Chapter 3 Discovery and development of drugs
• Preclinical drug development. Discovery of new drugs in the laboratory is an exercise in prediction. Selecting the target for a drug is probably the key decision.
• Techniques of discovery. Several approaches, including using small molecules, large proteins and nucleic-based approaches, broaden what we think of as a medicine.
• Studies in animals. Some are required by regulation (for safety), others give insight into the effect of the drug in the whole body, but none replaces the need for clinical testing.
• Experimental medicine. Getting the medicine into the clinic to test its properties and its effects on biological systems is a key step.
• Prediction. Failures of prediction occur and a drug may be abandoned at any stage, including after marketing. New drug development is a colossally expensive and commercially driven activity.
Making a new medicine
1. Selecting the molecular target you want the drug to act on to produce the desired effect.
2. Choosing the right chemical as the drug candidate. All the promise and all the faults become fixed at this point.
3. Designing the right clinical experiment to show that the medicine will really do what you want it to do.
Medicinal therapeutics rests on the two great supporting pillars of pharmacology:
1. Selectivity – the desired effect alone is obtained: ‘We must learn to aim, learn to aim with chemical substances’ (Paul Ehrlich).1
2. Dose – ‘The dose alone decides that something is no poison’ (Paracelsus).2
The huge increase in understanding of molecular signalling – both between cells and within cells – has opened many new opportunities to develop medicines that can target discrete steps in the body’s elaborate pathways of chemical reactions.3 The challenge, of course, is to do so in a way that produces benefit without harm. The more fundamental the pathway targeted the more likely there is to be a big effect, whether beneficial, harmful or both. No benefit comes without some risk.
1. More potential drugs and therapeutic targets were identified that could be experimentally validated in animals and humans. This ‘production line’ approach also led to a loss of integration of the established specialities (chemistry, biochemistry, pharmacology) and to an overall lack of understanding of how physiological and pathophysiological processes contribute to the interaction of drug and disease.
2. Theoretically, new drugs could be targeted at selected groups of patients based on their genetic make-up. This concept of ‘the right medicine for the right patient’ is the basis of pharmacogenetics (see p. 106), the genetically determined variability in drug response.
Pharmacogenetics has gained momentum from recent advances in molecular genetics and genome sequencing, due to:
• rapid screening for gene variants
• knowledge of the genetic sequences of target genes such as those coding for enzymes, ion channels, and other receptor types involved in drug response.
• The identification of subgroups of patients with a disease or syndrome based on their genotype. The most extreme of obvious examples of this are diseases caused by single gene defects. The ability accurately to subclassify based on common genetic variation is less clear.
• Targeting of specific drugs for patients with specific gene variants. This is most advanced in the field of cancer (usually targeting somatic changes in cancers) and increasingly in the field of the pharmacogenetics of safety (i.e. unwanted drug effects).
Consequences of these expectations include: smaller clinical trial programmes with well-defined patient groups (based on phenotypic and genotypic characterisation), better understanding of the pharmacokinetics and dynamics according to genetic variation, and improved monitoring of adverse events after marketing.
New drug development proceeds thus:
• Idea or hypothesis. ‘This protein causes this disease/these effects which I could stop by affecting protein function.’
• Design and synthesis of substances. ‘This molecule produces the wanted effect on protein function and has the physico-chemical characteristics that make it a potential medicine.’
• Studies on tissues and whole animal (preclinical studies). ‘When I test it in appropriate models it does what I expect and that allows me to believe it would do the same in humans.’
• Studies in humans (clinical studies) (see Ch. 4). ‘Initially I want to know whether the molecule has drug-like properties in terms of its kinetics. I then want to know that it does what I need it to do in terms of the effect on disease.’
• Granting of an official licence to make therapeutic claims and to sell (see Ch. 6). ‘Is my medicine better than than placebo? Does it do more than existing medicines and is it well tolerated.’
• Post-licensing (marketing) studies of safety and comparisons with other medicines. ‘Now many thousands of patients have taken it, am I still sure that it is safe.’
