[level-membership-for-pediatrics-category]
Chapter 650 Disorders of Keratinization
Disorders of Cornification
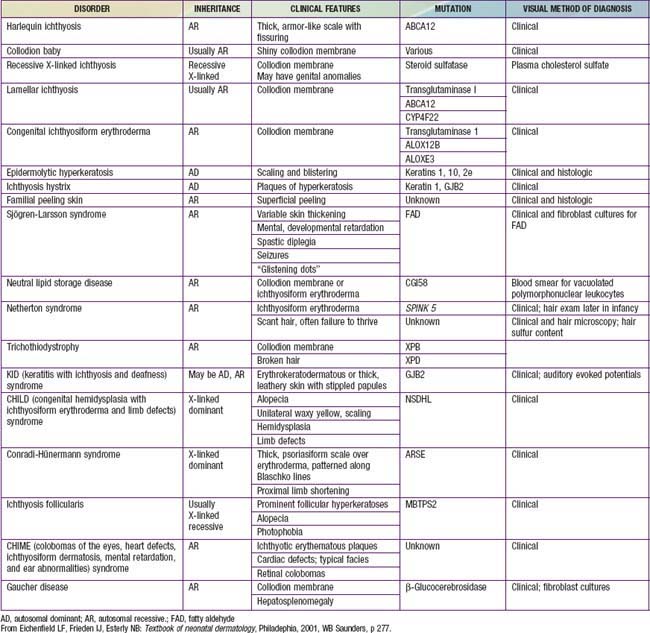
Disorders of cornification (ichthyoses) are a primary group of inherited conditions characterized clinically by patterns of scaling and histopathologically by hyperkeratosis. They are usually distinguishable on the basis of inheritance patterns, clinical features, associated defects, and histopathologic changes (Table 650-1).
Collodion Baby
Clinical Manifestations
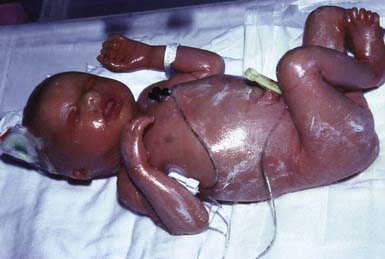
Collodion babies are covered at birth by a thick, taut membrane resembling oiled parchment or collodion (Fig. 650-1), which is subsequently shed. Affected neonates have ectropion, flattening of the ears and nose, and fixation of the lips in an O-shaped configuration. Hair may be absent or may perforate the abnormal covering. The membrane cracks with initial respiratory efforts and, shortly after birth, begins to desquamate in large sheets. Complete shedding may take several weeks, and a new membrane may occasionally form in localized areas.
Lamellar Ichthyosis and Congenital Ichthyosiform Erythroderma (Nonbullous Congenital Ichthyosiform Erythroderma)
Clinical Manifestations
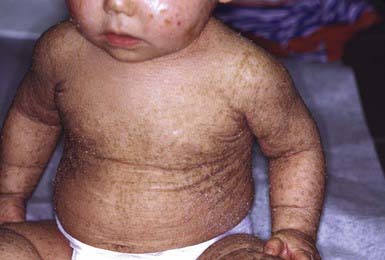
After shedding of the collodion membrane, if present, lamellar ichthyosis evolves into large, quadrilateral, dark scales that are free at the edges and adherent at the center. Scaling is often pronounced and involves the entire body surface, including flexural surfaces (Fig. 650-2). The face is often markedly involved, including ectropion and small, crumpled ears. The palms and soles are generally hyperkeratotic. The hair may be sparse and fine, but the teeth and mucosal surfaces are normal. Unlike in congenital ichthyosiform erythroderma, there is little erythema.
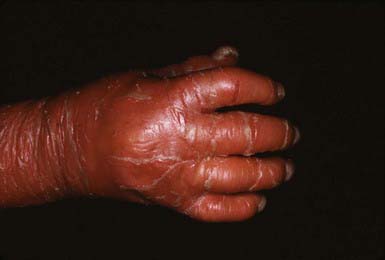
In congenital ichthyosiform erythroderma, erythroderma tends to be persistent, and scales, although they are generalized, are finer and whiter than in lamellar ichthyosis (Fig. 650-3). Hyperkeratosis is particularly noticeable around the knees, elbows, and ankles. Palms and soles are uniformly hyperkeratotic. Patients have sparse hair, cicatricial alopecia, and nail dystrophy. Neither form includes blistering.
