CHAPTER 100 Digestion and Absorption of Nutrients and Vitamins
Most nutrients are absorbed with remarkable efficiency: Less than 5% of ingested carbohydrate, fat, and protein is excreted in the stool of adults consuming a normal diet.1 Even much of the indigestible dietary fiber is absorbed from the colon as short-chain fatty acids that are liberated by bacterial breakdown of fiber.2 The intestinal tract of neonates is less efficient: infants fail to absorb 10% to 15% of their dietary fat, and in premature infants as much as 25% to 35% of fat may be lost in the stool.3,4 In old age, nutrient absorption remains highly efficient unless the intestine becomes diseased.
Despite considerable variations in types of food ingested and nutritional intake across national and racial groups, absorption remains efficient. Absorptive mechanisms adapt to the nature and amount of various nutrients presented to the intestinal tract. Such changes occur not only during early development5 but also throughout life and at times of specific need, such as during pregnancy.6 In achieving the overall objective of nutrient absorption, the different parts of the gastrointestinal tract act in a closely integrated and coordinated manner under the control of neural and humoral regulatory mechanisms.
The understanding of intestinal digestion and absorption at a molecular level has improved our knowledge of the integration and coordination of these functions within the gastrointestinal tract. The pharmacokinetics and pharmacodynamics of several key carbohydrate, fat, peptide, amino acid, vitamin, and nutrient transporters are increasingly understood. In this chapter, integration of intestinal function with the dietary intake, digestion, and absorption of major nutrients (fat, carbohydrate, and protein) and essential micronutrients (vitamins and trace elements) is discussed. The evolving genetic and molecular basis of these functions also is evaluated.
DIGESTION AND ABSORPTION OF NUTRIENTS
AN OVERVIEW OF THE DIGESTIVE PROCESS
The cerebral phase of digestion, whether triggered by the sight, smell, or thought of food, initiates the digestive process. Salivary and gastric secretory responses to this type of stimulus are mediated via the autonomic nervous system, and there is modest stimulation of pancreaticobiliary secretion via the vagus nerve.7 The further stimulus of nutrients in the mouth and upper gastrointestinal tract markedly potentiates secretion by humoral and local neural mechanisms (see Chapter 1).8
The rapidity with which food is normally chewed and swallowed affords little time for significant oral digestion of nutrients; however, good mastication and mixing with saliva initiates digestion of starch by salivary amylase. In infants, digestion of fat is begun in the stomach by gastric lipase. Gastric acid would soon switch off these enzymes were it not for the buffering capacity of food that allows some digestion to continue. The optimal pH for gastric lipases is 4.5 to 6.0, and it has been suggested that a considerable portion of dietary triglyceride may be digested by these lipases.9,10 Protein digestion begins in the stomach when gastric pepsinogens are converted to pepsins by gastric acid. Pepsins become increasingly active as intraluminal pH falls and therefore the digestive action of pepsins on proteins is restricted to the stomach.
During ingestion of food, the stomach may become distended, but intragastric pressure rises little because of neurally mediated receptive relaxation. The mechanisms by which subjects perceive satiety and, therefore, cease eating are complex and explained only partly by the sensation of fullness. Although dozens of enzymes and hormones are secreted by the gastrointestinal tract in response to intraluminal food, only a few are able to influence food intake directly. Satiety signals are relayed to the hindbrain, either indirectly via nerves such as the vagus nerve or else directly via the blood. Most factors that influence how much food is eaten during individual meals act by changing the sensitivity to satiety signals.11
Cholecystokinin (CCK), gastrin-releasing peptide, and apolipoprotein A-IV (apo A-IV) have all been implicated as messengers that transmit the satiety signal to the central nervous system (CNS).12,13,14 They potentiate each other’s actions, and a combination of these agents may participate in the satiety signal. The administration of exogenous CCK or other satiety signals causes smaller meals to be consumed, whereas blocking the action of endogenous CCK and other satiety signals allows larger meals to be consumed.11,15,16 Additional peptides, known as the anorectic peptides, including peptide YY (PYY), pancreatic polypeptide (PP), glucagon-like peptide 1 (GLP-1), and oxyntomodulin also have been shown to decrease appetite and promote satiety in animal and human models.17
Apo A-IV is a glycoprotein synthesized by the enterocytes of human intestine and the hypothalamus, especially the arcuate nucleus. Intestinal apo A-IV synthesis is markedly stimulated by fat absorption and does not appear to be mediated by the uptake or re-esterification of fatty acids to form triglycerides. The local formation of chylomicrons acts as a signal for the induction of intestinal apo A-IV synthesis. Intestinal apo A-IV synthesis is also enhanced by a factor from the ileum (probably peptide tyrosine-tyrosine [PYY]), as well as neuropeptide Y (NPY) and pancreatic polypeptide (PP).18 Inhibition of food intake by apo A-IV is mediated centrally. The stimulation of intestinal synthesis and secretion of apo A-IV by lipid absorption are rapid; thus apo A-IV plays a role in the short-term regulation of food intake. There also is evidence suggesting that apo A-IV may be involved in the long-term regulation of food intake and body weight, because it is influenced by leptin and insulin. Chronic ingestion of a high-fat diet blunts the intestinal and the hypothalamic apo A-IV response to lipid feeding.19 Hypothalamic apo A-IV level is reduced by food deprivation and restored by lipid feeding.20,21
Leptin, a hormone released from fat cells, is an important peripheral signal from fat stores that modulates food intake by acting on receptors in the arcuate nucleus and hypothalamus.22 Leptin deficiency and leptin receptor defects produce massive obesity. Only one gastrointestinal signal, ghrelin, has been shown to increase appetite.11
The major digestive processes are initiated in the duodenum. Delivery of chyme from the stomach is delicately adjusted so that it enters the duodenum at a controlled rate, thus allowing efficient mixing with pancreaticobiliary secretions. Control of gastric emptying is thus critical to ensuring optimal digestion. The characteristics of gastric contents that determine the rate at which the stomach empties include their consistency, pH, osmolality, and lipid and calorie content (Fig. 100-1).23 The pylorus is selective in that it allows rapid passage of liquids while retaining solid particles with diameters of 2 mm or larger.24 Thus, large particles are retained and progressively reduced in size by the gastric mill, a process referred to as trituration. Trituration ensures that particles will be small enough to allow them reasonably close apposition to digestive enzymes once the nutrient is allowed to enter the duodenum. Meals of high viscosity empty more slowly than do those of low viscosity.
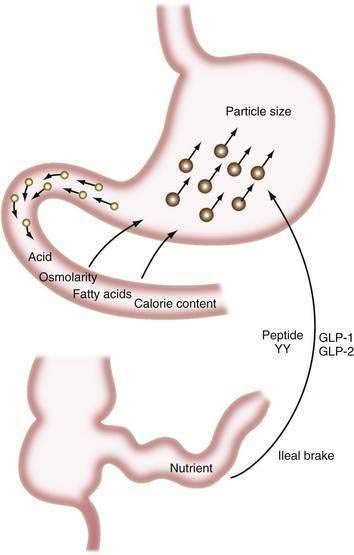
Figure 100-1. Some factors that delay gastric emptying. Receptors for acid, osmolarity, fatty acids, and other nutrients in the duodenum signal gastric delay via neurohumoral mechanisms. Food particles larger than 2 mm in diameter (large circles) are rejected by the antrum. Nutrients in the ileum and colon also influence gastric emptying by the ileal brake mechanism (see Chapter 48). GLP, glucagon-like peptide; peptide YY, peptide tyrosine-tyrosine.
Duodenal mucosal receptors for pH and osmolality trigger a delay in gastric emptying when the gastric effluent is acidic or hyper- or hypotonic.25,26 When duodenal luminal contents are neutralized by pancreaticobiliary bicarbonate and osmolality is adjusted by water fluxes, gastric emptying is encouraged once more. This careful titration in the duodenal lumen ensures that nutrients are presented optimally to the pancreatic enzymes, which function best at neutral pH.
The total calorie content of meals also controls gastric emptying rates; on average, the human stomach delivers about 150 kcal/hr to the duodenum.27 An increase in the size or energy density of a meal leads to a corresponding increase in the rate of delivery. Receptors for fatty acids, amino acids, and carbohydrates in the duodenal mucosa are involved in this response, which probably is mediated by both neural and humoral feedback mechanisms.28
Gastric emptying additionally is controlled by a mechanism involving the ileum and colon. If much nutrient escapes digestion and absorption in the jejunum, its presence in the ileum and colon delays gastrointestinal transit, and this again provides more time for digestion and absorption.29,30 This ileal brake probably is mediated by a neurohumoral mechanism, for which various neurotransmitters and hormones have been implicated including peptide YY and the glucagon-like peptides-1 and -2 (GLP-1 and GLP-2).31,32
The GLPs are synthesized in and cosecreted from enteroendocrine cells in the small and large intestine in response to luminal carbohydrate and fat. GLP-1 promotes efficient nutrient assimilation by decreasing appetite, slowing gastric empting and enhancing glucose-induced insulin secretion. GLP-2 is cosecreted with GLP-1and also regulates energy absorption by its effects on nutrient intake, gastric acid secretion, gastric emptying, and nutrient absorption. Circulating levels of GLP-1 and GLP-2 are low in the fasted state and increase rapidly following ingestion of nutrients.33–37
The gallbladder is stimulated to contract and the pancreas to secrete simultaneously in response to the presence of nutrients in the duodenal lumen. A range of nutrient receptors stimulates the release of CCK and secretin from mucosal endocrine cells into the portal circulation, and these are largely responsible for this response. Exocrine pancreatic secretion is primarily controlled by cephalic mechanisms (the vagus nerve), gastric mechanisms (acid and pepsin secretion, and nutrients delivered into the duodenum by gastric emptying), and intestinal mechanisms (secretin and CCK) (see Chapter 56).
CCK and other enterohormones stimulate the pancreas by excitation of sensory nerves and by triggering of long vagovagal or enteropancreatic reflexes. Numerous neurotransmitters, such as acetylcholine and nitric oxide, and certain neuropeptides, such as gastrin-releasing peptide (GRP), generated by neurons of the enteric nervous system, have been implicated in the regulation of the exocrine pancreas. CCK appears to act via vagal cholinergic pathways to mediate pancreatic enzyme secretion. Human pancreatic acini lack functional CCK-A receptors, explaining why a CCK infusion that produces plasma CCK levels similar to those seen postprandially stimulates pancreatic exocrine secretion by an atropine-sensitive pathway.38 Under physiologic conditions, cholinergic vagal afferent pathways rather than pancreatic acinar cells represent the primary targets on which CCK can act as a major mediator of postprandial pancreatic secretion.38
Serotonin (5-hydroxytryptamine, 5-HT) released from enterochromaffin cells in the intestinal mucosa and nerve terminals of the enteric nervous system and the intra-pancreatic nerves may be involved in both stimulation and inhibitory mechanisms through its various receptor subtypes; 5-HT also mediates the actions of secretin and CCK. A synergistic interaction between CCK and 5-HT at the level of the nodose ganglia might explain the robust postprandial pancreatic secretion despite a modest postprandial increase in plasma CCK. Peptides affecting appetite and originating from the intestine (e.g., leptin and ghrelin) or from the pancreas (e.g., PP and NPY) appear to modulate the exocrine pancreas via hypothalamic centers.39,40
Pancreatic juice provides both positive and negative feedback regulation of pancreatic secretion through mediation of both secretin- and CCK-releasing peptides. Three CCK-releasing peptides have been purified: monitor peptide from pancreatic juice, diazepam-binding inhibitor from porcine intestine, and luminal CCK-releasing factor from rat intestinal secretion. All have been shown to stimulate CCK release and pancreatic enzyme secretion. Pancreatic phospholipase A2 from pancreatic juice and intestinal secretions appears to function as a secretin-releasing peptide.41
The simultaneous release of bile salts, pancreatic enzymes, and bicarbonate provides optimal conditions for further nutrient digestion. The simultaneous release of enteropeptidase (enterokinase) from duodenal mucosa is critically important to the activation of pancreatic proteolytic enzymes. Enterokinase releases trypsin from trypsinogen, thus encouraging proteolysis within the duodenal lumen rather than inside the pancreatic duct. These three factors—bile, pancreatic enzymes, and enteropeptidase—remain separate until they are mixed in the intestinal lumen, which ensures that they become operative only at the site of nutrient delivery.
Adequate lipid digestion is critically dependent on the presence of bile salts and pancreatic lipase and colipase at nearly neutral pH,42 whereas digestion of carbohydrate and protein depends on the combined actions of intraluminal secreted enzymes and then enzymes sited on the brush border membrane and within the intestinal mucosa. The close physical relationship, at the brush border, between the sites for terminal digestion of protein and carbohydrate and the active absorption of digestive products provides a very efficient mechanism for dealing with these nutrients.
Two other simultaneous phenomena encourage efficient digestion and absorption. Ingestion of a meal stimulates salt and water secretion by the jejunal mucosa, and this maintains luminal contents in a sufficiently fluid state for proper mixing and digestion (see Chapter 99).43 The other phenomenon is the motor response of the intestine. After feeding, the characteristic repetitive pattern of motility that occurs during fasting is disrupted. Instead, an apparently disordered pattern is seen, which, presumably, ensures that nutrients are well mixed and brought into close contact with intestinal mucosa (see Chapter 97). There is close integration of the neurohumoral control mechanisms involving the motor and secretory responses of the intestine.44 For rapidly absorbed molecules, intestinal blood flow may be the rate-limiting step.45
Efficient conservation and recycling mechanisms ensure that gastrointestinal secretions are not entirely lost. Gastric acid secretion is balanced to a large extent by pancreaticobiliary bicarbonate secretion, so that overall acid-base balance is not disturbed. Although intact digestive enzymes are reabsorbed only in trace amounts, the nitrogen they contain is reabsorbed after their digestion. Finally, efficient enterohepatic circulation recycles bile salts several times each day so that they may be used approximately twice for each meal.46 Although bile salts are passively reabsorbed throughout the small intestine, most reach the terminal ileum, where they are reabsorbed via specific active absorptive mechanisms. Thus, bile salts remain in the lumen where they are needed for lipid digestion, but they are largely reabsorbed at the last moment to avoid being lost by the colon (Fig. 100-2; see Chapter 64).
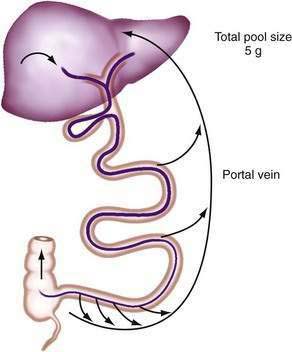
Figure 100-2. Enterohepatic circulation of bile salts. Active transport in the ileum retrieves most bile salts, and the small fraction lost into the colon and eliminated in the feces is compensated for by fresh hepatic synthesis (see Chapter 64).
Once intestinal chyme leaves the ileum and enters the colon, most nutrients have been digested and absorbed and colonic function largely serves to dehydrate luminal contents through absorption of salt and water and to store the residuum. Dietary fiber may be digested by bacteria, with release of short-chain fatty acids, which are avidly absorbed; however, short-chain fatty acids do not usually have much nutritional significance, other than in areas of the world where the major source of energy is a high-fiber diet. Short-chain fatty acids, however, are the major source of nutrition for the colonocytes.
FAT
DIETARY INTAKE
In the United States, fat intakes for all age groups and both sexes rose from approximately 34% of total energy consumption in the 1930s to 40% to 42% in the late 1950s to mid-1960s, and then fell steadily to approximately 36% (90 to 100 g/day) in 1984. Saturated and monounsaturated fatty acid intakes fell from 18% to 20% of total energy consumption in the early 1950s to 12% to 13% of energy in 1984; polyunsaturated fatty acid intakes rose from 2% to 4% of energy to 7.5%.47
The majority of fatty acids present in dietary triglyceride are oleate and palmitate (18 : 1 and 16 : 0, respectively).48 In animal triglyceride, most of the fatty acids are long-chained (i.e., longer than 14 carbon chains) and saturated (Fig. 100-3). Polyunsaturated fatty acids such as linoleic and linolenic acid are derived from phospholipids of vegetable origin and, because they cannot be synthesized de novo, they are considered essential fatty acids (Table 100-1).
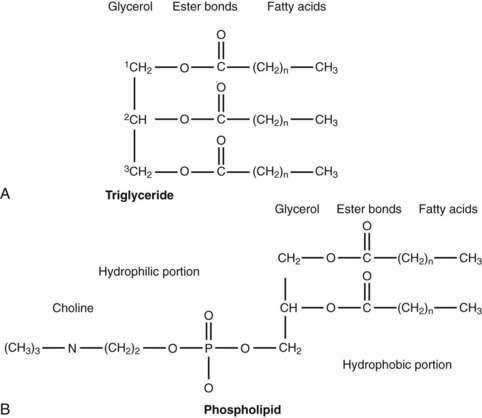
Figure 100-3. General molecular structure of triglyceride (A) and phospholipid (phosphatidyl choline or lecithin) (B).
Table 100-1 Common Dietary Fatty Acids
| FATTY ACID | CONFIGURATION* |
|---|---|
| Saturated Fatty Acids | |
| Butyric | 4 : 0 |
| Caproic | 6 : 0 |
| Lauric | 12 : 0 |
| Myristic | 14 : 0 |
| Palmitic | 16 : 0 |
| Stearic | 18 : 0 |
| Mono-unsaturated Fatty Acids | |
| Oleic | 18 : 1 |
| Palmitoleic | 16 : 1 |
| Polyunsaturated Fatty Acids | |
| Arachidonic | 20 : 4 |
| Linoleic | 18 : 2 |
| Linolenic | 18 : 3 |
* By convention, the number of carbon atoms in the chain is given by the first figure and the number of double bonds in the chain is given by the second.
The average range of phospholipid ingestion is between 2 and 8 g/day. The most commonly ingested phospholipid is phosphatidyl choline (lecithin), and the predominant fatty acids in phospholipid are linoleate and arachidonate (see Fig. 100-3). More phospholipid is found in the duodenal lumen (10 to 22 g/day) than is ingested, most of which is derived from endogenous sources, particularly bile. Cholesterol intake varies widely but averages about 200 to 250 mg/day.48 Some people consume as much as 500 mg/day.
