CHAPTER 62 Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is the most common cardiomyopathy and is responsible for significant morbidity and mortality. The etiology of DCM is quite heterogeneous, and the DCM phenotype likely represents a common final outcome in response to many different myocardial insults; however, it has been recognized more recently that genetic factors probably account for 35% to 50% of all cases of DCM.1,2 Advances in imaging have provided insight into mechanisms of pathology in DCM, and have allowed more confident noninvasive separation of patients with ischemic DCM from patients with nonischemic DCM.
DEFINITION
DCM is a disease of the myocardium that is characterized by dilation and impaired systolic function of the left ventricle or both ventricles.3
PREVALENCE AND EPIDEMIOLOGY
DCM is the most common of the cardiomyopathies, accounting for approximately 55% of cases, and responsible for greater than 90% of cases referred to specialty centers.3 Idiopathic DCM is the most common cause of congestive heart failure in young patients with an estimated prevalence of 36.5 per 100,000 individuals in the United States, and is responsible for more than 10,000 deaths per year.4 Depending on the diagnostic criteria applied, the annual incidence in adults is 5 to 8 cases per 100,000 population.5 The true incidence is likely underestimated because many asymptomatic cases are unrecognized. DCM is responsible for a high proportion of cases of heart failure and sudden cardiac death, and is a leading cause of cardiac transplantation. The mortality rate in the United States owing to cardiomyopathy is greater than 10,000 deaths per year, with DCM the major contributor to this mortality.6
ETIOLOGY AND PATHOPHYSIOLOGY
DCM has been linked to many different etiologies, including infection, hypertension, pregnancy, alcohol, autoimmune disease, nutritional deficiency, cardiotoxins (e.g., anthracycline, heavy metals, cocaine, methamphetamines), genetic inheritance (e.g., mitochondrial disorders), or any cardiovascular disease in which the degree of myocardial dysfunction is not explained by the abnormal load conditions (e.g., valvular dysfunction) or the extent of ischemic damage.3 The World Health Organization definition of DCM excludes patients with enlarged and dysfunctional ventricles secondary to ischemic or valvular dysfunction, which are categorized as their own specific cardiomyopathy (i.e., ischemic cardiomyopathy and valvular cardiomyopathy).3 Viral myocarditis may be an important etiology in childhood, but in adults the relationship between myocarditis and DCM is less clear, and inconsistent results have been achieved in the attempt to isolate specific disease pathogens.
Family screening has emphasized the importance of inheritance in the etiology of DCM. Genetic transmission is most often autosomal dominant, with a lesser number of autosomal recessive, or X-linked, cases. Echocardiography family screening studies have shown abnormalities in approximately 25% of relatives of patients with DCM, including DCM and isolated left ventricular enlargement. Of individuals with left ventricular enlargement, 10% to 25% develop clinical DCM with symptomatic heart failure, arrhythmias, or thromboembolism within 5 years.7 These observations suggest that familial DCM is a slowly progressive disorder, and that screening of first-degree relatives of patients with DCM should assume a similar role to that of screening in hypertrophic cardiomyopathy patients, with more well-established genetic linkages.
The autosomal forms of familial DCM can be grouped into forms with a pure DCM phenotype or DCM with cardiac conduction system disease. Genetic heterogeneity is the hallmark of autosomal dominant DCM with 15 loci mapped for pure DCM and 5 for DCM with cardiac conduction system disease. These mutations include genes encoding cardiac actin, desmin, δ-sarcoglycan, β-sarcoglycan, cardiac troponin T, and α-tropomyosin. Most genes identified to date encode either cytoskeletal or sarcomeric proteins. These proteins are important for structural integrity and for force transmission.2
X-linked DCM occurs in adolescent boys and young men with rapid progression from congestive heart failure to death or transplantation, and is characterized by mutations in the gene for cardiac dystrophin, a cytoskeletal protein providing structural support to the myocyte and linking it to the sarcolemma. The dystrophin gene is also responsible for Duchenne and Becker muscular dystrophies, which also have DCM as a prominent feature.8
Histologic changes associated with DCM are frequently nonspecific, and not all features may be present. DCM is characterized by progressive interstitial fibrosis with a reduced number of functional myocytes and, in advanced stages, relative wall thinning. Although atrophic changes predominate histologically, there is also myocyte elongation with an addition of newly formed sarcomeres, which is the major factor responsible for increased chamber size. Myocyte diameter increases, but is inadequate to preserve a normal ratio of wall thickness to chamber diameter. Myocyte nuclear hypertrophy and pleomorphism may also be seen. There is often an increase in interstitial T lymphocytes and focal accumulation of macrophages associated with individual myocyte death.8
MANIFESTATIONS OF DISEASE
Imaging Technique and Findings
Ultrasonography
Echocardiography reveals a dilated left ventricular cavity with reduced global function. End-systolic and end-diastolic diameter are increased, and ejection fraction, fractional shortening, stroke volume, and cardiac output are decreased (Figs. 62-1 and 62-2).
