amnesia such as viral limbic encephalitis, because the patient is otherwise alert, well and, more often than not, already in the recovery phase by the time of assessment. TGA typically occurs only once, and although cases of recurrent TGA are described, repeated episodes should alert the clinician to the possibility of transient epileptic amnesia (TEA).
Transient Epileptic Amnesia
Temporal lobe epilepsy is an increasingly recognized cause of transient memory impairment in middle-aged and elderly persons. The diagnosis is most secure when amnesic attacks are associated with a history of temporal lobe epilepsy or electroencephalographic evidence of temporal lobe discharges—which may be evident only on a during-sleep recording—and when the frequency of episodes is unequivocally improved by anticonvulsant therapy. Typical attacks of TEA differ in several ways from those of TGA, although the syndromes may be impossible to differentiate on the basis of the retrospective clinical account of an individual episode. The most useful distinguishing features of TEA are that the amnesic attacks are often briefer than those of TGA (less than 1 hour) and may occur on waking from sleep.10 In contrast to TGA, the patient may remember that he or she was unable to remember during the episode. It is not clear whether TEA results from an ictal or postictal state.
In addition to discrete attacks of TEA, some patients with temporal lobe epilepsy appear to have accelerated forgetting of new episodic memories. These patients do not have evidence of an anterograde amnesia over the half-hour period usually employed to test delayed recall during neuropsychological assessment. However, episodic memory recall over longer periods appears to be impaired.11 Typically, the problem becomes apparent to the patient when the patient is discussing specific events with family members, such as the details of a particular outing, or while he or she is looking at holiday photographs. Although the family members confirm that the patient appeared normal at the time the particular event took place, the patient subsequently claims no recollection of the episode. It is speculated that subclinical temporal spike activity—particularly during sleep—may interfere with long-term consolidation of these memories.
Fixed Memory Disorders
Strategic Lesions
Mesial temporal lobe
Lesions that damage the hippocampus and adjacent areas provide the archetypal substrate for amnesia. Possibly the most famous (or notorious, depending on one’s perspective) case in 20th century neuropsychology is that of H.M., a man who underwent bilateral mesial temporal lobectomy to control intractable epilepsy in the 1950s and was found postoperatively to have profound episodic memory impairment.12 Since H.M., numerous cases of hippocampal amnesia have been studied. Such cases typically have dense anterograde amnesia with variable retrograde amnesia. The degree of retrograde amnesia is approximately proportional to the extent of mesial temporal lobe lesions, whereas damage restricted to the cornu ammonis 1 field of the hippocampus may be associated with an almost pure anterograde amnesia.13
The hippocampus is particularly susceptible to anoxic damage; hence, bilateral focal hippocampal lesions are typically seen after resuscitation from cardiac arrest, carbon monoxide poisoning, and other states of hypoxemia or hypotension that cause inadequate cerebral perfusion. The blood supply of the hippocampus is predominantly via temporal branches of the posterior cerebral artery and, consequently, emboli to the vertebrobasilar system can also cause bilateral infarction. Unilateral infarcts (Fig. 4-4) from the same source tend to cause material-specific deficits; left- and right-sided lesions cause disproportionate verbal and visual memory deficits, respectively. The latter are usually evident in spatial memory tasks, although there is controversy regarding whether a lesion restricted to the right hippocampus is sufficient to cause the deficit. There is evidence to suggest that right parahippocampal damage may play a crucial role.14 Unilateral infarcts of the hippocampal head can also arise from anterior choroidal artery occlusion. Aside from vascular disease, the other major causes of focal mesial temporal lobe damage are viral and immune system–mediated limbic encephalitides (see sections on Immune System–Mediated Limbic Encephalitis and Viral Limbic Encephalitis).
Fornix
The fornix is the principal projection from the mesial temporal lobe to the mamillary body and thalamus. Most reports describe a relatively pure, anterograde amnesia. Iatrogenic lesions caused by surgical division in the course of removing colloid cysts from the third ventricle,15 as well as tumors and trauma, are recognized causes.