The (critical) phase of progress from the laboratory to humans is often termed translational science or experimental medicine. It was defined as ‘the application of biomedical research (pre-clinical and clinical), conducted to support drug development, which aids in the identification of the appropriate patient for treatment (patient selection), the correct dose and schedule to be tested in the clinic (dosing regimen) and the best disease in which to test a potential agent’.4
It will be obvious from the account that follows that drug development is an extremely arduous, highly technical and enormously expensive operation. Successful developments (1% of compounds that proceed to full test eventually become licensed medicines) must carry the cost of the failures (99%).5 It is also obvious that such programmes are likely to be carried to completion only when the organisations and the individuals within them are motivated overall by the challenge to succeed and to serve society, as well as to make money. A previous edition of this chapter included a quote from a paper I wrote from my time in academia and I leave it here:
Let us get one thing straight: the drug industry works within a system that demands it makes a profit to satisfy shareholders. Indeed, it has a fiduciary6 duty to do so. The best way to make a lot of money is to invent a drug that produces a dramatically beneficial clinical effect, is far more effective than existing options, and has few unwanted effects. Unfortunately most drugs fall short of this ideal.7
Techniques of discovery
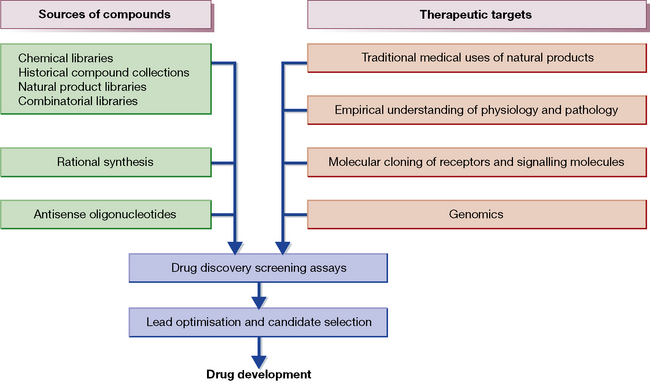
The newer technologies, the impact of which has yet to be fully felt, include the following:
Older approaches
to the discovery of new medicines that continue in use include:
• Animal models of human disease or an aspect of it of varying relevance to humans. It is always the case that the animal model is never a true model of human disease and can only model parts of the disease process. The trend in industry is to move fast to the clinic and not to rely too heavily on animal models.
• Natural products: modern technology for screening has revived interest and intensified the search. Multinational pharmaceutical companies now scour the world for leads from microorganisms (in soil or sewage or even from insects entombed in amber 40 million years ago), fungi, plants and animals. Developing countries in the tropics (with their luxuriant natural resources) are prominent targets in this search and have justly complained of exploitation (‘gene robbery’). Many now require formal profit-sharing agreements to allow such searches. Natural products have been particularly successful for finding antibiotics as evolution has done over a much longer time period what medicinal chemists struggle to do. The problem with natural product drug discovery is often that identifying the precise active ingredient is hard and the molecules usually cannot easily be synthesised.
• Traditional medicine, which is being studied for possible leads to usefully active compounds. This is particularly true in China where traditional medicines are being re-evaluated for effects. The most notable example in recent years is the rediscovery of artemisinin for malaria treatment.
• Modifications of the structures of known drugs: these are obviously likely to produce more agents with similar basic properties, but may deliver worthwhile improvements. It is in this area that the ‘me too’ and ‘me again’ drugs are developed. However, such approaches are very unlikely to be commercially successful in a world that rightly demands true advances in patient care as a prerequisite for making profits and charging high costs to cover the price of innovative discovery.
• Random screening of synthesised and natural products.
• New uses for drugs already in general use as a result of intelligent observation and serendipity,8 or advancing knowledge of molecular mechanisms, e.g. aspirin for antithrombotic effect. Once a good drug is on the market new uses are often discovered. A recent example is rituximab, a monocloncal antibody that kills B cells. Initially developed for the treatment of B cell lymphoma it was found to be effective for suppressing B cell autoimmunity in diseases such as rheumatoid arthritis.
Studies in animals9
Generally, the following are undertaken:
Toxicology
– to reveal physiological and/or histopathological changes induced by the drug, and to determine how these changes relate to dose.10 These involve:
• Acute toxicity: single-dose studies that allow qualitative and quantitative assessment of toxic reactions.
• Chronic and subchronic toxicity: repeat-dose studies to characterise the toxicological profile of a drug following repeated administration. This includes the identification of potential target organs and exposure–response relationships, and may include the potential for reversibility of effects.
