Chapter 21 Kidney diseases
COMMON CLINICAL PROBLEMS FROM KIDNEY DISEASE
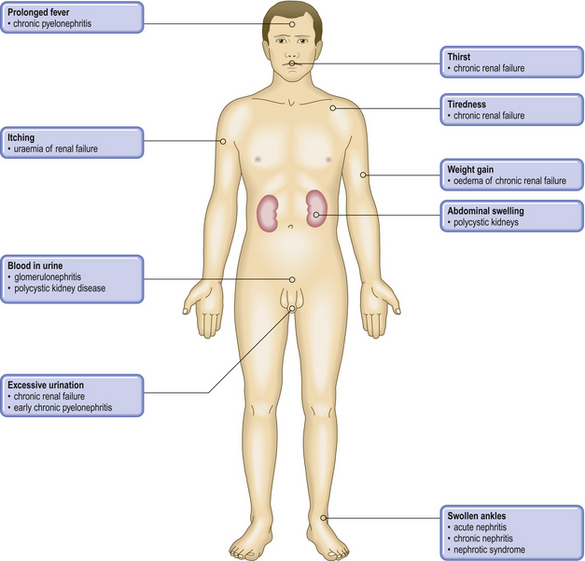
Pathological basis of renal symptoms and signs
| Symptom or sign | Pathological basis |
|---|---|
| Proteinuria | Increased permeability of the glomerular basement membrane |
| Uraemia | Renal failure |
| Haematuria |
Urinary casts
Hypertension
Oliguria or anuriaSevere renal failure (acute or chronic), obstruction or dehydrationPolyuria
Renal (ureteric) colicCalculus, blood clot or tumour in ureterOedemaPrimary renal abnormality leading to sodium and fluid retentionDysuriaStimulation of pain receptors in urethra due to inflammation
NORMAL STRUCTURE AND FUNCTION OF THE KIDNEYS
The kidneys contribute to the body’s biochemical homeostasis by:
The kidneys have a large functional reserve; the loss of one kidney produces no ill-effects. However, in renal disease waste products can accumulate, causing a condition known as uraemia. If the glomerular filters become excessively leaky, large protein molecules are lost in the urine—proteinuria. If the glomeruli are severely damaged, erythrocytes pass through causing haematuria.
The basic unit of the kidney is the nephron; each nephron comprises a glomerulus connected to a tubule. Each kidney contains approximately 1 million nephrons. These form in the embryonic metanephros, after the physiological involution of the pronephros and mesonephros. The ureter, calyceal system and collecting ducts form from the ureteric bud arising from the original duct of the pronephros—the Wolffian duct.
Glomerular structure and function
The formation of urine begins in the glomeruli, where the filtration of approximately 800 litres of plasma each day results in 140–180 litres of filtrate, most of which is reabsorbed by the tubules. Each glomerulus comprises a tuft of capillaries projecting into Bowman’s space (Fig. 21.1A).
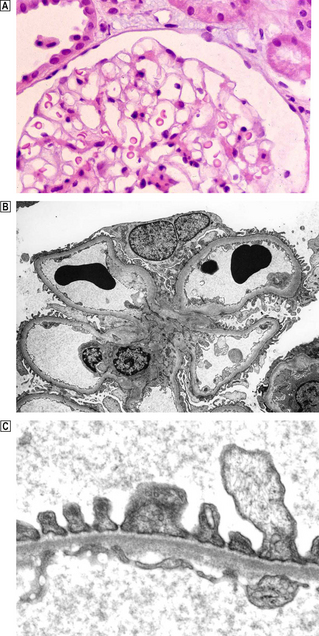
Fig. 21.1 Normal glomerulus.  Each glomerulus consists of capillaries invested by epithelial cells and is surrounded by Bowman’s space.
Each glomerulus consists of capillaries invested by epithelial cells and is surrounded by Bowman’s space.  Low-power electron micrograph showing capillary loops clustered around the mesangium. The basement membrane does not surround the capillary loop completely but is reflected onto the adjacent loop in the region of the mesangium, leaving the mesangial area covered only by the endothelium.
Low-power electron micrograph showing capillary loops clustered around the mesangium. The basement membrane does not surround the capillary loop completely but is reflected onto the adjacent loop in the region of the mesangium, leaving the mesangial area covered only by the endothelium.  High-power electron micrograph showing ultrastructure of the glomerular capillary. The wall of each glomerular capillary comprises an inner thin layer of fenestrated vascular endothelium, the basement membrane and an outer epithelial layer characterised by cytoplasmic (‘foot’) processes. Slit diaphragms span the gap between adjacent epithelial cells. In electron micrographs the basement membrane has a central dense zone, the lamina densa, surrounded on either side by the less densely packed lamina rara interna and externa.
High-power electron micrograph showing ultrastructure of the glomerular capillary. The wall of each glomerular capillary comprises an inner thin layer of fenestrated vascular endothelium, the basement membrane and an outer epithelial layer characterised by cytoplasmic (‘foot’) processes. Slit diaphragms span the gap between adjacent epithelial cells. In electron micrographs the basement membrane has a central dense zone, the lamina densa, surrounded on either side by the less densely packed lamina rara interna and externa.
Blood enters and leaves the glomerular capillaries by arterioles. In contrast to all other systemic capillaries in which there is a fall in pressure towards the venous end, the hydrostatic pressure within the glomerular capillary remains high throughout its length, thus enabling efficient filtration.
The glomerular capillary comprises:
All components of the capillary wall contribute to the filtration barrier, which is entirely extracellular and has two complementary aspects:
The integrity of the filtration barrier is disturbed in glomerular disease.
The attenuated endothelial cytoplasm lining the luminal aspect of the glomerular capillary has numerous holes or fenestrae, 70–100 nm diameter. Functionally the endothelial cells:
The basement membrane is a mesh of filamentous matrix proteins, including collagen IV, laminin and fibronectin, many of which are anionic, through which the ultrafiltate must pass.
The external aspect of the basement membrane bears epithelial cells with complex interdigitating cellular processes, termed foot processes, enveloping the capillary loops. Modified adherens-type junctions (filtration slit diaphragms) occur where the foot processes meet and are essential to the function of the epithelial cell (Fig. 21.1C). The integrity of the slit diaphragm is maintained by the complex inter-relationship of numerous proteins including nephrin, podocin and CD2-associated protein (CD2AP). Other proteins, such as integrins, span the membrane and anchor the actin cytoskeleton to the collagen IV in the lamina rara externa of the basement membrane. Therefore, changes in these proteins modify the configuration of the foot process, and defects in the genes encoding these proteins result in simplification of the foot processes.
The glomerular capillary tufts are supported centrally by mesangial cells, which proliferate and become more prominent in some diseases. Surrounded by a loose network of fibrillary material — the mesangial matrix — the mesangial cells comprise two types. One type of mesangial cell contains actin filaments and is contractile. They attach to the capillary basement membrane at the point where it is reflected over the mesangial matrix, thus anchoring the capillary to the central structure. Contraction of these cells therefore pulls on the glomerular basement membrane and will alter the shape and calibre of the capillary. Damage to this area reduces the strength of the capillary with the formation of a micro-aneurysm. The other type of mesangial cell resembles a monocyte and is analogous to a tissue macrophage. Both types of cell are involved in the mesangial reaction in glomerular disease by synthesising new matrix material and secreting cytokines responsible for cell proliferation and attraction of inflammatory cells.
The basement membrane is reflected over the mesangial area to extend onto the adjacent capillary, which means endothelium is attached to the mesangial matrix in the central core area (Fig. 21.1B). This allows access of immune complexes to the mesangium and the ability of mesangial cells to probe the capillary lumen.
Glomerular filtration rate
Blood flow through the kidneys produces 130–180 l/day of ultrafiltrate which is termed the glomerular filtration rate (GFR). The GFR reflects the permeability of the capillary wall, together with the hydrostatic and osmotic gradients between the capillary lumen and the Bowman’s capsular fluid. The hydrostatic pressure within the glomerular capillary is determined by the calibres of the feeder afferent and draining efferent arterioles.
The GFR is modified by three important mechanisms, all of which are closely inter-related and involve the juxtaglomerular apparatus (JGA):
Autoregulation and tubuloglomerular feedback
The JGA, situated at the hilum of the glomerulus, comprises the afferent and efferent arterioles and the modified tubular cells of the thick loop of Henle, the macula densa (Fig. 21.2), and enables autoregulation and tubuloglomerular feedback. The specialised cells of the macula densa monitor the level of chloride in the tubular luminal fluid, reflecting the amount of chloride reabsorbed by the tubule. A reduced GFR leads to a fall in the luminal chloride level. This results in dilatation of the afferent arteriole, together with constriction of the efferent arteriole, resulting from the release of renin. These two changes increase the hydrostatic pressure within the glomerular capillary and restore the GFR. Autoregulation and tubuloglomerular feedback are important for normal renal function, and are disturbed in patients with systemic hypertension due to renal artery stenosis.
Neurohormonal factors
Neurohormonal influences operate in patients who have systemic hypotension due to heart failure, or fluid depletion in gastroenteritis.
Both angiotensin II and noradrenaline (norepinephrine) from the adrenal are involved. Angiotensin II constricts the efferent more than the afferent arteriole, and noradrenaline constricts both vessels to the same degree, which preserves the GFR. Both angiotensin II and noradrenaline stimulate prostaglandin synthesis, which by reducing arteriolar tone prevents excessive renal ischaemia. Since non-steroidal anti-inflammatory drugs inhibit the synthesis of prostaglandin, in patients who are hypoperfused they block the potential protective effect of the prostaglandins and can result in acute renal failure.
By liberating angiotensin II, renin also causes the adrenal cortex to produce aldosterone which, in turn, leads to increased reabsorption of sodium by the distal tubular epithelium.
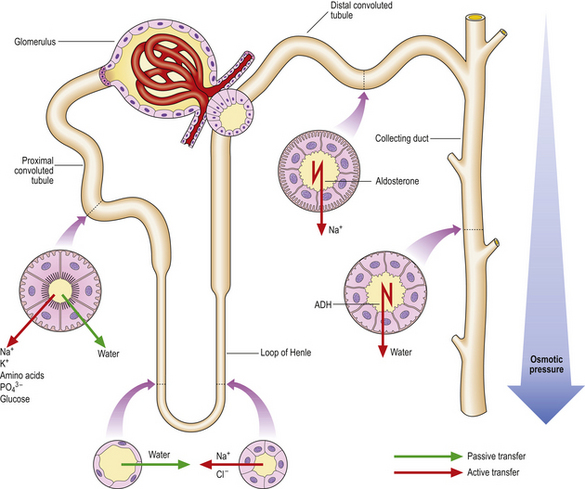
The glomerular filtrate, which is isotonic with the plasma, has to be substantially modified osmotically so that water and electrolytes are conserved and the waste metabolites are concentrated. This occurs as the filtrate flows through the tubules (Fig. 21.2).
Tubular structure and function
Epithelial cells modify the filtrate by transferring electrolytes and solutes aided by a series of carrier proteins or transporters within the apical (luminal) cell membrane. Transfer from the cytoplasm to the interstitial and peritubular fluid is performed by an energy-dependent ATPase pump situated on the basolateral membrane of the cell. The epithelial cells are separated from each other by tight junctions that contain claudins, membrane proteins that prevent the unregulated passage of electrolytes, water and solutes through the epithelial layer between the cells.
In the proximal tubule approximately 50–55% of the sodium in the filtrate is reabsorbed through selective sodium transporters, together with specific transmembrane co-transporters linked separately to glucose, phosphate or amino acids. In this way nearly all of the glucose, phosphate and amino acids are reabsorbed by the proximal tubule, thus altering the osmolality of the tubular fluid and causing water to flow into the cytoplasm through specialised water channels termed aquaporins. Some of the sodium transporters are linked with hydrogen exchange, whereby sodium is reabsorbed and hydrogen is excreted. Consequently, c. 80% of all the bicarbonate filtered is reabsorbed by the proximal tubules.
The loop of Henle, situated in the medulla and doubling back on itself, is the next part of the nephron through which the now reduced volume of the filtrate must pass. The two limbs have quite different physiological properties. The descending loop is permeable to water but not to ions, whereas the ascending limb is permeable to ions but, lacking aquaporins, is impermeable to water. Thus, the interstitium of the medulla becomes hypertonic. The filtrate in the loop lumen equilibrates with this, because of the permeability to water in the descending limb.