Diencephalon
The fornix, both directly and via the mamillary body, projects to the rostral thalamic nuclei and particularly to the anterior thalamic nucleus. Lesions in this region can cause dense anterograde and retrograde amnesia. Posteromedial central branches from the posterior cerebral arteries supply the anterior nuclei, and hence infarction caused by emboli in the vertebrobasilar system can cause bilateral lesions. This bilateral vulnerability is further increased because the central artery of Percheron, which arises from one or other posterior cerebral artery, divides to supply both mesial thalami; thus, a single vessel occlusion can lead to bilateral infarction. Bilateral lesions can also result from anoxic damage (deep watershed infarcts). As with mesial temporal lesions, unilateral thalamic lesions are associated with material-specific deficits. It is difficult to prove that amnesia may be caused by focal mamillary body lesions, but this seems the most parsimonious explanation in some cases.16 Patients with diencephalic amnesia may also have a degree of executive impairment, caused by disruption of frontostriatal networks.
Basal forebrain
Cases in which lesions in the region of the septal nuclei, the diagonal band of Broca, the nucleus basalis of Meynert, the substantia innominata, and the nucleus accumbens are associated with amnesia have been described. It is speculated that this may be a consequence of damage to cholinergic projections originating in the diagonal band of Broca and projecting to the hippocampus,17 although the specific anatomical mechanism is far from certain. Although this area lies in close proximity to the diencephalon, several reports indicate that the latter may be spared in such cases. The typical pathological process is rupture of an aneurysm in the anterior communicating artery.
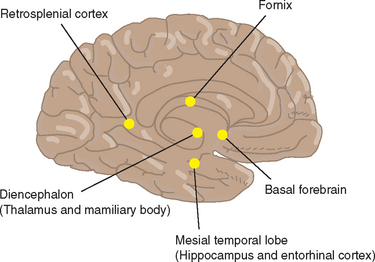
Retrosplenial cortex
As the name suggests, the retrosplenial cortex lies immediately adjacent to the splenium of the corpus callosum (Fig. 4-5). This cause of amnesia is rare, because focal lesions of the retrosplenial cortex are seldom encountered. Nevertheless, there have been several case reports in which amnesia was associated with damage to this area. Material-specific deficits are typical with unilateral lesions.18 Interestingly, the neural connections of the retrosplenial cortex are to the mesial temporal lobe and the dorsolateral prefrontal cortex, which place it in what is potentially a pivotal point in relation to structures involved in episodic memory processing. The usual pathological process is hemorrhage related to an underlying vascular malformation. Tumors of the splenium itself have also been reported to cause amnesia,19 and it may be that this is a consequence of retrosplenial cortex compression; however, the fornix lies subjacent to the ventrorostral surface of the splenium, so that damage to this structure may also be relevant to the genesis of the amnesic syndrome.
Korsakoff’s Psychosis
Korsakoff’s psychosis (or syndrome) is a severe, diencephalic amnesia caused by thiamine deficiency. It is typically seen in alcoholic patients with very poor diets, but it is important to remember that the critical factor is the dietary deficiency, rather than the alcohol. Thus, Korsakoff’s psychosis may occur in any disorder in which there is failure to maintain thiamine intake, such as gastrointestinal disorders (including gastric restriction surgery) and hyperemesis gravidarum. It usually follows from Wernicke’s encephalopathy, which consists of the classic triad of ataxia, ophthalmoplegia, and delirium, although presentation with delirium alone is common. It is therefore also referred to as Wernicke-Korsakoff syndrome. Korsakoff’s psychosis can be prevented at this encephalopathic stage by high-dose parenteral thiamine (given in combination with empirical multi–vitamin B group supplements, because multiple deficiencies may be present). Thiamine levels can be assessed by assay of erythrocyte transketolase activity, but this is seldom appropriate, because empirical therapy is straightforward and should not be delayed. Wernicke’s encephalopathy can be precipitated by glucose infusion in incipient thiamine deficiency. Of importance is that Mg2+ is a cofactor for thiamine activity, and patients may be resistant to thiamine therapy until concomitant magnesium deficiency is corrected.20 Unfortunately, if the patient has amnesia without delirium, which suggests that Korsakoff’s psychosis has already ensued, the prognosis for recovery is very poor. Nevertheless, suspected Korsakoff’s psychosis and limbic encephalitides (see next section) represent the most urgent management problems among the memory disorders, and thiamine deficiency should always be considered in cases with acute or subacute amnesia (Table 4-3).