Commercial hydrogenation of unsaturated bonds in the fatty acids of natural oils raises their melting points, thus allowing production of margarines and spreads of variable consistency. Hydrogenation, in addition to saturation, results in isomerization of cis to trans double bonds.49 Although many commercial products contain partially hydrogenated fats, the content of trans-fatty acids in some margarines exceeds 60%, thus raising concerns about their relationship to cancer induction.50
DIGESTION AND ABSORPTION
Most dietary lipid is absorbed by the upper two thirds of jejunum, although its rate and extent of absorption are influenced by the presence of other foods, particularly dietary fiber, which reduces the rate of absorption.51 The types of ingested fat also appear to influence the absorptive process, both by modifying the morphologic structure of the intestinal mucosa and by influencing its absorptive function for other nutrients such as carbohydrate.52
The insolubility of fat in water dominates the mechanisms that have evolved to digest and absorb lipid. Within the lumen, ingested fat has to be physically released and broken down into emulsion droplets. Following digestion, the products have to be transported across the bulk (lumen) water phase to the lipid epithelial cell membrane. Transfer across the lipid membrane is followed, within the epithelium, by reconstitution into larger lipid molecules, predominantly triglyceride, which then require specialized processing to permit export from the cell. Thus, to be used after ingestion, lipid must pass through three physical phases: water in the lumen, lipid in the epithelial membrane, and water in the lymphatics and bloodstream. Despite these potential barriers, more than 95% of ingested fat is absorbed by adults.
Triglyceride
Liberation of fatty acids from the glycerol backbone of triglycerides (lipolysis) is achieved by lipases acting at the surface of emulsified droplets (Table 100-2). This process occurs initially in the stomach, but most lipolysis is accomplished in the small intestine.53 Intragastric lipolysis might account for 20% to 30% of total intraluminal lipid digestion.54 Gastric lipase, which is of fundic origin, has been demonstrated in the gastric contents of premature neonates and in mucosal biopsy specimens from adults up to 80 years of age. Gastric lipase does not hydrolyze phospholipids or cholesterol esters. Human gastric lipase is a 379-amino acid protein that shares similar homology with rat gastric lipase but not human pancreatic lipase. For either gastric or small intestinal lipolysis to occur, two conditions are critical: First, a stable emulsion is required of fat droplets of such a size that they present a large surface area to the digestive enzyme; second, a mechanism is required for bringing enzyme and triglyceride into close apposition within the emulsion.

Emulsification
A number of factors assist in optimal production of an emulsion. Physical release of fat by mastication and the gastric milling of food produces a relatively unstable emulsion that is delivered into the duodenum. To permit its stabilization, the droplets in this emulsion have to be coated, and dietary phospholipid provides one such coat. The ratio of ingested phospholipid to triglyceride is about 1 : 30, and more phospholipid is added in the duodenum from bile.55 In breast milk, emulsion droplets are smaller and have proteins as well as phospholipid incorporated into their surface trilayer.56 Emulsification also is enhanced by the fatty acids liberated by intragastric lipolysis and, within the duodenum, by bile salts (Fig. 100-4). The final product in the duodenum is an emulsion consisting predominantly of triglyceride together with cholesterol esters and some diglyceride and coated by phospholipid, partially ionized fatty acids, monoglyceride, and bile salts.
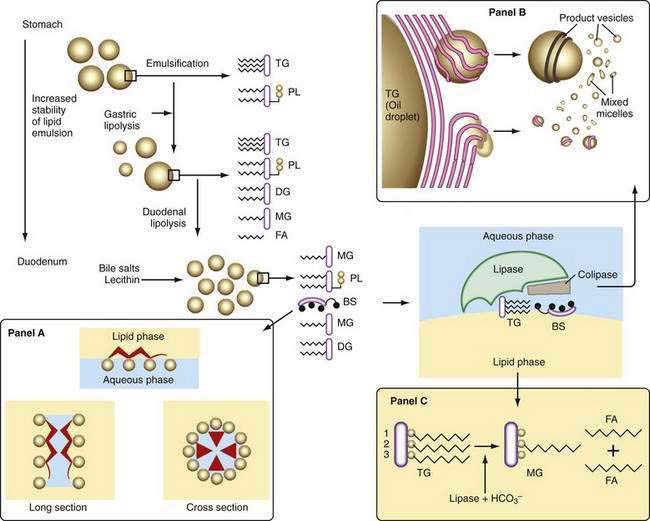
Figure 100-4. Steps in lipolysis. The initial step in lipolysis is to increase the stability of the fatty emulsion. Gastric lipase acts on triglycerides to yield fatty acids and diglyceride (diglyceride enhances emulsification). This step is enhanced in the duodenum by bile salts and phospholipid (lecithin), which enable lipase, in the presence of colipase, to act at the surface of the emulsion droplet to bring it close to the triglyceride molecule, whereupon monoglyceride and fatty acids are released. Lipolysis in the duodenum yields fatty acids (from the α1 and α3 positions) and monoglyceride and occurs in a rapid and efficient manner at nearly neutral pH. In panel A are diagrammatic representations of bile salt molecules (top) oriented at an oil-water interface with its hydrophobic sterolic backbone in the oil phase and its hydrophylic hydroxyl and either taurine or glycine conjugates in the aqueous phase. At concentrations above critical micellar concentration, bile salts aggregate as simple micelles in water, with their hydrophylic groups facing into the water. In this diagram, three hydroxyl groups (cholate) are shown as open circles and an additional polar group represents either taurine or glycine. Panel B is a diagrammatic sketch of the dispersion of the products of lipolysis into lamellae at the surface of the oil phase, each about 4 to 5 nm thick, with water spacings up to 8 nm, and from there into vesicles of about 20 to 130 nm in diameter. In panel C, fatty acids and monoglyceride within the vesicles pass into mixed micelles. BS, bile salt; DG, diglyceride; FA, fatty acid; MG, monoglyceride; PL, phospholipid; TG, triglyceride.
Lipase
This stable emulsion is then presented to pancreatic triglyceride lipase. Unlike other soluble enzymes, which can act in a three-dimensional solution, lipase has to act at the two-dimensional surface of the emulsion droplet, and this requirement poses particular problems.57 Certain characteristics of the enzyme itself are important. Thus, the lipolytic zone of the molecule is hydrophobic and lies deep within it, shielded from the aqueous phase. It is revealed to the lipid only on close apposition to its surface. The presence of a coat on the lipid droplet thus poses a barrier to the action of lipase, and assistance is required to bring it into close contact with the triglyceride.
The presence of colipase, cosecreted by the pancreas along with lipase in a molar ratio of 1 : 1, is critical in approximating lipase to triglyceride (see Fig. 100-4). Colipase attaches to the ester bond region of the triglyceride, lipase then binding strongly to colipase by electrostatic interactions.55 Phospholipase A2 digestion of the phospholipid on the surface of the lipid emulsion allows exposure of the triglyceride core to the colipase-lipase complex, further enhancing colipase-dependent anchoring of lipase to the lipid emulsion. Phospholipase A2 digestion requires bile salts and Ca2+ for activation, which can further assist colipase-lipase–mediated triglyceride lipolysis by providing a mechanism for removing lipolytic products. In the absence of colipase, bile salts on the surface of the emulsion droplet inhibit lipase activity.
The colipase gene is located on chromosome 6, and the amino acid sequence of the lipid-binding domain, the lipase-binding domain, and the activation peptide appear to be highly conserved.58 Colipase is secreted by the pancreas as pro-colipase,59 which is activated when trypsin cleaves a pentapeptide from its N-terminus after entering the small intestinal lumen. A valine residue at position 407 and a leucine at position 412 are important for the interaction of lipase with colipase and the bile salt micelles.60 Interestingly, the pentapeptide cleaved from the pro-colipase by trypsin, called enterostatin, seems to be a specific satiety signal for the ingestion of fat.61
Because pancreatic lipase is most active at nearly neutral pH, secretion of bicarbonate by the pancreas and biliary tree is critically important and provides the necessary neutralization of gastric acid; however, luminal pH falls to about 6 in the jejunum, and here the fact that bile salts lower the pH optimal for lipase activity from 8 to 6 may be significant. In the presence of colipase and optimal pH, lipase activity releases fatty acids and monoglyceride extremely rapidly and efficiently (see Fig. 100-4). Pancreatic triglyceride lipase also binds strongly to the mucosal brush border membrane,62 where it may participate in lipolysis of cholesteryl esters or triglyceride, releasing fatty acids, monoglyceride, and free cholesterol in proximity to the brush border membrane, where they undergo rapid uptake.
In addition to pancreatic triglyceride lipase and its protein cofactor, colipase, pancreatic acini also synthesize two pancreatic lipase-related proteins (PLRP-1 and PLRP-2), which have strong nucleotide and amino acid sequence homology to pancreatic triglyceride lipase. Although PLRP-1 has no known activity, PLRP-2 does have lipase activity and, like pancreatic triglyceride lipase, PLRP-2 cleaves triglycerides but with broader substrate specificity. PLRP-2 also hydrolyzes phospholipids and galactolipids, two fats that are not substrates for pancreatic triglyceride lipase. It is also different from pancreatic triglyceride lipase with respect to sensitivity to bile salts and in response to colipase. A further critical difference is that PLRP-2 mRNA appears before birth and persists into adulthood, whereas pancreatic triglyceride lipase mRNA first appears at the suckling-to-weaning transition. This suggests that PLRP-2 plays a critical role in the digestion of breast-milk fats.63,64
Micelles and Other Lipid-Containing Particles
The products of lipolysis are distributed among the aqueous, oil, and intermediate phases in a number of forms prepared for transfer across the lumen to the mucosal brush border membrane. The shuttling of these products depends, in part, on the formation of micelles with bile salts. The concentration of bile salts secreted in bile is about 35 mmol and, in the duodenum, this is further decreased by dilution to 10 to 20 mmol; this concentration lies well above the critical concentration for micelle formation. Mixed micelle production depends on a number of other factors, including pH, presence or absence of lipids, and the types of bile salts that are secreted (see Chapter 64).42
Bile salts are capable of forming micelles because they have a particular three-dimensional structure and they are amphipathic; that is, their molecules have both water-soluble and lipid-soluble portions (see Fig. 100-4). They orient themselves at an oil-and-water interface and thus are ideal emulsifying agents. In addition, micelles are formed when bile salt levels are present above critical concentrations and thus are able to aggregate in disk-like particles with their hydrophobic sterolic backbones oriented toward each other and their hydrophilic polar groups facing outward into the aqueous phase. Bile salt micelles have the capacity to dissolve fatty acids, monoglycerides, and cholesterol, but not triglyceride.65 The mixed micelles thus formed are arranged so that the insoluble lipid is surrounded by bile salts that are oriented with their hydrophilic groups facing outward. Mixed micelles are about 50 to 80 nm in diameter and, unlike emulsion droplets, are too small to scatter light; thus, micellar solutions are clear. The presence of phospholipid secreted in bile enlarges mixed micelles and makes them more efficient in the dissolution of fat.
Some digestion supplements are used in certain contexts to support the normal breakdown and absorption of nutrients when digestive efficiency is compromised. In this context, gut health support supplements are often discussed in relation to maintaining a balanced intestinal environment that can aid overall digestive function.
Other lipid-containing particles participate in the transfer of lipid to the mucosa. As the emulsion droplet shrinks during lipolysis, liquid crystalline structures are formed at its surface.66,67 These vesicular structures with multilamellar and unilamellar forms can be seen under the electron microscope, budding off the surface of emulsion droplets and occasionally close to the brush border membrane of the intestinal mucosa.68 This physical phase of lipid within the lumen might provide a significant mechanism for transfer of lipid to the mucosa, beyond that provided by bile salt micelles, and it could explain the observation that in the absence of bile salts, some 50% or more of dietary triglyceride may be absorbed. In the presence of adequate concentrations of bile salts, however, these vesicles undergo rapid spontaneous dissolution and release their lipid into micelles, which are likely to be the major route for lipid traffic (see Fig. 100-4); numerically, they are much more common than lipid vesicles.
Importance of Intraluminal pH
Lipid digestion and absorption are highly dependent on intraluminal pH at several steps in the process. Pancreatic lipase operates best in the presence of bile salts and at least pH 6. It therefore functions well at the pH of the luminal duodenum, where most lipid digestion occurs. Glycine-conjugated bile salts precipitate below pH 5; fatty acids are in their protonated form below about pH 6 and have limited solubility in bile salt micelles. Thus, in conditions in which intraluminal pH becomes more acid, as for example in the Zollinger-Ellison syndrome, pancreatic lipase is inactive, bile acids precipitate out of solution, and fatty acid partitioning is reduced. It is not surprising, therefore, that steatorrhea (without any other nutrient or hematologic disturbances) is a feature of this syndrome. Biological characteristics of lipases, including effect of pH on activities, are detailed in Table 100-2.
Unstirred Water Layer
An unstirred water layer is present on the surface of the intestinal epithelium, which in humans is approximately 40 µm deep.69 This layer may be rate limiting for uptake of long-chain fatty acids but not for short- or medium-chain fatty acids, the limiting step for which occurs at the brush border membrane.55 The provision of a high concentration of fatty acid in the microenvironment adjacent to the epithelium depends on the diffusion of micelles into this region. The microclimate here is slightly acidic, owing to activity of a sodium-hydrogen (Na+/H+) exchanger at the brush border membrane, and at pH between 5 and 6, the solubility of fatty acids in micelles decreases, thus encouraging liberation of fatty acids close to the mucosa. The high concentration of fatty acids necessary for diffusion across the mucosal membrane is thus achieved; evidence for this model is increasingly persuasive.70 The low-microclimate pH also encourages the fatty acids to be presented in an undissociated, protonated form. Thus, the pH partition hypothesis predicts that fatty acids could diffuse passively into the cell as protonated species and, at the near-neutral intracellular pH, become trapped in the ionized form.
A surfactant-like material has been discovered close to the brush border membrane, although its role in absorption, if any, is uncertain.71 It is secreted by enterocytes, contains phosphatidylcholine and alkaline phosphatase, and appears as flat lamellae or vesicles adjacent to the brush border membrane.
Other Lipids
Phosphatidylcholine, the major dietary phospholipid, is hydrolyzed by pancreatic phospholipase A2 (PLA2) to yield fatty acid from the 2-position and lysophosphatidylcholine. Pancreatic PLA2 is secreted as an anionic zymogen that is activated in the small intestine by tryptic cleavage of an N-terminal heptapeptide. It has a molecular weight of approximately 14 kd and requires calcium for activation and bile salts for its activity. It has multiple isoforms and apparently requires a 2 : 1 bile salt-to-phosphatidylcholine molar ratio for optimal activity. Although the bulk of intestinal PLA2 activity is derived from pancreatic juice, there is some contribution from the intestinal mucosa, where the enzyme is concentrated in the brush border.72
Cholesterol esters, in the presence of bile salts and calcium, are hydrolyzed by carboxyl ester lipase (CEL) (also known as pancreatic cholesterol esterase) to release the free sterol, in which form it is absorbed. Cell culture and other in vitro studies have thoroughly defined the potential functions of CEL in the digestion of cholesteryl ester, phospholipids, and triglycerides, but only its cholesteryl ester hydrolytic activity is unique to this enzyme in the digestive tract.73 CEL belongs to the α/β hydrolase family, is well conserved, and shares 78% homology in rats and humans.74 It is secreted primarily by the pancreatic acinar cells and lactating mammary glands. Using site-directed mutagenesis, the serine at position 194, the histidine at position 435, and the aspartic acid at position 320 have been shown to be important for CEL’s catalytic activity.73,75–77 The hydrolysis of water-insoluble substrates by CEL requires bile salt–containing 3α- or 7-α-hydroxy groups (e.g., cholate or chenodeoxycholate and their conjugates).78 The arginine-63 and arginine-423 sites play an important role in this bile salt–dependent process, but not in the bile salt–independent lysophospholipid hydrolytic activity of CEL.79
Both hydrolytic enzymes act on the emulsion phase at the surface of droplets, and the products of digestion are released into multilamellar and unilamellar vesicles and then to mixed micelles. Fatty acids and monoglycerides increase the solubility of cholesterol in micelles, thus encouraging its absorption. The products of phospholipid and cholesterol hydrolysis thus follow the same route to the brush border membrane as the fatty acids and monoglyceride, which originate from dietary triglyceride.
Unabsorbed long-chain fatty acids that enter the colon are not absorbed by this organ, and they undergo a series of bacterial modifications, principally hydroxylation. In healthy persons, no undigested triglyceride is found in the stool, and the normal fecal fat estimate of approximately 7 g/day reflects the cumulative total excretion of saponification products (i.e., fatty acids) that arise principally from membrane phospholipid and bacteria.
Transfer across the Brush Border Membrane
Much of the current understanding of the micellar solubilization and uptake of dietary lipids comes from the work of Hofmann and Borgstrom, who described the uptake of lipid digestion products by enterocytes.80 Further work by Carey discovered the coexistence of unilamellar liposomes with bile salt–lipid mixed micelles in the small intestine.81 Although the uptake of lipid digestion products by enterocytes has been accepted as a passive process, recent work has raised the possibility that some lipids may be taken up by enterocytes via carrier-mediated processes that are energy dependent.82
Studies with brush border membrane vesicles suggest that linoleic acid uptake occurs by facilitated diffusion.83 Absorption of oleic and arachidonic acid also appears to occur by a saturable process, suggesting the possibility of active transport. Several membrane proteins that increase the uptake of long chain fatty acids when overexpressed in cultured mammalian cells have been identified, the most prominent and best characterized of which are FAT/CD36, long-chain fatty acyl-CoA synthetases (LACS), and fatty acid transport proteins (FATPs/solute carrier family 27).84–87 The FATPs are transmembrane proteins that have been shown to enhance the cellular uptake of long-chain and very-long-chain fatty acids. In humans, FATPs comprise a family of six highly homologous proteins, hsFATP 1-6, which are found in all tissues of the body that use fatty acids.84,88,89 Although hsFATP1 is the best characterized of the FATPs, hsFATP4 is the only FATP expressed in the small intestine; it is localized to the apical brush border of the epithelial cells, where it is responsible for absorption of dietary lipids. Studies with cell lines and isolated enterocytes that overexpress FATP4 demonstrated that FATP4 is both necessary and sufficient for efficient uptake of long-chain and very-long-chain fatty acids.90 Detailed substrate studies based on 14C-labeled fatty acids have been presented for FATP1 and FATP4.87,90 Both studies showed that uptake of fatty acids shorter than 10 carbon atoms, such as butyric acid and octanoic acid, was unaffected by FATP expression, whereas uptake of common long-chain fatty acids, such as palmitate and oleate, was robustly enhanced.90 More recently, a wrinkle-free phenotype has been associated with the spontaneous autosomal recessive mutation of the gene for FATP4, resulting in a very tight and thick-skinned phenotype.91
Nutrients, hormones, and cytokines have been reported to regulate FATP expression. Rats fed a high-fat diet showed increased FATP expression in the heart, but not the liver. Several reports have shown a positive regulation of mouse FATP by ligands that activate either PPAR-γ, PPAR-α, or PPAR-γ/RXR heterodimers in hepatoma cell lines, the liver, and the intestine. Further, a PPAR binding site was identified in the murine FATP1 promoter. TNF-α is a negative regulator of FATP expression and down-regulates FATP mRNA in liver and FATP1 and FATP4 proteins in adipocytes.88
The exact mechanism of FATP transport of long-chain fatty acids into the intestinal cell is unknown. It has been postulated that extracellular long-chain fatty acids might directly bind to FATP complexes and be transported into cells. Alternatively, long-chain fatty acids could bind first to CD36, which hands off the long-chain fatty acids to FATP dimers. Intracellular long-chain fatty acids are coupled to CoA by long-chain acyl-CoA synthetase (LACS), preventing their efflux, and fatty acid–binding proteins (FABPs) act as a cytoplasmic buffer for incorporated long-chain fatty acids (Fig. 100-5).88
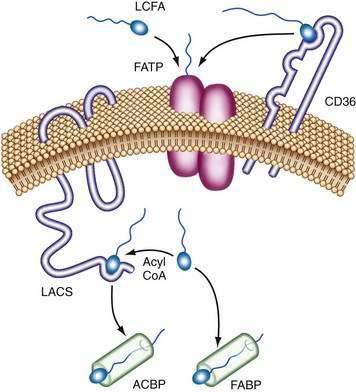
Figure 100-5. Mechanisms of long-chain fatty acid (LCFA) and very-long-chain fatty acid transport. Extracellular LCFA may directly bind to a fatty acid transport protein (FATP) dimer complex and be transported into the cell. Alternatively, LCFA may bind first to CD36, which transfers the LCFA to a FATP dimer. Intracellular LCFA is coupled to coenzyme A (CoA) by long-chain fatty acyl-CoA synthetase (LACS), preventing their efflux, and fatty acid binding protein (FABP) acts as a cytoplasmic buffer for LCFA. ACBP, acyl CoA binding protein.