Ichthyosis Vulgaris
Clinical Manifestations
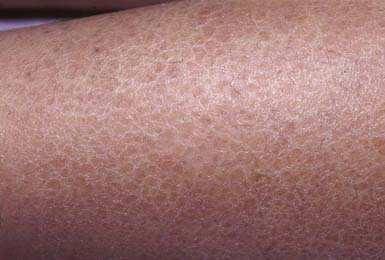
Ichthyosis vulgaris is the most common of the disorders of keratinization, with an incidence of 1/250 live births. Onset generally occurs in the 1st yr of life. In most cases, it is trivial, consisting only of slight roughening of the skin surface. Scaling is most prominent on the extensor aspects of the extremities, particularly the legs (Fig. 650-4). Flexural surfaces are spared, and the abdomen, neck, and face are relatively uninvolved. Keratosis pilaris, particularly on the upper arms and thighs, accentuated markings, and hyperkeratosis on the palms and soles, and atopy are relatively common. Scaling is most pronounced during the winter months and may abate completely during warm weather. There is no accompanying disorder of hair, teeth, mucosal surfaces, or other organ systems.
X-Linked Ichthyosis
Clinical Manifestations
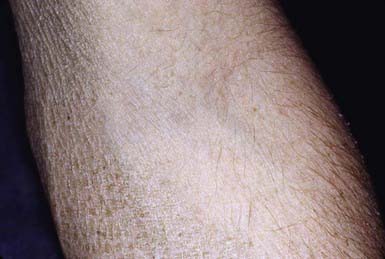
Skin peeling may be present at birth but typically begins at 3-6 mo of life. Scaling is most pronounced on the sides of the neck, lower face, preauricular areas, anterior trunk, and the limbs, particularly the legs. The elbow (Fig. 650-5) and knee flexures are generally spared but may be mildly involved. The palms and soles may be slightly thickened but are also usually spared. The condition gradually worsens in severity and extent. Keratosis pilaris is not present, and there is no increased incidence of atopy. Deep corneal opacities that do not interfere with vision develop in late childhood or adolescence and are a useful marker for the disease because they may also be present in carrier females. Some patients have larger deletions on the X chromosome that encompass neighboring genes, generating contiguous gene deletion syndromes. These include Kallmann syndrome (KAL1 gene), which consists of hypogonadotrophic hypogonadism and anosmia, X-linked chondroplasia punctata (ARSE gene), short stature, and ocular albinism. The rate of testicular cancer may be increased in patients with coexistent Kallmann syndrome. There is also an increased risk of attention deficit hyperactivity disorder and autism owing to a contiguous gene defect in neuroglin 4.
Epidermolytic Hyperkeratosis (Bullous Congenital Ichthyosiform Erythroderma)
Clinical Manifestations
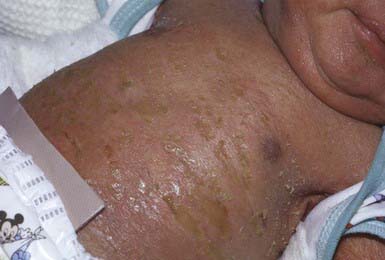
The clinical manifestations are initially characterized by the onset at birth of widespread blisters and erosions on a background of generalized erythroderma (Fig. 650-6). Recurrent blistering may be widespread in neonates and may cause diagnostic confusion with other blistering disorders. With time, the blister formation ceases, erythema decreases, and generalized hyperkeratosis develops. The scales are small, hard, and verrucous. Distinctive, parallel hyperkeratotic ridges develop over the joint flexures, including the axillary, popliteal, and antecubital fossae, and on the neck and hips. Palmoplantar keratoderma is associated with keratin 1 defects. The hair, nails, mucosa, and sweat glands are normal. Malodorous secondary bacterial infection is common and requires appropriate antibiotic therapy.