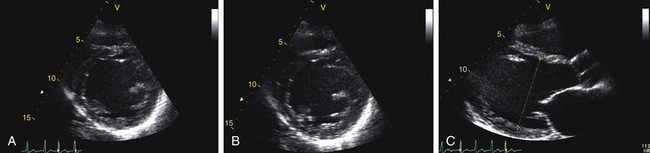
 FIGURE 62-1
FIGURE 62-1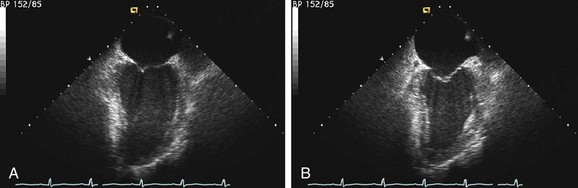
 FIGURE 62-2
FIGURE 62-2Evaluation of right ventricular function is also important. Patients with biventricular dysfunction have a lower New York Heart Association functional class, tend to have more severe left ventricular dysfunction, and have a worse long-term prognosis.9,10
Stress echocardiography may also play a role in assessment of patients with DCM. Patients with improvement in ejection fraction greater than 20% during stress echocardiography have a better prognosis. Drozd and colleagues11 showed that the incidence of cardiac death or transplantation is lower in patients with preserved contractile reserve. Another study assessed the prognostic significance of high-dose dobutamine stress echocardiography, and concluded that the change in wall motion score index is able to identify patients at greater risk for cardiac death during follow-up, and that change in wall motion score index had superior prognostic information to change in ejection fraction.12
Stress echocardiography with dipyridamole has also been used to assess coronary flow reserve in patients with DCM. Rigo and colleagues13 evaluated 129 patients with DCM and found that coronary flow reserve, assessed via Doppler velocity interrogation of the mid left anterior descending coronary artery, was often impaired, and that reduced coronary flow reserve was an independent marker of poor prognosis. Pratali and coworkers14 performed dobutamine and dipyridamole stress echocardiography in 87 patients with DCM and found that both tests have similar feasibility and prognostic accuracy.
Computed Tomography
In addition to assessing ventricular function, myocardial mass, and chamber size, CT can be used to screen for coronary artery disease. A few more recent investigations have compared the accuracy of coronary CT angiography with conventional angiography in assessing patients with ischemic or nonischemic DCM (Fig. 62-3). Andreini and associates15 studied 61 patients with idiopathic DCM and 139 patients with normal cardiac function and indications for coronary angiography. Using 16-row MDCT, the authors found excellent agreement with angiography, with sensitivity, specificity, and positive and negative predictive values for identification of greater than 50% stenosis of 99%, 96%, 81%, and 99%. Cornily and colleagues16 achieved similar results with a smaller group of 36 patients with DCM, also using a 16-row system and comparing results with conventional angiography.
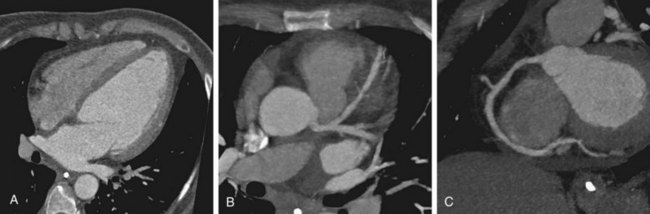
 FIGURE 62-3
FIGURE 62-3It has also been suggested that coronary calcification scoring alone may be adequate for distinguishing ischemic from nonischemic DCM (Fig. 62-4). Budoff and associates17 assessed 56 patients with cardiomyopathy using coronary angiography, nuclear exercise stress testing (Tc 99m sestamibi), and coronary calcification electron-beam CT. Nuclear stress testing had a sensitivity of 97% using the criteria of a reversible or fixed defect, but a low specificity of 18%. Using receiver operating curve analysis, the authors determined that a cutoff coronary calcification score of 100 yielded a sensitivity and specificity of 82%. CT has the potential to reduce or eliminate the use of conventional coronary angiography in distinguishing ischemic from nonischemic DCM. CT also effectively detects complications of DCM, such as atrial or ventricular thrombus.