TABLE 4-3 Checklist of Emergency Management of Management and Investigation of Acute Amnesia
|
2. Check blood biochemistry (including Mg) and hematology, and arrange cerebral imaging (MRI if possible). Is there:
|
CSF, cerebrospinal fluid; EEG, electroencephalogram; FDG-PET, fluorodeoxyglucose–positron emission tomography; HSV, herpes simplex virus; HSVE, herpes simplex virus encephalitis; IV, intravenous; Mg, magnesium; MRI, magnetic resonance imaging; PCR, polymerase chain reaction; VGKC-Ab, voltage-gated potassium channel antibodies.
Korsakoff’s psychosis typically causes a severe global amnesia. Anterograde amnesia is profound and accompanied by a severe retrograde amnesia. If the retrograde amnesia is temporally graded (worse for more recent retrograde memories) rather than complete, it usually spares only very remote memory. Typically, there is an associated dysexecutive syndrome. Neuronal loss is found in the mamillary body, the mediodorsal nucleus, and anterior thalamic nucleus; damage to the anterior thalamic nucleus appears most specific for Korsakoff’s psychosis–related amnesia.21
Immune System–Mediated Limbic Encephalitis
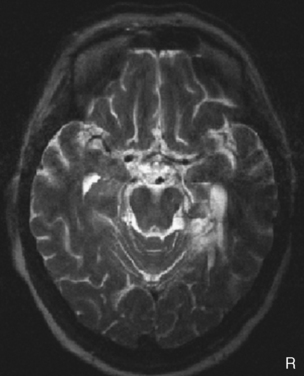
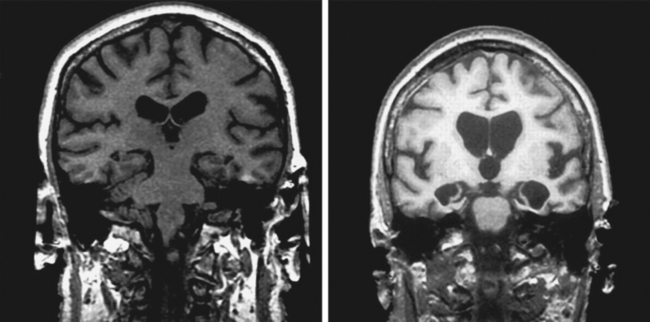
Paraneoplastic limbic encephalitis causes the subacute onset of amnesia, typically over days to weeks. Like other paraneoplastic neurological syndromes, antibodies raised in response to the tumor are thought to cross-react with neuronal epitopes. Patients typically present with memory impairment in association with confusion and, not infrequently, seizures. Because of these additional features, it is difficult to make appropriate generalizations about the type of memory deficit, but marked impairment of free recall of new information is typical. The electroencephalogram may show focal temporal lobe discharges. Magnetic resonance imaging (MRI) in the acute phase may reveal high signal in the hippocampi on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences. The presence of antineuronal antibodies in serum—most commonly anti-Hu—is supportive, but a negative result should not deter a search for malignancy if suspicion is high, particularly if there is a cerebrospinal fluid (CSF) pleocytosis.22 A two-pronged management strategy offers the best chance of improvement: treatment of the acute neurological disorder and definitive management of the tumor. Anecdotal reports suggest that immunomodulatory therapy (steroids, gamma globulin, or plasmapheresis) may be helpful in some cases and should be tried. The most commonly associated malignancy is small-cell lung cancer, with gonad, breast, and non-Hodgkin lymphoma also likely possibilities. Imaging of chest, abdomen, pelvis, and breasts should be performed. Paraneoplastic phenomena are often associated with very early tumors; therefore, if these investigations yield negative results, a full-body fluorodeoxyglucose positron emission tomographic scan to look for metabolic hot spots can increase yield. Cases in which hippocampal atrophy ensues have a poor prognosis for recovery. However, some patients with hippocampi that appear preserved on MRI may recover memory, although prognosis for such recovery is unpredictable (Fig. 4-6).23
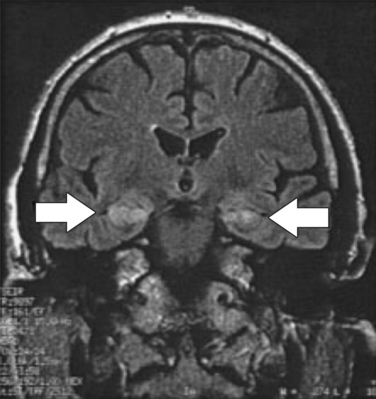
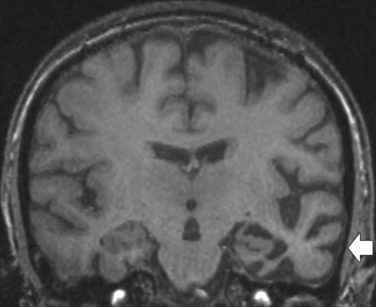
Nonparaneoplastic limbic encephalitis has a clinical onset and memory symptoms that may be indistinguishable from those of paraneoplastic limbic encephalitis.24 Temporal lobe seizures and confusion may also occur. The key feature is the presence of voltage-gated potassium channel antibodies (VGKC-Ab) in the serum, as are found in neuromyotonia, a condition that may coexist with the limbic encephalitis. Imaging studies indicate abnormal signal in the hippocampus on MRI scanning (Fig. 4-7), whereas the CSF is usually acellular.22 Anecdotal reports suggest that immunomodulatory therapy can improve amnesia and that the VGKC-Ab titer may be correlated with disease activity.
Viral Limbic Encephalitis