Erythrokeratoderma Variabilis
Clinical Manifestations
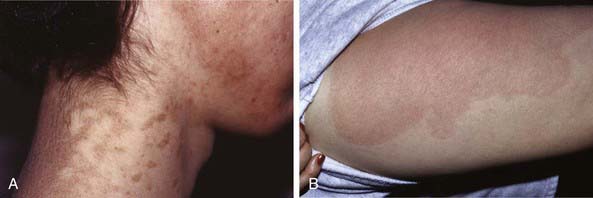
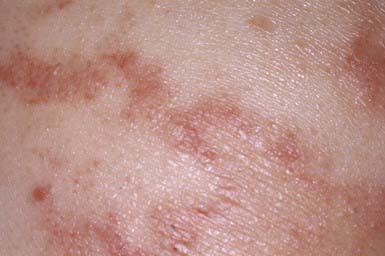
EKV usually manifests in the early months of life, progresses in childhood, and stabilizes in adolescence. It is characterized by two distinctive manifestations: sharply demarcated, hyperkeratotic plaques (Fig. 650-7A) and transient figurate erythema (Fig. 650-7B). The distribution is generalized but sparse; sites of predilection are the face, buttocks, axillae, and extensor surfaces of the limbs. The palms and soles may be thickened, but hair, teeth, and nails are normal.
Symmetric Progressive Erythrokeratoderma
Ichthyosiform Dermatoses
Several rare and distinct syndromes include ichthyosis as a constant feature.
Netherton Syndrome
Clinical Manifestations
Netherton syndrome is characterized by ichthyosis (usually ichthyosis linearis circumflexa but occasionally the lamellar or congenital types of ichthyosiform erythroderma), trichorrhexis invaginata and other hair shaft anomalies, and atopic diathesis. The disorder manifests at birth or in the first few months of life as generalized erythema and scaling. The trunk and limbs have diffuse erythema and superimposed migratory, polycyclic, and serpiginous hyperkeratotic lesions (Fig. 650-8), some with a distinctive double-edged margin of scale. Lichenification or hyperkeratosis tends to persist in the antecubital and popliteal fossae. The face and scalp may remain erythematous and scaling. Many hair shaft deformities, most notably, trichorrhexis invaginata, have been described in most patients with Netherton syndrome.
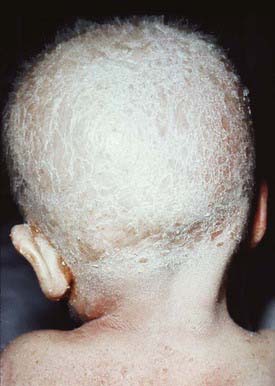
The ichthyosis is present in the first 10 days of life and may be especially marked around the eyes, mouth, and perineal area. The erythroderma is often intensified after infection. Infants may suffer from failure to thrive, recurrent bacterial and candidal infections, elevated serum immunoglobulin (Ig) E values, and marked hypernatremic dehydration. The most frequent allergic manifestations are urticaria, angioedema, atopic dermatitis, and asthma. Scalp hair is sparse and short and fractures easily (Fig. 650-9); eyebrows, eyelashes, and body hair are also abnormal. The characteristic hair abnormality can be identified with light microscopy; in the newborn, it may best be identified in eyebrow hair.
Other Syndromes with Ichthyosis
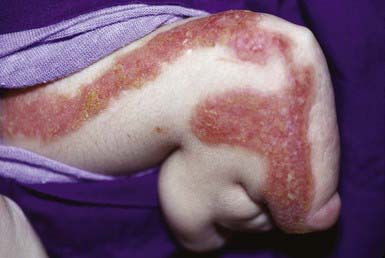
A number of other rare syndromes with ichthyosis as a consistent feature include the following: keratitis with ichthyosis and deafness (KID syndrome, connexin 26 gene), ichthyosis with defective hair having a banded pattern under polarized light and a low sulfur content (trichothiodystrophy), multiple sulfatase deficiency, neutral lipid storage disease with ichthyosis (Chanarin-Dorfman syndrome/CGI58 gene), and CHILD syndrome (Fig. 650-10; congenital hemidysplasia with ichthyosiform erythroderma and limb defects; NSDHL gene).