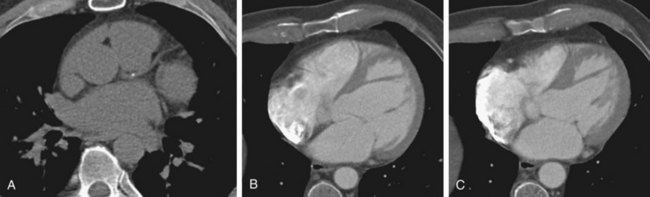
 FIGURE 62-4
FIGURE 62-4Magnetic Resonance Imaging
Infarcts on MDE images have a characteristic appearance, showing subendocardial or transmural enhancement in a distribution corresponding to the territory of the affected coronary artery. MDE imaging has a high sensitivity for detection of infarcts, and it has been shown that small infarcts seen using this technique are often missed with nuclear medicine perfusion imaging. MDE imaging is an excellent method for distinguishing nonischemic DCM from ischemic cardiomyopathy (Figs. 62-5 and 62-6). Soriano and colleagues18 evaluated 71 patients with heart failure and left ventricular systolic dysfunction without a prior history of myocardial infarction with coronary angiography and MDE imaging. Subendocardial or transmural hyperenhancement on MDE imaging characteristic of previous infarction was present in 81% of the angiography-positive group, whereas only 9% of the angiography-negative patients had an ischemic MDE pattern. McCrohon and coworkers19 evaluated 90 patients with heart failure with MRI and MDE imaging, and compared the results with coronary angiography. All angiography-positive patients showed an ischemic subendocardial or transmural MDE pattern. In the angiography-negative patients, 59% showed no hyperenhancement on MDE imaging, 13% had an ischemic hyperenhancement pattern (i.e., subendocardial or transmural), and 28% had a longitudinal or patchy mid-wall hyperenhancement pattern with subendocardial sparing on MDE imaging that was not restricted to distinct coronary territories.
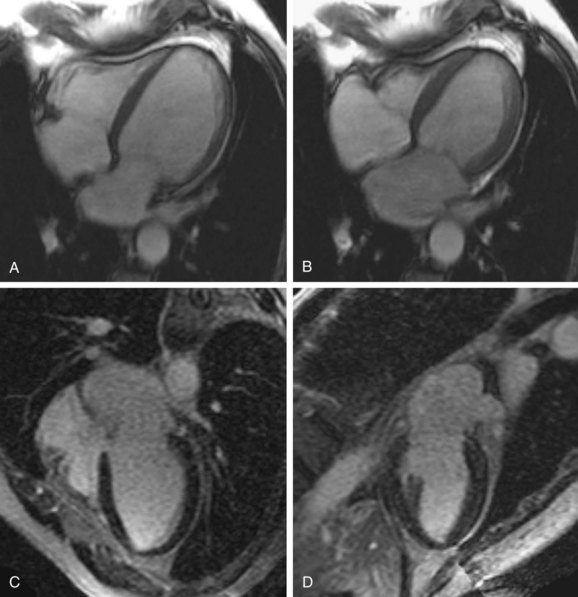
 FIGURE 62-5
FIGURE 62-5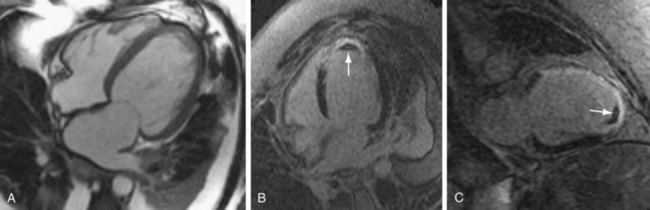
 FIGURE 62-6
FIGURE 62-6Although most patients with nonischemic DCM show no late gadolinium enhancement, there is more recent evidence that some patients have nonischemic enhancement patterns, in particular enhancement in the middle of the ventricular myocardium (Fig. 62-7), and that the presence of this late enhancement is an indicator of poor prognosis compared with patients without MDE. Assomull and colleagues20 evaluated 101 patients with DCM and found a mid-wall fibrosis pattern in 35% of patients. Mid-wall fibrosis was associated with a higher mortality and hospitalization for cardiovascular events and sudden cardiac death and ventricular tachycardia. Park and associates21 also evaluated 46 patients with nonischemic left ventricular systolic dysfunction with late enhancement MRI and showed that the absence of delayed hyperenhancement had excellent sensitivity and negative predictive value in predicting functional recovery of left ventricular systolic dysfunction.