The distal tubule is continuous with the ascending limb of the loop of Henle. The epithelial cells of this segment lack aquaporins, making this segment impermeable to water. Sodium and chloride are reabsorbed by a co-transporter, the activity of which is governed by the concentration of chloride in the luminal fluid. Transport of sodium and chloride in the loop of Henle and distal convoluted tubule is flow-dependent, an important concept in the context of understanding the action of loop diuretics which tend to increase the rate of flow. Calcium transport, under the influence of parathyroid hormone and 1,25-dihydroxycholecalciferol (vitamin D3), occurs in the distal convoluted tubule and adjacent segments.
The distal convoluted tubule continues into the collecting duct. Two main cell types are present:
Aldosterone increases the number of open sodium channels, thus increasing the reabsorption of sodium in the event of volume depletion. The principal cells of the collecting ducts are relatively impermeable to water due to the paucity of aquaporins on the apical membrane. However, under the influence of antidiuretic hormone (ADH) produced by the pituitary, a complex sequence of changes occurs within the cell. This culminates in the fusion of intracytoplasmic vesicles containing preformed aquaporins with the apical membrane so that water can be cleared into the circulation.
The intercalated cells are concerned with hydrogen ion excretion. The excreted hydrogen combines with ammonia in the lumen to form ammonium. Ammonia, formed in the proximal tubule by the metabolism of glutamine and by diffusion from the interstitial fluid, is freely diffusible in contrast to ammonium which is lipid insoluble and cannot pass back into the tubular cytoplasm.
The vasa recta is the delicate meshwork of capillaries that invests the tubules and is derived from the efferent glomerular arteriole. The configuration of the vascular network complements that of the tubule and plays an integral role in the functioning of the countercurrent mechanism.
Countercurrent mechanism
The countercurrent mechanism ensures urine of variable osmolarity forms in response to a variable water intake. The hairpin configuration of the loop of Henle, the complementary vasa recta, coupled with the selective permeabilities to ions and water of the different segments of the loop, the distal tubule and collecting tubules, are all pivotal to the countercurrent mechanism. The active transport of sodium by the thick ascending limb increases the osmolarity of the interstitium. As a result of this, water diffuses from the filtrate in the lumen of the descending limb, which is permeable to water but not to ions. With progress towards the tip of the loop, osmolarity of the filtrate and interstitium increases, particularly in the longer loops derived from the juxtamedullary glomeruli. The principal cells of the collecting tubules display a variable permeability to water under the influence of ADH, achieving urine of variable osmolarity by passing through this hyperosmolar environment on the way to the papillae.
Tamm–Horsfall protein (uromodulin) is a large mucoprotein produced exclusively by the cells of the thick ascending limb of the loop of Henle. It is the main constituent of all tubular casts and may help prevent infections of the urinary tract, but other functions are uncertain. Mutations in the gene encoding this protein are associated with medullary cysts, hyperuricaemia and progressive renal failure.
Renal papillae and urinary reflux
The collecting ducts open onto the surface of the renal papillae projecting into the calyces. The shape of the duct orifice is relevant to the development of reflux and pyelonephritis. Two patterns have been described:
Physiological changes
During pregnancy
During pregnancy the size and weight of the kidneys increases and the glomeruli enlarge. These changes are reflected in the raised GFR and renal plasma flow which reach a peak by 16 weeks and persist until the end of pregnancy.
Ageing
The size of the kidneys falls abruptly after the age of 60 years. Gradual shrinkage of the tubules commences at about 40 years. Arteries display intimal thickening, with progressive reduplication of elastic laminae. Small arteries develop medial hypertrophy and hyalinosis. The number of sclerosed or scarred glomeruli increases with age, thus reducing renal reserve. Drugs usually excreted by the kidneys must, therefore, be used cautiously in the elderly to avoid toxic accumulation.
RENAL DISEASE
Clinicopathological features
Diseases of the kidney can present with a variety of features, alone or in combination. As the kidneys are so often affected by a primary disease elsewhere in the body, a simple urine examination (e.g. colour, glucose, protein, haemoglobin) is routine practice in patients being investigated for a variety of disorders.
Investigation
The investigation of patients with renal disease is multidisciplinary (Table 21.1). Urine and blood analyses are essential; imaging, biopsies and cystoscopy are optional depending on the nature of the clinical problem. Tests with the greatest general clinical utility are urine testing for glucose (to exclude uncontrolled diabetes mellitus), protein (to determine the permeability characteristics of the glomerular basement membrane), and determination of blood concentrations of urea and/or creatinine, the latter being the more reliable indicator of renal function. The GFR is an important expression of renal function and is a useful parameter for monitoring the severity and progress of renal disease.
Table 21.1 Investigation of patients with urinary tract disease
| Investigation | Diagnostic utility |
|---|---|
Determination of urine production rate and concentrating power of the kidneys; investigation of urinary tract infections; urinary protein indicates integrity of glomerular filter; exclusion of diabetes mellitus; investigation of glomerular or tubular lesions
Determination of integrity of renal function; glomerular filtration rate can be calculated from urinary and plasma creatinine concentration and urine flow rate
Determination of kidney size and symmetry; investigation of suspected tumours, cysts, etc.; detection of calculi; position and integrity of uretersCystoscopyInvestigation of haematuria and other symptoms; biopsy of bladder lesions
Diagnosis of glomerular, tubular and interstitial renal diseases
Renal biopsy is performed only when clinically justified, because of the risk of haemorrhage. The biopsy is examined by light microscopy with additional information revealed by immunofluorescence and electron microscopy in some cases.
Accurate information about the incidence of diseases of the urinary tract is available from transplant and dialysis registries. However, two important factors conspire to make the true incidence of renal disease almost impossible to ascertain. First, not all countries have registries for the accurate recording of cases. Second, transplantation and dialysis registries record severe and end-stage disease only, making no allowance for mild and subliminal disease. Clinical experience suggests that the prevalence of post-infectious glomerulonephritis is much higher in Africa and India than in Europe and North America.
Pathophysiological basis of renal disease
Two main clinical syndromes occur in renal disease: nephritis (also referred to as the nephritic syndrome) and the nephrotic syndrome. As part of these, patients present with either acute or chronic renal failure.
Nephritis
Nephritis is characterised by an ‘active’ urinary sediment comprising red cells, white cells and urinary casts. The clinical manifestations are variable depending on the degree of damage ranging between intermittent painless (asymptomatic) haematuria, acute renal failure and rapidly progressive glomerulonephritis. The unifying feature of the diseases that cause nephritis is endothelial damage and inflammation. In cases of severe nephritis where the inflammatory infiltrate is intense and widespread (i.e. diffuse), there is a substantial reduction in the GFR. The diseases causing nephritis fall into three broad groups:
Nephrotic syndrome
The nephrotic syndrome is due to excessive leakiness of the glomerular filter and comprises:
The unifying abnormality in patients with the nephrotic syndrome is damage to the epithelial cells resulting in ‘effacement’, ‘fusion’ or ‘simplification’ of the foot processes. It results from molecular alterations at the base of the cells and the filtration slit diaphragms. Nephrin and podocin, amongst other proteins, span the cell membrane and effectively hold the cells together in the region of the filtration slit diaphragm. Similarly, integrins anchor the cell surface to the underlying lamina rara externa of the basement membrane, and these proteins in turn connect to the actin-based cytoskeleton of the cell. Thus, when these proteins are altered, through either mutation or damage, the cell cytoplasm retracts, resulting in simplification or effacement, and leading in some cases to focal epithelial separation from the basement membrane.
Diseases causing the nephrotic syndrome fall into four broad groups:
Proteinuria and oedema
Proteinuria
Three types of proteinuria occur in patients with renal disease emanating from:
Proteinuria arising from the glomeruli implies severe damage leading to loss of the slit diaphragms and effectively reducing the filtration surface. This results in a major change in the character of the resistance to macromolecules, with ensuing substantial leakage of proteins. A degreeof vasoconstriction of the efferent arteriole occurs in response to proteinuria and this serves to increase the intraglomerular hydrostatic pressure which exacerbates the passage of protein through the wall. Therefore, reducing the hydrostatic pressure in the capillary will have a beneficial effect and rationalises the use of antihypertensive therapy in chronic renal disease. The terms selective and non-selective proteinuria refer to the degree and composition of the proteinuria which corresponds to the degree of damage to the filter. Selective proteinuria is seen mostly in minimal change disease.
The tubular epithelial cells reabsorb most of the smaller low molecular weight proteins normally escaping from the glomeruli. Tubulo-interstitial diseases therefore will impede this reabsorption and result in loss of small proteins. In patients with multiple myeloma and other plasma cell dyscrasias, excessive quantities of immunoglobulin light chains are produced and appear in the urine.
Detection of microalbuminuria is particularly important in the management of diabetes and indicates very early increases in permeability. The urinary loss of albumin in these cases amounts to 30–300 mg/day. Routine ‘dipstick’ testing of urine will not detect microalbuminaemia and is therefore unsuitable for managing diabetics.
Oedema
Oedema is common in renal disease, especially in patients with the nephrotic syndrome, and also in some severe forms of nephritis. Sodium retention by the damaged kidney is the fundamental event, but the mechanism is not known. Oedema in renal disease relates therefore to sodium retention and fluid retention rather than alterations in plasma proteins and plasma osmotic pressure.
Renal failure
Renal failure, ‘impairment’ or ‘insufficiency’ are terms used when the kidneys are unable to excrete waste products and fail to manage electrolytes and water in the usual way.
Acute renal failure
Acute renal failure is said to occur when the plasma creatinine concentration has increased by at least 0.5 mg/dl within the past month. The causes of acute renal failure fall into three main groups:
Chronic renal failure
Chronic renal failure (CRF) leads to clinical features often referred to as uraemia, which reflect a severe reduction in functioning renal mass. The development of the clinical changes in uraemia results from:
Alterations in electrolyte excretion are minimal initially because of adaptive mechanisms involving reducing tubular reabsorption and do not become evident until the GFR reduces by c. 80%. Conversely, organic solutes are excreted primarily by glomerular filtration and will accumulate when the GFR falls. Although individual proteins and metabolites cannot be linked to specific uraemic symptoms, they are known collectively as uraemic toxins.
Hormone production by the severely damaged kidney is impaired. The consequences include renal osteodystrophy and secondary hyperparathyroidism, hypertension and anaemia.
Renal osteodystrophy and secondary hyperparathyroidism
Phosphate retention occurs with mild reductions of GFR and is important in CRF. Whilst calcium and phosphate are inversely related, the role of phosphate retention in CRF is pivotal. By attaching to specific receptors, calcitriol (vitamin D3), produced predominantly by the proximal tubular epithelial cells, appears to be the main regulator of phosphate and exerts a negative feedback on the parathyroids, reducing both production and release of parathyroid hormone (PTH). In CRF, a rise in phosphate inhibiting the secretion of calcitriol may in turn reduce the inhibitory effect on PTH release. The reduced level of calcitriol continues as renal damage proceeds and GFR falls. Eventually the inhibitory effect by PTH on phosphate reabsorption by the proximal tubule will be saturated and hyperphosphataemia and hyperparathyroidism will persist, with two important clinical consequences:
The effects of hyperparathyroidism in patients with CRF can be minimised by a low phosphate diet or the administration of a phosphate-binding agent. Calcitriol is also used but is associated with hypercalcaemia due to the effect on bone. Calcimimetics, which bind to the receptors on the surface of the parathyroid cell, reduce the synthesis and release of PTH. However, oversuppression of PTH may result in adynamic bone disease.
Hypertension
Hypertension occurs in c. 90% of patients with CRF, due mostly to fluid retention; it responds to diuretics. Autoregulation in response to the reduced GFR releases renin and consequently angiotensin II. Renal scarring and subsequent focal renal ischaemia is thought to be a cause of increased renin secretion. The management of hypertension in patients with chronic renal disease is very important since persistent high blood pressure will exacerbate glomerular damage and further reduce GFR.
Anaemia
Anaemia is common in patients with CRF and accounts for their lethargy and general lack of well-being which worsen with deteriorating renal function. Anaemia results from inadequate renal production of erythropoietin (EPO). The precise site of EPO formation is uncertain but the endothelial cells of the peritubular capillaries have been suggested. Red cell survival is reduced; bleeding tendencies consequent on altered platelet function and iron deficiency are also associated with CRF.
Recombinant EPO treatment corrects most of these changes with marked symptomatic improvement. Iron supplements are required to keep pace with the red cell production as stores can become depleted. Importantly, about one-third of patients develop hypertension, which may be severe. Increased viscosity of the blood and reversal of the peripheral vasodilatory effect of the anaemia have both been implicated.
MECHANISMS OF GLOMERULAR DAMAGE
Glomeruli can be damaged by immunological or non-immunological mechanisms.