Palmoplantar Keratodermas
Diffuse Hyperkeratosis of Palms and Soles (Unna-Thost, Vorner)
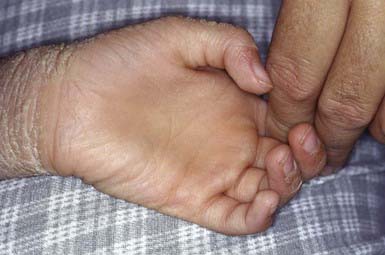
Unna-Thost and Vorner type palmoplantar keratodermas (PPKs), although clinically inseparable, were thought to be separate entities. They were separated histologically by the presence (Vorner) or absence (Unna-Thost) of epidermolytic hyperkeratosis. They represent the clinical spectrum of the same disease caused by mutations in keratin (KRT1 and KRT9 genes). This autosomal dominant disorder manifests in the first few months of life as erythema that gradually progresses to sharply demarcated, hyperkeratotic, scaling plaques over the palms (Fig. 650-11) and soles. The margins of the plaques often remain red; plaques may extend along the lateral aspects of the hands and feet and onto the volar wrists and the heels. Hyperhidrosis is usually present, but hair, teeth, and nails are usually normal. Striate (DSG1, DSP, KRT1 genes) and punctate forms of palmar and plantar hyperkeratosis represent distinct entities.
Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129:1319-1321.
Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
Lane EB, McLean WH. Keratins and skin disorders. J Pathol. 2004;204:355-366.
Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol. 2006;16:349-359.
Rajpopat S, Moss C, Mellerio J, et al. Harlequin ichthyosis. Arch Dermatol. Feb 2011. [Epub ahead of print]
Sandilands A, Sutherland C, Irvine AD, et al. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122(Pt 9):1285-1294.
Shwayder T. Disorders of keratinization: diagnosis and management. Am J Clin Dermatol. 2004;5:17-29.
Smith FJD, Irvine AD, Terron-Kwiatkiwski A, et al. Loss -of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38:337-342.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 650 Disorders of Keratinization
Disorders of Cornification
Disorders of cornification (ichthyoses) are a primary group of inherited conditions characterized clinically by patterns of scaling and histopathologically by hyperkeratosis. They are usually distinguishable on the basis of inheritance patterns, clinical features, associated defects, and histopathologic changes (Table 650-1).
Collodion Baby
Clinical Manifestations
Collodion babies are covered at birth by a thick, taut membrane resembling oiled parchment or collodion (Fig. 650-1), which is subsequently shed. Affected neonates have ectropion, flattening of the ears and nose, and fixation of the lips in an O-shaped configuration. Hair may be absent or may perforate the abnormal covering. The membrane cracks with initial respiratory efforts and, shortly after birth, begins to desquamate in large sheets. Complete shedding may take several weeks, and a new membrane may occasionally form in localized areas.
Lamellar Ichthyosis and Congenital Ichthyosiform Erythroderma (Nonbullous Congenital Ichthyosiform Erythroderma)
Clinical Manifestations
After shedding of the collodion membrane, if present, lamellar ichthyosis evolves into large, quadrilateral, dark scales that are free at the edges and adherent at the center. Scaling is often pronounced and involves the entire body surface, including flexural surfaces (Fig. 650-2). The face is often markedly involved, including ectropion and small, crumpled ears. The palms and soles are generally hyperkeratotic. The hair may be sparse and fine, but the teeth and mucosal surfaces are normal. Unlike in congenital ichthyosiform erythroderma, there is little erythema.
In congenital ichthyosiform erythroderma, erythroderma tends to be persistent, and scales, although they are generalized, are finer and whiter than in lamellar ichthyosis (Fig. 650-3). Hyperkeratosis is particularly noticeable around the knees, elbows, and ankles. Palms and soles are uniformly hyperkeratotic. Patients have sparse hair, cicatricial alopecia, and nail dystrophy. Neither form includes blistering.
Ichthyosis Vulgaris
Clinical Manifestations
Ichthyosis vulgaris is the most common of the disorders of keratinization, with an incidence of 1/250 live births. Onset generally occurs in the 1st yr of life. In most cases, it is trivial, consisting only of slight roughening of the skin surface. Scaling is most prominent on the extensor aspects of the extremities, particularly the legs (Fig. 650-4). Flexural surfaces are spared, and the abdomen, neck, and face are relatively uninvolved. Keratosis pilaris, particularly on the upper arms and thighs, accentuated markings, and hyperkeratosis on the palms and soles, and atopy are relatively common. Scaling is most pronounced during the winter months and may abate completely during warm weather. There is no accompanying disorder of hair, teeth, mucosal surfaces, or other organ systems.