Limbic encephalitis also occurs with herpes simplex virus encephalitis (HSVE); typically, herpes simplex virus (HSV) type 1 is the causative organism, although HSV type 2 can produce the same illness. The clinical features are similar to those of immune system–mediated limbic encephalitis, with memory impairment, confusion, and seizures, although the presentation is typically more fulminant. There is a significant mortality rate, and memory sequelae are common in survivors. HSVE is a medical emergency, in that the best chance for full recovery is with early treatment. Empirical acyclovir should be commenced in all suspected cases as first-line management (i.e., before the diagnostic workup is started; see Table 4-3). Cerebral imaging with MRI, to look for signal changes and hemorrhage in the rostral temporal lobes, often provides supportive evidence. If MRI is unavailable, computed tomography can be utilized, although it is considerably less sensitive.25 Periodic temporal lobe discharges on electroencephalography also provide good supportive evidence. The CSF study shows a lymphocytic pleocytosis, but a very high erythrocyte count—suggestive of a traumatic tap—may also result from cortical hemorrhagic necrosis. Definitive diagnosis is usually established by demonstration of a positive CSF polymerase chain reaction assay for HSV.
In the acute phase, memory assessment is usually superfluous because patients are typically delirious, often requiring sedation and mechanical ventilation. Various combinations of semantic and episodic memory impairments are seen in survivors, depending on the precise location and extent of cerebral damage. Mesial temporal damage is associated with anterograde amnesia, and because the distribution of pathological process is often asymmetrical, material-specific deficits are common. Variable degrees of retrograde amnesia may occur in association with the anterograde memory deficit, and HSVE can occasionally give rise to a relatively focal retrograde amnesia.26 This phenomenon appears to occur when there is disproportionate temporal isocortical damage (as opposed to hippocampal and entorhinal lesions). Temporal isocortical damage can also cause semantic memory impairments. Unlike semantic dementia (see later discussion), these are often quite patchy, giving rise to what are referred to as category-specific deficits.27 Semantic knowledge can be considered in categories defined by their semantic relatedness. For instance, varieties of land animals, breeds of dogs, varieties of birds, body parts, tools, and musical instruments can all be considered as belonging to definable semantic categories and tested for accordingly. The commonest reported category-specific deficit is the dissociation of synthetic artifacts from living creatures, but various other permutations are also described. Category-specific deficits are of research interest because they offer insights into the neural structuring of semantic memory. The basic principle is that as the degree of similarity between semantic exemplars increases, so too does the overlap in the neural networks that represent the items. The attributes of an item give rise to a stable neural representation—or “attractor” in computational models—that identifies it as a specific, nameable entity. For semantically related items (e.g., land animals), these attractors share common features and thus may share common vulnerability if part of the network is damaged. See Rogers and Plaut (2002) for a detailed review of lesion evidence, computational models and proposed theories of category-specific deficits.28
Progressive Memory Disorders
Alzheimer’s Disease
Alzheimer’s disease is the commonest cause of memory impairment. In established Alzheimer’s disease, the diagnosis is usually straightforward: Patients have severe impairment of episodic memory with variable degrees of impairment in other cognitive domains, including semantic and working memory. However, the insidious onset and slowly progressive nature of the impairment make early diagnosis more difficult because, in contrast to the disorders already discussed, patients do not start out with a dense amnesia. It is increasingly recognized that relatively isolated episodic memory impairment can precede full-blown dementia by several years.29 Because the clinical diagnostic criteria for Alzheimer’s disease include impairment in multiple cognitive domains, early symptomatic cases that have not crossed