[level-membership-for-pediatrics-category]
Chapter 686 Disorders Involving Cartilage Matrix Proteins
Spondyloepiphyseal Dysplasias
Lethal Spondyloepiphyseal Dysplasias
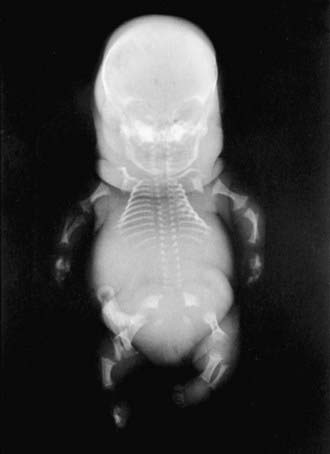
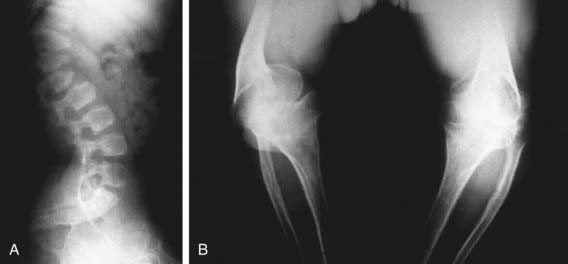
The severity of radiographic changes correlates with the clinical severity (Fig. 686-1). Both conditions produce short, broad tubular bones with cupped metaphyses. The pelvic bones are hypoplastic, and the cranial bones are not well mineralized. The vertebral bodies are poorly ossified in the entire spine in achondrogenesis type II and in the cervical and sacral spine in hypochondrogenesis. The pedicles are ossified in both.
Spondyloepiphyseal Dysplasia Congenita
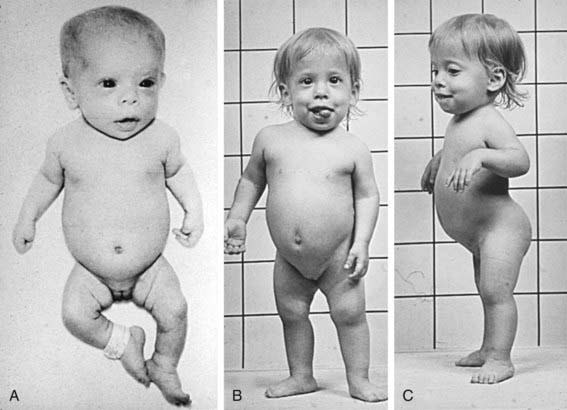
The phenotype of this group, SED congenita (OMIM 183900), is apparent at birth. The head and face are usually normal, but a cleft palate is common. The neck is short and the chest is barrel shaped (Fig. 686-2). Kyphosis and exaggeration of the normal lumbar lordosis are common. The proximal segments of the limbs are shorter than the hands and feet, which often appear normal. Some infants have clubfoot or exhibit hypotonia.
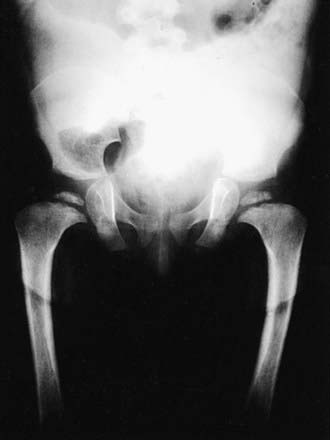
Skeletal radiographs of the newborn reveal short tubular bones, delayed ossification of vertebral bodies, and proximal limb bone epiphyses (Fig. 686-3). Hypoplasia of the odontoid process, a short, square pelvis with a poorly ossified symphysis pubis, and mild irregularity of metaphyses are apparent.
Stickler Dysplasia (Hereditary Osteoarthro-Ophthalmopathy)
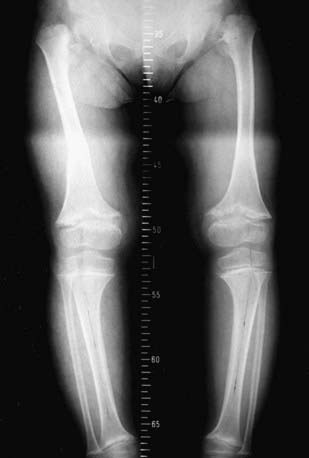
Short stature is not a feature of Stickler dysplasia (OMIM 184840). It resembles SED because of its joint and eye manifestations. Mutations of genes encoding type XI collagen, which functionally interacts with type II collagen, have been identified in Stickler-like disorders (OMIM 184840, OMIM 215150). Stickler dysplasia is often identified in the newborn because of cleft palate and micrognathia (Pierre Robin anomaly, Chapter 303). Infants typically have severe myopia and additional ophthalmologic complications, including choroidoretinal and vitreous degeneration; retinal detachment is common during childhood (Fig. 686-4). Sensorineural hearing loss can arise during adolescence, which is when symptoms of osteoarthritis can also begin. Special attention must be given to the eye complications even in childhood.

