[level-membership-for-pediatrics-category]
73 Disorders of Growth
Etiology and Pathogenesis
Longitudinal Growth and Mediating Factors
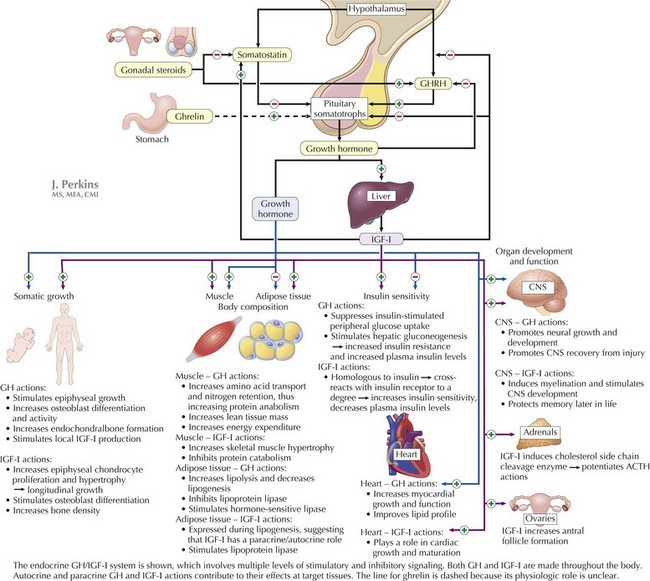
Growth hormone (GH) is the most important growth-regulating hormone; its pulsatile secretion from the anterior pituitary somatotrophs is regulated by two hypothalamic peptide hormones, GH-releasing hormone (GHRH) and somatostatin, which, respectively, stimulate and inhibit its release (Figure 73-1). Circulating GH is bound to GH-binding protein (GHBP), which corresponds to the extracellular domain of the GH receptor. Many, although not all, of GH’s actions are mediated through insulin-like growth factor (IGF)-I (i.e., somatomedin-C). Serum IGF-I is produced principally in the liver in response to GH and circulates in the bloodstream bound to IGF-binding proteins (especially IGFBP-3) that control its bioavailability. IGF-I acts at target tissues by binding cell-surface IGF receptors and triggering multiple downstream effects that include cellular hypertrophy and proliferation. IGF-I and free fatty acids also inhibit GH secretion at the level of the pituitary and hypothalamus.
Clinical Presentation
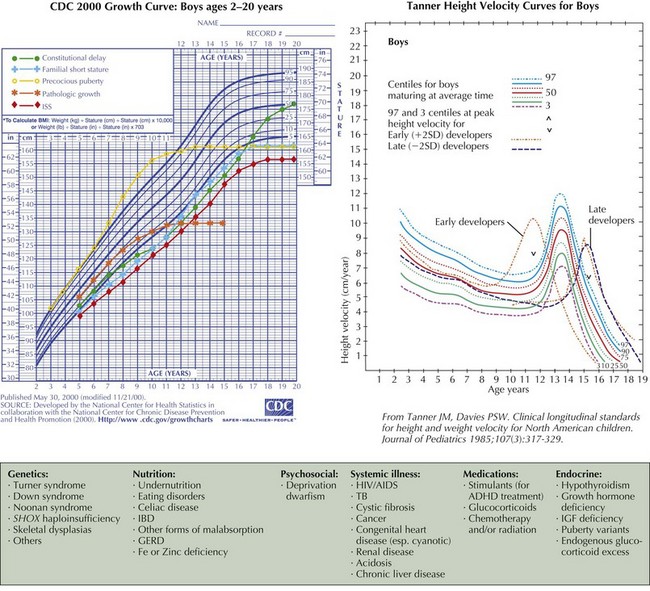
The causes of short stature can be divided into three general categories: chronic disease (including undernutrition), familial short stature (FSS), and constitutional delay of growth and puberty (CDGP) (Figure 73-2). Endocrine diseases are rare causes of short stature and are distinguished from other causes of short stature by linear growth failure that is more significant than weight loss. In developed countries, most short children have FSS, constitutional growth delay, or both. Short stature and constitutional growth delay are diagnoses of exclusion. The following is a focused review of the clinical presentations of selected normal variants and causes of short stature; see Box 73-1 for a comprehensive list of growth disorders.