this threshold cannot be labeled “Alzheimer’s disease” if the consensus diagnostic criteria are strictly applied.30 This had led to the introduction of the term amnestic mild cognitive impairment (MCIa) to describe isolated impairment of episodic memory.31 In spite of the current vogue for “diagnosing” MCIa, it must be emphasized that this term does not denote a pathological entity but rather a degree of severity. If symptomatic Alzheimer’s disease begins with isolated episodic memory impairment, why not revise the diagnostic criteria so that MCIa is called Alzheimer’s disease? The problem is that in the clinic, the more subtle the cognitive impairment, the less certain it is that symptoms will progress. Many patients with what appears to be isolated memory impairment may have a psychiatric illness or simply have “a bad memory” that will not worsen over time. Annual progression rates from the MCIa stage to probable Alzheimer’s disease are usually reported to be in the vicinity of 15% per annum. However, there is considerable variability in progression rates between cohorts, which most likely relates to differences in clinical features at baseline; within the designation of MCIa, there is scope for inclusion of cases that vary from minimal impairment to being on the cusp of a consensus classification of Alzheimer’s disease. Therefore, it is not surprising that MCIa patients who exhibit features suggestive of Alzheimer’s disease—such as subtle non-mnestic cognitive deficits, hippocampal atrophy visible on MRI, and temporoparietal hypometabolism evident on positron emission tomography—are at highest risk of early deterioration to dementia.
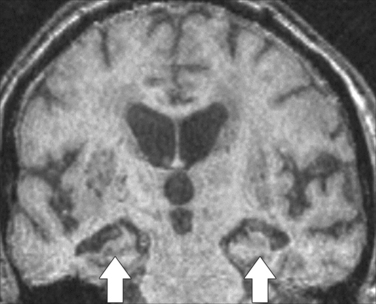
The cause of the episodic memory deficit in Alzheimer’s disease is complex. Early neurofibrillary tangle deposition (one of the hallmarks of Alzheimer’s disease) in the mesial temporal lobe suggests that memory impairment may be underpinned by damage to this area. However, although structural imaging may reveal hippocampal atrophy in many individuals (Fig. 4-8), there is considerable overlap in this feature between Alzheimer’s disease and healthy aging. Likewise, although cholinergic depletion is well recognized in Alzheimer’s disease, pathological32 and cholinergic ligand-based functional imaging studies33 suggest that this is not a major feature of the very early stage of the disease. Positron emission tomography suggests that the deficit may result from the combined effect of a degeneration of the functional network, including the mesial temporal lobe, diencephalon, and retrosplenial/posterior cingulate cortex.34
Dementia with Lewy Bodies
Dementia with Lewy bodies (DLB) is probably the second commonest cause of dementia in elderly persons, after Alzheimer’s disease. It is characterized by fluctuating confusion, hallucinations (typically of the formed visual type), spontaneous extrapyramidal features, and a neuropsychological profile of prominent working memory and visuoperceptual deficits.35 Nevertheless, the presenting complaint is often stated to be “memory problems.” The key difference from Alzheimer’s disease is that for the degree of episodic memory impairment, there is disproportionate impairment of the aforementioned domains. The memory impairment itself is also qualitatively different from that seen in Alzheimer’s disease and, indeed, from amnesic syndromes in general. In keeping with the working memory deficit, patients have marked deficits in encoding. For instance, in three-trial learning of a seven-element name and address, many patients with very mild Alzheimer’s disease can repeat all seven elements after the first trial and, if not, performance generally improves over the subsequent trials. In contrast, DLB patients tend to have marked impairment of registration after the first trial and little or no improvement over subsequent trials. However, whereas delayed recall in early Alzheimer’s disease is markedly impaired in spite of good registration (e.g., learning trial scores are 6/7, 7/7, and 7/7; delayed-recall score is 0/7), many patients with DLB recall a large proportion of what they managed to encode (e.g., learning trial scores are 3/7, 4/7, and 4/7; delayed-recall score is 3/7).
