[level-membership-for-pediatrics-category]
69 Disorders of Calcium and Bone Metabolism
Regulation of Serum Calcium and Phosphorus
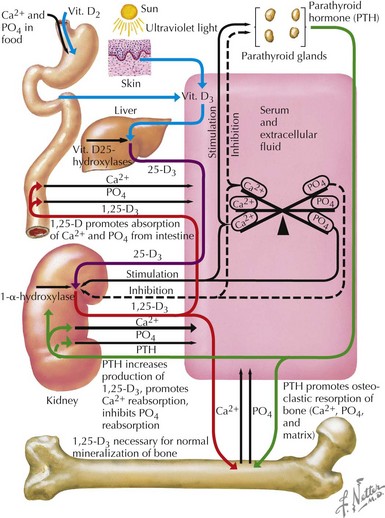
Most (99%) of the body’s calcium exists as hydroxyapatite in bone, with the remaining 1% present in extracellular fluids. Serum calcium exists in three fractions: 50% to 55% is free (ionized) calcium; about 10% is complexed with low-molecular-weight anions; and 35% to 40% is bound to proteins, mainly albumin and, to a lesser extent, globulins. The calciotropic hormones calcitriol (the fully active form of vitamin D) and parathyroid hormone (PTH) act on their target organs, kidney, intestines, and bone to regulate mineral homeostasis (Figure 69-1). Phosphatonins such as FGF23 also play important regulatory roles in mineral metabolism and complement the actions of other calciotropic hormones; phosphatonins decrease renal phosphorus reabsorption while reducing synthesis of calcitriol and secretion of PTH.
Dynamics of Bone Homeostasis
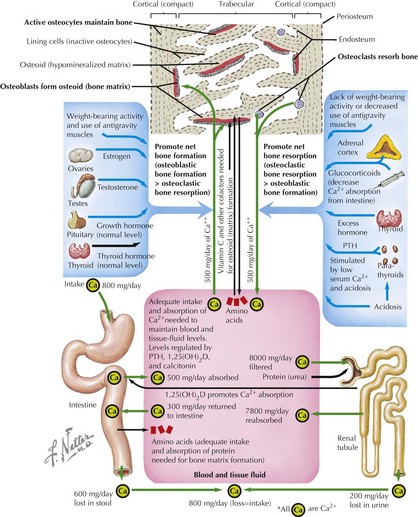
Bone modeling and remodeling are regulated by a variety of factors such as biomechanical loading, hormonal balance, acid–base status, and drug exposures (Figure 69-2). Bone adapts its strength in response to the magnitude and direction of the forces to which it is subjected. Mechanical forces on the skeleton arise primarily from muscle contraction. This capacity of bone to respond to mechanical loading with increased bone size and strength is greatest during growth, especially during puberty and adolescence. Increased production of estrogen and testosterone in addition to increased pulsatile secretion of growth hormone are the hormonal hallmarks of puberty. These events act in an anabolic manner on bone to promote net bone formation.
Osteomalacia and Rickets
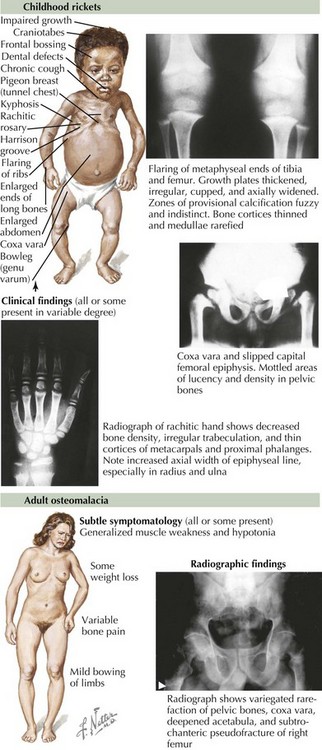
Osteomalacia is characterized by a defect in bone mineralization and occurs in both adults and children (Figure 69-3). By contrast, rickets represents a defect in mineralization of cartilage in the growth plate and therefore occurs only in children. Rickets and osteomalacia are classified as calcipenic or phosphopenic, depending on whether the defect in mineralization results from a primary deficiency of calcium and vitamin D or phosphorus. There are many forms of osteomalacia and rickets, both acquired and genetic, but the most common cause is nutritional deficiency of vitamin D. Other causes of vitamin D deficiency include chronic use of anticonvulsants, chronic kidney failure, hepatic disease, and malabsorption syndromes. Phosphopenic rickets can occur as a result of chronic use of medications that absorb phosphorus in the intestine or decreased renal phosphate reabsorption. The symptoms of osteomalacia may be subtle, with patients typically complaining of diffuse bone pain, proximal muscle weakness, and generalized fatigue. In children, rickets can cause growth failure and skeletal deformity. Over time, a waddling gait may result from the hip pain and thigh muscle atrophy. Biochemical abnormalities in patients with vitamin D deficiency include elevated serum levels of alkaline phosphatase and PTH, low serum phosphate, low or normal serum calcium, and low serum concentrations of 25(OH)D.