Immunological mechanisms
Immune glomerular injury
Immunological damage causes most human glomerular disease. There are two mechanisms:
The resulting disease is called glomerulonephritis (in some cases, glomerulopathy). Genetic factors influence susceptibility and prognosis.
Nephrotoxic antibody
In anti-GBM disease, an IgG antibody forms against the alpha-3 chain in the collagenase-resistant component of collagen IV within the basement membrane, binds to it and activates the complement cascade. Renal biopsy reveals, in glomeruli, a linear pattern of immunofluorescent staining for IgG and granular staining for C3. Polymorphs are attracted and a florid proliferative glomerulonephritis results (Fig. 21.3).
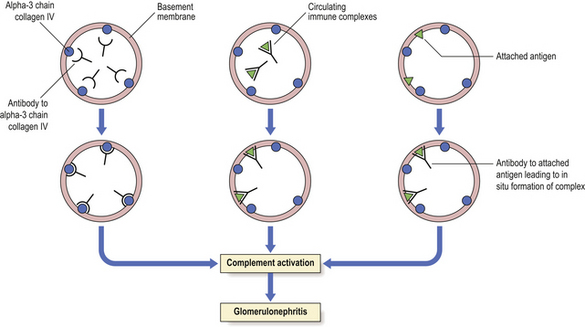
Fig. 21.3 Immunological mechanisms of glomerulonephritis. Glomerulonephritis is due to an antibody reaction to glomerular antigen (alpha-3 chain of collagen IV), to circulating immune complexes that become deposited in the glomeruli, or to antibody reacting to a foreign antigen that has become attached to the basement membrane. The final common pathway of glomerular injury is complement activation resulting in varying degrees of cell damage and inflammatory cell recruitment.
Anti-GBM disease is an uncommon cause of glomerulonephritis and in some cases is associated with pulmonary haemorrhages (Goodpasture’s syndrome), because the antigen also occurs in the alveolar basement membrane. Clinical and histological features of anti-GBM disease are discussed below.
Immune complex deposition/activation
An immune complex develops when an antibody binds to its soluble specific antigen. The antigen may be extrinsic, e.g. derived from an infective agent, or endogenous, e.g. DNA in lupus; the latter is said to be autoimmune. Some immune complexes form large lattice structures within the circulation and are eliminated by the reticuloendothelial system; others are smaller and initiate glomerular damage by either deposition or in-situ formation (Fig. 21.3). The complexes seen in the glomeruli are termed ‘deposits’. The glomeruli are vulnerable because the kidneys filter large volumes of blood. Immune complex damage occurs by:
The interaction of antigen and antibody within the deposit activates the complement cascade with the production of the C5b-9 membrane attack complex. In addition, C3a and C5a are chemotactic and recruit polymorphonuclear leukocytes, macrophages and monocytes.
The site of the interaction determines the type of glomerular lesion and the clinical features. Thus, deposits within the mesangium or subendothelial lamina rara interna have access to the circulating blood, and complement activation elicits a proliferative reaction and an active nephritis with haematuria. Both membrano-proliferative glomerulonephritis and IgA disease have this pattern.
In contrast, deposits within the subepithelial lamina rara externa will also activate complement, but the reactants are sequestered from the circulation by the basement membrane. There is therefore no evidence of inflammatory reaction; an example of this pattern is seen in membranous glomerulonephritis (glomerulopathy).
Participation by glomerular cells in glomerular disease
Cells within the glomerulus participate in the reaction and consequently to the development of the lesion and the fate of the glomerulus. These cells produce a variety of cytokines and locally influence the coagulation cascade.
Epithelial cells overlying subepithelial deposits are stimulated to produce basement membrane material, mostly laminin. This overproduction results in irregular projections which separate and then partially surround the deposits. These projections, called ‘spikes’, are characteristic of membranous glomerulopathy. The accumulation of extracellular matrix material is an important aspect of the evolution of glomerular lesions having a wide aetiological background. This accumulation of matrix material is a balance between production, degradation and remodelling and it contributes to the development of glomerulosclerosis.
Endothelial cells lose their natural thromboresistance leading to platelet deposition on the endothelial cell surface and further damage. This is relevant in conditions such as hypertension, diabetes and the inflammatory diseases of blood vessels (vasculitis).
Mediators of glomerular damage
Glomerular cells are affected by a wide variety of substances:
The contribution of the individual mediators varies in each case.
Non-immunological mechanisms
Non-immunological mechanisms include:
CONGENITAL DISEASES
Approximately 10% of individuals have a congenital abnormality of the urinary tract, some of which are hereditary:
Developmental abnormalities
Conditions affecting the volume of renal tissue
Renal agenesis (absence of the kidney) may be unilateral or bilateral.
Bilateral agenesis results from failure of initiation of the pronephros–metanephros sequence; the ureteric bud fails to develop. It occurs in c. 0.04% of all pregnancies and is associated with a severe reduction in amniotic fluid (oligohydramnios) resulting in neonatal death due to lung hypoplasia. The majority of cases are stillborn. There are also developmental abnormalities of other tissues derived from the mesonephros, e.g. bladder and genitalia. Accompanying spinal cord abnormalities with involvement of the hind gut and fused lower extremities is termed sirenomelia (mermaid syndrome).
Unilateral agenesis of a kidney is common, affecting up to 0.1% of the population. The opposite kidney undergoes marked hypertrophy and is prone to trauma and to reflux nephropathy since it is often associated with pelvi-ureteric or ureterovesical junction obstruction. The relatively high incidence of a solitary kidney in the general population means it is imperative to confirm the presence of a second kidney before renal biopsy or nephrectomy.
Disorders of differentiation
Renal dysplasia may present in childhood as an abdominal mass simulating a tumour. It is characterised by islands of undifferentiated mesenchyme or cartilage within the parenchyma. The pathogenesis is failure of induction of both the ureteric bud and mesenchymal tissues leading to abnormal development of the collecting ducts and subsequent loss of potential nephrons with the formation of cysts. The prognosis is good if the lesion is unilateral with no obstruction.
Anatomical abnormalities
Ectopic kidneys form in an abnormal site, usually the pelvis, and may be associated with intestinal malrotation. They may present as a suspicious pelvic mass and there is a risk of infection due to the ureteric kinking that often accompanies this condition.
Horseshoe kidney results from fusion of the two nephrogenic blastemas during fetal life. Most are fused at the lower pole. The condition is not rare and renal function is usually normal.
Genetic abnormalities leading to enzyme abnormalities
Cystinuria
Cystinuria (Ch. 7) results from defective tubular reabsorption of several dibasic amino acids including cystine, lysine, ornithine and arginine. There is mutation in the genes for the amino acid transporters. Cystine crystals are found in the urine; stones may develop by the second or third decade.
Renal tubular acidosis
Two forms of renal tubular acidosis affect different tubular segments. Type 1 (‘distal’) causes metabolic acidosis and a persistent urinary pH of 5.5 because the hydrogen-ATPase pump in the apical membrane is deficient due to acquired or genetic factors. Type 2 (‘proximal’) causes a metabolic acidosis associated with a low plasma bicarbonate; there is impaired reabsorption of bicarbonate by the proximal tubular cells.
Genetic abnormalities leading to abnormal proteins
Congenital nephrotic syndrome
This is a rare autosomal recessive condition. There are mutations of the gene for nephrin (NPHS1) on chromosome 19. Consequently, the nephrin formed is defective and, being an integral part of the filtration slit diaphragm of the podocyte, there is effacement of the foot processes and proteinuria which is present before birth. The kidneys are enlarged and appear microcystic due to dilatation of the proximal tubules and Bowman’s capsule. Affected infants have additionally the characteristic Potter facies and marked motor and mental retardation, and are particularly vulnerable to pneumococcal infection. There is no treatment, but the lesion does not recur in a transplanted kidney.
Hereditary nephritis
Hereditary nephritic syndromes result from mutations in the genes encoding alpha chains in the collagen IV molecule and result in abnormal basement membranes which feature collagen IV. The least severely affected patients have an autosomal recessive form of inheritance. Two forms are recognised: thin basement membrane disease and persistent haematuria.
Thin basement membrane disease
Thin basement membrane disease is identified ultrastructurally when patients are investigated for chronic haematuria.Also known as benign familial haematuria, the condition is common, occurring in some 25% of patients investigated for haematuria, and may be present in about 1% of the population. The clinical importance lies in excluding more serious causes of haematuria and differentiating it from the early stages of Alport’s disease.
Alport’s disease
Alport’s disease is recognised clinically by the triad of:
The most common form (90%) is X-linked, due to a mutation of the gene encoding the alpha-5 chain of collagen IV. The mutation causes an inability to form normal basement membranes which feature collagen IV, notably in the kidney, eye and ear, thus explaining the clinical triad. Males are affected more frequently and more severely; the eventual outcome is renal failure by the second decade. In contrast, renal function may be preserved in females until the fifth decade. The deafness, which is for high frequencies, is often difficult to demonstrate. Ocular disease occurs in severely affected patients and involves dislocation of the lens, cataracts and corneal dystrophy.
Microscopic changes are most noticeable in the late stages when glomerulosclerosis develops. Electron microscopy reveals an irregular thinned and reduplicated basement membrane giving rise to the ‘basket weave’ pattern characteristic of the disease.
CYSTIC DISEASE




Cystic disease of the kidney comprises a heterogeneous group of conditions which are genetic or acquired. Accurate diagnosis is essential so that, where the disease is genetically transmitted, the appropriate genetic counselling can be given to patients and their relatives.
Formerly, renal cystic disease was classified morphologically, but with improved imaging and characterisation of genetic factors the pathogenesis of these conditions is being clarified. The current classification of cystic disease, correlating morphological features and genetic factors, will almost certainly be revised as further information becomes available (Table 21.2).
Table 21.2 Cystic disease: classification
| Simple cysts |
| Polycystic kidney disease |
Renal medullary cysts
Renal cysts associated with genetic diseases affecting multiple systems
Acquired (dialysis related) cysts
Pathogenesis of cysts
The development of cysts is thought to involve several processes involving the development and polar differentiation of the epithelial cells. Reversal of the polarity of transporter proteins results in transport of fluid via the sodium pump into the tubular lumen which then becomes cystic. There are also changes in the primary cilia where all of the major proteins relevant to cystic disease are normally expressed.
Autosomal dominant polycystic kidney disease (ADPKD)
ADPKD is the primary renal disease in 8% of adult patients in the European Dialysis and Transplantation Registry. The condition is always bilateral; the kidneys are grossly enlarged, each commonly weighing 1000g or more (Fig. 21.4). The kidneys are distorted by numerous cysts, from a few millimetres up to c. 100 mm in diameter, with thin bands of renal parenchyma compressed between them. Most cysts contain clear fluid, but previous haemorrhage can result in the contents being brown due to haemosiderin. The cysts are formed at all levels of the nephron.
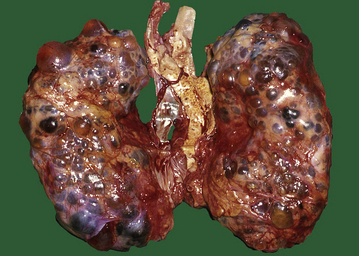
Fig. 21.4 Autosomal dominant polycystic disease (ADPKD). Both kidneys are greatly enlarged by numerous cysts of varying sizes.
ADPKD is inherited as an autosomal dominant trait with a high degree of penetrance; this is relevant to genetic counselling and screening. Most cases are linked to the alpha-globin cluster on the short arm of chromosome 16 (PKD 1) which encodes polycystin-1; the remainder are due to mutations on chromosome 4 (PKD 2) which encodes polycystin-2. This condition presents at any age from childhood to late adult life. Renal function is maintained until the enlarging cysts press on the adjacent parenchyma, causing ischaemic changes leading to hypertension or renal failure. There is an association with berry aneurysms of the circle of Willis, a frequent source of often fatal subarachnoid haemorrhage (Ch. 26). Cysts also occur within the liver, pancreas and lungs, but have no functional significance in these organs.
Autosomal recessive polycystic kidney disease (ARPKD)
Autosomal recessive polycystic disease (ARPKD) is a rare condition which has a recessive pattern of inheritance. All of these patients have mutations in a single gene known as the ‘polycystic kidney and hepatic disease gene 1’ (PKHD1) on chromosome 6 which encodes for the protein fibrocystin/polyductin. Fibrocystin is a transmembrane receptor located in the cilia of the renal epithelial cells and is involved in the differentiation of renal collecting ducts and bile ducts.