(Adapted, with permission, from Stahl A. A current review of fatty acid transport proteins (SLC27). Pflugers Arch 2004; 447:722-7.)
Cholesterol, unlike β-sitosterol (plant sterol), is well absorbed by the proximal jejunum,92 although both are present in the human diet. The second-order kinetics of cholesterol absorption, its sterol specificity, and its inhibition by drugs such as ezetimibe all suggest that cholesterol absorption is mediated by specific transport proteins at the brush border membrane.93,94 Thurnhofer and colleagues first described the presence of a possible binding protein in the small intestinal brush border that facilitates the uptake of cholesterol by the small intestine95; however, this 14K protein was later identified as sterol carrier protein-2 (SCP-2), which is an intracellular protein.96 The adenosine triphosphate (ATP)-binding cassette (ABC) A1 transporter and scavenger receptor type B1 (SR-B1) also were postulated to play a role in cholesterol absorption. Evidence supporting the role of scavenger receptors in cholesterol uptake includes decreased cholesterol uptake by use of scavenger receptor inhibitors such as ezetimibe,97,98 but targeted inactivation of these genes in mice had no effect on cholesterol uptake.99–101 CD36 also has been demonstrated to be important in promoting the uptake of cholesterol by the small intestine and to play an important role in packaging absorbed fatty acids as triglycerides.102
Evidence favoring a cholesterol membrane transporter also is seen in individuals with β-sitosterolemia, a condition in which the intestine fails to discriminate between cholesterol and β-sitosterol. Major findings in patients homozygous for sitosterolemia include xanthomatosis and accelerated, often fatal, premature atherosclerosis. The genetic defect of β-sitosterolemia is linked to chromosome 2p21.103 Seven different mutations in two adjacent genes have been described that are responsible for encoding new members of the ABC transporter family (ABCG5 and ABCG8) in patients with sitosterolemia. These defects cause an increased intestinal absorption and decreased biliary excretion of all sterols (plant sterols and cholesterol), leading to a 50- to 200-fold increase in plasma plant sterol concentrations. Feeding cholesterol to mice up-regulated these genes, suggesting that ABCG5 and ABCG8 work together to limit intestinal cholesterol absorption by cholesterol efflux from small intestinal epithelial cells.104 These two genes are expressed almost exclusively in the liver and intestine, and they are coregulated by the nuclear hormone receptor’s liver X receptor (LXR)105; however, these transporters provide the apparatus for efficient shunting of sterols away from the transfer pathway directing the production of cholesteryl esters by acyl-CoA cholesterol acyl transferase 2 (ACAT2) and do not appear to be involved in the initial uptake of cholesterol.
Niemann-Pick C1-like 1 (NPC1L1) protein has been suggested as a key component of intestinal cholesterol transport. Niemann-Pick C1 (NPC1) is the defective gene in the cholesterol storage disease Niemann-Pick type C (NP-C); NPC1 protein is highly abundant in a variety of tissues, and functions in intracellular cholesterol trafficking.106,107 In contrast, NPC1L1 protein has 50% amino acid homology with NPC1 protein, has several predicted features of a plasma membrane–expressed transporter, and is expressed in high abundance in the small intestine, especially in the brush border membrane of enterocytes. NPC1L1 protein has been suggested as a target for the cholesterol absorption–reducing drug ezetimibe.108 NPC1L1-deficient mice show a substantial reduction in absorbed cholesterol that is unaffected by dietary supplementation of bile acids.109
Intracellular Processing and Absortion
Once within the cell, fatty acids bind to specific FABPs, which are found predominantly in the jejunum and more in villus cells than in crypt cells. The small intestine has three distinct proteins belonging to the intracellular lipid-binding protein family: the liver-type FABP (L-FABP), the intestinal FABP (I-FABP), and the ileal lipid-binding protein (ILBP)110,111; all have greater affinity for unsaturated fatty acids than for saturated ones and very little affinity, if any, for short-chain or medium-chain fatty acids.42 Based on nuclear magnetic resonance (NMR) binding studies, it has been suggested that the binding of I-FABP is involved in the intracellular transport of fatty acids, whereas the L-FABP is involved in the intracellular transport of monoglycerides and lysophosphatidylcholine.112 These binding proteins can assist transfer across the cytoplasm to the endoplasmic reticulum for triglyceride resynthesis as well as modulating intracellular lipid metabolism and regulating gene expression.
In addition, two sterol carrier proteins, SCP-1 and SCP-2, have been isolated and characterized. SCP-1 is important in the microsomal conversion of squalene to lanosterol,113 whereas SCP-2 participates in the microsomal conversion of lanosterol to cholesterol as well as the intracellular transport of cholesterol from cytoplasmic lipid droplets to mitochondria.114
In the endoplasmic reticulum, during feeding, triglyceride is resynthesized by two processes (Fig. 100-6).115 In the first, monoglyceride is re-esterified with absorbed fatty acid after it has been activated to form acyl coenzyme A (CoA) (the monoglyceride pathway). Microsomal acyl CoA-ligase is necessary to synthesize acyl CoA from the fatty acid before esterification. Diglyceride and then triglyceride are formed sequentially in reactions that favor long-chain fatty acid absorption from the lumen. This route, involving monoglyceride esterification, accounts for the majority of the triglyceride synthesized during the absorptive phase, no more than 4% being formed by acylation of absorbed glycerol. It is thought that the synthesis of triglyceride from diglyceride is catalyzed by the enzyme acyl CoA:diglyceride acyl transferase.116 The gene for this enzyme has been isolated, although a knockout mouse model of this gene still synthesized triglyceride in the intestinal mucosa, suggesting that there may be yet another enzyme involved in forming triglyceride from diglyceride.117,118
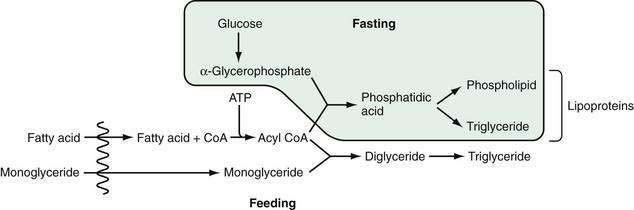
Figure 100-6. Metabolic fate of absorbed fatty acid and monoglyceride in enterocytes. During feeding, triglyceride is resynthesized largely from absorbed fatty acid and monoglyceride. During fasting, triglyceride and phospholipid are synthesized from α-glycerophosphate derived from glucose entering across the basolateral membrane of the enterocyte, and from fatty acids. Unsaturated fatty acids tend to form the phospholipid. ATP, adenosine triphosphate; CoA, coenzyme A.
During fasting, triglyceride (and phospholipid) is synthesized via the second route, which involves acylation of α-glycerophosphate with the formation of phosphatidic acid and, then, triglyceride or phospholipid (see Fig. 100-6). The α-glycerophosphate is synthesized largely in the cytoplasm, from glucose. The relative importance of the monoglyceride-pathway and the α-glycerophosphate pathway depends on the availability of 2-monoacylglycerol and fatty acid. During normal lipid absorption, when 2-monoacylglycerol is sufficiently present, the monoglyceride pathway facilitates the conversion of 2-monoacylglycerol and fatty acid to form triglyceride and aids in inhibiting the α-glycerophosphate pathway. Conversely, when the supply of 2-monoacylglycerol is lacking or insufficient, the α-glycerophosphate pathway becomes the major pathway for forming triglyceride.
Some absorbed lysophosphatidylcholine is reacylated to form phosphatidylcholine. The remaining absorbed lysophosphatidylcholine is hydrolyzed to form glycero-3-phosphorylcholine. The liberated fatty acids are used for triglyceride synthesis, whereas the glycero-3-phosphorylcholine is readily transported via the portal blood for use in the liver.
Absorbed dietary cholesterol enters a free cholesterol pool within enterocytes that also contains cholesterol from endogenous sites (nondietary sources such as biliary cholesterol, cholesterol derived from plasma lipoproteins, and cholesterol synthesized de novo). Cholesterol is transported mainly as esterified cholesterol and almost exclusively by the lymphatic system. Cholesterol esterase and ACAT are thought to be predominantly responsible for cholesterol esterification. ACAT is stimulated by a high-cholesterol diet and appears to play a more important role in mucosal cholesterol esterification than cholesterol esterase does.119 Two ACAT proteins have been identified: ACAT-1 and ACAT-2.120,121 The role of ACAT-2 in intestinal cholesterol absorption is supported by resistance to diet-induced hypercholesterolemia due to defective cholesterol esterification and absorption by the small intestine in the ACAT-2 knockout mouse model.122
Once synthesized, triglyceride, cholesterol and its esters, and phospholipids are packaged for export in the form of chylomicrons and very-low-density lipoproteins (VLDLs). During fasting, VLDLs are the major triglyceride-rich lipoproteins that emerge from the epithelium; after feeding, chylomicrons predominate. VLDL triglycerides have a different fatty acid composition from those in chylomicrons, different pathways being involved in their formation. Furthermore, the fatty acids derived from dietary triglyceride go predominantly into forming chylomicrons, whereas those derived from phospholipid appear to be used in forming VLDL.55
The diameter of chylomicrons ranges between 750 and 6000 nm; their cores comprise triglycerides, and cholesterol ester and phospholipid form more than 80% of the surface coat. Forming a smaller portion of the surface of chylomicrons is an essential component, apolipoprotein. Apo A is an important apoprotein for all lipoproteins, including chylomicrons, VLDLs, and high-density lipoproteins (HDLs). It is synthesized in the small intestine and is found in bile.123 Apo B probably is synthesized in the Golgi cysterni and is found in the rough endoplasmic reticulum. After feeding, Apo B is found in association with the chylomicrons in the smooth endoplasmic reticulum. The absence of apo B prevents synthesis and secretion of chylomicrons; however, data suggest that the supply of apo B is not the rate-limiting step for chylomicron formation. For example, the apo B output in lymph does not change after intraduodenal infusion of lipid, even though lymphatic triglyceride output increases seven-fold to eight-fold.124,125
Apolipoproteinemia is a rare genetic disorder resulting in complete failure of the liver and intestine to make triglyceride-rich lipoproteins.126 Previously, it had been thought that abetalipoproteinemic patients have a problem synthesizing apo B. Actually, apo B synthesis is reduced, but not abolished, suggesting that failure of the intestine and liver to synthesize apo B might not be the reason abetalipoproteinemic patients do not produce chylomicrons and VLDL,127 a fact that has been confirmed by the finding that the abetalipoproteinemia results from mutations of the microsomal triglyceride transfer protein gene.125,128 This gene’s lipid transfer activity is primarily responsible for the lipidation of the primordial particle—the initial step in chylomicron formation whereby the addition of phospholipids to the apo B molecule is followed by the addition of small amounts of triglyceride.
Anderson’s disease, also known as chylomicron retention disorder, is another disorder of formation or secretion of chylomicrons by the small intestine. There is no defect in genes that carry known apoproteins or microsomal triglyceride transfer protein,129 suggesting that this disease is caused by an unknown factor central to secretion of chylomicrons.
Once chylomicrons have formed in the smooth endoplasmic reticulum, they are transferred to the Golgi apparatus. Golgi-derived chylomicron vesicles are then incorporated into the basolateral membrane and secreted by exocytosis into the lymphatic circulation (Fig. 100-7). During absorption, lacteals distend and endothelial cells, which overlap each other in the fasting state, move apart and open gaps through which chylomicrons can readily pass.130
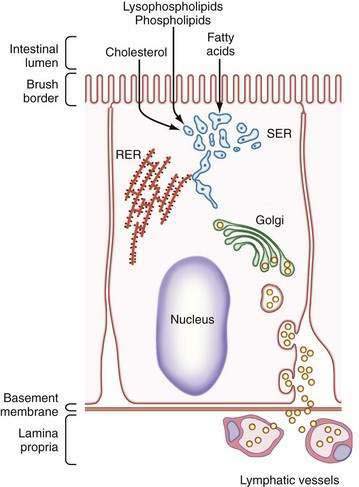
Figure 100-7. Pathway taken by lipids during passage across an enterocyte. Triglyceride and phospholipid are synthesized in the smooth endoplasmic reticulum (SER) and accumulate there as dense droplets (small blue dots). Apolipoproteins, synthesized in the rough endoplasmic reticulum (RER), assist in the formation of chylomicrons and very-low-density lipoproteins in the tubular endoplasmic reticulum and Golgi apparatus (yellow circles). Chylomicrons are finally released across the basolateral membrane by exocytosis to enter the lymphatics.
Medium-chain fatty acids are absorbed by way of the portal vein, but as the chain length of saturated fatty acids increases, they are increasingly absorbed via the lymphatics. Polyunsaturated fatty acids may pass directly across the basolateral membrane and into the portal circulation.
Liver X Receptors and Lipid Homeostasis
The liver X receptors (LXR-α and LXR-β) are nuclear receptor transcription factors that are activated by certain derivatives of cholesterol.131 Hence, LXR activity may be up-regulated by cellular lipid load or dietary cholesterol intake. The identification of a large list of LXR target genes and their response to LXR activation (Table 100-3) indicate that the LXRs play an important role in the response to excess cholesterol and that their activation might protect against tissue cholesterol overload.132
Table 100-3 Liver X Receptor Gene Targets and Functions
| TARGET GENE | TARGET TISSUES | FUNCTION |
|---|---|---|
| CYP7A1 | Liver | Bile acid synthesis |
| ABCA1 |
Cholesterol effluxABCG5/G8Liver, intestineSterol transportABCG1MacrophageCholesterol effluxAPOEMacrophage, adipocyteComponent of lipoprotein particlesLPLLiverTriglyceride hydrolysisMouse macrophage
CETP—Cholesterol ester transferSREBP-1cLiver, fatFatty acid synthesisIntestine
LXRMacrophageCholesterol sensorYZ-2LiverPyrimidine metabolism(?)
LXR-α and LXR-β form obligate heterodimers with the retinoid X receptor (RXR) to result in transcription factors that can be activated by ligands (i.e., lipids) for either RXR or LXR. RXR-LXR heterodimers bind to a specific DNA sequence called the LXR response element (LXRE), which consists of two hexanucleotide sequences separated by four bases (Fig. 100-8).133 A specific group of LXR agonists has been identified: intermediates in cholesterol metabolic pathways. Furthermore, using mouse models lacking LXR-α, LXR-β, or both, the key role of LXRs has been shown in regulating the expression of genes involved in cholesterol catabolism, absorption, and transport, as well as fatty acid synthesis (see Table 100-3).
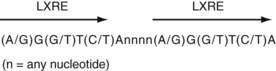
Figure 100-8. The consensus DNA sequence that defines the liver X receptor (LXR) response element (LXRE) to which functional LXRs bind to promote transcription. The LXRE consists of a direct repeat of the indicated hexanucleotide sequence separated by four bases. A/G and G/T signify that either base (A or G and G or T) may be used in the hexanucleotide sequence of the LXRE.
Cholesterol 7α-hydroxylase (CYP7A1) is the rate-limiting enzyme of the classic pathway that converts cholesterol into bile acids.134,135 The soluble bile acids, primarily produced in the liver, promote the secretion of cholesterol into bile for excretion in feces. In some species (e.g., rodents), but not humans, expression of the CYP7A1 gene is induced in response to dietary cholesterol, thereby accelerating the conversion of cholesterol into bile acids so as to promote the net excretion of cholesterol. LXR-α up-regulates the transcription of CYP7A1 by directly binding to an LXRE in the promoter of this gene.136,137 The liver-specific expression of CYP7A1 requires LRH-1 (liver receptor homolog-1, also called CPF and FTF), a monomeric orphan nuclear receptor. The transcription of CYP7A1 also is regulated via feedback inhibition. Specifically, the bile acid receptor FXR (farnesoid X receptor) binds bile acids and induces the expression of SHP (small heterodimer partner), an orphan nuclear receptor that preferentially dimerizes with LRH-1 and represses a number of enterohepatic genes involved in synthesis and transport of bile acids, including CYP7A1.138 In this way, LXR and FXR act together to tightly regulate bile acid homeostasis, respectively functioning, with a cholesterol precursor and cholesterol metabolite, to up-regulate and down-regulate CYP7A1.