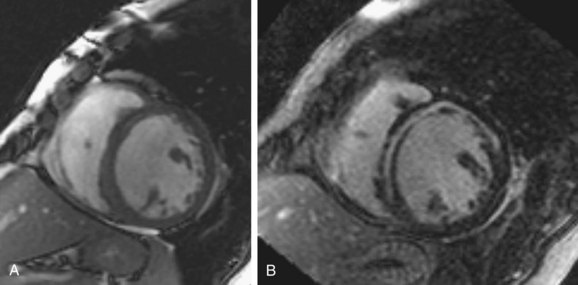
 FIGURE 62-7
FIGURE 62-7Nuclear Medicine
Nuclear medicine techniques have an important role in evaluation of patients with DCM. More recent development of ECG gated single photon emission computed tomography (SPECT) techniques allows assessment of myocardial perfusion and ventricular function, and SPECT would seem to be an ideal tool for distinguishing ischemic and nonischemic DCM (Fig. 62-8). Danias and colleagues22 assessed 37 patients with severely reduced left ventricular function with exercise Tc 99m sestamibi gated SPECT and found that the summed stress and rest perfusion defect scores were widely separated between the two groups, and completely distinguished ischemic from nonischemic patients. A larger trial from the same group using the same technique assessed 164 patients with ejection fraction less than 40% and without known coronary artery disease. Using a combined analysis of stress perfusion, reversibility, and regional wall motion deficits, Danias and colleagues23 achieved a high sensitivity (94%) but relatively low specificity (45%) for detection of ischemic cardiomyopathy. Generally, patients with ischemic cardiomyopathy have more severe perfusion defects than patients with nonischemic cardiomyopathy. Fixed defects are occasionally encountered in nonischemic cardiomyopathy, and may be related to attenuation associated with the severely dilated left ventricular cavity and supine imaging.
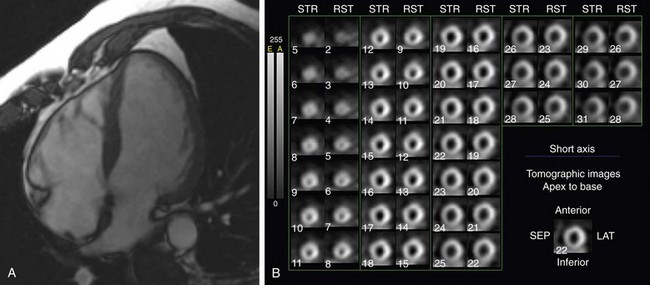
 FIGURE 62-8
FIGURE 62-8Dobutamine stress myocardial perfusion imaging has been used to predict patient response to β blocker therapy in DCM. Kasama and coworkers24 found that the change in ejection fraction measured by Tc 99m tetrofosmin gated SPECT was significantly higher in patients who responded to therapy than in nonresponders.
PET can also be used to assess DCM patients. O’Neill and colleagues25 examined 44 patients with PET, echocardiography, and radionuclide ventriculography, and found that myocardial scarring (defined as a matched perfusion and metabolic defect) was very common, occurring in 91% of patients, and that the extent of scar correlated with the QRS duration.