Nutritional Rickets
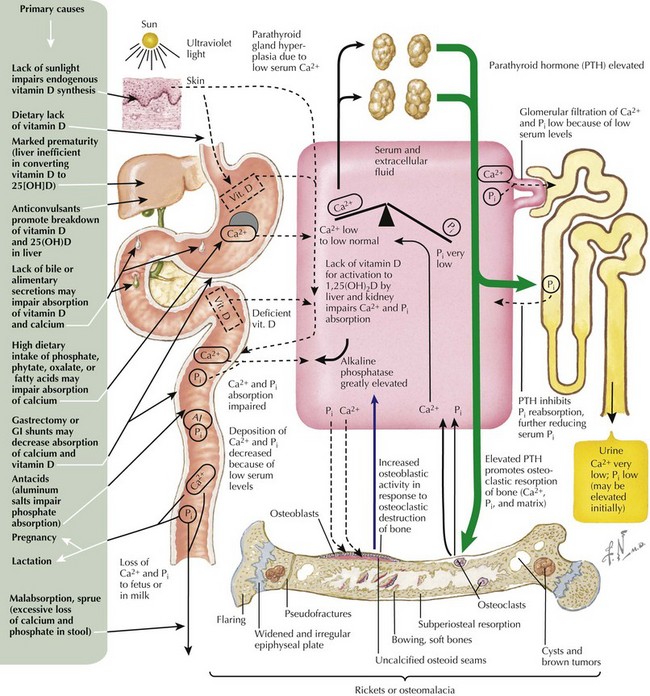
Nutritional rickets secondary to vitamin D deficiency is common throughout the world and reflects inadequate exposure to sunlight and poor intake of dietary vitamin D. Vitamin D deficiency is easily prevented, and the prevalence of this condition can be reduced by adequate nutritional intake of vitamin D or vitamin D–fortified foods (Figure 69-4). This form of rickets has a peak incidence between 3 and 18 months of age. Additional risk factors for vitamin D deficiency include dark skin, protracted exclusive breastfeeding, use of sunscreens or conservative clothing, fat malabsorption, use of anticonvulsants that induce hepatic P450 enzymes, marked prematurity, and lack of biliary secretions that may impair absorption of vitamin D and calcium. Mild to moderate vitamin D deficiency may be present for months before rickets is obvious on physical examination, and severe vitamin D deficiency may manifest as hypocalcemic seizures, growth failure, lethargy, irritability, and a predisposition to respiratory infections. Although relatively simple to prevent, vitamin D deficiency continues to be a significant problem worldwide, and vitamin D deficiency rickets continues to be a public health problem.
Hypophosphatemic Rickets
X-Linked Hypophosphatemic Rickets
This X-linked, dominant disorder is caused by mutations in the PHEX gene that encodes a specialized metalloprotease enzyme (Figure 69-5). X-linked hypophosphatemic rickets (XLHR) is the most common form of genetic rickets, with an estimated prevalence of one in 15,000. Loss-of-function mutations in PHEX are associated with reduced degradation and clearance of FGF23; in turn, elevated circulating levels of FGF23 reduce expression of renal sodium–phosphate cotransporters and inhibit 1-α-hydroxylase activity. Patients with XLHR have normal serum levels of 1,25(OH)2D3, which are inappropriate in the context of hypophosphatemia. In general, males and females are similarly affected.