DLB is associated with a marked cholinergic deficit and, although cholinesterase inhibitors were introduced primarily for Alzheimer’s disease, it appears from anecdotal reports and small trials that they are more useful in improving cognition in DLB.36 In addition to the positive effects that these drugs have on the neuropsychiatric features of DLB, they seem particularly helpful in improving working memory and the related encoding deficit.
Frontotemporal Dementia
There are three clinical presentations of frontotemporal dementia: a neuropsychiatric syndrome, nonfluent progressive aphasia, and semantic dementia.37 Of these, semantic dementia is of most interest with respect to impainment of declarative memory.38 As the name suggests, these patients have progressive semantic memory impairment (factual knowledge, word meanings, and object knowledge), which gives rise to comprehension deficits and fluent aphasia. Unlike post-HSVE cases, significant category-specific deficits in semantic knowledge are rarely encountered, possibly because HSVE is more likely to result in a patchy distribution of cortical damage. In fascinating contrast to amnesic syndromes, patients with semantic dementia have relative preservation of episodic memory, as evinced by their often remarkably rich ability to recount anecdotes from the recent past. An important caveat for the neuropsychological assessment of episodic memory in semantic dementia is that the semantic knowledge deficit confounds performance on verbal memory tasks. Word-list learning or story recall is aided under normal circumstances by the ability to make semantic associations; if semantic knowledge is degraded, then word-list learning is analogous to normal subjects’ learning a list of unfamiliar foreign-language words. Nevertheless, because degeneration is usually maximal in the left temporal lobe, a degree of verbal episodic memory impairment is difficult to rule out. However, on nonverbal tests that minimize semantic associative knowledge (e.g., the Rey-Osterrieth Complex Figure Copy Test [Fig. 4-9]), delayed recall scores are often within the normal range.
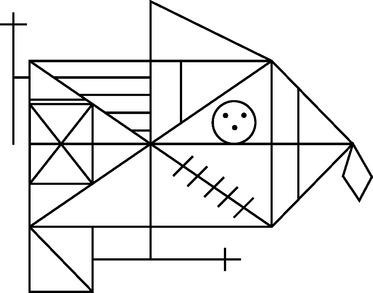
The remote memory profile also provides an interesting contrast to that seen in classic amnesic syndromes. Recent episodic memory (dating back weeks to months) is relatively preserved in comparison with memory from more remote time periods,39 a finding that is also true of semantic facts that can be dated to a specific time period (such as knowledge of famous people and events).40 These findings suggest that remote autobiographical memories may actually be supported by similar mechanisms to those involved in semantic memory.
The distribution of degenerative change in semantic dementia is typically asymmetrical and involves the anterior temporal lobes (especially the poles and inferior surfaces) (Fig. 4-10). The preservation of episodic memory was initially thought result from sparing of the hippocampi; however, volumetric MRI has shown that hippocampal atrophy in semantic dementia is at least as severe as that seen in Alzheimer’s disease, which suggests that the amnesia in the latter may be more a consequence of damage to other areas. A final contentious issue in semantic dementia is whether the asymmetrical atrophy of the temporal lobes can give rise to material-specific semantic memory deficits. Some authors have suggested that patients with greater right-sided atrophy have more problems with nonverbal semantics41 (such as prosopagnosia for famous faces). The alternative view is that semantic knowledge is bilaterally distributed but naming is lateralized to the left. Consequently, verbal semantics are more impaired with greater degrees of left temporal atrophy, because this weights word-processing ability.