Generally, it is desirable that tests be performed in two relevant species, based on the pharmacokinetic profile, one a rodent and one a non-rodent. The duration of the studies depends on the conditions of clinical use and is defined by Regulatory Agencies (Tables 3.1 and 3.2).
Table 3.1 Single and repeated dose toxicity requirements to support studies in healthy normal volunteers (Phase 1) and in patients (Phase 2) in the European Union (EU), and Phases 1, 2 and 3 in the USA and Japan1
| Minimum duration of repeated-dose toxicity studies | ||
|---|---|---|
| Duration of clinical trial | Rodents | Non-rodents |
| Single dose | 2 weeks2 | 2 weeks |
| Up to 2 weeks | 2 weeks | 2 weeks |
| Up to 1 month | 1 month | 1 month |
| Up to 3 months | 3 months | 3 months |
| Up to 6 months | 6 months | 6 months |
| > 6 months | 6 months | Chronic3 |
1 In Japan, if there are no Phase 2 clinical trials of equivalent duration to the planned Phase 3 trials, conduct of longer-duration toxicity studies is recommended as given in Table 3.2.
2 In the USA, specially designed single-dose studies with extended examinations can support single-dose clinical studies.
3 Regulatory authorities may request a 12-month study or accept a 6-month study, determined on a case-by-case basis.
Table 3.2 Repeated-dose toxicity requirements to support Phase 3 studies in the EU, and marketing in all regions1
| Minimum duration of repeated-dose toxicity studies | ||
|---|---|---|
| Duration of clinical trial | Rodents | Non-rodents |
| Up to 2 weeks | 1 month | 1 month |
| Up to 1 month | 3 months | 3 months |
| Up to 3 months | 6 months | 3 months |
| > 3 months | 6 months | Chronic2 |
1 When a chronic non-rodent study is recommended if clinical use more than 1 month.
2 Regulatory authorities may request a 12-month study or accept a 6-month study, determined on a case-by-case basis.
Ethics and legislation
Controversy surrounding the use of animals in scientific research is not new. The renowned Islamic physician Avicenna (980–1037) was aware of the issues for he held that ‘the experimentation must be done with the human body, for testing a drug on a lion or a horse might not prove anything about its effect on man’.11 Leonardo da Vinci (1452–1519) predicted that one day experimentation on animals would be judged a crime, but Descartes12 asserted that ‘Animals do not speak, therefore they do not think, therefore they do not feel’. Later, Jeremy Bentham (1748–1832), the founding father of utilitarian philosophy, asked of animals: ‘The question is not, Can they reason? nor Can they talk? but Can they suffer?’.
In our present world, billions of animals are raised to provide food and many to be used for scientific experiments. The arguments that evolve from this activity centre on the extent to which non-human animals can be respected as sentient beings of moral worth, albeit with differences between species. In recent years, a boisterous animal rights movement, asserting the moral status of animals, has challenged their use as experimental subjects.13 Mainstream medical and scientific opinion around the world accepts that animal research continues to be justified, subject to important protections. This position is based on the insight that research involving animals has contributed hugely to advances in biological knowledge that have in turn allowed modern therapeutics to improve human morbidity and mortality. However, each experiment must be justified and its results must be expected to deliver insight into safety or efficacy. Animal models do contribute to the understanding of human physiology and disease because we share so many biological characteristics and a medicine when introduced into the organism is exposed to a vast array of conditions that we do not fully understand and are unable to reproduce outside the living body. The study of a drug in the whole organism remains an essential step in the process of discovery and development of medicines.
• Animals are only used as a last resort.
• Every practical step is taken to avoid distress or suffering.
• The smallest possible number of animals is used.
• The potential benefits have to be weighed against the cost to the animals; the simplest or least sentient species is used.
• The work is realistic and achievable, and the programme designed in the way most likely to produce satisfactory results.
• The results must impact the decision-making process for discovery or development of the medicine.
Dollery C.T. Beyond genomics. Clin. Pharmacol. Ther.. 2007;82(2):366–370.
Evans W.E., Relling M.V. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–468.
Garattini S., Chalmers I. Patients and the public deserve big changes in the evaluation of drugs. Br. Med. J.. 2009;338:804–806.