Clinical presentation depends on the severity of involvement and occurs in neonates, children and young adults. The antenatal or perinatal patient has the characteristic Potter facies associated with oligohydramnios and is either stillborn or develops respiratory distress soon after birth. The kidneys are enlarged and show a characteristic radial pattern of fusiform cysts which are dilated collecting ducts. The enlarged kidneys are usually palpable and may impair birth.
Patients who survive to childhood or adult life develop abnormalities of the liver, ranging from bile duct proliferation and cysts, to substantial hepatic fibrosis and cirrhosis which will eventually interfere with hepatic function and produce portal hypertension (Ch. 16). In addition, chronic lung disease, growth retardation and urinary tract infections are recorded. The median survival is 27 years for patients who survive the first month.
Renal medullary cysts
Medullary cystic diseases (nephronophthisis and medullary cystic kidney disease) have been categorised and clarified using genetic and molecular biological features. Clinical and pathological features are shared, but they have different patterns of inheritance and are associated with different extrarenal conditions. Medullary cysts develop as the diseases progress so they are not reliable early diagnostic features.
Nephronophthisis (NPH) encompasses a group of autosomal recessive inherited conditions. These are related to mutations in different NPH genes, which encode important proteins for renal development. They affect different age groups.
Medullary cystic kidney disease (MCKD) is rare, usually presents in adults and has an autosomal dominant inheritance. These patients fail to concentrate urine and lose salt, with polyuria and polydipsia in most cases. Two genes are involved: the more damaging, MCKD2, is situated on chromosome 16 and encodes for uromodulin (Tamm–Horsfall protein). Patients with this anomaly develop renal failure during the third decade of life.
Medullary sponge kidney
Medullary sponge kidney is a disease of adults resulting from dilated collecting ducts in the medulla, causing cysts mainly in the papillae. Renal function is usually normal but large calculi form within the cysts, causing pain, haematuria, infections and obstruction.
Dialysis-associated cysts
Dialysis-associated cysts occur in the kidneys of patients in chronic renal failure who have been dialysed long term. The cysts vary in size and number, frequently show intracystic haemorrhage and contain oxalate crystals. The epithelial lining forms micropapillary proliferations and c. 5% of patients with acquired cystic disease develop renal cell carcinoma; therefore patients with acquired cystic disease should be monitored.
GLOMERULAR DISEASE


 Immunological injury may be due to anti-glomerular basement membrane antibody (anti-GBM) or to immune complex deposition
Immunological injury may be due to anti-glomerular basement membrane antibody (anti-GBM) or to immune complex deposition


Classification
Classification of glomerular disease usually presents difficulties for students of renal disease. As with many aspects of human disease, it is impossible to provide a satisfactory classification of glomerular disease on the basis of one set of features. There are three parallel and complementary classifications:
Clinicopathologically, diseases affecting the glomeruli are best grouped initially into two broad categories:
Nomenclature of glomerular injury
Glomeruli show a limited range of reactions to injury. The following nomenclature is used to describe the pattern of injury:
Additional terms are used to describe the character of the light microscopic changes within the glomeruli:
Recognition of these changes and the use of this nomenclature have important clinical implications; for example, the presence of crescents in >50% of glomeruli corresponds to rapidly progressive glomerulonephritis, which heralds a poor prognosis unless it is treated effectively.
Morphology alone gives little indication of the precise nature of the underlying or primary abnormality. Similar morphological features occur in entirely different conditions. To establish an accurate diagnosis, immunological and ultrastructural assessment is needed.
Clinical presentations
Patients with glomerular disease usually present with one of five conditions.
There is considerable overlap of the conditions within these clinical states (Table 21.3). Several diseases, therefore, may give rise to the same clinical picture; conversely, many diseases fall into more than one of the presenting clinical conditions.
Table 21.3 Glomerular disease: clinical presentations
| Recurrent painless haematuria: |
Asymptomatic proteinuria:
Acute nephritis:
Nephrotic syndrome:
Chronic renal failure:
PRIMARY GLOMERULAR DISEASES
Anti-glomerular basement membrane disease
Anti-glomerular basement membrane (anti-GBM) disease is uncommon and occurs predominantly in young men. The mechanism of this disease is discussed above. The usual presenting feature is rapidly progressive renal failure. Haemoptysis may be present as part of the pulmonary–renal (Goodpasture’s) syndrome. The prognosis is poor without treatment, but plasma exchange (plasmapheresis) substantially improves the outlook, particularly in patients treated early.
Histologically, the characteristic lesion is a focal and segmental glomerulonephritis. With increasing severity, segmental necrosis with fibrin deposition, and a florid crescentic glomerulonephritis occurs (Fig. 21.5).
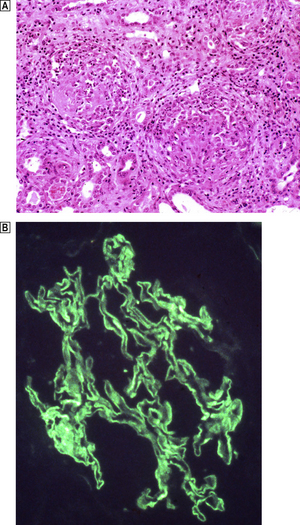
Fig. 21.5 Anti-glomerular basement membrane disease. The glomerular injury in this case is due to anti-glomerular basement membrane antibody.  The resulting damage causes obliteration of the Bowman’s space by macrophages, inflammatory cells and epithelial cells forming a crescent.
The resulting damage causes obliteration of the Bowman’s space by macrophages, inflammatory cells and epithelial cells forming a crescent.  Immunofluorescence reveals linear deposition of immunoglobulin on the glomerular basement membrane.
Immunofluorescence reveals linear deposition of immunoglobulin on the glomerular basement membrane.
Immune complex-mediated lesions
Immune complex-mediated glomerular injury (for mechanism, see above) results in a variety of glomerular reactions depending on the nature and site of the complexes.
Diffuse proliferative glomerulonephritis
Acute diffuse proliferative glomerulonephritis is one of the patterns of glomerular damage associated with a post-infective aetiology. It is usually an acute lesion following a transient infection. For many years it has been associated with a preceding beta-haemolytic streptococcal infection (post-streptococcal glomerulonephritis). However, a variety of other causative organisms produce this change, including:
Post-streptococcal glomerulonephritis
Beta-haemolytic streptococci of Lancefield group A, Griffith’s subtypes 12, 4 and 1, cause post-streptococcal glomerulonephritis. The primary infection usually causes pharyngitis but may also involve the middle ear or skin. There are considerable geographical differences in the incidence andseverity of post-streptococcal glomerulonephritis; for example, it is the commonest renal disease in India, but its incidence is falling in the UK.
Clinical features
Post-streptococcal glomerulonephritis affects all ages, but children are more commonly affected, with the onset of malaise, fever and nausea 7–14 days after a sore throat. There is oliguria (a significant reduction in urine volume), and the urine is dark or ‘smoky’ due to the presence of microscopic haematuria. Facial oedema, often periorbital, and a mild degree of hypertension are apparent on examination. A raised anti-streptolysin O (ASO) titre and a significant reduction in complement (C3) levels are present. Urine analysis reveals haematuria, together with white cells and casts, with a variable degree of proteinuria.
Histological features
The glomeruli are distended and hypercellular (Fig. 21.6A). All of the glomeruli are involved, hence the use of the term ‘diffuse’. The increase in cellularity is due to the proliferation and swelling of mesangial, endothelial and epithelial cells, together with polymorphonuclear leukocytes. Some glomeruli may show proliferation of cells lining Bowman’s capsule to form a crescent; the presence of crescents in 60% or more of the glomeruli indicates a rapidly progressive disease and heralds a poor prognosis. There is usually interstitial oedema and a variable inflammatory cell infiltrate. The tubules contain red cell casts and the tubular epithelial cells may show degenerative changes.
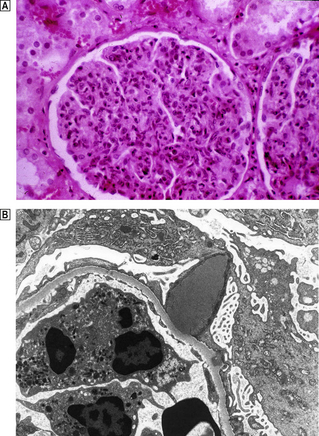
Fig. 21.6 Diffuse proliferative glomerulonephritis.  The glomerular injury in this case, characterised by hypercellularity owing to cellular proliferation and acute inflammatory cell infiltration, is due to immune complex deposition.
The glomerular injury in this case, characterised by hypercellularity owing to cellular proliferation and acute inflammatory cell infiltration, is due to immune complex deposition.  Ultrastructure of diffuse proliferative glomerulonephritis. The principal abnormality is the presence of electron-dense immune complexes deposited between the epithelial cells and the basement membrane. The cells with the granular cytoplasm in the lumen are neutrophil polymorphs attracted by the activation of complement in the loose subendothelial material.
Ultrastructure of diffuse proliferative glomerulonephritis. The principal abnormality is the presence of electron-dense immune complexes deposited between the epithelial cells and the basement membrane. The cells with the granular cytoplasm in the lumen are neutrophil polymorphs attracted by the activation of complement in the loose subendothelial material.
Using immunofluorescence techniques, granular deposits of IgG and C3 are identified on the peripheral basement membranes. Ultrastructurally, subepithelial deposits correspond to the granular IgG and C3 demonstrated by the immunological techniques (Fig. 21.6B).
Pathogenesis
The precise streptococcal antigen is not yet known—one is antigenically similar to streptokinase, others are cationic: streptococcal M protein, for example, cross-reacts with basement membrane antigens. Current evidence suggests an in-situ antigen–antibody reaction in post-streptococcal glomerulonephritis.
Prognosis
This is good in children, but only 60% of adults recover fully. Acute post-streptococcal glomerulonephritis is usually treated conservatively; renal biopsy is not usual unless the anticipated improvement fails to occur. In most cases the characteristic morphological changes will have resolved by 6–8 weeks after the onset of the illness. All that remains is mild mesangial hypercellularity, which may persist for many months or even years. The relationship between post-streptococcal glomerulonephritis and subsequent chronic glomerulonephritis remains controversial.
Focal proliferative glomerulonephritis
‘Focal’ implies uneven involvement of the glomeruli with some affected and the majority normal. In addition, involvement of only parts of individual glomeruli is usual, and the term ‘segmental’ is also appropriate. Thus, focal and segmental lesions are often found together. A focal glomerulonephritis is a fairly common pattern of reaction which is found in a variety of systemic diseases including:
The unqualified term ‘focal glomerulonephritis’ is not helpful clinically. The immunological and ultrastructural profile of the glomerular lesion in the renal biopsy must be ascertained for a more precise diagnosis.
There is one significant condition having a focal pattern that affects the kidney primarily: IgA disease. IgA deposition also occurs systemically, giving rise to Henoch–Schönlein purpura, a systemic vasculitis with involvement of the kidney.
IgA disease
IgA disease is now recognised as a major cause of chronic renal failure and insufficiency. The features include:
Histological features
A wide spectrum of changes occurs. Mild focal mesangial proliferation is associated with a good prognosis. In contrast, at the other end of the spectrum, a mesangiocapillary pattern, sometimes with segmental necrosis, is associated with a more rapid deterioration of renal function. IgA disease is often accompanied by a brisk interstitial inflammatory component, and established interstitial fibrosis is an ominous feature.
Membranous glomerulopathy
Membranous glomerulopathy (membranous glomerulonephritis, MGN), a chronic immune complex-mediated disorder, has a distinctive histological picture but many causes. It is an important cause of nephrotic syndrome.
Aetiology
In approximately 85% of patients with membranous glomerulopathy there is no identifiable cause; it is idiopathic. Other cases have an identifiable cause; these are secondary membranous glomerulopathies:
These patients are important to identify because the renal lesions may subside when the causative factor is removed. Approximately 10% of patients with systemic lupus erythematosus have membranous change.
Clinical features and prognosis
MGN affects all ages, but more commonly adults, with the highest incidence in the fifth to seventh decades. Males are affected more frequently. The presenting feature is either proteinuria or nephrotic syndrome. Hypertension occurs in about half of the patients. Treatment is controversial; most cases are unresponsive to steroids. Adult patients progress over a variable and unpredictable period; the 10-year survival is c. 75% and c. 30% will develop CRF due to glomerulosclerosis. The prognosis in children is much better, particularly when there is proteinuria alone. In these cases, only 10% develop renal failure, and some 50% remit.