Box 73-1 Differential Diagnosis of Short Stature
Constitutional Delay of Growth and Puberty
CDGP is the second most common cause of (transient) relative short stature in childhood. Children with CDGP usually have a normal birth length, begin crossing height percentiles by age 2 years, and settle in the lower percentiles, where they track until the onset of puberty in their peers, when they appear to drop further away from the growth curve. This growth pattern is associated with delayed puberty, which is defined as the absence of secondary sexual characteristics at an age beyond +2 SD for the population (see Chapter 67), that is, the absence of thelarche (breast development) in girls older than age 13 years, no signs of puberty in boys older than age 14 years, or no menarche in girls by 4 years after thelarche or by age 16 years (primary amenorrhea). CDGP represents the late end of the spectrum of pubertal development. Because children with CDGP enter puberty (and thus accelerate their growth velocity) later than most of their peers, they develop a transient relative height and sexual maturation gap relative to their age-matched peers. However, children with CDGP continue to grow after their peers cease growth, and thus they eventually attain an adult height within the normal range. Family history often reveals relatives with similarly delayed puberty (“late blooming”). There are no other abnormalities present other than short stature and delayed puberty. A detailed history and physical examination and thorough review of the child’s historical growth curve are essential steps to establishing this diagnosis; an isolated point on the growth curve at the time of the visit is insufficient. The differential diagnosis of CDGP includes Kallmann’s syndrome and isolated gonadotropin deficiency; lack of anosmia can exclude the former, but only time or genetic analyses can help distinguish between CDGP and the latter. Bone age is significantly delayed, which provides reassurance that sufficient time for growth remains for the child to reach the predicted adult height. Any physical or biochemical abnormalities should suggest another diagnosis. When CDGP occurs superimposed on FSS, the child’s short stature can appear severe; however, the child is still healthy, and his or her growth is still a normal variant.
Poor Nutrition
Optimal statural growth requires optimal nutrition; malnutrition is the most common cause of poor growth globally (see Chapter 13). Total calorie and/or protein-calorie malnutrition suppresses hepatic IGF-I production, decreasing negative feedback to the hypothalamus and pituitary. This results in increased GH production and secretion and thus elevated basal and stimulated serum GH levels. Inadequate food intake and malabsorption are both important causes of malnutrition. Decreased nutrient intake can result from oropharyngeal malformations (e.g., Pierre Robin sequence, cleft lip or palate), abnormal oral-motor function (e.g., pervasive developmental delay), as well as loss of appetite caused by use of certain medications (e.g., stimulants for treatment of attention deficit hyperactivity disorder or chemotherapeutic agents). Malabsorption can also cause malnutrition, and disorders such as celiac disease, inflammatory bowel disease, and cystic fibrosis can present as growth failure (see Chapter 111). Poor weight gain generally precedes the decrease, and eventual failure, of linear growth. Bone age and puberty are often delayed. Taking a detailed dietary history is essential to establishing this diagnosis. A 3-day diet log is a useful tool; this record can be analyzed for intake of total calories, macronutrients, and micronutrients. Laboratory evaluations should include all those involved in the evaluation for chronic illness.
Chronic Systemic Illnesses
Many chronic illnesses are associated with short stature (see Box 73-1). General mechanisms include anorexia, nutrient malabsorption, chronic acidosis or hypoxemia, anemia, increased energy requirements, and medical therapy (e.g., glucocorticoids). In most children with short stature caused by chronic illness, weight tends to be depressed to a greater extent than height or there is a lag between the onset of weight deceleration and subsequent height deceleration. Bone age is usually delayed and approximates the height age. The growth curve typically shows a period of normal growth followed by growth deceleration or cessation consistent with the onset of illness.
Hypothyroidism
Growth failure is one of the most significant manifestations of hypothyroidism in children (see Chapter 68). Because most congenital hypothyroidism is detected with neonatal screening programs, most cases of hypothyroidism-associated growth failure are attributable to acquired hypothyroidism, most commonly from autoimmune Hashimoto (chronic lymphocytic) thyroiditis or from iodine deficiency. Growth retardation caused by acquired hypothyroidism can take several years to clearly manifest, but when it is clinically significant, it tends to be severe and progressive. Puberty is often delayed, but precocious puberty (still with a paradoxically delayed bone age) can be seen as well.
Evaluation and Management
Measurement Techniques and Growth Charts
Measurement of length or stature must be accurate and reproducible to correctly assess the degree of interval growth. Children younger than 2 years should be measured in a supine position, children older than 3 years of age should be measured standing, and children in between can be measured either way depending on their ability to stand erect for the duration of measurement. Whether supine or standing, proper measuring technique is crucial; both processes involve fixed measurement equipment and proper positioning (Figure 73-3). Recumbent lengths should be plotted on the length (ages birth-36 months) growth charts and standing heights on the height (age 2-20 years) charts because a person’s length usually exceeds his or her height. A child’s current height and overall growth pattern should be interpreted within the context of standards typical for the local population. In the United States, most practitioners use the Centers for Disease Control and Prevention’s growth charts, which are percentile curves that illustrate the distribution by age, in U.S. children, of length or height, weight, head circumference (for ages birth-3 years), and BMI (BMI = Weight (kg)/Height (m)2). The data used to construct the updated 2000 growth charts were derived from cross-sectional population surveys rather than longitudinal studies following the same children over time. Thus, an individual’s growth pattern may differ somewhat from the standardized pattern, especially during periods of rapid growth when timing is important (infancy and the pubertal growth spurt).