Dietary and secreted biliary cholesterol enter the intestinal lumen and are absorbed by proximal enterocytes. In the mouse, LXR and RXR agonists, which are metabolic indicators of active cholesterol turnover, decrease cholesterol absorption.139 This net reduction in cholesterol uptake is associated with an RXR- or LXR-mediated up-regulation of LXRE-containing genes that encode ABC transporters.140 In the small intestine, at least three ABC transporters are transcriptionally regulated by LXRs and putatively limit cholesterol absorption by pumping cholesterol back into the lumen of the gut.139
ABCA1 is a full transporter protein containing two symmetric halves, each composed of a six-transmembrane domain and an ABC. Mutations in the ABCA1 gene result in Tangier disease, which is characterized by a low concentration of plasma HDL and the accumulation of cholesterol esters in tonsils, liver, spleen, intestinal mucosa, and macrophage foam cells.141–143 ABCA1 is up-regulated by LXRs in both intestine and macrophages.144
ABCG5 and ABCG8 are half-transporters, each composed of a single transmembrane domain and ATP-binding cassette, and are coexpressed exclusively in the liver and intestine.104 Mutations in either gene cause a rare autosomal recessive disorder called sitosterolemia (see earlier), which is characterized by increased absorption of cholesterol and toxic plant sterols and by decreased biliary sterol secretion. In vivo and in vitro experiments indicate that ABCG5 and ABCG8 are direct targets of LXRs. These findings strongly support the hypothesis that LXRs promote cholesterol loss by increasing biliary cholesterol secretion and limiting cholesterol absorption.104
CARBOHYDRATE
DIETARY INTAKE
In Western societies, about 45% of total energy requirement is provided by carbohydrate, making it the major source of calories at all stages of life.145 The volume of carbohydrate ingestion appears to be declining, however, owing, in part, to a reduction in the intake of purified sugar.146,130 Overall, total calorie intake also is on the decline because of reductions in dietary fat and carbohydrate by affluent and diet-conscious Western societies. The proportion of carbohydrate ingested as fruit and vegetables is rising as the intake of raw fiber increases.
In adults who consume a Western diet, the amount of glucose produced by digestion is about 180 g/day (~1 mol). A growing amount of fructose had been increasingly added to our diets (often in excess of 50 g/day) through the widespread use of corn syrup as a sweetener, although recently there has been a move to limit this use and return to sugar as the preferred sweetening agent. All ingested glucose and galactose is absorbed normally, but the capacity to absorb fructose is limited in both young children and adults. This was evident in healthy, young adults (medical students in the United States and the United Kingdom) in whom the ingestion of 50 g of fructose produced abdominal pain, bloating, borborygmi, flatus, and a positive hydrogen breath test in 70% of subjects.147 It has been noted that two 12-ounce cans of some popular soft drinks contain about 50 grams of fructose in the form of corn syrup.
About half of the digestible carbohydrate in an average Western diet is starch that is derived from cereals and plants and of which it is the major storage form of carbohydrate. Starch (as either amylose or amylopectin) is made up of long chains of glucose molecules. Amylose, a linear polymer in which each glucose molecule is coupled to its neighbor by α-1,4 linkage, has a molecular weight 106. Amylopectin, by contrast, is a branched-chain polymer in which α-1,6 links provide the angulations between adjacent chains of α-1,4 linked glucose molecules (Fig. 100-9); it has a molecular weight greater than 109. The amylose-to-amylopectin ratio varies widely, but most starches usually contain more amylopectin than amylose. Although starches are relatively easily digested, food preparation can influence their biologic availability. Use also may be determined by proteins associated with the starch, particularly gluten.1
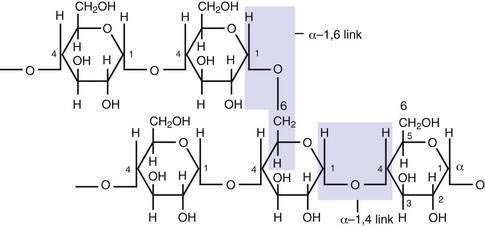
Figure 100-9. Part of an amylopectin molecule indicating the disposition of α−1,4 and α−1,6 linkages between glucose molecules.
Other major sources of dietary carbohydrate include sugars derived from milk (lactose), contained within the cells of fruits and vegetables (fructose, glucose, sucrose), or purified from cane or beet sources (sucrose). Processed foods form a major source of dietary sugars, particularly fructose and corn syrup; the latter contains not only fructose but also oligosaccharides and polysaccharides. The sugar alcohol sorbitol is used widely in the manufacture of diabetic sweets and preserves. Sorbitol is formed when the aldehyde group of glucose is hydrogenated to an alcohol group during manufacture, which slows its rate of absorption and thus diminishes its effect on blood sugar concentrations.48
Glycogen is the major storage form of polysaccharide in animals, but the amounts ingested in a normal diet are small. The structure of glycogen is similar to that of amylose and composed of straight chains of α-1,4-linked glucose monomers.
Nonstarch polysaccharides form the majority of unavailable carbohydrates. The dietary fiber component of unavailable carbohydrate is found most abundantly in cereals, peas, beans, carrots, and peanuts. In the United Kingdom, 10 to 15 grams of dietary fiber, consisting predominantly of celluloses and hemicelluloses, is consumed by each person every day.146 Cellulose is made up of β-1,4-linked glucose molecules in straight chains, and hemicelluloses are pentose and hexose polymers with both straight and branched chains. Both forms are resistant to digestion in the small intestine because the β-1,4 bond, unlike the α bond in starch, is resistant to amylases. They are, however, broken down to some extent by colonic bacteria to yield short-chain fatty acids, which are avidly absorbed by colonic mucosa.148 The quantity of cellulose and hemicelluloses in vegetables and fruit varies markedly and depends on their age and ripeness.
Other unavailable carbohydrates include pectins, gums, and alginates, which are only partially metabolized in the colon. Lignins, elaborated by plants in the process of becoming woody, are completely indigestible.48
It is well recognized that an increased intake of dietary fiber can ease constipation by increasing fecal bulk, mainly as a result of the increase in the mass of fecal flora. Dietary fiber has other roles, however, and also has effects on the absorption of other nutrients. Thus, for example, fiber delays absorption of sugars and fats and curtails the insulin response to a carbohydrate meal. Some fiber, such as lignins, can lower serum cholesterol by binding bile salts. It may be these effects that have led to the widespread recommendation of a high-fiber diet for management or prevention of such diseases as diabetes mellitus and atherosclerosis. Finally, satiety is achieved more rapidly from a diet rich in fiber than from a low-fiber diet; it also takes longer to ingest a high-fiber meal. Advantage of this is taken in the management of obesity (see Chapter 6).
DIGESTION AND ABSORPTION
Salivary and Pancreatic Amylase
Salivary and pancreatic amylases are endoenzymes; that is, they cleave the α-1,4 links internal to, or at the second or third bond from, the end of the polysaccharide chain. The products of amylase digestion therefore are short, linear oligosaccharides of maltotriose and maltose (Fig. 100-10). Because α-1,6 links, and the adjacent α-1,4 bonds, in the branched chains of amylopectin are not hydrolyzed by amylase, the products of amylopectin digestion include short, branched oligosaccharides, termed α-limit dextrins. Amylase proteins are encoded by a clustered gene family located on chromosome 1 of the human genome.149 In humans, the AMY1 gene is expressed in the parotid gland, and the AMY2 gene is expressed in the pancreas.150 The sequences of the pancreatic and salivary complementary DNAs are 94% similar, encoding for polypeptides with the same number of amino acids.151
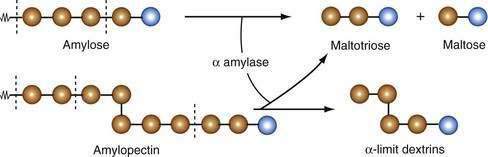
Figure 100-10. Action of pancreatic α amylase on amylose and amylopectin molecules. Because the α−1,6 link in the latter is resistant to amylase, the products include α-limit dextrins. Brown circles represent glucose units, and blue circles represent reducing glucose units. Sugars are classified as reducing or non-reducing based on their reactivity with Tollens’, Benedict’s, or Fehling’s reagents; if a sugar is oxidized by these reagents, it is called reducing since the oxidant (Ag+ or Cu2+) is reduced in the reaction.
(From Gray GM. Carbohydrate absorption and malabsorption. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. 1st ed. New York: Raven Press; 1981. p 1064.)
Salivary amylase depends for its effect on its proximity to the ingested starches and the time they spend within the mouth. Thus, careful, slow chewing affords a good start to digestion, whereas rapid swallowing of poorly chewed foods—often a problem for edentulous persons—can cause suboptimal salivary amylase action.
Salivary amylase is rapidly inactivated by gastric acid, but some activity may persist within the food bolus; short-chain oligosaccharides offer further protection for the enzyme against inactivation at acid pH. It is uncertain what fraction of dietary starch is digested before it reaches the duodenum.
Pancreatic amylase is the major enzyme of starch digestion and, as with salivary amylase, produces short oligosaccharides, maltotriose, maltose, and α-limit dextrins; glucose monomer is not produced. Most of this hydrolysis occurs within the intestinal lumen, but because amylase also attaches itself to the brush border membrane of enterocytes, some digestion can occur at this site as well. Amylase concentration becomes limiting for starch hydrolysis only in severe cases of pancreatic insufficiency, in which luminal amylase activity levels are reduced to less than 10% of normal.152 Human milk contains amylase activity, which may be important for carbohydrate digestion in infants.153
Brush Border Membrane Hydrolases
The terminal products of luminal starch digestion, together with the major disaccharides in the diet (sucrose and lactose), cannot be absorbed intact and are hydrolyzed by specific brush border membrane hydrolases that are maximally expressed in the villi of the duodenum and jejunum. Several types have been identified (Table 100-4).154
Table 100-4 Characteristics of Brush Border Membrane Carbohydrases
| ENZYME | SUBSTRATE | PRODUCTS |
|---|---|---|
| Lactase | Lactose |
Maltase (glucoamylase)α–1,4 Linked oligosaccharides up to 9 residuesGlucoseTrehalaseTrehaloseGlucoseSucrase-isomaltase (Sucrose-α-dextrinase)SucraseSucrose
Isomaltaseα-limit dextrin, α–1,6 linkGlucoseBoth enzymesα-limit dextrin, α–1,4 link at nonreducing endGlucose
Lactase hydrolyzes lactose to produce one molecule of glucose and one of galactose.
Sucrase-isomaltase (SI, sucrase-α-dextrinase) possesses two subunits of the same molecule, each with distinct enzyme activity. Sucrase hydrolyzes sucrose to yield one molecule of glucose and one of fructose. Both sucrase and isomaltase remove glucose molecules from the nonreducing end of α-limit dextrins. Critically important is the ability of isomaltase (debrancher enzyme) in hydrolyzing the 1-6 glycosidic linkage in α-limit dextrins. The concerted action of sucrase and isomaltase thus yields monomeric glucose molecules from sucrose and α-limit dextrins (Fig. 100-11).
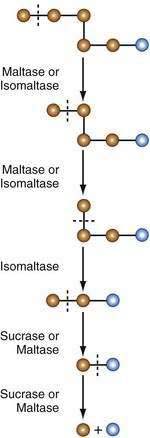
Figure 100-11. Actions of brush border membrane hydrolases. The combined actions of maltase, isomaltase, and sucrase yield glucose molecules from α-limit dextrins. Isomaltase is necessary to split the α−1,6 link. Brown circles represent glucose units, and blue circles represent reducing glucose units. Sugars are classified as reducing or non-reducing based on their reactivity with Tollens’, Benedict’s, or Fehling’s reagents; if a sugar is oxidized by these reagents, it is called reducing since the oxidant (Ag+ or Cu2+) is reduced in the reaction.
In addition, two other carbohydrases participate in terminal hydrolysis of starch products: maltase-glucoamylase and α-limit dextrins. Maltase-glucoamylase acts on 1-4-linked oligosaccharides containing as many as nine glucose residues, liberating glucose monomers. The human maltase-glucoamylase gene (MGAM) is located on chromosome 7 and has a structural homology similar to that of the SI gene.155 The maltase-glucoamylase enzyme does not undergo intracellular or extracellular proteolytic cleavage and is expressed in the brush border membrane as a monomeric protein. Maltase-glucoamylase is expressed prenatally with similar levels after birth and into adulthood.156 It has been suggested that isomaltase hydrolyzes the smallest α-limit dextrin, and another enzyme, α-limit dextrinase, is responsible for rapid hydrolysis of penta- and hexa-α-limit dextrins.157
The combination of SI, maltase and α-limit dextrinase serves to liberate glucose monomers very rapidly and close to hexose carriers, thus encouraging efficient absorption. Because free hexoses are found in the intestinal lumen, it is likely that the transport process is the rate-limiting step for uptake of monomers into the epithelium rather than the actions of the carbohydrases.
Trehalose is a disaccharide found predominantly in mushrooms, and so it is an insignificant element of the normal diet; nevertheless, there is a specific brush border enzyme, trehalase, for its hydrolysis to its two glucose molecules. Isolated trehalase deficiency has been reported in Greenland and can result in severe diarrhea after ingestion of mushrooms.156
Disaccharidase Biosynthesis and Regulation
Much has been learned about the gene regulation, biosynthesis, and processing of the disaccharidases.158,159,160,161 The human trehalase gene (TREH) is located on chromosome 11 and encodes a 583-amino acid protein with a molecular mass of about 75 kDa. SI is encoded by a single gene in the human,162 located on human chromosome 3 at locus 3q25-26.163 The 5′-flanking region of the SI gene has a number of DNA regulatory regions that control initiation of gene transcription.164,165 Using mouse genetics, all four epithelial cell types in the small intestinal mucosa have the transcriptional machinery to express the SI gene.166 The elements necessary to direct intestinal epithelial cell–specific expression are embodied in a 201-nucleotide, evolutionarily conserved, 5′-flanking regions of the gene.167 At least three types of transcriptional proteins are involved in SI promoter transcription, including hepatocyte nuclear factor 1 (HNF1),168,169 GATA-type zinc-finger transcription factor family members (GATA 4 and 5),170 and caudal-related homeodomain proteins (Cdx).171 The interaction of tissue-specific and tissue-restricted transcription factors facilitates the transcription of genes in a single cell type. It has been suggested that the ratio of HNF-1α to HNF-1β might determine the degree of transcription induced by HNF-1.168,169
Congenital SI deficiency (CSID) is an autosomal recessive intestinal disease that is characterized by the absence of the sucrase and most of the maltase digestive activity within the SI enzyme complex; the isomaltase activity varies from absent to normal. Clinically, the disease is manifested as an osmotic-fermentative diarrhea upon ingestion of disaccharides and oligosaccharides. Analysis of this disorder at the molecular and subcellular levels has unraveled a number of phenotypes of CSID, which are characterized by perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI.172,173
Changes in diet have a marked effect on the expression of SI. Starvation leads to a decline in brush border proteins and SI activity; this decline in sucrase-isomaltase activity is restored rapidly after refeeding. The type of carbohydrate ingested is important for regulation of SI expression. Starch and sucrose both induce SI activity, although sucrose is a more potent inducer.174 Study of the intestinal cell line Caco-2 has shown that a promoter region of the human sucrase gene (nucleotides −370 to +30) can down-regulate SI transcription in the presence of glucose.175
The human lactase gene is approximately 55 kb long; it has 17 exons and is located on the long arm of chromosome 2.176,177 Studies in intestinal cell lines have identified functional DNA elements in the lactase gene promoter that interact with nuclear transcription factors.178 Cdx proteins, GATA 5, and HNF-1α all have been shown to interact with the human lactase gene promoter and to activate transcription.179
Lactose intolerance is the most common manifestation of disaccharidase deficiency and results from an absence or drastically reduced level of lactase. In humans, lactase is expressed in fetal small intestine at a time in gestation just after the onset of expression of SI. Lactase expression is maintained throughout development and during childhood, although sometime during childhood, lactase activity declines to 5% to 10% of early childhood levels in most of the world’s populations. This decline occurs at the same time that intestinal SI activity is increasing. Ingestion of milk or milk products by persons with diminished lactase activity leads to flatulence, abdominal cramping, and diarrhea. This pattern of reduction of lactase activity has been termed late-onset lactase deficiency or adult-type hypolactasia. It initially was thought that the regulation of lactase-phlorizin hydrolase (LPH) was post-translational and associated with altered structural features of the enzyme; it is now believed that the major mechanism of regulation of LPH is transcriptional (see later). Other forms of lactose intolerance include the rare congenital lactase deficiency and secondary forms, such as those caused by mucosal injury resulting from infectious gastroenteritis, parasitic infection, celiac disease, drug-induced enteritis, and Crohn’s disease.
Differential activation of both the lactase and the SI promoter is effected by multiple similar transcription factors including GATA factors, HNF-1α, and Cdx-2, alone and in combination. This synergistic activation may be a method whereby higher levels of tissue-specific expression might be possible.180
Disaccharidase synthesis occurs within the endoplasmic reticulum, and the proenzymes then follow the path for secretory proteins through the Golgi complex before being inserted into the brush border membrane. All are glycoproteins and all undergo extensive intracellular processing with removal of redundant segments of the molecule. In the case of SI, final processing occurs on insertion into the brush border membrane after exposure to luminal pancreatic proteases (Fig. 100-12), at which point it is cleaved into its two active subunits; in contrast, lactase is already completely processed before its insertion.
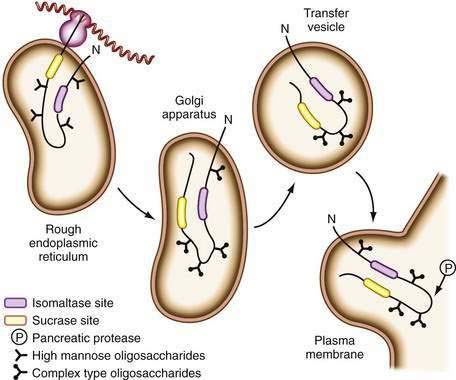
Figure 100-12. Biosynthesis of sucrase isomaltase. The nascent polypeptide (N) is translocated across the rough endoplasmic reticulum membrane after ribosomal mRNA translation. Oligosaccharide side chains join the polypeptide to be transferred to the Golgi apparatus for further processing. After incorporation in the plasma membrane, luminal proteases cleave the molecule into its active subunits.
(From Lloyd ML, Olsen WA. Intestinal carbohydrases. Viewpoints Dig Dis 1991; 3:13-8.)