Histological features
Capillary wall thickening, without proliferation or inflammation, and characteristic spikes occur in all glomeruli (Fig. 21.7). There are granular immune deposits of IgG and C3 in the thickened capillary walls. Electron microscopy reveals immune complexes deposited on the outer aspect of the basement membrane beneath the epithelial cells and there is effacement of the foot processes. The epithelial cells overlying the deposits produce new basement membrane, forming the spikes seen light microscopically; this encircles the deposits which undergo degradation and lysis. Eventually the affected glomeruli become sclerosed.
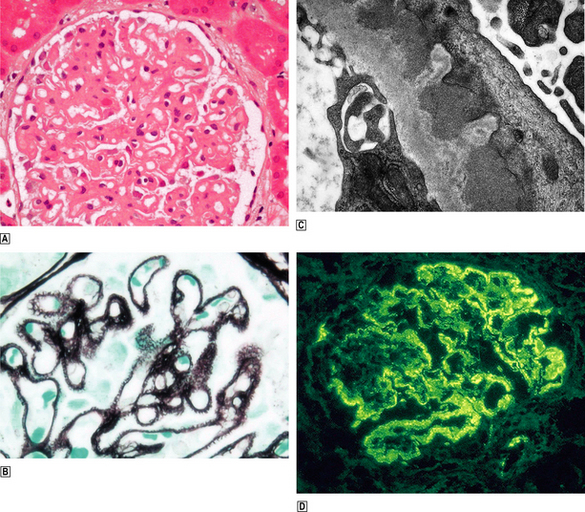
Fig. 21.7 Membranous glomerulopathy.  Membranous glomerulopathy is characterised by thickened glomerular capillary walls.
Membranous glomerulopathy is characterised by thickened glomerular capillary walls.  Histological staining by a silver impregnation method reveals minute ‘spikes’ on the outer aspects of the capillary wall.
Histological staining by a silver impregnation method reveals minute ‘spikes’ on the outer aspects of the capillary wall.  An electron micrograph shows the capillary wall to be thickened by the deposition of electron-dense deposits beneath the epithelial cells and between them new basement membrane corresponding to the ‘spikes’ seen histologically.
An electron micrograph shows the capillary wall to be thickened by the deposition of electron-dense deposits beneath the epithelial cells and between them new basement membrane corresponding to the ‘spikes’ seen histologically.  Immunofluorescence microscopy reveals granular deposition of IgG in the glomerular basement membranes, corresponding to the electron-dense deposits ultrastructurally.
Immunofluorescence microscopy reveals granular deposition of IgG in the glomerular basement membranes, corresponding to the electron-dense deposits ultrastructurally.
Membrano-proliferative glomerulonephritis
Membrano-proliferative glomerulonephritis (MPGN), a glomerular lesion with many causes, features both membrane thickening and proliferation (Fig. 21.8A). In addition, there is accentuation of the lobular architecture of the glomerulus. The thickened capillary walls, which have a duplicated ‘tram track’ appearance of the basement membrane revealed by silver staining (Fig. 21.8B), prompt the alternative name of mesangiocapillary glomerulonephritis (MCGN). This duplication is due to a new layer of basement membrane laid down by cell processes interposed between the endothelium and the basement membrane (Fig. 21.8C). Two main types are recognised.
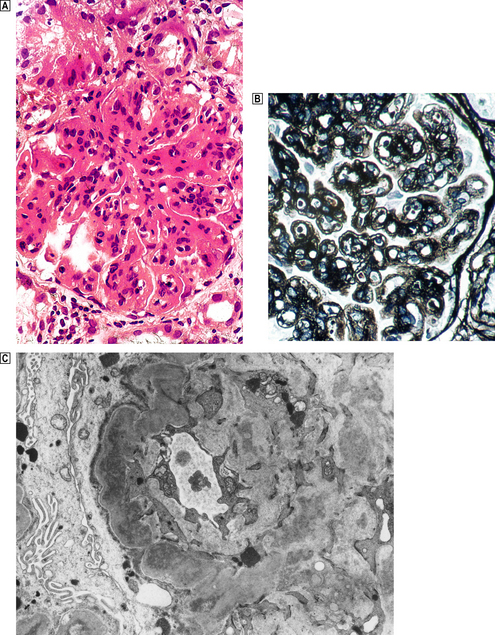
Fig. 21.8 Membrano-proliferative glomerulonephritis.  The combination of mesangial cell proliferation and basement membrane thickening exaggerates the lobular architecture of the glomerulus.
The combination of mesangial cell proliferation and basement membrane thickening exaggerates the lobular architecture of the glomerulus.  MST stain shows the new layer of basement membrane giving rise to the reduplicated peripheral profiles characteristic of mesangiocapillary glomerulonephritis.
MST stain shows the new layer of basement membrane giving rise to the reduplicated peripheral profiles characteristic of mesangiocapillary glomerulonephritis.  Electron microscopy shows the wall between the capillary lumen and urinary space to be thickened by the interposition of cellular processes and electron-dense deposits in relation to them.
Electron microscopy shows the wall between the capillary lumen and urinary space to be thickened by the interposition of cellular processes and electron-dense deposits in relation to them.
Type I MPGN
Type I MPGN is an immune complex-mediated lesion. Patients with this condition fall into two groups:
Most patients present with nephrotic syndrome, but some have haematuria. A persistently low serum complement C3 (hypocomplementaemia) occurs in two-thirds of patients. There is progressive deterioration over 10 or more years.
Type II MPGN
Type II MPGN is characterised by markedly thickened capillary walls expanded by a discontinuous linear deposition of altered basement membrane material and C3. Ultrastructurally, large electron-dense ribbon-like deposits give rise to the preferable alternative name—dense deposit disease (Fig. 21.9).
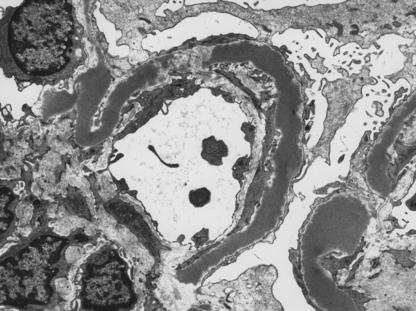
Fig. 21.9 Dense deposit disease. Electron micrograph showing the irregular ribbon-like deposits in dense deposit disease.
The lesion results from self-perpetuating activation of the alternative pathway of the complement cascade by a C3 nephritic factor (C3NeF), now termed NeFa, and explains the hypocomplementaemia.
Crescentic glomerulonephritis
Crescentic glomerulonephritis, which equates clinically to rapidly progressive glomerulonephritis (RPGN), is a manifestation of severe glomerular damage characterised by the presence of >50% cellular crescents that eventually compress the glomeruli (Fig. 21.10). There is a florid necrotising glomerulonephritis with fibrinoid necrosis and breaks in the basement membrane.
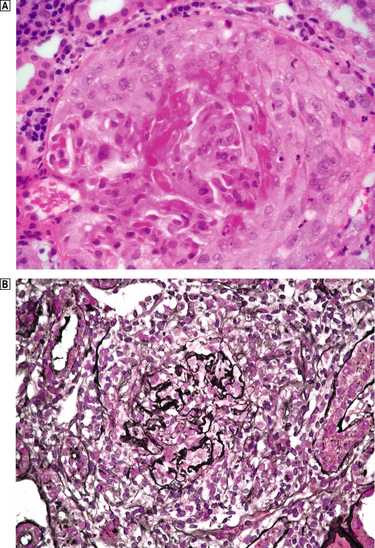
Fig. 21.10 Crescentic glomerulonephritis, in this case due to pauci-immune ANCA-associated glomerulonephritis.  The glomerulus, which is partially necrotic, is surrounded by an exuberant crescent.
The glomerulus, which is partially necrotic, is surrounded by an exuberant crescent.  Silver stains emphasise the disruption of the Bowman’s capsule and the compression of the glomerulus by the crescent.
Silver stains emphasise the disruption of the Bowman’s capsule and the compression of the glomerulus by the crescent.
Crescentic glomerulonephritis may be limited to the kidneys or be part of a systemic small vessel vasculitis. Three main categories lead to this histological appearance:
Immune complex-mediated represents a heterogeneous group in which crescentic nephritis complicates a known form of glomerulonephritis, e.g. IgA, Henoch–Schönlein purpura (HSP), lupus nephritis, MPGN, etc.
Pauci-immune glomerulonephritis is characterised by few or no immunoreactants on immunofluorescent staining. Some 85% of these patients have demonstrable antineutrophil cytoplasmic antibody (ANCA). ANCA has been shown to damage the glomerular capillaries and small renal vessels by interacting with and activating neutrophils.
Anti-GBM disease has already been discussed (p. 577). In c. 50% of cases the lesion is restricted to the kidneys. When the renal disease is accompanied by haemoptysis and pulmonary haemorrhages it is part of the pulmonary–renal (Goodpasture’s) syndrome, which accounts for the remainder. This disease is commoner in young men; the causative factor is unknown although both a recent flu-like illness and exposure to hydrocarbons feature in the clinical history in some patients.
Anti-GBM disease is the most aggressive lesion and requires vigorous immunosuppression augmented by plasma exchange (plasmapheresis) at a very early stage. Nevertheless, there is a high risk of permanent scarring of the kidney with subsequent hypertension.
Minimal change disease
Minimal change disease (MCD) is an important cause of nephrotic syndrome, particularly in children. With a light microscope the glomeruli are normal; the extensive effacement of the epithelial foot processes is evident only by electron microscopy.
Pathogenesis
This is still not clear. The association of MCD with drug sensitivities, stings, venoms and Hodgkin’s disease has suggested immune mechanisms influencing the permeability of the capillary wall.
Clinical features and prognosis
Minimal change disease affects all ages, but is much more common in children, with a peak age incidence below 5 years and with a male preponderance. In a few patients it follows an upper respiratory infection or prophylactic immunisation. Nephrotic syndrome responsive to steroid therapy is the classical presentation. The prognosis in children is good with no permanent renal damage, but in adults the outlook is variable.
Focal glomerulosclerosis
Focal segmental glomerulosclerosis (FSGS) is an important cause of nephrotic syndrome; the incidence is increasing in adults. It accounts for c. 10% of children with nephrotic syndrome. This pattern of glomerular damage results from a variety of pathogenic and aetiological factors such as in diabetic nephropathy, HIV infection, heroin abuse and reflux nephropathy. It occurs not uncommonly in the late stages of immune complex-induced lesions, e.g. IgA nephropathy.
Aetiology and pathogenesis
There is good evidence (e.g. recurrent FSGS in transplanted kidneys) for a circulating factor having an aetiological role in FSGS but this has not yet been fully characterised.
Congenital nephrotic syndrome features focal and segmental glomerulosclerosis, one cause of which has been identified in the mutation of the gene for nephrin (NPHS1). Gene aberrations for the associated proteins in the slit diaphragm are associated with familial FSGS, each family showing an individual mutation.
Histologically, sclerosis is recognised by expansion of the matrix together with obliteration of the capillary lumen, sometimes involving only part of the glomerulus (segmental). Hyalinosis of the capillary loops is present in some cases of FSGS. Vascular hyalinosis is also seen and there is interstitial fibrosis and tubular atrophy. Electron microscopy shows foot process effacement, but there are no deposits to correspond to the non-specific entrapment of IgM and C3 seen immunohistochemically.
SECONDARY GLOMERULAR DISEASES




Secondary glomerular disease implies renal damage occurring as part of a systemic condition. These systemic conditions may be:
Immune complex-mediated conditions
Immune complex-mediated systemic conditions that may involve renal damage include:
Systemic lupus erythematosus
Renal involvement occurs in about 70% of lupus patients. The lesions are varied and represent a spectrum with increasing degrees of damage. The glomeruli contain granular immune deposits typically comprising all of the immunoglobulins (IgG, IgM, IgA) with C3 and C1q. A diffuse proliferative glomerular reaction is the most frequently occurring histological picture. Membranous change occurs in c. 20% of cases and is associated with marked proteinuria or nephrotic syndrome.
Henoch–Schönlein purpura
Henoch–Schönlein purpura (HSP) is a systemic vasculitis affecting the skin, joints, intestine and kidneys and occurs most commonly in childhood before the age of 10 years. A purpuric rash typically affects the extensor aspects of the arms and legs and buttocks. This is due to leukocytoclastic angiitis in the dermal capillaries and is related to IgA deposits within them. Joint pains and abdominal pains, with or without intestinal haemorrhage, are also related to involvement of small blood vessels. Most patients with HSP have one episode which completely resolves. However, recurrences occur but are usually milder and shorter than the initial episode.
The proportion of patients with renal involvement is difficult to assess. Significant renal damage occurs in over one-third of cases, ranging from proteinuria with nephrotic syndrome, to rapidly progressive glomerulonephritis.