Another group has used PET to assess global and regional myocardial oxygen consumption (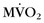 ) and blood flow in patients with DCM and left bundle branch block.26 Complete left bundle branch block is a common finding in severe DCM and is a strong predictor of mortality. Global and regional
) and blood flow in patients with DCM and left bundle branch block.26 Complete left bundle branch block is a common finding in severe DCM and is a strong predictor of mortality. Global and regional 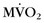 and myocardial blood flow were assessed using acetate C 11 PET. Patients with severe DCM and left bundle branch block exhibited a significantly lower (impaired) global
and myocardial blood flow were assessed using acetate C 11 PET. Patients with severe DCM and left bundle branch block exhibited a significantly lower (impaired) global 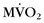 and reduced myocardial blood flow at rest than patients with mild or moderate disease without left bundle branch block. Analysis of regional differences in
and reduced myocardial blood flow at rest than patients with mild or moderate disease without left bundle branch block. Analysis of regional differences in 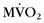 and blood flow revealed more heterogeneous distribution of
and blood flow revealed more heterogeneous distribution of 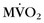 and myocardial blood flow in DCM patients with left bundle branch block.
and myocardial blood flow in DCM patients with left bundle branch block.
CRT has been advocated in patients with poor left ventricular function and conduction delays for symptomatic improvement and prolonging survival. Lindner and colleagues27 studied patients with nonischemic cardiomyopathy before and 4 months after CRT using acetate C 11 PET, and showed that CRT induces changes of 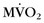 and myocardial blood flow leading to a more uniform distribution with less regional heterogeneity. More novel applications of nuclear medicine include the use of Tc 99m–labeled annexin A5 to identify focal, regional, or global uptake of annexin A5, a marker of cellular apoptosis.28
and myocardial blood flow leading to a more uniform distribution with less regional heterogeneity. More novel applications of nuclear medicine include the use of Tc 99m–labeled annexin A5 to identify focal, regional, or global uptake of annexin A5, a marker of cellular apoptosis.28



Agricola E, Oppizzi M, Pisani M, et al. Stress echocardiography in heart failure. Cardiovasc Ultrasound. 2004;2:11-25.
Jackson E, Bellenger N, Seddon M, et al. Ischaemic and non-ischaemic cardiomyopathies—cardiac MRI appearances with delayed enhancement. Clin Radiol. 2007;62:395-403.
Kim DH, Choi SI, Chang HJ, et al. Delayed hyperenhancement by contrast-enhanced magnetic resonance imaging: clinical applications for various cardiac diseases. J Comput Assist Tomogr. 2006;30:226-232.
Rochitte CE, Tassi EM, Shiozaki AA. The emerging role of MRI in the diagnosis and management of cardiomyopathies. Curr Cardiol Rep. 2006;8:44-52.
Soler R, Rodriguez E, Remuinan C, et al. Magnetic resonance imaging of primary cardiomyopathies. J Comput Assist Tomogr. 2003;27:724-734.
Thiene G, Basso C, Calabrese F, et al. Twenty years of progress and beckoning frontiers in cardiovascular pathology: cardiomyopathies. Cardiovasc Pathol. 2005;14:165-169.
Wood MJ, Picard MH. Utility of echocardiography in the evaluation of individuals with cardiomyopathy. Heart. 2004;90:707-712.
1 Murphy RT, Starling RC. Genetics and cardiomyopathy: where are we now? Cleve Clin J Med. 2005;72:465-483.
2 Hughes SE, McKenna WJ. New insights into the pathology of inherited cardiomyopathy. Heart. 2005;91:257-264.
3 Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation. 1996;93:841-842.
4 Gillum RF. Idiopathic dilated cardiomyopathy in the United States, 1970-1982. Am Heart J. 1986;111:752-755.
5 Codd MB, Sugrue DD, Gersh BJ, et al. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy: a population-based study in Olmsted County, Minnesota, 1975-1984. Circulation. 1989;80:564-672.
6 Gillum RF. Idiopathic cardiomyopathy in the United States, 1970-1982. Am Heart J. 1986;111:752-755.
7 Baig MK, Golman JH, Caforio AL, et al. Familial DCM: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195-201.
8 Towbin JA, Bowles NE. Dilated cardiomyopathy: a tale of cytoskeletal proteins and beyond. J Cardiovasc Electrophysiol. 2006;17:919-926.
9 LaVecchia L, Paccanara M, Bonanno C, et al. Left ventricular versus biventricular dysfunction in idiopathic dilated cardiomyopathy. Am J Cardiol. 1999;83:120-122.