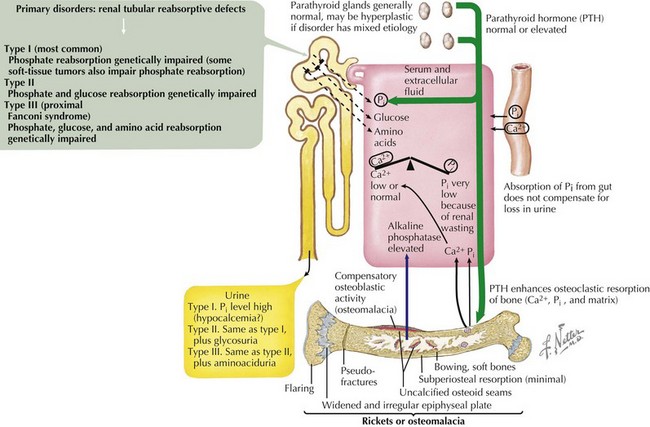
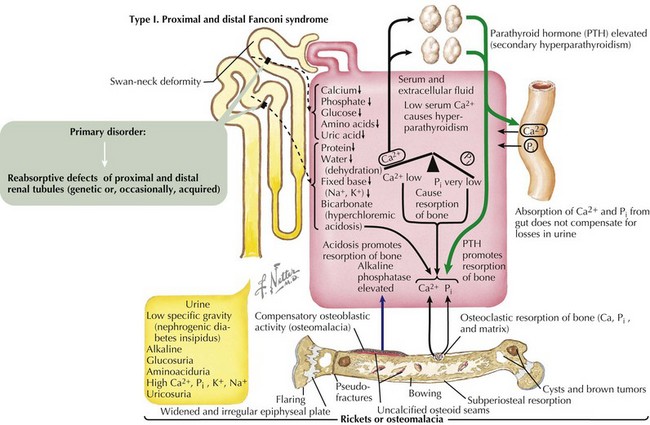
Fanconi Syndrome
Fanconi syndrome (FS) is associated with a variety of genetic defects (e.g., tyrosinemia) or can occur as a result of acute tubular necrosis, heavy metal and drug exposure, or protein malnutrition (Figure 69-6). Type 1 FS is the most common form and is the result of global tubular malfunction leading to urinary losses of bicarbonate, calciuria, phosphaturia, glycosuria, and proteinuria. Calcium losses in the urine cause secondary hyperparathyroidism; however, this increase in PTH cannot overcome the calcium and phosphate losses in the urine. Prolonged acidosis coupled with increased levels of PTH promoted bone resorption over time leads to rickets in children and osteomalacia in adults. Treatment consists of correcting any underlying primary disorder contributing to the development of FS, correction of acidosis, phosphate supplementation, and 1,25(OH)2D3 replacement.
Consortium A. Autosomal dominant hypophosphatemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345-348.
Goji K, Ozaki K, Sadewa AH, et al. Somatic and germline mosaicism for a mutation of the PHEX gene can lead to genetic transmission of X-linked hypophosphatemic rickets and mimics an autosomal dominant trait. J Clin Endocrinol Metab. 2006;91:365-370.
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
Institute of Medicine 2011 Dietary reference intakes for calcium and vitamin D. Washington, DC; The National Academies Press.
Lifshitz F. Pediatric Endocrinology, ed 5. New York: Informa Healthcare USA; 2007.
Misra M, Pacaud D, Petryk A, et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. on behalf of the Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Pediatrics. 2008;122(2):398-417.
Wagner C, Greer FR, American Academy of PediatricsSection on Breastfeeding Medicine and Committee on Nutrition. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. 2008;122(5):1142-1152.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
69 Disorders of Calcium and Bone Metabolism
Regulation of Serum Calcium and Phosphorus
Most (99%) of the body’s calcium exists as hydroxyapatite in bone, with the remaining 1% present in extracellular fluids. Serum calcium exists in three fractions: 50% to 55% is free (ionized) calcium; about 10% is complexed with low-molecular-weight anions; and 35% to 40% is bound to proteins, mainly albumin and, to a lesser extent, globulins. The calciotropic hormones calcitriol (the fully active form of vitamin D) and parathyroid hormone (PTH) act on their target organs, kidney, intestines, and bone to regulate mineral homeostasis (Figure 69-1). Phosphatonins such as FGF23 also play important regulatory roles in mineral metabolism and complement the actions of other calciotropic hormones; phosphatonins decrease renal phosphorus reabsorption while reducing synthesis of calcitriol and secretion of PTH.
Dynamics of Bone Homeostasis
Bone modeling and remodeling are regulated by a variety of factors such as biomechanical loading, hormonal balance, acid–base status, and drug exposures (Figure 69-2
[/not-level-membership-for-pediatrics-category]