Lean M.E.J., Mann J.I., Hoek J.A., et al. Translational research. Br. Med. J.. 2008;337:705–706.
Lesko L.J. Personalized medicine: elusive dream or imminent reality? Clin. Pharmacol. Ther.. 2007;81(6):807–816.
Meyer U.A. Pharmacogenetics – five decades of therapeutic lessons from genetic diversity. Nat. Rev. Genet.. 2004;5(9):669–676.
Pound P., Ebrahim S., Sandercock P., et al. Where is the evidence that animal research benefits humans? Br. Med. J.. 2004;328:514–517.
Vallance P., Levick M. Drug discovery and development in the age of molecular medicine. Clin. Pharmacol. Ther.. 2007;82(2):363–365.
Weinshilboum R., Wang L. Pharmacogenomics: bench to bedside. Nat. Rev. Drug Discov.. 2004;3(9):739–748.
1 Paul Ehrlich (1845–1915), a German scientist, who pioneered the scientific approach to drug discovery. The 606th organic arsenical that he tested against spirochaetes (in animals) became a successful medicine (Salvarsan 1910); it and a minor variant were used against syphilis until superseded by penicillin in 1945.
2 Paracelsus (1493–1541) was a controversial figure who has been portrayed as both ignorant and superstitious. He had no medical degree; he burned the classical medical works (Galen, Avicenna) before his lectures in Basle (Switzerland) and had to leave the city following a dispute about fees with a prominent churchman. He died in Salzburg (Austria), either as a result of a drunken debauch or because he was thrown down a steep incline by ‘hitmen’ employed by jealous local physicians. But he was right about the dose.
3 Culliton B J 1994 Nature Medicine 1:1 [editorial].
4 Johnstone D 2006 pA2 online (E-journal of the British Pharmacological Society) 4(2). Available at: http://www.pa2online.org/articles/article.
5 The cost of development of a new chemical entity (NCE) (a novel molecule not previously tested in humans) from synthesis to market (general clinical use) is estimated at over $1.6 billion; the process may take as long as 15 years (including up to 10 years for clinical studies), which is relevant to duration of patent life and so to ultimate profitability; if the developer does not see profit at the end of the process, the investment will not be made. The drug may fail at any stage, including the ultimate, i.e. at the official regulatory body, after all the development costs have been incurred. It may also fail (due to adverse effects) within the first year after marketing, which constitutes a catastrophe (in reputation and finance) for the developer as well as for some of the patients. Pirated copies of full regulatory dossiers have substantial black market value to competitor companies, who have used them to leapfrog the original developer to obtain a licence for their unresearched copied molecule. Dossiers may be enormous, even a million pages or the electronic equivalent, the latter being very convenient as it allows instant searching.
6 Held or given in trust (OED).
7 Vallance P 2005 Developing an open relationship with the drug industry. Lancet 366:1062–1064.
8 Serendipity is the faculty of making fortunate discoveries by general sagacity or by accident; the word derives from a fairytale about three princes of Serendip (Sri Lanka) who had this happy faculty.
9 Mouse, rat, hamster, guinea-pig, rabbit, cat, dog, and sometimes monkey are used (but not all for any one drug). Non-clinical (pharmacotoxicological) studies must be carried out in conformity with the provisions of internationally agreed standards known as Good Laboratory Practice (GLP). In Europe, regulations ensure that all tests on animals are conducted in accordance with Council Directive 86/609/EEC. Certain studies in animals can be substituted by validated in vitro tests provided that the test results are of comparable quality and usefulness for the purpose of safety evaluation. The pharmacological and toxicological tests must demonstrate the potential toxicity of the product and any dangerous or undesirable toxic effects that may occur under the proposed conditions of use in human beings; these should be evaluated in relation to the pathological condition concerned. The studies must also demonstrate the pharmacological properties of the product, in both qualitative and quantitative relationship to the proposed use in human beings.
10 Details can be found at: http://www.ema.europa.eu/.
11 Bull J P 1959 The historical development of clinical therapeutic trials. Journal of Chronic Diseases 10:218–248.
12 René Descartes (1596–1650), French philosopher, mathematician and scientist, acknowledged as one of the chief architects of the modern age.
13 The publication of Animal Liberation (New York: New York Review/Random House) by Peter Singer in 1975 is widely regarded as having provided its moral foundation.