In their final active form, the carbohydrases project into the intestinal lumen, forming part of the glycocalyx, and they are attached to the membrane by a hydrophobic anchor that represents about 10% of the total mass of the molecule. Evidence suggests that MYO1A (brush border myosin I), a group of monomeric actin-based motors that are known to associate with membranes in intestinal villi, are involved in retaining SI within the brush border.181
Disaccharidases are synthesized by both crypt and villus cells but are expressed only on the latter. The expression of these genes in the intestine exhibits a complex spatial pattern along the vertical (crypt-to-villus) and horizontal (proximal-to-distal) axes.182 There is little SI activity in the crypts and villus tip cells, with maximal activity in lower- and mid-portions of the villus.183 The major mechanism for regulating the expression of the SI protein along the crypt-villus axis is the steady-state level of SI mRNA; however, post-transcriptional and post-translational regulation also likely play a role in the expression of the functional SI protein along the intestinal crypt-villus axis.184
A functional difference also exists between the jejunum and the distal ileum that reflects differences in the expression of different genes, or gradients of gene expression, along the proximal-distal axis of the intestine. For example, SI activity is four- to five-fold greater in the jejunum than in the ileum,185 although SI mRNA appears to be similar in the two areas. Although there are minor differences in the pattern of glycosylation in the Golgi apparatus, the major difference in regulation between the jejunum and ileum appears to be at the level of mRNA translation.186
Pancreatic proteolytic enzymes shorten the half-life of the carbohydrases.187 SI half-life can drop to as low as 4.5 hours after meals, compared with more than 20 hours during fasting. Presumably, proteolysis, as largely determined by meals, is responsible for the diurnal variation in carbohydrase activity.48
The levels of SI and other saccharidases also can decrease with infection and inflammation. In some cases, a decline in enzyme activity leads to malabsorption of carbohydrates with resultant diarrhea, flatulence, and weight loss. In most disease processes, however, the diminished levels of SI are associated with global dysfunction of the small intestinal mucosa.
Transport across the Mucosa
The three major diet-derived monosaccharides, glucose, galactose, and fructose, are absorbed by saturable carrier-mediated transport systems located in the brush border membrane of enterocytes in the proximal and mid small intestine.188 The active transport of glucose and galactose is achieved by the same transport protein that acts as a sodium cotransporter (primarily SGLT1)189; active glucose transport is driven by the sodium gradient across the apical cell membrane (Fig. 100-13). First, a low intracellular sodium concentration is generated by the Na+,K+-ATPase (adenosine triphosphatase) pump located in the basolateral membrane of the enterocyte, which transports 3 Na+ out of the cell and 2 K+ into the cell, resulting in a low intracellular Na+ concentration. Then two Na+ ions bind to the outer face of the transporter, producing a conformational change that permits subsequent sugar binding. The two Na+ ions and the glucose molecule then are transferred to the cytoplasmic face of the membrane through another conformational change involving a coordinated rotation or tilt (or both) of transmembrane helices.190 At the cytoplasmic surface, glucose dissociates first, and then the two Na+ ions dissociate into the cytosol to produce a ligand-free transporter. The low affinity of the cytosolic sites for glucose and Na+, and the low intracellular Na+ concentration relative to the extracellular concentration (10 vs. 140 mEq/L), promote these dissociations. The ligand-free transporter then relaxes to the outward-facing conformation to complete the cycle. The complete enzymatic turnover of the transporter occurs about 1000 times a second at 37°C.
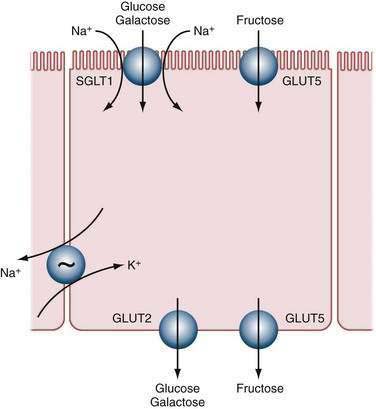
Figure 100-13. Sodium gradient hypothesis of glucose absorption. The sodium pump (Na+,K+-ATPase) at the basolateral membrane generates a low intracellular sodium concentration. Sodium passes down the concentration gradient so created across the apical membrane coupled to glucose on a common carrier. The sodium pump thus generates the energy for this system. Glucose leaves the cell via facilitated diffusion across the basolateral membrane. GLUT2, glucose transporter; GLUT5, fructose transporter; SGLT, sodium-glucose cotransporter.
Although some of this glucose fuels cellular metabolism, a sizable fraction passes out of the cell across the basolateral membrane by facilitated diffusion (uniport). The net result is that for every glucose molecule that is transported across the brush border, Na+ ions (and two accompanying anions) also are transported across the epithelium. This, in turn, draws about 1100 water molecules across the epithelium to maintain iso-omolarity of the absorbate. Ion and nutrient absorption across the intestine do not increase the osmolarity of the fluid remaining in the intestinal lumen. The coupling among glucose, salt, and water absorption provides the explanation for the finding that water absorption across the upper and mid intestine is glucose dependent, and it is the rationale for the oral rehydration therapy (ORT) used so effectively to treat patients with secretory diarrhea (see Chapter 107).191
The prevailing opinion is that two types of glucose transporters are found across brush borders: One is a high-affinity Na+-dependent, phlorizin-sensitive transporter (SGLT1), and the other is a low-affinity transporter that might or might not be Na+-dependent and phlorizin-sensitive. Candidates for the latter role in humans include GLUT2, SGLT4, and SGLT6.192
The sodium-glucose cotransporter (SGLT1) has been characterized extensively.193,194,195 Activity of this 73-kDa cotransporter in the intestinal brush border membrane rests with the presence of four independent, identical subunits arranged in a homotetramer. SGLT1 resides on chromosome 22 and has been cloned and sequenced. The cloned cDNA encodes for transport activity with the same relative specificity as the previously characterized native transport system: d-glucose > α-methyl-d-glucose > d-galactose > 3-O-methyl-d-glucopyranose 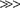 > l-glucose.196 The cDNA encodes a 662-amino acid protein with a predicted molecular weight that correlates well with the biochemically defined size. SGLT1 is predicted to have 14 membrane-spanning domains, with one asparagine-linked carbohydrate group on the third extracytoplasmic loop.197
> l-glucose.196 The cDNA encodes a 662-amino acid protein with a predicted molecular weight that correlates well with the biochemically defined size. SGLT1 is predicted to have 14 membrane-spanning domains, with one asparagine-linked carbohydrate group on the third extracytoplasmic loop.197
The expression and activity of glucose transport in the intestinal brush border are regulated by short-term and longer-term processes. In the short term, activity of glucose transport is increased by both protein kinase A- and C-dependent processes.198 The mechanism of this enhanced activity is an increase in the number of membrane transporters, mediated by changes in exocytosis and endocytosis of membrane vesicles that contain the transport protein. Longer-term regulation of glucose transport is mediated by changes in the expression of SGLT, which is controlled by changes in the nutrient environment.199
Glucose-galactose malabsorption is characterized by the neonatal onset of severe diarrhea upon the newborn’s ingestion of breast milk or regular infant formula. Several distinct mutations in the SGLT1 gene have been identified (e.g., a missense mutation resulting in a change of amino-acid residue 28 from an aspartate to an asparaginase),9 and most of these mutations are responsible for defective passage of the SGLT1 through the biosynthetic machinery from the endoplasmic reticulum or for poor trafficking from the Golgi apparatus to the brush border membrane. Rarely do mutant SGLT1 proteins reach the brush border at a normal rate, in which case, the glucose transport is defective.192
Fructose absorption occurs by facilitated diffusion: Transport occurs not against a concentration gradient but with a carrier protein to achieve transport rates greater than one would expect from simple diffusion. This process is completely independent of glucose absorption. Studies in humans have shown that there is a saturable, facilitative transport system for fructose in the intestinal epithelium that has a lower activity than that for transport of glucose and galactose. The protein responsible for most apical membrane fructose transport is a member of the facilitative monosaccharide transporter family called GLUT5, encoded by the gene SLC2A5. This 501-amino acid protein in humans has 12 membrane-spanning domains, as do other GLUT molecules, and transports fructose exclusively.200 GLUT2, however, might assist in absorption of excess luminal fructose. Little fructose is metabolized in the enterocytes, and fructose is transported across the basolateral membrane (by GLUT2 and GLUT5) and is taken up and metabolized rapidly by the liver, resulting in low postabsorptive blood levels of fructose. There may be more than one type of fructose transport system.
Malabsorption of fructose in humans can be prevented by the simultaneous administration of glucose, suggesting that another glucose-responsive system may be present in the enterocytes. No inherited disorders of fructose transport (GLUT5) have been reported.201 Overall, fructose is not as well absorbed as glucose. High levels of dietary fructose can lead to dietary intolerance. Ingestion of large amounts of fructose can cause diarrhea, excessive intestinal gas, and recurrent abdominal pain. Fructose malabsorption has been associated with similar symptoms.202
Debate has developed over the mechanism of the passive or diffusive component of intestinal glucose absorption and, indeed, whether it exists.203 Pappenheimer and colleagues proposed that paracellular solvent drag contributes a passive component, which, at high concentrations of sugars similar to those in the jejunal lumen immediately after a meal, is several-fold greater than the active component mediated by the Na+-glucose cotransporter SGLT1.204 Other investigators have argued that the kinetics of glucose absorption can be explained solely in terms of SGLT1 and that a passive or paracellular component plays little, if any, part.205 More-recent data suggest that the passive component of glucose absorption exists but that it is facilitated because it is mediated by the rapid glucose-dependent activation and recruitment of the facilitative glucose transporter GLUT2 to the brush-border membrane. This is regulated through a protein kinase C–dependent pathway activated by glucose transport through SGLT1 and also involves mitogen-activated protein kinase (MAP kinase) signaling pathways.206
Exit from the Epithelium
Most hexoses are exported from the epithelial cell by way of the basolateral membrane, although small amounts are used for intracellular metabolism. Exit across the basolateral membrane depends on facilitated diffusion (not requiring energy) via a specific carrier. Two genes that are expressed in the small intestine—GLUT2, the basolateral-membrane–associated glucose transporter, and GLUT5, an apical membrane fructose transporter—encode these facilitative sugar transport proteins.207 GLUT2 has molecular structural characteristics similar to those of the other members of this family of genes. The protein has 500 amino acids with many hydrophobic residues that predict a total of 12 membrane-spanning domains. There is one long extracellular loop between membrane-spanning domains 1 and 2 that contains an asparagine that is N-glycosylated and one long cytoplasmic loop between membrane-spanning domains 6 and 7. Once the hexoses have entered the interstitial space, they pass onward by diffusion into the portal circulation.
A congenital defect in glucose transport by GLUT2 has been identified (Fanconi-Bickel syndrome). Because GLUT2 is normally expressed in the liver, pancreas, and kidney as well as in the intestine, defects in this transporter are expected to have a widespread effect on glucose homeostasis. Indeed, patients with the Fanconi-Bickel syndrome exhibit tubular nephropathy, fasting hypoglycemia, rickets, stunted growth, and hepatomegaly secondary to glycogen accumulation.208,209
The accepted dogma of intestinal glucose absorption at the basolateral membrane by glucose transporters has been challenged by studies of intestinal glucose absorption in GLUT2 null mice and in patients with GLUT2 deficiency; in both cases, glucose absorption was not impaired. Additional work has suggested that there are two separate pathways for the exit of sugar from enterocytes: one that involves GLUT2 and another that requires glucose phosphorylation, the transfer of glucose-6-phosphate into the endoplasmic reticulum and the release of free glucose into the blood. The release mechanism is unclear, but it has been proposed to involve vesicle trafficking. This postulate is supported by oral tolerance tests on a patient with congenital deficiency in glucose-6-phosphate translocase1, in whom glucose absorption was impaired but not eliminated.209
Not all potentially digestible carbohydrate is absorbed in the small intestine. As much as 20% of dietary starch escapes into the colon, particularly that derived from cereals and potatoes.2 Most of this unclaimed carbohydrate, however, is metabolized by colonic bacteria, and the short-chain fatty acids thus derived are readily absorbed; hydrogen and methane also are generated and contribute to production of flatus.
PROTEIN
DIETARY INTAKE
Dietary proteins are the major source of amino acids, and in the average Western diet they provide about 10% to 15% of energy intake. Affluent populations ingest more protein than they need to maintain their normal balance. An average adult in a Western country consumes 70 to 100 g of protein per day, whereas the poor in Asia and Africa consume 50 g or less per day.48 Recommended dietary requirements vary from 0.75 to 1 g/kg of body weight per day, but deficiency states are rare even with intakes of 0.5 g/kg per day or less. In the United Kingdom, protein intake has remained fairly steady since the mid-1970s, but with the marked decline in fat and carbohydrate ingestion, the ratio of protein to nonprotein energy intake has risen.48 Little harm appears to occur in the unusual subgroups of society who consume very large amounts of protein, although renal function can be impaired by this dietary habit. The Masai tribes of Africa and the Gaucho of South America, who consume 250 to 300 g (largely of animal origin) per day, suffer no obvious untoward effects from this consumption.48
The variety of types of animal and plant proteins is enormous. Generally, plant proteins are less digestible than those derived from animals, but some fibrous animal proteins, such as keratin and collagen, also are relatively indigestible. High-proline proteins such as the glutenins are less thoroughly digested than are others. The quality of proteins depends largely on their amino acid composition; proteins rich in essential amino acids are regarded as being of high quality. Proteins from animal sources have a high content of essential amino acids, unlike proteins from certain specific plant sources, which are said to be incomplete because they lack or contain only certain essential amino acids. Such deficiencies in essential amino acids typically are overcome in a mixed diet; however, the relative contribution of animal and plant protein varies according to geographic region. In developed countries, such as in North America and Europe, animal protein contributes about 70% of the total protein compared with developing nations in the Middle East and Africa where the animal protein contribution can be as low as 20%.
Food processing, by heat for example, can cause inter- and intramolecular bonding in the proteins to produce polymeric forms that are relatively resistant to hydrolysis.1 Other constituents of the diet also can interfere with protein digestion; for example, starch and reducing sugars have the potential to impair digestion.48 Despite these interferences, digestion and absorption of proteins are remarkably complete, and only about 3% to 5% of ingested nitrogen is lost in the stool, probably because of the resistance of some peptide bonds to hydrolysis.1 A few selected proteins are resistant to proteolysis in the small intestine, including secretory immunoglobulin (Ig) A and intrinsic factor. Among the 20 common amino acids that form animal and plant proteins, 8 cannot be synthesized by animals: leucine, isoleucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. These eight essential amino acids have to be ingested, usually in plant-derived foods. Histidine also is required for growth in infants.
PROTEINS FROM ENDOGENOUS SOURCES
Almost half of all protein that enters the intestine is derived from endogenous sources. Of this, about 20 to 30 g/day are derived from secretions of salivary, gastric, biliary, pancreatic, and mucosal origin. Another 30 g/day of protein are provided by epithelial cells desquamated from the villus tips, and 2 g of plasma proteins are delivered into the intestinal lumen each day.
DIGESTION AND ABSORPTION
Pepsins
Digestion of proteins begins in the stomach with the action of pepsins. Pepsins are released from their precursor pepsinogens with the loss of a small basic peptide by autoactivation in an acid pH. At neutral or alkaline pH, the pepsinogen amino-terminal region is folded in such a way as to mask a catalytic site. In the acidic environment of the stomach, the catalytic site is uncovered and then proceeds to remove the amino-terminal region, which consists of 40 amino acids, thereby generating the active form of the molecule, pepsin. Pepsinogen release from chief cells is stimulated by gastrin, histamine, and cholinergic stimulation and closely mirrors acid secretion.210
The pepsins are a family of endoproteases that hydrolyze internal peptide bonds in proteins. They act preferentially on peptide bonds formed by the aromatic amino acids phenylalanine and tyrosine and by the branched-chain amino acid leucine. There are two immunologically distinct groups of pepsins (groups 1 and 2), although eight fractions are identified electrophoretically. Both of the immunologically separated species are secreted by chief cells, but group 2 isoforms also are present in the mucus cells in the oxyntic and pyloric areas of the stomach and in Brunner’s glands of the duodenum. Their substrate specificities vary little, but their pH optima differ slightly (between 1.8 and 3.5); all are irreversibly inactivated in alkali.
Pepsins remain active at the acid pH of gastric contents to produce a mixture of peptides with a small portion of amino acids. Pepsin activity is therefore confined to the stomach. The completeness of gastric proteolysis depends, in part, on the rate of gastric emptying, the pH of intragastric contents, and the types of protein ingested. Moreover, the products of protein digestion by pepsins in the stomach can further influence acid and pepsinogen secretion as well as gastric emptying. Subjects who are achlorhydric or who have lost control of gastric emptying as a result of pyloroplasty or partial gastrectomy do not appear to have a problem with assimilation of protein, suggesting that gastric proteolysis is not an essential component of digestion.
Pancreatic Proteases
In contradistinction to amylase and lipase, which are secreted in their active forms, each of the pancreatic proteases is secreted as a proenzyme and therefore must be activated within the intestinal lumen. Enterokinase (enteropeptidase) plays a key role in proteolysis. It is liberated from its superficial position in the brush border membrane by the action of bile acids,211 its action being to convert trypsinogen to trypsin by removing its hexapeptide NH2 terminus. Trypsin in turn activates the other proteases and continues to split more trypsin from trypsinogen (Fig. 100-14).
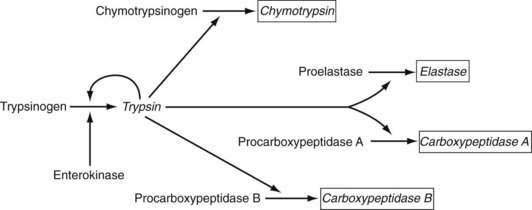
Figure 100-14. Activation of pancreatic proteolytic enzymes. Enterokinase (enteropeptidase) plays a critical role in activating trypsinogen to form trypsin. Trypsin in turn activates not only more trypsinogen but also the other proteolytic enzyme precursors. Products of enzymatic actions are shown in italics.
The proteases are classified as endo- and exopeptidases, according to the sites of the peptide bonds against which they are most active. Endopeptidases include trypsin, chymotrypsin, and elastase, and exopeptidases include carboxypeptidase A and B (Table 100-5).