Specific Elements of the Physical Examination
When performing the physical examination, the practitioner should look for signs of underlying genetic abnormalities (e.g., dysmorphic features), midline defects suggesting possible hypothalamic or pituitary malformations (e.g., a central maxillary incisor), or systemic illness (e.g., papilledema, aphthous ulcers, rachitic rosary). The degree of dental maturation, which correlates with skeletal maturation (delayed dentition can imply delayed bone age), should also be assessed. Determining the child’s pubertal status using the Tanner staging criteria is a critical part of the examination (see Chapter 67). Disproportionate growth can be ascertained via measurement of the upper-to-lower body segment ratio (upper segment length is the distance from top of the head to the top of the symphysis pubis, and lower segment length is the distance from the top of the pubic symphysis to the floor; normal ratios are 1.7 : 1 at birth, 1.3 : 1 at 3 years of age, and 1 : 1 by age 7 years) and arm span (distance between the tips of the middle digits with both arms fully extended; normally arm span is less than standing height before age 8 years, equal to height at ages 8-12 years, and greater than height after age 12 years).
Management
Management of abnormal growth depends on the underlying cause:
AACE Growth Hormone Task Force. American Association of Clinical Endocrinologists: medical guidelines for clinical practice for growth hormone use in adults and children—2003 update. Endocrine Pract. 2003;9(1):64-76.
Centers for Disease Control and Prevention. CDC Growth Charts, United States. Available at http://www.cdc.gov/growthcharts
Collett-Solbert PF, Petryk A, on behalf of the Members of the Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Changes in Recombinant Human Growth Hormone (rhGH) Prescribing Information. Updated 2008. Available at http://www.lwpes.org/policyStatements/ChangesrhGHprescribing_information_31408.pdf
Clayton PE. Management of the child born small for gestational age through to adulthood: a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J Clin Endocrinol Metab. 2007;92(3):804-810.
Greulich WW, Pyle SI. Radiographic Atlas of Skeletal Development of the Hand and Wrist. Stanford: Stanford University Press; 1959.
Grimberg A, Kutikov JK, Cucchiara AJ. Sex differences in patients referred for evaluation of poor growth. J Pediatr. 2005;146:212-216.
Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocrinol Metab. 2000;85(11):3990-3993.
Hall SS, Size Matters. How Height Affects the Health, Happiness, and Success of Boys—And the Men They Become. Boston: Houghton Mifflin; 2006.
Tanner JM, Goldstein H, Whitehouse RH. Standards for children’s height at ages 2-9 years allowing for height of parents. Arch Dis Child. 1970;45:755-762.
Tanner JM, Whitehouse RH, Marshall WA, et al. Assessment of Skeletal Maturity and Prediction of Adult Height (TW2 Method). New York: Academic Press; 1975.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
73 Disorders of Growth
Etiology and Pathogenesis
Longitudinal Growth and Mediating Factors
Growth hormone (GH) is the most important growth-regulating hormone; its pulsatile secretion from the anterior pituitary somatotrophs is regulated by two hypothalamic peptide hormones, GH-releasing hormone (GHRH) and somatostatin, which, respectively, stimulate and inhibit its release (Figure 73-1). Circulating GH is bound to GH-binding protein (GHBP), which corresponds to the extracellular domain of the GH receptor. Many, although not all, of GH’s actions are mediated through insulin-like growth factor (IGF)-I (i.e., somatomedin-C). Serum IGF-I is produced principally in the liver in response to GH and circulates in the bloodstream bound to IGF-binding proteins (especially IGFBP-3) that control its bioavailability. IGF-I acts at target tissues by binding cell-surface IGF receptors and triggering multiple downstream effects that include cellular hypertrophy and proliferation. IGF-I and free fatty acids also inhibit GH secretion at the level of the pituitary and hypothalamus.
Clinical Presentation
The causes of short stature can be divided into three general categories: chronic disease (including undernutrition), familial short stature (FSS), and constitutional delay of growth and puberty (CDGP) (Figure 73-2). Endocrine diseases are rare causes of short stature and are distinguished from other causes of short stature by linear growth failure that is more significant than weight loss. In developed countries, most short children have FSS, constitutional growth delay, or both. Short stature and constitutional growth delay are diagnoses of exclusion. The following is a focused review of the clinical presentations of selected normal variants and causes of short stature; see Box 73-1 for a comprehensive list of growth disorders.
Box 73-1 Differential Diagnosis of Short Stature
[/not-level-membership-for-pediatrics-category]