10 La Vecchia L, Varotto L, Zanolla L, et al. Right ventricular function predicts transplant-free survival in idiopathic dilated cardiomyopathy. J Cardiovasc Med. 2006;7:706-710.
11 Drozd J, Krzeminska-Pakula M, Plewka M, et al. Prognostic value of low-dose dobutamine echocardiography in patients with dilated cardiomyopathy. Chest. 2002;121:216-222.
12 Pratali L, Picano E, Otasevic P, et al. Prognostic significance of the dobutamine echocardiography test in idiopathic dilated cardiomyopathy. Am J Cardiol. 2001;88:1374-1378.
13 Rigo F, Gherardi S, Galderisi M, et al. The independent prognostic value of contractile and coronary flow reserve determined by dipyridamole stress echocardiography in patients with idiopathic dilated cardiomyopathy. Am J Cardiol. 2007;99:1154-1158.
14 Pratali L, Otasevic P, Nekovic A, et al. Prognostic value of pharmacologic stress echocardiography in patients with idiopathic dilated cardiomyopathy: a prospective head-to-head comparison between dipyridamole and dobutamine test. J Cardiac Fail. 2007;13:836-842.
15 Andreini D, Pontone G, Pepi M, et al. Diagnostic accuracy of multidetector computed tomography coronary angiography in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2007;49:2044-2050.
16 Cornily JC, Gilard M, Le Gal G, et al. Accuracy of 16-detector multislice spiral computed tomography in the initial evaluation of dilated cardiomyopathy. Eur J Radiol. 2007;61:84-90.
17 Budoff MJ, Jacob B, Rasouli ML, et al. Comparison of electron beam computed tomography and technetium stress testing in differentiating cause of dilated versus ischemic cardiomyopathy. J Comput Assist Tomogr. 2005;29:699-703.
18 Soriano CJ, Ridocci F, Estornell J, et al. Noninvasive diagnosis of coronary artery disease in patients with heart failure and systolic dysfunction of uncertain etiology, using late gadolinium-enhanced cardiovascular magnetic resonance. J Am Coll Cardiol. 2005;45:473-478.
19 McCrohon JA, Moon JC, Prasad SK, et al. Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance. Circulation. 2003;108:54-59.
20 Assomull RG, Prasad SK, Lyne J, et al. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol. 2006;48:1977-1985.
21 Park S, Choi BW, Rim SJ, et al. Delayed hyperenhancement magnetic resonance imaging is useful in predicting functional recovery of nonischemic left ventricular systolic dysfunction. J Card Fail. 2006;12:93-99.
22 Danias PG, Ahlberg AW, Clark BA, et al. Combined assessment of myocardial perfusion and left ventricular function with exercise technetium-99m sestamibi gated single-photon emission computed tomography can differentiate between ischemic and nonischemic dilated cardiomyopathy. Am J Cardiol. 1998;82:1253-1258.
23 Danias PG, Papaioannou GI, Ahlberg AW, et al. Usefulness of electrocardiographic-gated stress technetium-99m sestamibi single-photon emission computed tomography to differentiate ischemic from nonischemic cardiomyopathy. Am J Cardiol. 2004;94:14-19.
24 Kasama S, Toyama T, Kumakura H, et al. Dobutamine stress 99mTc-tetrofosmin quantitative gated SPECT predicts improvement of cardiac function after carvedilol treatment in patients with dilated cardiomyopathy. J Nucl Med. 2004;45:1878-1884.
25 O’Neill JO, McCarthy PM, Brunken RC, et al. PET abnormalities in patients with nonischemic cardiomyopathy. J Card Fail. 2004;10:244-249.
26 Lindner O, Vogt J, Baller D, et al. Global and regional myocardial oxygen consumption and blood flow in severe cardiomyopathy with left bundle branch block. Eur J Heart Fail. 2005;7:225-230.
27 Lindner O, Vogt J, Kammeier A, et al. Effect of cardiac resynchronization therapy on global and regional oxygen consumption and myocardial blood flow in patients with non-ischaemic and ischaemic cardiomyopathy. Eur Heart J. 2005;26:70-76.
28 Kietselaer BL, Reutelingsperger CP, Boersma HH, et al. Noninvasive detection of programmed cell loss with 99mTc-labeled annexin A5 in heart failure. J Nucl Med. 2007;48:562-567.