Table 100-5 Pancreatic Proteolytic Enzymes, Their Sites of Action, and Their Products
| ENZYME | ACTION | PRODUCTS |
|---|---|---|
| Trypsin | Endopeptidase; cleaves internal bonds at lysine or arginine residues; cleaves other pancreatic proenzymes | Oligopeptides |
| Chymotrypsin | Endopeptidase; cleaves bonds at aromatic or neutral amino acid residues | Oligopeptides |
| Elastase | Endopeptidase; cleaves bonds at aliphatic amino acid residues | Oligopeptides |
| Carboxypeptidase A | Exopeptidase; cleaves aromatic amino acids from carboxy terminal end of protein and peptides | Aromatic amino acids and peptides |
| Carboxypeptidase B | Exopeptidase; cleaves arginine or lysine from carboxy terminal end of proteins and peptides | Arginine, lysine, and peptides |
Trypsin, chymotrypsin, and elastase have specificity for peptide bonds adjacent to certain specific amino acids. They split peptide bonds within the protein molecule, whereas exopeptidases remove a single amino acid from the carboxyl terminal end of the peptide. Trypsin produces short-chain oligopeptides that are further hydrolyzed by the exopeptidases, carboxypeptidase A acting on aromatic and aliphatic carboxyl terminals, and carboxypeptidase B acting on peptides containing basic carboxyl terminals. The final products of intraluminal digestion thus are produced by cooperative activity of endo- and exopeptidases and consist of a number of neutral and basic amino acids together with peptides of two to six amino acids in length. About 30% of luminal amino nitrogen is found in amino acids and about 70% is found in oligopeptides.212
In addition to nutrient protein hydrolysis, pancreatic proteases have other functions: They split vitamin B12 from the R protein to which it is linked so that it then can bind intrinsic factor; they increase the turnover of brush border membrane hydrolytic enzymes, and, as discussed earlier, they initiate the final steps in the processing of the SI complex; finally, they may have a role in the inactivation of some organisms.1
Digestion at the Brush Border Membrane and in the Cytoplasm
In contrast to the absorption of carbohydrate, which is largely restricted to uptake of hexose monomers across the brush border membrane, amino acids can be absorbed either as monomers or as di- or tripeptides. Indeed, amino acid absorption is achieved more efficiently in the form of peptides than as single amino acids (Fig. 100-15).213 The fact that the vast majority of the end-products of protein digestion that reach the portal circulation are amino acids, however, speaks strongly in favor of the presence of peptidases in the epithelium.
Figure 100-15. Rates of glycine absorption (mean ± standard error of mean) from perfusion solutions containing equivalent amounts of glycine in free or peptide (diglycine, triglycine) form. Results are from studies in the jejunums of four normal humans.
(From Adibi SA, Morse EL, Masilamani SS, Amin PM. Evidence for two different modes of tripeptide disappearance in human intestine. Uptake by peptide carrier systems and hydrolysis by peptide hydrolases. J Clin Invest 1975; 56:1355-63.)
Patients with cystinuria and Hartnup’s disease, who have specific defects in the absorption of basic and neutral amino acids, respectively, do not develop protein deficiency states because the absorption of peptides in these patients is normal.214 The discovery that di- and tripeptides are actively transported by the brush border membrane of enterocytes has been valuable in explaining this observation, and it emphasizes the need for critical evaluation of the supposed nutritional advantage provided by elemental diets that consist only of free amino acids.
A range of peptidases is present in the brush border membrane and in the cytoplasm of villus epithelial cells for the hydrolysis of oligopeptides up to approximately eight amino acid residues in length (Table 100-6).215–217 The peptidases on the brush border membrane differ in several important respects from those within the cytoplasm (Table 100-7). About 90% of the dipeptidases are found in the cytoplasm and only about 10% in the brush border, whereas the distribution of hydrolases for tetrapeptides is the reverse of this. Peptidases for pentapeptides and larger molecules are confined almost entirely to the brush border membrane. Cytoplasmic enzymes are much more heat labile than those in the brush border, and there are differences in the electrophoretic mobility patterns for the two sets of enzymes.75
Table 100-6 Peptidases Found on the Brush Border Membrane and in the Cytoplasm of Villus Epithelial Cells
| PEPTIDASE | ACTION | PRODUCTS |
|---|---|---|
| Brush Border Membrane Peptidases | ||
| Amino-oligopeptidases (at least two types) | Cleave amino acids from carboxy terminus of 3-8 amino acid peptides | Amino acids and dipeptides |
| Aminopeptidase A | Cleaves dipeptides with acidic amino acids at amino terminus | Amino acids |
| Dipeptidase I | Cleaves dipeptides containing methionine | Amino acids |
| Dipeptidase III | Cleaves glycine-containing dipeptides | Amino acids |
| Dipeptidyl aminopeptidase IV | Cleaves proline-containing peptides with free α-amino groups | Peptides and amino acids |
| Carboxypeptidase P | Cleaves proline-containing peptides with free carboxy terminus | Peptides and amino acids |
| Gamma-glutamyl transpeptidase | Cleaves γ-glutamyl bonds and transfers glutamine to amino acid or peptide acceptors | Gamma glutamyl amino acid or peptide |
| Folate conjugase | Cleaves pteroyl polyglutamates | Monoglutamate |
| Cytoplasmic Peptidases | ||
| Dipeptidases (several types) | Cleave most dipeptides | Amino acids |
| Aminotripeptidase | Cleaves tripeptides | Amino acids |
| Proline dipeptidase | Cleaves proline-containing dipeptides | Proline and amino acids |
Table 100-7 Distribution of Peptidase Activity
| SUBSTRATE | BRUSH BORDER MEMBRANE (%) | CYTOPLASM (%) |
|---|---|---|
| Dipeptides | 5-10 | 80-95 |
| Tripeptides | 10-60 | 30-60 |
| Tetrapeptides | 90 | 1-10 |
| Higher peptides | 98 | 0 |
Most oligopeptidases appear to be aminopeptidases; that is, they act by removing residues from the amino terminus of the peptide. The chain length of the peptides is an important factor that determines not only whether the site at which hydrolysis occurs is at the brush border or within the cell but also its rate. Thus, rates of brush border membrane hydrolysis for tripeptides are most rapid and for dipeptides is least rapid, whereas tetra- and pentapeptide hydrolysis rates occupy an intermediate position.213
Distinct from the amino oligopeptidases are at least three other peptidases. Aminopeptidase A has specificity for peptides with acidic amino acids at their amino termini. Aminopeptidases 1 and 3 (distinguished on electrophoretic mobility) have specificities for different substrates with different amino acid peptide bonds.1
Proline-containing oligopeptides are not readily hydrolyzed by most proteases, although many proteins—including collagen, gliadin, and casein—are rich in proline. Two proline-specific carboxypeptidases, however, have been demonstrated in the brush border membrane. They have slightly different substrate specificities218 and together with a cytoplasmic proline dipeptidase, they are likely to be responsible for hydrolysis of proline-rich peptides.
A number of other brush border membrane peptidases need to be mentioned. Gamma glutamyl transpeptidase hydrolyzes gamma glutamyl peptide bonds, with the transfer of the gamma glutamyl group to another amino acid to form a gamma glutamyl amino acid or peptide derivative.1 The role of this brush border membrane in the intestine is not yet clear. Folate conjugase, an enzyme concerned with hydrolysis of dietary folate, will be considered later. The recent demonstration of angiotensin I-converting enzyme (ACE) in intestinal mucosa suggests that it, too, might hydrolyze dietary peptides.219 Indirect evidence suggests that endopeptidases also may be present on the brush border membrane, because protein digestion occurs, even in the complete absence of pancreatic function; these enzymes have yet to be isolated.
As with other proteins, synthesis of each specific peptidase occurs in the rough endosplasmic reticulum, and following transfer to the Golgi apparatus, the proteins are transported to the brush border membrane, where they are inserted by exocytic fusion.220,221 They are attached to the basement membrane by short anchoring pieces in a manner analogous to the attachment of disaccharidases222; however, unlike the latter enzymes, there is little post-translational processing, either within the cytoplasm or by pancreatic enzymes on the brush border.
Of the cytoplasmic dipeptidases, the most abundant appears to be one with broad specificity for neutral amino acid-containing dipeptides. The tripeptidase isolated has broad specificity for amino-terminal residues and high specificity toward tripeptides containing proline as the amino-terminal residue, which distinguishes it from the brush border membrane amino oligopeptidase. Other characteristics of the tripeptide that are required for rapid hydrolysis include a free α amino group, an α carboxyl group, and an l-configuration for the two amino acid residues.223
Absorption of Peptides
Substrate inhibition studies indicate that tri- and dipeptides inhibit uptake of either peptide from the lumen, but neither is affected by single amino acids. Such evidence suggests that small peptides use a separate transporter system from those used by single amino acids. By contrast, tetrapeptide absorption is inhibited by single amino acids but not by di- and tripeptides, suggesting that tetrapeptides are split before absorption.
The advantage of dipeptide absorption over single amino acid absorption has been largely demonstrated experimentally with single peptides containing a single amino acid, usually glycine.213 Several studies, however, have demonstrated the kinetic advantage of peptides over amino acids, even in complex mixtures of partial digests of proteins.224,225 Absorption was greater from tryptic hydrolysates of proteins than from a mixture of amino acids. Furthermore, the wide variation in rates of absorption seen with different individual amino acids was reduced when they were presented as a tryptic hydrolysate.
A number of other factors influence digestion and absorption. The presence of amino acids in the lumen inhibits peptide hydrolysis (product inhibition), whereas luminal glucose and luminal acidification each inhibit amino acid and peptide absorption.213 There is good evidence to suggest that di- and tripeptides are taken up by a single type of transporter with some stereospecificity because the length of the amino acid side chains on the di- or tripeptides is important; the longer the side chain the more preferred the substrate for the absorption site (Table 100-8).226 The l-isomers of the amino acids in dipeptides are much preferred to the d-isomers, whereas the presence of acidic and basic amino acid residues in dipeptides reduces affinity for the transport system, compared with neutral amino acid residues. Affinity is also greater for dipeptides than for tripeptides, at least in the example of peptides that contain glycine. The transporter for peptides is not dependent on sodium, but cotransport with protons may occur instead.227
Table 100-8 Relative Specificities of Intestinal Peptide Transporters
| DIPEPTIDES | TRIPEPTIDES |
|---|---|
| L-form of amino acids | d-form |
| Neutral amino acids | Acidic or basic amino acids |
| Long side chains | Short side chains |
The peptide transporter for human small intestine has been cloned228,229 and is a member of a superfamily of H+-coupled peptide transporters. The human protein consists of 708 amino acids, with a predicted core molecular size of 79 kDa that contains 12 transmembrane domains. The gene is located on chromosome 13. In humans, it is expressed in the small intestine (duodenum, jejunum, and ileum), but not in the esophagus, stomach, or colon. In the small intestine it is expressed only on absorptive epithelium. It recognizes a variety of neutral, anionic, and cationic dipeptides as substrates,230,231 which explains the broad substrate specificity of the intestinal peptide transport system.
The most interesting feature of this transport process is that it uses a transmembrane electrochemical H+-gradient rather than a transmembrane electrochemical Na+-gradient as its driving force.232 There is an acid pH microclimate on the luminal surface of the intestinal brush border membrane that creates a H+-gradient across the brush border membrane in vivo. This acid pH microclimate is generated and maintained by the combined action of the Na+-H+ exchanger in the brush border membrane and Na+,K+-adenosine triphosphatase (ATPase) in the basolateral membrane of the enterocyte. The mechanism of the transport process is a simultaneous translocation of H+ and peptide substrate involving a single H+ binding site on the protein (Fig. 100-16).233,234
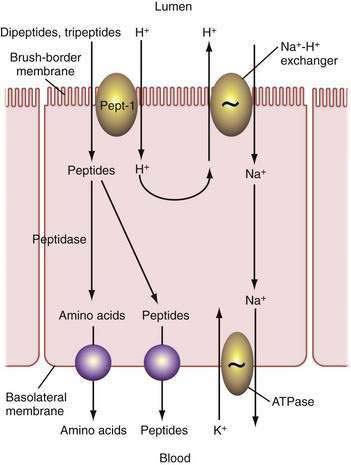
Figure 100-16. Peptide transport across the intestinal epithelium. This transport process uses a transmembrane H+-gradient rather than a transmembrane electrochemical Na+-gradient as the driving force. The acid pH microclimate on the luminal surface of the intestinal brush border membrane is generated and maintained by the combined action of the Na+-H+ exchanger in the brush border membrane and Na+,K+-ATPase (adenosine triphosphatase) in the basolateral membrane of the enterocyte. The mechanism of the transport process is a simultaneous translocation of H+ and peptide substrate involving a single H+ binding site on the protein, Pept-1.
A multitude of processes are involved in the absorption of peptides. The well-established processes include a Na+-H+ exchanger located in the brush-border membrane that maintains an intracellular alkaline pH, a Na+,K+-ATPase located in the basolateral membrane that maintains an inside negative membrane potential, and several cytoplasmic peptidases that prevent intracellular accumulation of absorbed peptides. These enzymes convert most of the absorbed oligopeptides to amino acids that are either used by the absorbing cells or are released into the portal circulation via the amino acid transporters located on the basolateral membrane of these cells. The oligopeptides that escape hydrolysis by the cytoplasmic peptidases are transported across the basolateral membrane into the portal circulation by a peptide transporter that appears to be different from the Pept-1 transporter. Oligopeptide transport could be regulated by alteration in activity or abundance of Pept-1, Na+-H+ exchanger, Na+,K+-ATPase, cytoplasmic peptidases, and basolateral oligopeptide transporter.235,236
Studies of individual substrates and hormones in cell culture have shown that the membrane population of Pept-1 is increased by dipeptides, certain amino acids, insulin, and leptin and decreased by epidermal growth factor (EGF) and triiodothyronine. In the case of dipeptides, EGF, and thyroid hormone, there are parallel changes in the gene expression brought about by alteration of transcription or stability of Pept-1 mRNA. In contrast, treatment with insulin and leptin does not induce any alteration in the Pept-1 gene expression, and the mechanism of increased protein expression appears to be increased trafficking from a preformed cytoplasmic pool to the apical membrane.235,236
Transport of Amino Acids
Whereas there appears to be only one type of dipeptide transporter in the brush border membrane for the 400 possible dipeptides, there is a multiplicity of transport mechanisms for the 20 amino acids. In adults, these are situated on villus enterocytes and involve carrier-mediated active transport or facilitated diffusion processes, which typically depends on the Na+ gradient as the driving force; a small portion may be absorbed by simple diffusion, independent of any ion gradient. There has been some difficulty in defining the number and types of transporters because of their overlapping specificities; several amino acids use a number of different transport systems (Table 100-9). On the basis of kinetic studies, at least four active processes have been identified for transport of neutral amino acids across the apical cell membrane. Each is electrogenic and sodium dependent. One has broad specificity for a number of neutral amino acids (NBB system); a second provides another route for phenylalanine and methionine (PHE system); a third provides a mechanism for imino acid absorption (IMINO system); and the fourth transports beta amino acids. Separate sodium-dependent, active transport processes for basic and acidic amino acids also have been demonstrated, and some evidence suggests that facilitated diffusion of these types of amino acids also occurs, although this is likely to be a minor pathway.
Table 100-9 Major Amino Acid Transport Systems Detected in Intestinal Epithelial Cells
| TRANSPORT SYSTEM | SUBSTRATES |
|---|---|
| Brush Border Membrane | |
| Neutral Amino Acids | |
| SLC6A19 | Neutral amino acids |
| SLC36A1 | Imino acids; proline, hydroxyproline |
| SLC6A20 | Imino acids |
| SLC6A14 | Neutral and cationic amino acids |
| SLC1A5 | Alanine, serine, cysteine, glycine, asparagine |
| SLC7A9/SLC3A1 | Neutral amino acids, cationic amino acids, cysteine |
| Basic Amino Acids | |
| Lysine, cysteine, basic amino acids | |
| Acidic Amino Acids | |
| SLC1A1 (X-GA−) | Glutamate, aspartate |
| Basolateral Membrane | |
| L | Broad selectivity |
| A | Broad selectivity |
| SLC1A5 (ASC) | Neutral amino acids, alanine, serine, cysteine |
| N | Glutamine, histidine, asparagine |
Genomic advances have allowed most mammalian amino acid transport functions to be attributed to specific gene products: At least 52 amino acid transporter-related gene products are grouped within 12 solute carrier families, with their own new nomenclature.237 The classic Na+-dependent imino acid transporter has been identified as the human PAT1 (human proton-coupled amino acid transporter 1) or solute carrier SLC36A1. This high-capacity imino acid carrier has been localized to the small intestinal luminal membrane and transports imino and amino acids (glycine, proline, alanine, taurine).238,239 Human PAT1 mediates 1 : 1 symport of protons and small neutral amino acids. The acid microclimate of the brush border membrane drives transport of the amino acids into the cytosol. Transport activity is independent of Na+ and Cl− (Fig. 100-17). In addition, the IMINO system is a Na+-dependent transporter with specificity toward the imino acids proline and hydroxyproline. The protein responsible for this transport activity is SIT1(Na+-coupled imino acid transporter 1).240,241
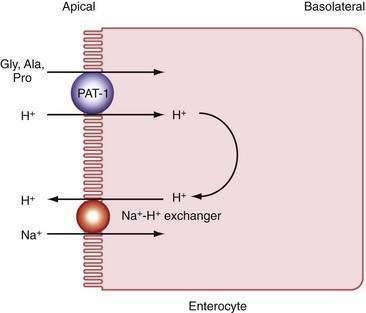
Figure 100-17. Intestinal amino acid transport. The human protein-coupled amino acid transporter (PAT-1) is involved in the absorption of small amino acids across the apical membrane. The acid microenvironment generated by a Na+-H+ exchanger provides the electrochemical proton gradient that drives amino acids to the cytosol. Ala, alanine; Gly, glycine; Pro, proline.
(Boll M, Daniel H, Gasnier B. The SLC36 family: Proton-coupled transporters for the absorption of selected amino acids from extracellular and intracellular proteolysis. Pflugers Arch 2004; 447[5]:776-9).
System B0,+, also present on the brush border membrane, mediates the Na+ and Cl− coupled electrogenic transport of neutral as well as cationic amino acids across the brush border membrane. The gene encoding the protein responsible for this activity, ATB0,+ is located on human chromosome X. A separate Na+-dependent transport system, X−AG, (SLC1A1) is specific for anionic amino acid, aspartate, and glutamate. The glutamate transporter expressed in the intestinal brush border membrane is known as the EAAT3. This transporter is defective in the inherited amino acid transport defect known as dicarboxylic aciduria.
Hartnup’s disease is a disorder of renal and gastrointestinal neutral amino acid transport that is inherited as an autosomal recessive trait. The gene causing Hartnup’s disease has been localized to chromosome 5p (it had previously been localized to chromosome 19), and a new gene, SLC6A19, a sodium-dependent and chloride-independent neutral amino acid transporter, has been suggested as the defective gene by two separate groups.242,243 This transporter has been shown to be expressed in the intestine and has properties of system B0. System B0 refers to a broad range of amino acids with neutral (0) charge. SLC1A5 is the proposed ASC carrier for the neutral amino acids alanine, serine, and cysteine.244 Whereas the SLC1A1 carrier cotransports 3 Na+ and 1 H+ with countertransport of 1 K+, the SLC1A5 transporter mediates Na+-dependent transport.