Memory Impairment in Psychiatric Disorders
Hysterical Amnesia
True hysterical amnesia is a fairly uncommon disorder but one that most commonly manifests with a characteristic profile: focal retrograde amnesia. The patient presents with a dense amnesia for a discrete period, with apparently normal memory for events from before and after the amnesic period and normal performance on tests of new learning. Alternatively, there may be a complete retrograde amnesia, including loss of personal identity. Psychological precipitants for this so-called psychogenic fugue state, such as marital problems, criminal fraud, and bereavement, are often reported. One caveat is that cases of organic focal retrograde amnesia are also described, particularly in connection with focal temporal isocortical damage (such as after HSVE or severe head injury).42 Nevertheless, in organic cases, the border zone between preservation and loss in remote memory is often rather blurred. Furthermore, in organic cases, anterograde memory is usually not entirely normal, particular in the early stages. In other words, focal retrograde amnesia tends to be relative in organic disorders but appears absolute in hysterical amnesia.43
Dudai Y. Memory from A to Z. Keywords, Concepts and Beyond. Oxford, UK: Oxford University Press, 2002.
Kopelman MD. Disorders of memory. Brain. 2002;125:2152-2190.
Nestor PJ, Scheltens P, Hodges JR. Advances in the early detection of Alzheimer’s disease. Nat Med. 2004;5:S34-S41.
Rogers TT, Plaut DC. Connectionist perspectives on category-specific deficits. In: Forde E, Humphreys G, editors. Category Specificity in Brain and Mind. Brighton, UK: Psychology Press; 2002:251-289.
Squire LR, Zola SM. Episodic memory, semantic memory, and amnesia. Hippocampus. 1998;8:205-211.
Tulving E, Craik FM, editors. The Oxford Handbook of Memory. New York: Oxford University Press, 2000.
1 Tulving E, Kapur S, Craik FI, et al. Hemispheric encoding/retrieval asymmetry in episodic memory: positron emission tomography findings. Proc Natl Acad Sci U S A. 1994;91:2016-2020.
2 Baddeley A. Working memory. Science. 1992;255:556-559.
3 Hayashi Y, Majewska AK. Dendritic spine geometry: functional implication and regulation. Neuron. 2005;46:529-532.
4 Nadel L, Moscovitch M. Memory consolidation, retrograde amnesia and the hippocampal complex. Curr Opin Neurobiol. 1997;7:217-227.
5 Vargha-Khadem F, Gadian DG, Watkins KE, et al. Differential effects of early hippocampal pathology on episodic and semantic memory. Science. 1997;277:376-380.
6 Manns JR, Hopkins RO, Squire LR. Semantic memory and the human hippocampus. Neuron. 2003;38:127-133.
7 Kensinger EA, Schacter DL. Emotional content and reality-monitoring ability: fMRI evidence for the influences of encoding processes. Neuropsychologia. 2005;43:1429-1443.
8 Moscovitch M. Memory and working-with-memory: a component process model based on modules and central systems. J Cogn Neurosci. 1992;4:257-267.
9 Jovin TG, Vitti RA, McCluskey LF. Evolution of temporal lobe hypoperfusion in transient global amnesia: a serial single photon emission computed tomography study. J Neuroimaging. 2000;10:238-241.
10 Zeman AZ, Boniface SJ, Hodges JR. Transient epileptic amnesia: a description of the clinical and neuropsychological features in 10 cases and a review of the literature. J Neurol Neurosurg Psychiatry. 1998;64:435-443.
11 Blake RV, Wroe SJ, Breen EK, et al. Accelerated forgetting in patients with epilepsy: evidence for an impairment in memory consolidation. Brain. 2000;123(Pt 3):472-483.
12 Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957;20:11-21.
13 Zola-Morgan S, Squire LR, Amaral DG. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci. 1986;6:2950-2967.
14 Bohbot VD, Kalina M, Stepankova K, et al. Spatial memory deficits in patients with lesions to the right hippocampus and to the right parahippocampal cortex. Neuropsychologia. 1998;36:1217-1238.
15 Hodges JR, Carpenter K. Anterograde amnesia with fornix damage following removal of IIIrd ventricle colloid cyst. J Neurol Neurosurg Psychiatry. 1991;54:633-638.