Infective endocarditis
Renal complications of infective endocarditis (Ch. 13) are:
The last two complications are due to immune complex deposition.
In cases with a focal and segmental glomerulonephritis the kidney shows multiple haemorrhagic foci throughout the cortex and beneath the capsule; it is one of the causes of a ‘flea-bitten’ kidney. The renal lesions subside when the bacterial source of the antigen is removed by intensive antibiotic therapy or removal of the valve.
Metabolic conditions
Metabolic conditions involving renal damage include:
Diabetes mellitus
Diabetes mellitus is associated with damage to large and small vessels throughout the body. Severe atheroma involving the renal artery may cause renal ischaemic lesions and hypertension. Involvement of the microcirculation, in addition to causing lesions in the retina, nerves and skin, significantly affects the kidneys, leading to glomerulopathy, arteriolar hyalinosis and tubulo-interstitial lesions. The combination of changes that occur in individual cases is varied and referred to collectively as diabetic nephropathy.
Diabetic glomerulopathy
Glomerular disease in diabetics causes proteinuria, which becomes heavier as the disease progresses, leading to nephrotic syndrome and chronic renal failure. The clinical importance of diabetic glomerulopathy is exemplified by the fact that diabetes is the commonest cause of end-stage renal failure in the United States, Europe and Japan. In comparison to patients with insulin-dependent diabetes (type 1), those with non-insulin-dependent diabetes (type 2) have a shorter course to end-stage renal failure and a higher mortality. Being older they have more cardiovascular disease—myocardial infarcts and congestive cardiac failure. In contrast, patients with type 1 diabetes have a higher incidence of microvascular lesions causing retinopathy and neuropathy.
Histologically, three types of glomerular lesion occur—a spectrum of increasing severity:
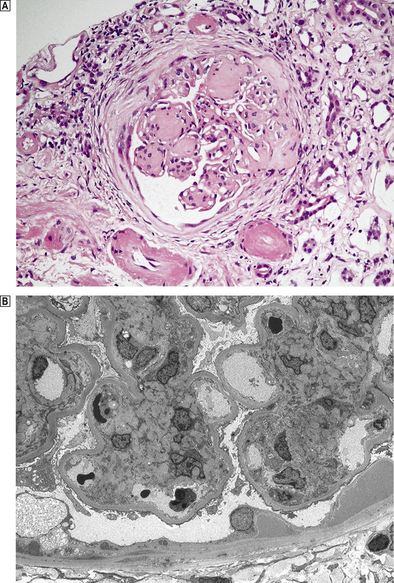
Fig. 21.11 Diabetic glomerulopathy (nodular glomerulosclerosis).  This lesion features hyaline sclerotic nodules, thickened capillary walls and hyaline arteriolar change.
This lesion features hyaline sclerotic nodules, thickened capillary walls and hyaline arteriolar change.  An electron micrograph shows an early mesangial nodule and the thickened basement membrane characteristic of this condition.
An electron micrograph shows an early mesangial nodule and the thickened basement membrane characteristic of this condition.
The glomerular lesions are accompanied by arteriolar hyalinosis affecting both the afferent and efferent arterioles. Tubular basement membrane thickening, analagous to the glomerular changes with tubular atrophy and interstitial fibrosis, completes the picture of diabetic nephropathy.
Pathogenesis of diabetic nephropathy
Hyperglycaemia has a central role in the pathogenesis of diabetic nephropathy and is prominent when control is poor. The effects of hyperglycaemia are mediated by several metabolic pathways, the most important of which are the accumulation of advanced glycation end products (AGEs) and increased glucose flux through changes in the polyol–inositol pathway. Hyperglycaemia thereby induces morphological changes and matrix protein production. The resultant imbalance between synthesis and degradation explains the accumulation of matrix material. The basement membrane becomes more permeable.
Renal papillary necrosis
Renal papillary necrosis occurs frequently in diabetics with acute pyelonephritis. The blood supply to the renal papillae via the vasa recta is tenuous, and the vasculopathy together with the effects of the inflammation result in ischaemia of the papillae, which become infarcted. The necrotic papillae may then become detached (Fig.12.12) and either cause an obstruction or are passed in the urine.
Accumulation of abnormal proteins in the basement membrane
Renal amyloidosis
Renal involvement is present in 80–90% of cases of secondary amyloidosis (Ch. 7). The affected patients have heavy proteinuria or the nephrotic syndrome. Renal involvement is the presenting feature in over 50% of patients, and leads inevitably to chronic renal failure, with extensive glomerulosclerosis within 1–2 years.
Multiple myeloma
Renal damage occurs frequently in patients with multiple myeloma. Renal failure may be the presenting feature. The most significant lesions are tubulo-interstitial, characterised by proteinaceous casts to which there is a giant cell reaction. Glomerular involvement is uncommon in myeloma, but includes amyloid infiltration in about 10% of cases and the deposition of monoclonal cryoglobulin in a smaller proportion.
Vascular damage
Diseases causing vascular damage resulting in glomerular lesions include:
Vasculitides
The vasculitides are a group of conditions in which there is acute or chronic inflammation of the blood vessel wall. There is considerable overlap between these conditions and, so that an appropriate diagnosis is made, the clinical, immunological and pathological findings are all relevant. Glomerular changes occur in those cases involving small blood vessels and are considered in detail in the section on crescentic glomerulonephritis and other ANCA-associated lesions.
Polyarteritis nodosa
Polyarteritis nodosa (Chs 13 and 25) involves medium-sized vessels throughout the body. Involvement of renal vessels involves the interlobular to arcuate arteries mainly and does not cause small vessel involvement or glomerulonephritis. When these arteries are inflamed, patients experience haematuria and loin pain due to renal infarcts. Hypertension is a common feature.
Wegener’s granulomatosis
Wegener’s granulomatosis is a rare necrotising vasculitis affecting the nose and upper respiratory tract (Ch. 14) in addition to the kidneys. It occurs more commonly in males in their fourth and fifth decades, although it can occur at any age. The clinical indicators of renal involvement vary from microscopic haematuria to rapidly progressive renal failure. If the condition remains untreated, progressive renal impairment is inevitable, but cyclophosphamide induces a remission in the majority of patients, with complete resolution of the glomerular lesions if treated early enough.
Thrombotic microangiopathies
Thrombotic microangiopathies are characterised by the deposition of thrombi within the capillaries, arterioles and small arteries of a variety of organs. There are two major conditions in this group: haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura, with considerable clinical overlap.
The extensive deposition of thrombi within the microvasculature leads a microangiopathic haemolytic anaemia. The circulating erythrocytes are deformed and fragmented (helmet cells, burr cells, schistocytes) by the strands of fibrin. The haemolytic anaemia is accompanied by a thrombocytopenia due in part to the excessive deposition of platelets, but damage to the platelets also occurs.
Haemolytic uraemic syndrome
Haemolytic uraemic syndrome (HUS) is a complex condition in which there is:
The classical or epidemic form of haemolytic uraemic syndrome is accompanied by diarrhoea, often due to verotoxin-producing Escherichia coli infections from eating contaminated meat or dairy products. Patients without diarrhoea (diarrhoea-negative) have the atypical form of haemolytic uraemic syndrome; these cases are a heterogeneous group which is further subdivided according to aetiology; for example, infections with non-verotoxin-producing organisms, secondary to systemic diseases (particularly autoimmune disorders), drugs and pregnancy.
Thrombosis occurs in the renal arteries more frequently in patients with the atypical form and consequently there is a higher mortality.
Histological features of HUS
The glomeruli contain thrombi within the capillary lumen and there is swelling and separation of the endothelial cells. Mesangial interposition is also seen with double contouring of the basement membrane. Arteriolar changes are more often seen in diarrhoea-negative cases with swelling of the endothelial cells and subendothelial space. The lumen of the vessel is compromised and fibrin and fragmented red cells are seen in the vessel walls; this is termed ‘fibrinoid necrosis’. Similar changes are seen in the interlobular arteries which may severely narrow the lumen. This results in ischaemic changes in the glomeruli supplied by the vessel and explains the poor prognosis seen in patients with diarrhoea-negative HUS.
Thrombotic thrombocytopenic purpura
The histological changes in the kidney are less marked than in haemolytic uraemic syndrome. Thrombi are present in the glomerular capillaries. The afferent arterioles characteristically feature eosinophilic granular thrombus and may become aneurysmally dilated. Cellular proliferation may be a prominent feature in some cases.
Disseminated intravascular coagulation
Disseminated intravascular coagulation (DIC) (Ch. 23) is a systemic problem involving activation of the coagulation cascade by a variety of extraneous factors.
Fibrin thrombi are identified within the glomerular capillaries, but the condition is characterised by a marked reduction in the levels of fibrinogen and factors V and VII, and is termed a consumptive coagulopathy, in contrast with the thrombotic microangiopathies where endothelial damage is the initiating factor.
RENAL DISEASE IN ASSOCIATION WITH SPECIFIC INFECTIONS
Viral infections involving the kidney are becoming increasingly important. They involve the glomeruli, tubules and interstitium by a variety of mechanisms.
Hepatitis B virus (HBV) is associated with several renal diseases. It is important to diagnose these forms of glomerulonephritis since the usual treatment for idiopathic forms is not appropriate and may be detrimental in HBV-induced disease.The renal conditions include:
Hepatitis C virus (HCV) is associated with mixed cryoglobulinaemia, membranoproliferative glomerulonephritis and membranous nephropathy with or without cryoglobulinaemia. HCV-containing immune complexes are identified in the capillary walls and mesangium of the glomeruli. The recommended treatment is with interferon-alpha and ribaflavin, although the later is not tolerated when there is renal impairment. Rituximab is used in patients who do not respond or for whom other treatments are inappropriate.
HIV-associated nephropathy occurs in patients before the development of AIDS and is therefore distinct from any infective aspects of the latter disease. Clinically, in untreated individuals, there is an abrupt onset of heavy non-selective proteinuria and progressive deterioration in renal function to renal failure within months. Histologically, all of the major compartments of the kidney are involved:
Polyomavirus-induced nephropathy is endemic and causes disease in immunocompromised individuals. The two polyoma viruses known (BK and JC) share 70% of their sequence homology with the SV40 virus. They have been implicated in a variety of conditions including Guillain–Barré syndrome, hepatitis and SLE. BK virus displays tropism for the urogenital epithelium and causes interstitial nephritis in HIV-infected patients and renal transplants. It is an important cause of graft failure.
RENAL TRANSPLANTATION
Patients in chronic renal failure, who in the past would have died, are now effectively maintained on either peritoneal dialysis or haemodialysis, both of which place restrictions on the patient’s lifestyle. For young and otherwise fit patients with domestic and occupational responsibilities, renal transplantation offers freedom from the restrictions of regular dialysis and has transformed the quality of their lives.
Cadaveric kidneys are most frequently used, but because suitable donors are scarce there is increasing use of living donors, both related and unrelated. The long-term survival of the grafts from unrelated donors, e.g. spouse or friend, is almost as good as from parent or sibling. Graft survival is affected by the source of the kidney, histocompatibility match, the number of episodes of acute rejection, delayed graft function, pre-sensitisation, recipient race (poor in blacks) and age of the recipient.
Changes resulting from procedural events
Events during the process of renal transplantation can lead to delayed graft function. Preservation injury results from renal anoxia during harvesting or transport. Perfusion injury occurs as a result of the graft being perfused with storage solution which is too cold during preparation; this causes endothelial damage and consequent microthrombi form in the glomerular capillaries and small arteries.
Allograft rejection
Two distinct types of graft rejection can damage the kidney during the first days or weeks after transplantation. Both are immunologically mediated and involve different aspects of the immune system: acute cellular rejection involves the cell-mediated system and features a T-cell response; acute antibody-mediated rejection involves activation of the complement cascade.
Chronic allograft rejection—seen after some years and responsible for long-term graft loss—involves both mechanisms.
Graft rejection must be distinguished from graft ischaemia (e.g. due to stenosed or occluded vascular anastomosis) and from drug-induced pathology (e.g. due to immunosuppressive agents such as ciclosporin and tacrolimus).
Acute cellular rejection (ACR)
A gradual rise in the serum creatinine, weight gain, fever and tenderness over the grafted kidney alert to acute cellular rejection, the most common form of rejection, usually in the first 6 weeks. The interstitium, tubules, arteries and glomeruli are all affected. A brisk interstitial infiltrate of activated lymphoblasts and T-cells features most frequently in ACR (Fig. 21.13). Damage to arterial and arteriolar endothelium occurs in more severe cases. In most cases, glomerular damage is minimal and limited to occasional segmental mononuclear cells. The T-cell infiltrate is thought to be promoted by a concurrent cytomegalovirus infection. Acute cellular rejection usually responds to pulses of steroids, ciclosporin or OKT3 treatment.