The B0,+ is a Na+-independent transport system that recognizes neutral and cationic amino acids in addition to the disulfide amino acid cystine. It is a heterodimer, consisting of a light and heavy chain, with the genes responsible for each chain being found on chromosomes 2 and 19, respectively. This transporter is defective in cystinuria.
Several hormones have been shown to alter the amino acid and peptide transport process in the intestine. Somatostatin and vasoactive intestinal polypeptide decrease these transport processes, whereas EGF, neurotensin, cholecystokinin, and secretin enhance them. Human Pept1 appears to be inhibited by protein kinase C245 and cyclic adenosine monophosphate (cAMP).246 The expression of the intestinal peptide transporter is also modulated by dietary protein content.247 Even though the peptide transporter is expressed along the entire small intestine, diet-induced changes in the expression of the transporter are specific to certain regions. A high-protein diet increases the steady state levels of the transporter-specific messenger RNA in the middle and distal regions of the small intestine. The expression of the brush border peptidases dipeptidylcarboxypeptidase and dipeptidylaminopeptidase IV, which release dipeptides from oligopeptides, also are enhanced by a high-protein diet.
Exit from the Epithelium
Exit through the basolateral membrane operates via a number of different mechanisms that involve active transport and diffusion of both facilitated types.248 Active, sodium-dependent processes exist at this membrane for the uptake of neutral amino acids, which presumably supply nutrients for crypt cells and for villus enterocytes during fasting when a luminal source is unavailable (see Table 100-9). Villus enterocytes normally receive the amino acids necessary for production of their own protein from luminal nutrients; crypt cells obtain their supply from the portal circulation. Of all the amino acids, glutamine appears to be a unique and major source of energy for enterocytes; ammonia is an important metabolic byproduct of this process. Active uptake of glutamine at the basolateral membrane, as well as via apical membrane processes, is therefore of particular importance.
It has been estimated that approximately 10% of amino acids are used in the production of enterocyte protein. Some of these proteins are secreted across the basolateral membrane specifically by villus enterocytes, including apo A-I and apo A-IV, secretion of which increases many-fold after a fatty meal.48
The intestinal basolateral membrane possesses a set of amino acid transport systems that differ from those in the brush border membrane. The amino acid transport systems in the basolateral membrane function to export amino acids from the enterocytes into the portal circulation during feeding. They also participate in the import of amino acids from the portal circulation into the enterocyte for cellular metabolism when amino acids are not available from the intestinal lumen, such as between meals. The intestinal basolateral membrane also possesses a peptide transporter system that is probably identical to that in the brush border membrane and that facilitates the exit of hydrolysis-resistant small peptides from the enterocyte into the portal circulation.
Several well documented amino acid transport systems have been described in the basolateral membrane. System y+L is the amino acid exchanger that permits Na+-independent efflux of cationic amino acids from intestinal cells into the blood coupled to the Na+-dependent influx of neutral amino acids from the blood into intestinal cells. System A is a Na+-coupled transport system for neutral amino acids, including glutamine, that plays a role in the entry of amino acids from the blood into intestinal cells for cellular metabolism. This Na+-coupled neutral amino acid transporter (SNAT) consists of three subtypes, SNAT1, 2, and 4; SNAT2 is expressed in the small intestine.249
Very small amounts of dipeptides have been detected in the portal circulation after a meal, but the great majority of absorbed products of protein digestion that reach the circulation are in the form of single amino acids. A somewhat surprising finding is that digestion of protein continues into the ileum, with approximately 40% of ingested protein undergoing transport in this segment of small intestine.250
CHAPTER 100 Digestion and Absorption of Nutrients and Vitamins
Most nutrients are absorbed with remarkable efficiency: Less than 5% of ingested carbohydrate, fat, and protein is excreted in the stool of adults consuming a normal diet.1 Even much of the indigestible dietary fiber is absorbed from the colon as short-chain fatty acids that are liberated by bacterial breakdown of fiber.2 The intestinal tract of neonates is less efficient: infants fail to absorb 10% to 15% of their dietary fat, and in premature infants as much as 25% to 35% of fat may be lost in the stool.3,4 In old age, nutrient absorption remains highly efficient unless the intestine becomes diseased.
Despite considerable variations in types of food ingested and nutritional intake across national and racial groups, absorption remains efficient. Absorptive mechanisms adapt to the nature and amount of various nutrients presented to the intestinal tract. Such changes occur not only during early development5 but also throughout life and at times of specific need, such as during pregnancy.6 In achieving the overall objective of nutrient absorption, the different parts of the gastrointestinal tract act in a closely integrated and coordinated manner under the control of neural and humoral regulatory mechanisms.
The understanding of intestinal digestion and absorption at a molecular level has improved our knowledge of the integration and coordination of these functions within the gastrointestinal tract. The pharmacokinetics and pharmacodynamics of several key carbohydrate, fat, peptide, amino acid, vitamin, and nutrient transporters are increasingly understood. In this chapter, integration of intestinal function with the dietary intake, digestion, and absorption of major nutrients (fat, carbohydrate, and protein) and essential micronutrients (vitamins and trace elements) is discussed. The evolving genetic and molecular basis of these functions also is evaluated.
DIGESTION AND ABSORPTION OF NUTRIENTS
AN OVERVIEW OF THE DIGESTIVE PROCESS
The cerebral phase of digestion, whether triggered by the sight, smell, or thought of food, initiates the digestive process. Salivary and gastric secretory responses to this type of stimulus are mediated via the autonomic nervous system, and there is modest stimulation of pancreaticobiliary secretion via the vagus nerve.7 The further stimulus of nutrients in the mouth and upper gastrointestinal tract markedly potentiates secretion by humoral and local neural mechanisms (see Chapter 1).8
The rapidity with which food is normally chewed and swallowed affords little time for significant oral digestion of nutrients; however, good mastication and mixing with saliva initiates digestion of starch by salivary amylase. In infants, digestion of fat is begun in the stomach by gastric lipase. Gastric acid would soon switch off these enzymes were it not for the buffering capacity of food that allows some digestion to continue. The optimal pH for gastric lipases is 4.5 to 6.0, and it has been suggested that a considerable portion of dietary triglyceride may be digested by these lipases.9,10 Protein digestion begins in the stomach when gastric pepsinogens are converted to pepsins by gastric acid. Pepsins become increasingly active as intraluminal pH falls and therefore the digestive action of pepsins on proteins is restricted to the stomach.
During ingestion of food, the stomach may become distended, but intragastric pressure rises little because of neurally mediated receptive relaxation. The mechanisms by which subjects perceive satiety and, therefore, cease eating are complex and explained only partly by the sensation of fullness. Although dozens of enzymes and hormones are secreted by the gastrointestinal tract in response to intraluminal food, only a few are able to influence food intake directly. Satiety signals are relayed to the hindbrain, either indirectly via nerves such as the vagus nerve or else directly via the blood. Most factors that influence how much food is eaten during individual meals act by changing the sensitivity to satiety signals.11
Cholecystokinin (CCK), gastrin-releasing peptide, and apolipoprotein A-IV (apo A-IV) have all been implicated as messengers that transmit the satiety signal to the central nervous system (CNS).12,13,14 They potentiate each other’s actions, and a combination of these agents may participate in the satiety signal. The administration of exogenous CCK or other satiety signals causes smaller meals to be consumed, whereas blocking the action of endogenous CCK and other satiety signals allows larger meals to be consumed.11,15,16 Additional peptides, known as the anorectic peptides, including peptide YY (PYY), pancreatic polypeptide (PP), glucagon-like peptide 1 (GLP-1), and oxyntomodulin also have been shown to decrease appetite and promote satiety in animal and human models.17
Apo A-IV is a glycoprotein synthesized by the enterocytes of human intestine and the hypothalamus, especially the arcuate nucleus. Intestinal apo A-IV synthesis is markedly stimulated by fat absorption and does not appear to be mediated by the uptake or re-esterification of fatty acids to form triglycerides. The local formation of chylomicrons acts as a signal for the induction of intestinal apo A-IV synthesis. Intestinal apo A-IV synthesis is also enhanced by a factor from the ileum (probably peptide tyrosine-tyrosine [PYY]), as well as neuropeptide Y (NPY) and pancreatic polypeptide (PP).18 Inhibition of food intake by apo A-IV is mediated centrally. The stimulation of intestinal synthesis and secretion of apo A-IV by lipid absorption are rapid; thus apo A-IV plays a role in the short-term regulation of food intake. There also is evidence suggesting that apo A-IV may be involved in the long-term regulation of food intake and body weight, because it is influenced by leptin and insulin. Chronic ingestion of a high-fat diet blunts the intestinal and the hypothalamic apo A-IV response to lipid feeding.19 Hypothalamic apo A-IV level is reduced by food deprivation and restored by lipid feeding.20,21
Leptin, a hormone released from fat cells, is an important peripheral signal from fat stores that modulates food intake by acting on receptors in the arcuate nucleus and hypothalamus.22 Leptin deficiency and leptin receptor defects produce massive obesity. Only one gastrointestinal signal, ghrelin, has been shown to increase appetite.11
The major digestive processes are initiated in the duodenum. Delivery of chyme from the stomach is delicately adjusted so that it enters the duodenum at a controlled rate, thus allowing efficient mixing with pancreaticobiliary secretions. Control of gastric emptying is thus critical to ensuring optimal digestion. The characteristics of gastric contents that determine the rate at which the stomach empties include their consistency, pH, osmolality, and lipid and calorie content (Fig. 100-1).23 The pylorus is selective in that it allows rapid passage of liquids while retaining solid particles with diameters of 2 mm or larger.24 Thus, large particles are retained and progressively reduced in size by the gastric mill, a process referred to as trituration. Trituration ensures that particles will be small enough to allow them reasonably close apposition to digestive enzymes once the nutrient is allowed to enter the duodenum. Meals of high viscosity empty more slowly than do those of low viscosity.
Figure 100-1. Some factors that delay gastric emptying. Receptors for acid, osmolarity, fatty acids, and other nutrients in the duodenum signal gastric delay via neurohumoral mechanisms. Food particles larger than 2 mm in diameter (large circles) are rejected by the antrum. Nutrients in the ileum and colon also influence gastric emptying by the ileal brake mechanism (see Chapter 48). GLP, glucagon-like peptide; peptide YY, peptide tyrosine-tyrosine.
Duodenal mucosal receptors for pH and osmolality trigger a delay in gastric emptying when the gastric effluent is acidic or hyper- or hypotonic.25,26 When duodenal luminal contents are neutralized by pancreaticobiliary bicarbonate and osmolality is adjusted by water fluxes, gastric emptying is encouraged once more. This careful titration in the duodenal lumen ensures that nutrients are presented optimally to the pancreatic enzymes, which function best at neutral pH.
The total calorie content of meals also controls gastric emptying rates; on average, the human stomach delivers about 150 kcal/hr to the duodenum.27 An increase in the size or energy density of a meal leads to a corresponding increase in the rate of delivery. Receptors for fatty acids, amino acids, and carbohydrates in the duodenal mucosa are involved in this response, which probably is mediated by both neural and humoral feedback mechanisms.28
Gastric emptying additionally is controlled by a mechanism involving the ileum and colon. If much nutrient escapes digestion and absorption in the jejunum, its presence in the ileum and colon delays gastrointestinal transit, and this again provides more time for digestion and absorption.29,30 This ileal brake probably is mediated by a neurohumoral mechanism, for which various neurotransmitters and hormones have been implicated including peptide YY and the glucagon-like peptides-1 and -2 (GLP-1 and GLP-2).31,32
The GLPs are synthesized in and cosecreted from enteroendocrine cells in the small and large intestine in response to luminal carbohydrate and fat. GLP-1 promotes efficient nutrient assimilation by decreasing appetite, slowing gastric empting and enhancing glucose-induced insulin secretion. GLP-2 is cosecreted with GLP-1and also regulates energy absorption by its effects on nutrient intake, gastric acid secretion, gastric emptying, and nutrient absorption. Circulating levels of GLP-1 and GLP-2 are low in the fasted state and increase rapidly following ingestion of nutrients.33–37
The gallbladder is stimulated to contract and the pancreas to secrete simultaneously in response to the presence of nutrients in the duodenal lumen. A range of nutrient receptors stimulates the release of CCK and secretin from mucosal endocrine cells into the portal circulation, and these are largely responsible for this response. Exocrine pancreatic secretion is primarily controlled by cephalic mechanisms (the vagus nerve), gastric mechanisms (acid and pepsin secretion, and nutrients delivered into the duodenum by gastric emptying), and intestinal mechanisms (secretin and CCK) (see Chapter 56).
CCK and other enterohormones stimulate the pancreas by excitation of sensory nerves and by triggering of long vagovagal or enteropancreatic reflexes. Numerous neurotransmitters, such as acetylcholine and nitric oxide, and certain neuropeptides, such as gastrin-releasing peptide (GRP), generated by neurons of the enteric nervous system, have been implicated in the regulation of the exocrine pancreas. CCK appears to act via vagal cholinergic pathways to mediate pancreatic enzyme secretion. Human pancreatic acini lack functional CCK-A receptors, explaining why a CCK infusion that produces plasma CCK levels similar to those seen postprandially stimulates pancreatic exocrine secretion by an atropine-sensitive pathway.38 Under physiologic conditions, cholinergic vagal afferent pathways rather than pancreatic acinar cells represent the primary targets on which CCK can act as a major mediator of postprandial pancreatic secretion.38
Serotonin (5-hydroxytryptamine, 5-HT) released from enterochromaffin cells in the intestinal mucosa and nerve terminals of the enteric nervous system and the intra-pancreatic nerves may be involved in both stimulation and inhibitory mechanisms through its various receptor subtypes; 5-HT also mediates the actions of secretin and CCK. A synergistic interaction between CCK and 5-HT at the level of the nodose ganglia might explain the robust postprandial pancreatic secretion despite a modest postprandial increase in plasma CCK. Peptides affecting appetite and originating from the intestine (e.g., leptin and ghrelin) or from the pancreas (e.g., PP and NPY) appear to modulate the exocrine pancreas via hypothalamic centers.39,40
Pancreatic juice provides both positive and negative feedback regulation of pancreatic secretion through mediation of both secretin- and CCK-releasing peptides. Three CCK-releasing peptides have been purified: monitor peptide from pancreatic juice, diazepam-binding inhibitor from porcine intestine, and luminal CCK-releasing factor from rat intestinal secretion. All have been shown to stimulate CCK release and pancreatic enzyme secretion. Pancreatic phospholipase A2 from pancreatic juice and intestinal secretions appears to function as a secretin-releasing peptide.41
The simultaneous release of bile salts, pancreatic enzymes, and bicarbonate provides optimal conditions for further nutrient digestion. The simultaneous release of enteropeptidase (enterokinase) from duodenal mucosa is critically important to the activation of pancreatic proteolytic enzymes. Enterokinase releases trypsin from trypsinogen, thus encouraging proteolysis within the duodenal lumen rather than inside the pancreatic duct. These three factors—bile, pancreatic enzymes, and enteropeptidase—remain separate until they are mixed in the intestinal lumen, which ensures that they become operative only at the site of nutrient delivery.
Adequate lipid digestion is critically dependent on the presence of bile salts and pancreatic lipase and colipase at nearly neutral pH,42 whereas digestion of carbohydrate and protein depends on the combined actions of intraluminal secreted enzymes and then enzymes sited on the brush border membrane and within the intestinal mucosa. The close physical relationship, at the brush border, between the sites for terminal digestion of protein and carbohydrate and the active absorption of digestive products provides a very efficient mechanism for dealing with these nutrients.
Two other simultaneous phenomena encourage efficient digestion and absorption. Ingestion of a meal stimulates salt and water secretion by the jejunal mucosa, and this maintains luminal contents in a sufficiently fluid state for proper mixing and digestion (see Chapter 99).43 The other phenomenon is the motor response of the intestine. After feeding, the characteristic repetitive pattern of motility that occurs during fasting is disrupted. Instead, an apparently disordered pattern is seen, which, presumably, ensures that nutrients are well mixed and brought into close contact with intestinal mucosa (see Chapter 97). There is close integration of the neurohumoral control mechanisms involving the motor and secretory responses of the intestine.44 For rapidly absorbed molecules, intestinal blood flow may be the rate-limiting step.45
Efficient conservation and recycling mechanisms ensure that gastrointestinal secretions are not entirely lost. Gastric acid secretion is balanced to a large extent by pancreaticobiliary bicarbonate secretion, so that overall acid-base balance is not disturbed. Although intact digestive enzymes are reabsorbed only in trace amounts, the nitrogen they contain is reabsorbed after their digestion. Finally, efficient enterohepatic circulation recycles bile salts several times each day so that they may be used approximately twice for each meal.46 Although bile salts are passively reabsorbed throughout the small intestine, most reach the terminal ileum, where they are reabsorbed via specific active absorptive mechanisms. Thus, bile salts remain in the lumen where they are needed for lipid digestion, but they are largely reabsorbed at the last moment to avoid being lost by the colon (Fig. 100-2; see Chapter 64).
Figure 100-2. Enterohepatic circulation of bile salts. Active transport in the ileum retrieves most bile salts, and the small fraction lost into the colon and eliminated in the feces is compensated for by fresh hepatic synthesis (see Chapter 64).
Once intestinal chyme leaves the ileum and enters the colon, most nutrients have been digested and absorbed and colonic function largely serves to dehydrate luminal contents through absorption of salt and water and to store the residuum. Dietary fiber may be digested by bacteria, with release of short-chain fatty acids, which are avidly absorbed; however, short-chain fatty acids do not usually have much nutritional significance, other than in areas of the world where the major source of energy is a high-fiber diet. Short-chain fatty acids, however, are the major source of nutrition for the colonocytes.
FAT
DIETARY INTAKE
In the United States, fat intakes for all age groups and both sexes rose from approximately 34% of total energy consumption in the 1930s to 40% to 42% in the late 1950s to mid-1960s, and then fell steadily to approximately 36% (90 to 100 g/day) in 1984. Saturated and monounsaturated fatty acid intakes fell from 18% to 20% of total energy consumption in the early 1950s to 12% to 13% of energy in 1984; polyunsaturated fatty acid intakes rose from 2% to 4% of energy to 7.5%.47
The majority of fatty acids present in dietary triglyceride are oleate and palmitate (18 : 1 and 16 : 0, respectively).48 In animal triglyceride, most of the fatty acids are long-chained (i.e., longer than 14 carbon chains) and saturated (Fig. 100-3). Polyunsaturated fatty acids such as linoleic and linolenic acid are derived from phospholipids of vegetable origin and, because they cannot be synthesized de novo, they are considered essential fatty acids (Table 100-1).