16 Dusoir H, Kapur N, Byrnes DP, et al. The role of diencephalic pathology in human memory disorder. Evidence from a penetrating paranasal brain injury. Brain. 1990;113(Pt 6):1695-1706.
17 Abe K, Inokawa M, Kashiwagi A, et al. Amnesia after a discrete basal forebrain lesion. J Neurol Neurosurg Psychiatry. 1998;65:126-130.
18 McDonald CR, Crosson B, Valenstein E, et al. Verbal encoding deficits in a patient with a left retrosplenial lesion. Neurocase. 2001;7:407-417.
19 Rudge P, Warrington EK. Selective impairment of memory and visual perception in splenial tumours. Brain. 1991;114:349-360.
20 McLean J, Manchip S. Wernicke’s encephalopathy induced by magnesium depletion. Lancet. 1999;353:1768.
21 Harding A, Halliday G, Caine D, et al. Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain. 2000;123(Pt 1):141-154.
22 Darnell RB, Posner JB. A new cause of limbic encephalopathy. Brain. 2005;128:1745-1746.
23 Bak TH, Antoun N, Balan KK, et al. Memory lost, memory regained: neuropsychological findings and neuroimaging in two cases of paraneoplastic limbic encephalitis with radically different outcomes. J Neurol Neurosurg Psychiatry. 2001;71:40-47.
24 Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127:701-712.
25 Jha S, Patel R, Yadav RK, et al. Clinical spectrum, pitfalls in diagnosis and therapeutic implications in herpes simplex encephalitis. J Assoc Physicians India. 2004;52:24-26.
26 O’Connor M, Butters N, Miliotis P, et al. The dissociation of anterograde and retrograde amnesia in a patient with herpes encephalitis. J Clin Exp Neuropsychol. 1992;14:159-178.
27 Warrington EK, Shallice T. Category specific semantic impairments. Brain. 1984;107:829-854.
28 Rogers TT, Plaut DC. Connectionist perspectives on category-specific deficits. In: Forde E, Humphreys G, editors. Category Specificity in Brain and Mind. Brighton, UK: Psychology Press; 2002:251-289.
29 Amieva H, Jacqmin-Gadda H, Orgogozo JM, et al. The 9 year cognitive decline before dementia of the Alzheimer type: a prospective population-based study. Brain. 2005;128:1093-1101.
30 McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease. Neurology. 1984;34:939-944.
31 Petersen RC, Stevens JC, Ganguli M, et al. Practice parameter: early detection of dementia: mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:1133-1142.
32 DeKosky ST, Ikonomovic MD, Styren SD, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145-155.
33 Rinne JO, Kaasinen V, Jarvenpaa T, et al. Brain acetylcholinesterase activity in mild cognitive impairment and early Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:113-115.
34 Nestor PJ, Fryer TD, Smielewski P, et al. Limbic hypometabolism in Alzheimer’s disease and mild cognitive impairment. Ann Neurol. 2003;54:343-351.
35 McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113-1124.
36 Grace J, Daniel S, Stevens T, et al. Long-Term use of rivastigmine in patients with dementia with Lewy bodies: an open-label trial. Int Psychogeriatr. 2001;13:199-205.
37 Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546-1554.
38 Hodges JR, Patterson K, Oxbury S, et al. Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain. 1992;115:1783-1806.
39 Nestor PJ, Graham KS, Bozeat S, et al. Memory consolidation and the hippocampus: further evidence from studies of autobiographical memory in semantic dementia and frontal variant frontotemporal dementia. Neuropsychologia. 2002;40:633-654.
40 Hodges JR, Graham KS. A reversal of the temporal gradient for famous person knowledge in semantic dementia: implications for the neural organisation of long-term memory. Neuropsychologia. 1998;36:803-825.
41 Snowden JS, Thompson JC, Neary D. Knowledge of famous faces and names in semantic dementia. Brain. 2004;127:860-872.
42 Kapur N, Ellison D, Smith MP, et al. Focal retrograde amnesia following bilateral temporal lobe pathology. A neuropsychological and magnetic resonance study. Brain. 1992;115(Pt 1):73-85.
43 Kopelman MD. Focal retrograde amnesia and the attribution of causality: An exceptional critical view. Cogn Neuropsychol. 2000;17:585-621.