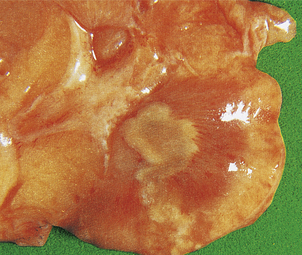
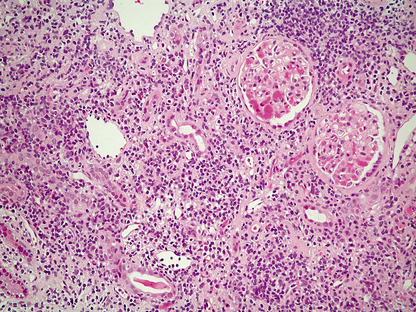
Acute humoral rejection (AHR) or acute antibody-mediated rejection
The clinical features of AHR are more dramatic and acute than with ACR. Acute and severe graft dysfunction due to AHR may occur at any time after transplantation. AHR is less responsive to therapy and carries a worse prognosis for the kidney than ACR. Histological changes range from acute tubular injury with no cellular infiltrate, through infiltration of the peritubular capillaries by neutrophils, to frank necrosis of the arteries. Complement activation is an integral part of the immunological response in AHR.
Presensitisation (e.g. previous transplantation) increases the risk of AHR. Antibodies to donor-specific human leukocyte class I antigens are responsible for AHR due to either undetected presensitisation or an occult antigen.
Hyperacute rejection
Hyperacute rejection is due to preformed complement-fixing antibodies in the blood stream of the recipient, usually to human leukocyte or ABO antigens. The immune damage is directed at the endothelial cells of the graft, and the speed of onset is related to the antibody concentration. In some cases the reaction is immediate, and is apparent to the surgeon on establishing a flow of blood through the graft, when the kidney becomes flaccid, cyanosed and mottled, suggesting intrarenal vasoconstriction. This compares with an onset at 1–2 days when the levels are low. Thrombi form in arteries and glomeruli accompanied by interstitial haemorrhages. Cortical infarcts occur as a result of vascular thrombosis.
Chronic rejection
Chronic rejection, an important cause of graft failure months or years after transplantation, is heralded by an insidious increase in creatinine, often associated with proteinuria and hypertension. Vascular changes dominate the histology with marked fibrous intimal thickening of arteries by myointimal cells and a variable T-cell infiltrate and foamy macrophages. The glomerular changes are less impressive with mild mesangial thickening. Ultrastructurally, however, there is foot process effacement and loss of endothelial fenestrae. The basement membrane of the peritubular capillaries is reduplicated. There is progressive interstitial fibrosis and tubular atrophy.
Banff classification
Transplant rejection is classified according to two working schemes, the first devised internationally at Banff, the second the Collaborative Clinical Trials in Transplantation. The two are congruent and seek to grade each histological aspect of rejection. Adoption of these schemes aims to improve the consistency of reporting between centres and thus enable more meaningful conclusions on a larger group of patients, particularly in the context of multicentre therapeutic trials.
DISEASES AFFECTING BLOOD VESSELS
In addition to the conditions mentioned above, the renal vasculature may be damaged in:
Renal infarction
Two mechanisms of infarction are recognised:
Embolic infarction
Most renal infarcts result from embolisation of:
Many renal infarcts are clinically silent, but some result in haematuria and loin pain.
Renal infarcts are pale or white, and have a characteristic wedge shape with the apex directed towards the hilum.
Diffuse cortical necrosis
Diffuse cortical necrosis is a rare condition complicating pregnancy or trauma associated with severe haemorrhage or severe sepsis. Profound hypotension occurs in these situations, but there is considerable controversy relating to the pathogenesis of this condition. Vasoconstriction is important because infarction can be avoided by the use of angiotensin antagonists.
Diffuse cortical necrosis is a cause of acute anuric renal failure. The prognosis is poor when the infarction is generalised, but is less ominous when the infarction is focal.
Macroscopically, the appearance is striking: the external surface bears irregular yellowish areas with intervening congestion and haemorrhage. The cut surface shows the infarction to be confined to the cortex.
RENAL DISEASE IN PREGNANCY
The kidneys undergo morphological and functional changes during pregnancy. Some of these changes affect the renal response to disease and influence the interpretation of function tests. Whatever the cause, renal impairment at the beginning of pregnancy has important implications; the risks to the mother and fetus are significant. The risk of deteriorating renal function and hypertension increases and fetal morbidity and mortality rise. Pregnancy is likely to exacerbate active and proliferative systemic lupus. Maternal diabetes is associated with prematurity and neonatal complications when there is established nephropathy and renal impairment. The risk of pre-eclampsia is also increased in these patients. In general, however, there are no long-term adverse effects of pregnancy on diabetic nephropathy.
Infection
Infection is the most frequent urinary tract abnormality in pregnant women and is usually detected on routine antenatal urine testing. Asymptomatic bacteriuria occurs in up to 10% of pregnant women, and such patients are at risk of acute pyelonephritis. Prompt diagnosis and early treatment are essential. Renal abnormalities such as the coarse polar scarring of vesico-ureteric reflux are detected radiologically in a high proportion of these patients. In addition, physiological changes occur in the smooth muscle cells of the lower urinary tract in pregnancy, with slower ureteric peristaltic activity and pelvi-ureteric dilatation. This leads to stasis which, combined with infection, particularly with urea-splitting organisms, predisposes to stone formation; existing stones may enlarge considerably during pregnancy.
Hypertension
Hypertension is an important complication of pregnancy. The patient may have latent essential hypertension or concurrent renal disease. Alternatively, pre-eclampsia or eclampsia may be present. Pre-eclampsia is characterised by hypertension, proteinuria and oedema; eclampsia supervenes when fits occur. In severe cases the glomeruli are large and relatively bloodless due to marked swelling of the endothelial and mesangial cells. Arteries and arterioles display endothelial swelling and myointimal proliferation. The vascular and glomerular lesions are reversible when the hypertension has been corrected. However, a thrombotic microangiopathy may be superimposed during the course of pre-eclampsia, and persistent hypertension may ensue when fibrinoid necrosis of the arterioles has occurred.
TUBULO-INTERSTITIAL DISORDERS
In tubulo-interstitial conditions there is damage to the tubular epithelial cells and the interstitium. These disorders account for a significant proportion of patients presenting with impaired renal function.
Infections of the kidney affect the tubules and the interstitium, and may present as acute renal failure if there are complications such as tubular or papillary necrosis. These are discussed separately because of their clinical importance.
Acute tubular epithelial cell damage causes the condition known as acute tubular necrosis.
Acute tubular necrosis



Acute tubular necrosis (ATN) is a very important cause of acute renal failure; patients often present with extreme oliguria (less than 100 ml of urine each 24 h). The importance of ATN is that it is fully recoverable if the patient is given adequate supportive fluid and electrolyte therapy. Following the initial oliguria, there is a later diuretic phase due to the loss of urinary concentration. In the oliguric phase, hyperkalaemia with the risk of cardiac arrhythmias presents a serious threat to life. This situation contrasts with hypokalaemia which can occur in the early diuretic phase.
The histological features range from sublethal injury, which is often difficult to identify morphologically, to necrosis of the epithelial cells. In most cases a severe metabolic disturbance is the essential change and the term ‘necrosis’ is not entirely appropriate. The oliguria comes about in these cases because the cells of the proximal tubules and loops of Henle are rendered unable to reabsorb sodium chloride. Thus the luminal fluid at the macula densa contains too much chloride and the tubuloglomerular feedback system is activated, reducing the GFR.
The principal causes of acute tubular necrosis are:
Ischaemic ATN
Ischaemic ATN follows a variety of clinical situations, such as trauma, burns and infections in which the patient becomes shocked. Profound hypotension is usually responsible for the hypoperfusion of the peritubular circulation. The kidneys are pale and swollen. Histology reveals patchy epithelial cell injury along the entire length of the tubules; the cells are flattened and vacuolated, making distinction between proximal and distal tubules very difficult histologically. Inflammatory cells pack the vasa recta in response to the necrotic cells; the interstitium is oedematous. Casts occur frequently in the distal tubules and collecting ducts; they are composed of cellular debris and protein, including Tamm–Horsfall protein. In the case of ATN resulting from a crush injury, myoglobin is present in the casts. Following a mismatched blood transfusion, haemoglobin would be present.
Toxic ATN
Toxic ATN results from a wide variety of substances, the kidney being vulnerable because of the high metabolic activity of the epithelial cells and the high perfusion rate. The causes are:
The kidneys are swollen and red. Histologically, there is often marked vacuolation of the tubular epithelial cytoplasm. The damage is characteristically restricted to the proximal tubular cells, those of the distal tubule being spared. This situation contrasts with the picture in ischaemic ATN in which the tubular cells along the entire length of the tubule are affected. Recovery is indicated in biopsies by the presence of mitotic figures within the flattened cells.
Interstitial nephritis
Interstitial nephritis is a heterogeneous group of conditions involving the interstitium that have a profound effect on renal function. The interstitium contains a complex network of capillaries, derived from the efferent arterioles of the glomeruli. In addition, the blood supply to the medulla emanates from the vasa recta arising from the efferent arterioles of the juxtamedullary glomeruli. Thus, damage to the glomerular vessels will cause tubular dysfunction. Therefore, damage to the interstitium, by either inflammation or fibrosis, will affect the peritubular capillary network and cause profound disturbance in renal function.
Acute interstitial nephritis
Acute interstitial nephritis is an important cause of acute renal failure which is often oliguric; c. 60% require dialysis initially. Acute interstitial nephritis is a complication of severe systemic bacterial infections. Most cases of acute renal failure due to acute interstitial nephritis are due to drugs, most commonly penicillins, sulphonamides and non-steroidal anti-inflammatory drugs (NSAIDs). The kidneys are large and swollen due to oedema. The interstitium is oedematous and infiltrated by lymphocytes, macrophages and eosinophils with infiltration of the tubules (tubulitis). This explains the urinary finding of white cell casts in these cases. The renal changes will resolve completely if the drug is discontinued, often with little lasting damage. The reaction is idiosyncratic and not dose-related, but will recur if the patient is re-exposed to the offending drug.
Vesico-ureteric reflux
Vesico-ureteric reflux is the commonest cause of chronic interstitial nephritis (Fig. 21.14). Organisms enter the kidney from the bladder. The primary abnormality is the angle at which the terminal ureteric segment traverses the bladder wall. Normally the ureter is at an acute angle to the mucosal surface, so that contraction of the bladder wall during micturition closes the orifice. In patients with reflux, the terminal portion is short and oriented at approximately 90˚ to the mucosal surface; contraction of the bladder holds the orifice open and facilitates reflux of urine. Reflux into the kidney itself occurs through compound papillae (Fig. 21.14).
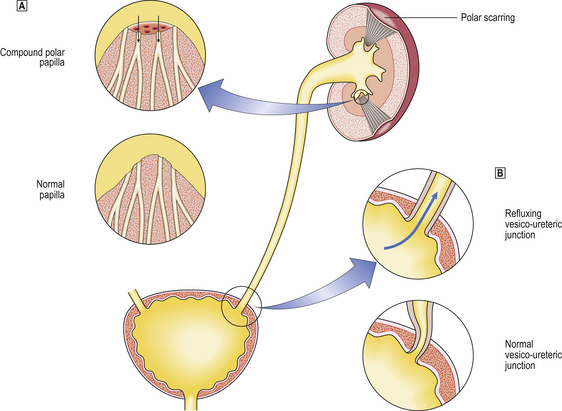
Fig. 21.14 Chronic interstitial nephritis due to urinary reflux. Urinary reflux can occur due to either obstruction of urinary flow or, as shown in this diagram, congenital anomalies.  Compound polar papillae. Contrasting with the normal convex papilla, compound papillae have concave tips without valve-like openings of the collecting ducts which enable intraparenchymal reflux.
Compound polar papillae. Contrasting with the normal convex papilla, compound papillae have concave tips without valve-like openings of the collecting ducts which enable intraparenchymal reflux.  Refluxing vesico-ureteric junction. Normally, the terminal ureter runs through the bladder wall beneath the surface and enters the lumen at an angle forming a valve; refluxing junctions result from the absence of the valve-like structure.
Refluxing vesico-ureteric junction. Normally, the terminal ureter runs through the bladder wall beneath the surface and enters the lumen at an angle forming a valve; refluxing junctions result from the absence of the valve-like structure.