Schmid Metaphyseal Dysplasia
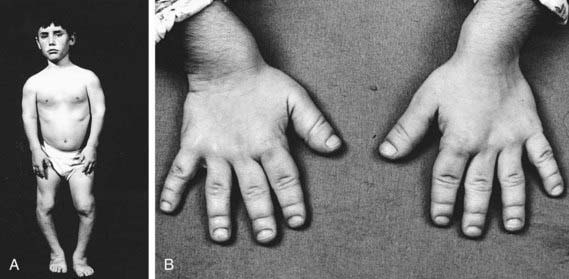
Schmid metaphyseal dysplasia (OMIM 156500) is one of several chondrodysplasias in which metaphyseal abnormalities dominate the radiographic features. It typically manifests in early childhood with mild short stature, bowing of the legs, and a waddling gait (Fig. 686-5). Joints, such as the wrist, may be enlarged. Radiographs show flaring and irregular mineralization of the metaphyses of tubular bones of the proximal limbs (Fig. 686-6). Coxa vara is usually present and can require surgical correction. Short stature becomes more evident with age and affects the lower extremities more than the upper extremities; the manifestations are limited to the skeleton.

Pseudoachondroplasia and Multiple Epiphyseal Dysplasia
Newborns with pseudoachondroplasia are average in size and appearance. Gait abnormalities and short stature mainly affect the limbs and become apparent in late infancy. The short stature becomes marked as the child grows and is associated with generalized joint laxity (Fig. 686-7). The hands are short, broad, and deviated in an ulnar direction; the forearms are bowed. Developmental milestones and intelligence are usually normal. Lumbar lordosis and deformities of the knee develop during childhood; the latter often requires surgical correction. Pain is common in weight-bearing joints during childhood and adolescence, and osteoarthritis develops late in the 2nd decade of life. Adults range in height from 105 to 128 cm.
Skeletal radiographs show distinctive abnormalities of vertebral bodies and of both epiphyses and metaphyses of tubular bones (Fig. 686-8).
The more severe clinical phenotype has its onset during childhood, with mild short-limbed short stature, pain in weight-bearing joints, and a waddling gait. Radiographs show delayed and irregular ossification of epiphyses. In more mildly affected patients the disorder might not be recognized until adolescence or adulthood. Radiographic changes may be limited to the capital femoral epiphyses. In the latter case, mild MED must be distinguished from bilateral Legg-Calvé-Perthes disease (Chapter 670.3). Precocious osteoarthritis of hips and knees is the major complication in adults with MED. Adult heights range from 136 to 151 cm.
Boerkoel CF, O’Neill S, Andre JL, et al. Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literature. Eur J Pediatr. 2000;159:1-7.
Horton WA, Hecht JT. Chondrodysplasias: disorders of cartilage matrix proteins. In: Royce PM, Steinmann B, editors. Connective tissue and its heritable disorders. New York: Wiley-Liss; 2002:909-938.
Kennedy J, Jackson G, Ramsden S, et al. Comp mutation screening as an aid for the clinical diagnosis and counselling of patients with a suspected diagnosis of pseudoachondroplasia or multiple epiphyseal dysplasia. Eur J Hum Genet. 2005;13:547-555.
Kennedy J, Jackson GC, Barker FS, et al. Novel and recurrent mutations in the c-terminal domain of comp cluster in two distinct regions and result in a spectrum of phenotypes within the pseudoachondroplasia—multiple epiphyseal dysplasia disease group. Hum Mutat. 2005;25:593-594.
Tompson SW, Merriman B, Funari VA, et al. A recessive skeletal dysplasia, semd aggrecan type, results from a missense mutation affecting the c-type lectin domain of aggrecan. Am J Hum Genet. 2009;84:72-79.
Unger S, Bonafe L, Superti-Furga A. Multiple epiphyseal dysplasia: clinical and radiographic features, differential diagnosis and molecular basis. Best Pract Res Clin Rheumatol. 2008;22:19-32.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 686 Disorders Involving Cartilage Matrix Proteins
Spondyloepiphyseal Dysplasias
Lethal Spondyloepiphyseal Dysplasias
The severity of radiographic changes correlates with the clinical severity (Fig. 686-1). Both conditions produce short, broad tubular bones with cupped metaphyses. The pelvic bones are hypoplastic, and the cranial bones are not well mineralized. The vertebral bodies are poorly ossified in the entire spine in achondrogenesis type II and in the cervical and sacral spine in hypochondrogenesis. The pedicles are ossified in both.
Spondyloepiphyseal Dysplasia Congenita
The phenotype of this group, SED congenita (OMIM 183900), is apparent at birth. The head and face are usually normal, but a cleft palate is common. The neck is short and the chest is barrel shaped (Fig. 686-2). Kyphosis and exaggeration of the normal lumbar lordosis are common. The proximal segments of the limbs are shorter than the hands and feet, which often appear normal. Some infants have clubfoot or exhibit hypotonia.
Skeletal radiographs of the newborn reveal short tubular bones, delayed ossification of vertebral bodies, and proximal limb bone epiphyses (Fig. 686-3). Hypoplasia of the odontoid process, a short, square pelvis with a poorly ossified symphysis pubis, and mild irregularity of metaphyses are apparent.
[/not-level-membership-for-pediatrics-category]