The appearance of reflux nephropathy is characteristic. Deep irregular scars occur towards the poles of the kidney. Involvement may be unilateral or, if bilateral, is characteristically asymmetrical. Microscopically, there is interstitial fibrosis with atrophic and dilated tubules containing eosinophilic casts (‘thyroidisation’ of the kidney, so called because of the histological resemblance to thyroid).
Analgesic nephropathy
Analgesic nephropathy is an adverse reaction to analgesics which is now less frequent since combined aspirin and phenacetin preparations are no longer available. The phenacetin and its toxic metabolites concentrate in the medulla. The aspirin is thought to induce medullary ischaemia by inhibiting the synthesis of vasodilatory prostaglandins. The effect is on the medullary capillaries and vasa recta which develop thickening of the basement membranes. The toxic and ischaemic effects are synergistic, leading eventually to renal papillary necrosis. The prognosis is good if the analgesic abuse is terminated at an early stage; however, if continued, progressive fibrosis ensues.
Transitional cell carcinomas of the renal pelvis and ureter occur more frequently in patients with analgesic nephropathy.
Lesions associated with metabolic disorders
A variety of metabolic diseases and disturbances affect the tubules and interstitium. These include:
Hypokalaemic nephropathy
Persistently low plasma levels of potassium occur in:
Hypokalaemia causes coarse vacuolation of the tubular epithelial cells principally in the proximal tubules, but in severe states those of the distal tubules are also involved; the medullary tubules are spared.
Urate (gouty) nephropathy
Renal damage due to elevated levels of uric acid in the blood occurs in gout (Ch. 7). Additionally, patients with chronic renal damage due to pyelonephritis or glomerulonephritis have impaired filtration and reduced tubular secretion of uric acid; this leads to retention of uric acid and its subsequent deposition in the kidney. Uric acid crystallises in an acid environment such as that found in the distal tubules, collecting ducts and interstitium of the papillae.
Acute urate nephropathy
Acute urate nephropathy presents as acute renal failure. It occurs principally in patients with myeloproliferative or solid tumours and is precipitated by chemotherapy, when extensive breakdown of cells releases vast quantities of nucleic acids. The cut surface of affected kidneys displays yellow streaks within the medulla due to precipitation of urate crystals filling the tubular lumina; this precipitation causes obstruction and tubular dilatation.
Chronic urate nephropathy
Chronic urate nephropathy is more insidious and occurs in patients with persistently elevated uric acid levels, as in gout. Urate crystals precipitate in the tubular lumina causing chronic obstruction and tubular damage with a granulomatous tubulo-interstitial nephritis. The kidneys become reduced in size and the cortex becomes atrophic and thinned.
Urate stones may occur in both acute and chronic nephropathy, and there is an increased incidence of pyelonephritis.
Hypercalcaemic nephropathy (nephrocalcinosis)
Calcium deposition in the kidneys occurs when there is hypercalcaemia: this is ‘metastatic’ calcification (Ch. 7). The onset of renal symptoms is insidious; the tubular disturbance results in an inability to concentrate urine, with consequent polyuria often causing the patient to complain of nocturia. The kidney is often scarred and focally calcified. The cut surface of the kidneys reveals stones within the pelvicalyceal system, and linear white streaks and flecks. There is interstitial fibrosis, a non-specific inflammatory infiltrate and tubular atrophy in relation to the calcification.
Oxalate nephropathy
Calcium oxalate deposition occurs systemically in the tissues when blood levels are high. Increased urinary excretion of oxalate also occurs and the kidneys may be damaged. Hyperoxalaemia and the resulting hyperoxaluria are either primary or secondary. The primary form is due to deficiencies of hepatic enzymes, which are concerned with the decarboxylation of glyoxylate, which accumulates and is oxidised by an alternative pathway to oxalate. Hepatic together with renal transplantation are required to replace the deficient hepatic enzyme. Oxalate accumulates in many tissues (e.g. myocardium), causing systemic problems after the onset of renal failure, and transplantation before that stage is recommended. Secondary hyperoxaluria is seen in poisoning with ethylene glycol (antifreeze) or the anaesthetic agent methoxyflurane, and in pyridoxine (vitamin B6) deficiency. Oxalate deposition is also seen in the kidneys in a variety of chronic renal disorders.
In advanced cases, the kidneys are small, granular and scarred, the thinned cortex reflecting the fibrosis and tubular disruption that occurs with the deposition of oxalate. Stones are often present in the pelvis and calyceal system.
Lesions due to physical agents
Obstructive uropathy
Obstructive uropathy is an important cause of interstitial nephritis; the causes include:
Clinical features
The signs, symptoms and prognosis depend on the level of the obstruction. Thus, a renal calculus or a fragment of sloughed papilla will cause renal colic, whereas obstruction due to carcinoma of the bladder or benign prostatic hyperplasia will be accompanied by bladder symptoms. Acute obstruction in the lower urinary tract will result in anuria and pain, and if not relieved is incompatible with survival.
Pathogenesis
The pelvis and calyceal system become dilated due to back pressure; the dilatation is mild in cases of acute obstruction. The peristaltic activity of the ureters is increased in obstruction; this raises the intrapelvic pressure, which in turn is transmitted into the renal parenchyma. Initially, the filtrate formed is reabsorbed through lymphatic and vascular channels. The continued rapid rise in pressure, however, reduces glomerular and medullary blood flow and eventually impairs glomerular filtration.
Gross dilatation occurs as a result of prolonged back pressure. The kidney becomes a dilated sac-like structure: this is hydronephrosis (Fig. 21.15).
Pyelonephritis




Pyelonephritis is an infection in the kidney that may arise by haematogenous or retrograde ureteric routes. Due to their rich blood supply, the kidneys are often involved in severe systemic infections by direct spread of organisms through septicaemia. The commonest infecting organisms are bacteria. Urinary tract infections are common within the community, second only to upper respiratory infections. However, not all urinary tract infections are associated with pyelonephritis; organisms can gain access to the kidney only if there is vesico-ureteric reflux.
The incidence of pyelonephritis parallels that of obstructive uropathy. In infancy, boys are mainly affected because of anatomical abnormalities. From puberty to middle age, females show the highest incidence, related to urethral trauma and pregnancy. After 40 years, prostatic disease provides an obstructive aetiology in ageing men. Other factors include instrumentation (e.g. catheterisation, cystoscopy) and diabetes mellitus.
The clinical distinction between acute and chronic pyelonephritis is quite clear.
Acute pyelonephritis
Acute pyelonephritis, due to infection of the kidney by pyogenic organisms, presents with malaise and fever, and pain and tenderness in the loins. Dysuria and urgency of micturition indicate an associated infection in the lower urinary tract. Finding pus cells in the urine (pyuria) is helpful, but white cell casts provide unequivocal evidence of pyelonephritis. Urine culture in suspected cases is imperative, but bacteriuria is regarded as significant only when in excess of 105 culture-forming units/ml; this result eliminates cases of extraneous bacterial contamination.
Pathogenesis
The pathogenesis of acute pyelonephritis is either:
Haematogenous spread can occur in a patient with infective endocarditis or bacteraemia from other sources; the spectrum of organisms can be wide, including bacteria, fungi, rickettsia and viruses. Previous renal damage or structural abnormality predisposes to organisms localising in the kidney.
More commonly, pyelonephritis results from organisms gaining access from the lower urinary tract, known as an ascending infection, in association with reflux of urine. In these cases the infecting organisms are Gram-negative bacilli (e.g. E. coli, Proteus spp. and Enterobacter) from the patient’s faecal flora. This occurs frequently in young women; predisposing factors include the short urethra, urethral trauma during sexual intercourse, and pregnancy. Instrumentation of the urinary tract in both sexes increases both the incidence and the variety of infecting organisms.
Morphology
There are either abscesses throughout the cortex and medulla or wedge-shaped confluent areas of suppuration. Minute abscesses are randomly distributed when the infection is blood-borne, but tend to be located at the upper and lower poles when associated with urinary reflux. A lower urinary tract infection may be associated with inflammation of the pelvic and calyceal mucosa, and pus may be present in the pelvis.
Histology reveals intratubular polymorphs together with interstitial oedema and inflammation (Fig. 21.16). These white cell ‘granular’ casts pass into the bladder and are evident on microscopy of the urine. With healing, fibrosis occurs in the interstitium and the inflammatory infiltrate becomes dominated by lymphocytes and plasma cells.
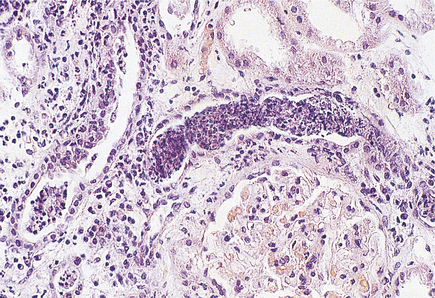
Complications
Three important complications may develop in acute pyelonephritis:
Chronic pyelonephritis
Chronic pyelonephritis is associated with obstruction developing during adulthood, the site of which determines whether or not one or both kidneys are involved. In contrast to reflux nephropathy, the obstruction leads to dilatation of the renal pelvis and calyceal system with widespread atrophy of the overlying parenchyma. The papillae are stretched and distorted, enabling intraparenchymal reflux to occur. Histologically, atrophic changes with ‘thyroidisation’ are seen together with inflammatory cells. Obstruction is the predominant aetiological factor together with persistent bacteria and an altered host response.
Renal tuberculosis
The kidneys can be affected by tuberculosis as part of generalised miliary spread from an active tuberculous lesion elsewhere (usually in the lungs); the kidneys become dotted with numerous minute white granulomas.
Solitary tuberculous lesions occur in the kidneys of adults. These may or may not be associated with other active tuberculous lesions elsewhere; they may represent reactivation of a dormant lesion. The kidney contains an irregular white mass filled with caseous material. This arises within the renal parenchyma but may eventually rupture into the calyceal system, leaving an open, ragged cavity and enabling tubercle bacilli to seed along the ureter and into the bladder. Severe and longstanding tuberculosis may produce a tuberculous pyelonephrosis with complete destruction of the kidney. ‘Sterile’ pyuria is an important feature in renal tuberculosis and should stimulate an active search for the acid–alcohol-fast bacilli in the urine.
Commonly confused conditions and entities relating to renal and urinary tract pathology
| Commonly confused | Distinction and explanation |
|---|---|
| Glomerulonephritis and glomerulopathy | Glomerulonephritis is strictly inflammation (hence ‘-itis’) of glomeruli. However, the term is also used for some forms of glomerular injury (e.g. membranous glomerulonephritis) lacking inflammatory features; glomerulopathy is a better term, and is becoming more widely used. |
| Creatinine and creatine | Creatinine is relevant to renal pathology; its blood concentration is used to assess glomerular function. Creatinine is the anhydride of creatine (N-methyl-guanidinoacetic acid); when phosphorylated, it is an important storage form of high-energy phosphate. |
| Membranous and proliferative | Both terms are used to describe the histology of glomerulonephritis (GN). In membranous GN the dominant feature is basement membrane thickening. In proliferative GN the dominant feature is glomerular hypercellularity. |
| Focal, segmental, global and diffuse | These terms are used to describe, first, whether most (diffuse) or some (focal) glomeruli are abnormal, and, second, whether the abnormality affects the entire glomerulus (global) or only a segment (segmental). Recognition of these patterns is useful in diagnosis and prognosis. |
| Haematuria and haemoglobinuria | Haematuria is the presence of red blood cells in the urine, indicating severe glomerular injury (in which case red cell casts, formed in the tubules, may be found on urine microscopy) or a tumour or trauma in the urinary tract. Haemoglobinuria is due to intravascular haemolysis and can occur in the absence of urinary tract disease. |
Avner E.D., Sweeney W.E.. Renal cystic disease: new insights for the clinician. Pediatric Clinics of North America. 2006;53:889-909.
Blumenthal I.. Vesicoureteric reflux and urinary tract infection in children. Postgraduate Medical Journal. 2006;82:31-35.
Eaton D.C., Pooler J.. Vander’s renal physiology, 6th edn. Columbus, Ohio: McGraw-Hill Medical; 2004.
Pannu N., Klarenbach S., Wiebe N., et al. Renal replacement therapy in patients with acute renal failure: a systematic review. Journal of the American Medical Association. 2008;299:793-805.
Rennke H.G., Denker B.M., Burton D.R.. Renal pathophysiology, 2nd edn.. Philadelphia: Lippincott Williams & Wilkins, 2006.
Steddon S., Ashman N., Cunningham J., Chesser A.. Oxford handbook of nephrology and hypertension. Oxford: Oxford University Press; 2006.