Figure 100-3. General molecular structure of triglyceride (A) and phospholipid (phosphatidyl choline or lecithin) (B).
Table 100-1 Common Dietary Fatty Acids
| FATTY ACID | CONFIGURATION* |
|---|---|
| Saturated Fatty Acids | |
| Butyric | 4 : 0 |
| Caproic | 6 : 0 |
| Lauric | 12 : 0 |
| Myristic | 14 : 0 |
| Palmitic | 16 : 0 |
| Stearic | 18 : 0 |
| Mono-unsaturated Fatty Acids | |
| Oleic | 18 : 1 |
| Palmitoleic | 16 : 1 |
| Polyunsaturated Fatty Acids | |
| Arachidonic | 20 : 4 |
| Linoleic | 18 : 2 |
| Linolenic | 18 : 3 |
* By convention, the number of carbon atoms in the chain is given by the first figure and the number of double bonds in the chain is given by the second.
The average range of phospholipid ingestion is between 2 and 8 g/day. The most commonly ingested phospholipid is phosphatidyl choline (lecithin), and the predominant fatty acids in phospholipid are linoleate and arachidonate (see Fig. 100-3). More phospholipid is found in the duodenal lumen (10 to 22 g/day) than is ingested, most of which is derived from endogenous sources, particularly bile. Cholesterol intake varies widely but averages about 200 to 250 mg/day.48 Some people consume as much as 500 mg/day.
Commercial hydrogenation of unsaturated bonds in the fatty acids of natural oils raises their melting points, thus allowing production of margarines and spreads of variable consistency. Hydrogenation, in addition to saturation, results in isomerization of cis to trans double bonds.49 Although many commercial products contain partially hydrogenated fats, the content of trans-fatty acids in some margarines exceeds 60%, thus raising concerns about their relationship to cancer induction.50
DIGESTION AND ABSORPTION
Most dietary lipid is absorbed by the upper two thirds of jejunum, although its rate and extent of absorption are influenced by the presence of other foods, particularly dietary fiber, which reduces the rate of absorption.51 The types of ingested fat also appear to influence the absorptive process, both by modifying the morphologic structure of the intestinal mucosa and by influencing its absorptive function for other nutrients such as carbohydrate.52
The insolubility of fat in water dominates the mechanisms that have evolved to digest and absorb lipid. Within the lumen, ingested fat has to be physically released and broken down into emulsion droplets. Following digestion, the products have to be transported across the bulk (lumen) water phase to the lipid epithelial cell membrane. Transfer across the lipid membrane is followed, within the epithelium, by reconstitution into larger lipid molecules, predominantly triglyceride, which then require specialized processing to permit export from the cell. Thus, to be used after ingestion, lipid must pass through three physical phases: water in the lumen, lipid in the epithelial membrane, and water in the lymphatics and bloodstream. Despite these potential barriers, more than 95% of ingested fat is absorbed by adults.
Triglyceride
Liberation of fatty acids from the glycerol backbone of triglycerides (lipolysis) is achieved by lipases acting at the surface of emulsified droplets (Table 100-2). This process occurs initially in the stomach, but most lipolysis is accomplished in the small intestine.53 Intragastric lipolysis might account for 20% to 30% of total intraluminal lipid digestion.54 Gastric lipase, which is of fundic origin, has been demonstrated in the gastric contents of premature neonates and in mucosal biopsy specimens from adults up to 80 years of age. Gastric lipase does not hydrolyze phospholipids or cholesterol esters. Human gastric lipase is a 379-amino acid protein that shares similar homology with rat gastric lipase but not human pancreatic lipase. For either gastric or small intestinal lipolysis to occur, two conditions are critical: First, a stable emulsion is required of fat droplets of such a size that they present a large surface area to the digestive enzyme; second, a mechanism is required for bringing enzyme and triglyceride into close apposition within the emulsion.
Emulsification
A number of factors assist in optimal production of an emulsion. Physical release of fat by mastication and the gastric milling of food produces a relatively unstable emulsion that is delivered into the duodenum. To permit its stabilization, the droplets in this emulsion have to be coated, and dietary phospholipid provides one such coat. The ratio of ingested phospholipid to triglyceride is about 1 : 30, and more phospholipid is added in the duodenum from bile.55 In breast milk, emulsion droplets are smaller and have proteins as well as phospholipid incorporated into their surface trilayer.56 Emulsification also is enhanced by the fatty acids liberated by intragastric lipolysis and, within the duodenum, by bile salts (Fig. 100-4). The final product in the duodenum is an emulsion consisting predominantly of triglyceride together with cholesterol esters and some diglyceride and coated by phospholipid, partially ionized fatty acids, monoglyceride, and bile salts.
Figure 100-4. Steps in lipolysis. The initial step in lipolysis is to increase the stability of the fatty emulsion. Gastric lipase acts on triglycerides to yield fatty acids and diglyceride (diglyceride enhances emulsification). This step is enhanced in the duodenum by bile salts and phospholipid (lecithin), which enable lipase, in the presence of colipase, to act at the surface of the emulsion droplet to bring it close to the triglyceride molecule, whereupon monoglyceride and fatty acids are released. Lipolysis in the duodenum yields fatty acids (from the α1 and α3 positions) and monoglyceride and occurs in a rapid and efficient manner at nearly neutral pH. In panel A are diagrammatic representations of bile salt molecules (top) oriented at an oil-water interface with its hydrophobic sterolic backbone in the oil phase and its hydrophylic hydroxyl and either taurine or glycine conjugates in the aqueous phase. At concentrations above critical micellar concentration, bile salts aggregate as simple micelles in water, with their hydrophylic groups facing into the water. In this diagram, three hydroxyl groups (cholate) are shown as open circles and an additional polar group represents either taurine or glycine. Panel B is a diagrammatic sketch of the dispersion of the products of lipolysis into lamellae at the surface of the oil phase, each about 4 to 5 nm thick, with water spacings up to 8 nm, and from there into vesicles of about 20 to 130 nm in diameter. In panel C, fatty acids and monoglyceride within the vesicles pass into mixed micelles. BS, bile salt; DG, diglyceride; FA, fatty acid; MG, monoglyceride; PL, phospholipid; TG, triglyceride.
Lipase
This stable emulsion is then presented to pancreatic triglyceride lipase. Unlike other soluble enzymes, which can act in a three-dimensional solution, lipase has to act at the two-dimensional surface of the emulsion droplet, and this requirement poses particular problems.57 Certain characteristics of the enzyme itself are important. Thus, the lipolytic zone of the molecule is hydrophobic and lies deep within it, shielded from the aqueous phase. It is revealed to the lipid only on close apposition to its surface. The presence of a coat on the lipid droplet thus poses a barrier to the action of lipase, and assistance is required to bring it into close contact with the triglyceride.
The presence of colipase, cosecreted by the pancreas along with lipase in a molar ratio of 1 : 1, is critical in approximating lipase to triglyceride (see Fig. 100-4). Colipase attaches to the ester bond region of the triglyceride, lipase then binding strongly to colipase by electrostatic interactions.55 Phospholipase A2 digestion of the phospholipid on the surface of the lipid emulsion allows exposure of the triglyceride core to the colipase-lipase complex, further enhancing colipase-dependent anchoring of lipase to the lipid emulsion. Phospholipase A2 digestion requires bile salts and Ca2+ for activation, which can further assist colipase-lipase–mediated triglyceride lipolysis by providing a mechanism for removing lipolytic products. In the absence of colipase, bile salts on the surface of the emulsion droplet inhibit lipase activity.
The colipase gene is located on chromosome 6, and the amino acid sequence of the lipid-binding domain, the lipase-binding domain, and the activation peptide appear to be highly conserved.58 Colipase is secreted by the pancreas as pro-colipase,59 which is activated when trypsin cleaves a pentapeptide from its N-terminus after entering the small intestinal lumen. A valine residue at position 407 and a leucine at position 412 are important for the interaction of lipase with colipase and the bile salt micelles.60 Interestingly, the pentapeptide cleaved from the pro-colipase by trypsin, called enterostatin, seems to be a specific satiety signal for the ingestion of fat.61
Because pancreatic lipase is most active at nearly neutral pH, secretion of bicarbonate by the pancreas and biliary tree is critically important and provides the necessary neutralization of gastric acid; however, luminal pH falls to about 6 in the jejunum, and here the fact that bile salts lower the pH optimal for lipase activity from 8 to 6 may be significant. In the presence of colipase and optimal pH, lipase activity releases fatty acids and monoglyceride extremely rapidly and efficiently (see Fig. 100-4). Pancreatic triglyceride lipase also binds strongly to the mucosal brush border membrane,62 where it may participate in lipolysis of cholesteryl esters or triglyceride, releasing fatty acids, monoglyceride, and free cholesterol in proximity to the brush border membrane, where they undergo rapid uptake.
In addition to pancreatic triglyceride lipase and its protein cofactor, colipase, pancreatic acini also synthesize two pancreatic lipase-related proteins (PLRP-1 and PLRP-2), which have strong nucleotide and amino acid sequence homology to pancreatic triglyceride lipase. Although PLRP-1 has no known activity, PLRP-2 does have lipase activity and, like pancreatic triglyceride lipase, PLRP-2 cleaves triglycerides but with broader substrate specificity. PLRP-2 also hydrolyzes phospholipids and galactolipids, two fats that are not substrates for pancreatic triglyceride lipase. It is also different from pancreatic triglyceride lipase with respect to sensitivity to bile salts and in response to colipase. A further critical difference is that PLRP-2 mRNA appears before birth and persists into adulthood, whereas pancreatic triglyceride lipase mRNA first appears at the suckling-to-weaning transition. This suggests that PLRP-2 plays a critical role in the digestion of breast-milk fats.63,64
Micelles and Other Lipid-Containing Particles
The products of lipolysis are distributed among the aqueous, oil, and intermediate phases in a number of forms prepared for transfer across the lumen to the mucosal brush border membrane. The shuttling of these products depends, in part, on the formation of micelles with bile salts. The concentration of bile salts secreted in bile is about 35 mmol and, in the duodenum, this is further decreased by dilution to 10 to 20 mmol; this concentration lies well above the critical concentration for micelle formation. Mixed micelle production depends on a number of other factors, including pH, presence or absence of lipids, and the types of bile salts that are secreted (see Chapter 64).42
Bile salts are capable of forming micelles because they have a particular three-dimensional structure and they are amphipathic; that is, their molecules have both water-soluble and lipid-soluble portions (see Fig. 100-4). They orient themselves at an oil-and-water interface and thus are ideal emulsifying agents. In addition, micelles are formed when bile salt levels are present above critical concentrations and thus are able to aggregate in disk-like particles with their hydrophobic sterolic backbones oriented toward each other and their hydrophilic polar groups facing outward into the aqueous phase. Bile salt micelles have the capacity to dissolve fatty acids, monoglycerides, and cholesterol, but not triglyceride.65 The mixed micelles thus formed are arranged so that the insoluble lipid is surrounded by bile salts that are oriented with their hydrophilic groups facing outward. Mixed micelles are about 50 to 80 nm in diameter and, unlike emulsion droplets, are too small to scatter light; thus, micellar solutions are clear. The presence of phospholipid secreted in bile enlarges mixed micelles and makes them more efficient in the dissolution of fat.
Other lipid-containing particles participate in the transfer of lipid to the mucosa. As the emulsion droplet shrinks during lipolysis, liquid crystalline structures are formed at its surface.66,67 These vesicular structures with multilamellar and unilamellar forms can be seen under the electron microscope, budding off the surface of emulsion droplets and occasionally close to the brush border membrane of the intestinal mucosa.68 This physical phase of lipid within the lumen might provide a significant mechanism for transfer of lipid to the mucosa, beyond that provided by bile salt micelles, and it could explain the observation that in the absence of bile salts, some 50% or more of dietary triglyceride may be absorbed. In the presence of adequate concentrations of bile salts, however, these vesicles undergo rapid spontaneous dissolution and release their lipid into micelles, which are likely to be the major route for lipid traffic (see Fig. 100-4); numerically, they are much more common than lipid vesicles.
Importance of Intraluminal pH
Lipid digestion and absorption are highly dependent on intraluminal pH at several steps in the process. Pancreatic lipase operates best in the presence of bile salts and at least pH 6. It therefore functions well at the pH of the luminal duodenum, where most lipid digestion occurs. Glycine-conjugated bile salts precipitate below pH 5; fatty acids are in their protonated form below about pH 6 and have limited solubility in bile salt micelles. Thus, in conditions in which intraluminal pH becomes more acid, as for example in the Zollinger-Ellison syndrome, pancreatic lipase is inactive, bile acids precipitate out of solution, and fatty acid partitioning is reduced. It is not surprising, therefore, that steatorrhea (without any other nutrient or hematologic disturbances) is a feature of this syndrome. Biological characteristics of lipases, including effect of pH on activities, are detailed in Table 100-2.
Unstirred Water Layer
An unstirred water layer is present on the surface of the intestinal epithelium, which in humans is approximately 40 µm deep.69 This layer may be rate limiting for uptake of long-chain fatty acids but not for short- or medium-chain fatty acids, the limiting step for which occurs at the brush border membrane.55 The provision of a high concentration of fatty acid in the microenvironment adjacent to the epithelium depends on the diffusion of micelles into this region. The microclimate here is slightly acidic, owing to activity of a sodium-hydrogen (Na+/H+) exchanger at the brush border membrane, and at pH between 5 and 6, the solubility of fatty acids in micelles decreases, thus encouraging liberation of fatty acids close to the mucosa. The high concentration of fatty acids necessary for diffusion across the mucosal membrane is thus achieved; evidence for this model is increasingly persuasive.70 The low-microclimate pH also encourages the fatty acids to be presented in an undissociated, protonated form. Thus, the pH partition hypothesis predicts that fatty acids could diffuse passively into the cell as protonated species and, at the near-neutral intracellular pH, become trapped in the ionized form.
A surfactant-like material has been discovered close to the brush border membrane, although its role in absorption, if any, is uncertain.71 It is secreted by enterocytes, contains phosphatidylcholine and alkaline phosphatase, and appears as flat lamellae or vesicles adjacent to the brush border membrane.
Other Lipids
Phosphatidylcholine, the major dietary phospholipid, is hydrolyzed by pancreatic phospholipase A2 (PLA2) to yield fatty acid from the 2-position and lysophosphatidylcholine. Pancreatic PLA2 is secreted as an anionic zymogen that is activated in the small intestine by tryptic cleavage of an N-terminal heptapeptide. It has a molecular weight of approximately 14 kd and requires calcium for activation and bile salts for its activity. It has multiple isoforms and apparently requires a 2 : 1 bile salt-to-phosphatidylcholine molar ratio for optimal activity. Although the bulk of intestinal PLA2 activity is derived from pancreatic juice, there is some contribution from the intestinal mucosa, where the enzyme is concentrated in the brush border.72
Cholesterol esters, in the presence of bile salts and calcium, are hydrolyzed by carboxyl ester lipase (CEL) (also known as pancreatic cholesterol esterase) to release the free sterol, in which form it is absorbed. Cell culture and other in vitro studies have thoroughly defined the potential functions of CEL in the digestion of cholesteryl ester, phospholipids, and triglycerides, but only its cholesteryl ester hydrolytic activity is unique to this enzyme in the digestive tract.73 CEL belongs to the α/β hydrolase family, is well conserved, and shares 78% homology in rats and humans.74 It is secreted primarily by the pancreatic acinar cells and lactating mammary glands. Using site-directed mutagenesis, the serine at position 194, the histidine at position 435, and the aspartic acid at position 320 have been shown to be important for CEL’s catalytic activity.73,75–77 The hydrolysis of water-insoluble substrates by CEL requires bile salt–containing 3α- or 7-α-hydroxy groups (e.g., cholate or chenodeoxycholate and their conjugates).78 The arginine-63 and arginine-423 sites play an important role in this bile salt–dependent process, but not in the bile salt–independent lysophospholipid hydrolytic activity of CEL.79
Both hydrolytic enzymes act on the emulsion phase at the surface of droplets, and the products of digestion are released into multilamellar and unilamellar vesicles and then to mixed micelles. Fatty acids and monoglycerides increase the solubility of cholesterol in micelles, thus encouraging its absorption. The products of phospholipid and cholesterol hydrolysis thus follow the same route to the brush border membrane as the fatty acids and monoglyceride, which originate from dietary triglyceride.
Unabsorbed long-chain fatty acids that enter the colon are not absorbed by this organ, and they undergo a series of bacterial modifications, principally hydroxylation. In healthy persons, no undigested triglyceride is found in the stool, and the normal fecal fat estimate of approximately 7 g/day reflects the cumulative total excretion of saponification products (i.e., fatty acids) that arise principally from membrane phospholipid and bacteria.
Transfer across the Brush Border Membrane
Much of the current understanding of the micellar solubilization and uptake of dietary lipids comes from the work of Hofmann and Borgstrom, who described the uptake of lipid digestion products by enterocytes.80 Further work by Carey discovered the coexistence of unilamellar liposomes with bile salt–lipid mixed micelles in the small intestine.81 Although the uptake of lipid digestion products by enterocytes has been accepted as a passive process, recent work has raised the possibility that some lipids may be taken up by enterocytes via carrier-mediated processes that are energy dependent.82
Studies with brush border membrane vesicles suggest that linoleic acid uptake occurs by facilitated diffusion.83 Absorption of oleic and arachidonic acid also appears to occur by a saturable process, suggesting the possibility of active transport. Several membrane proteins that increase the uptake of long chain fatty acids when overexpressed in cultured mammalian cells have been identified, the most prominent and best characterized of which are FAT/CD36, long-chain fatty acyl-CoA synthetases (LACS), and fatty acid transport proteins (FATPs/solute carrier family 27).84–87 The FATPs are transmembrane proteins that have been shown to enhance the cellular uptake of long-chain and very-long-chain fatty acids. In humans, FATPs comprise a family of six highly homologous proteins, hsFATP 1-6, which are found in all tissues of the body that use fatty acids.84,88,89 Although hsFATP1 is the best characterized of the FATPs, hsFATP4 is the only FATP expressed in the small intestine; it is localized to the apical brush border of the epithelial cells, where it is responsible for absorption of dietary lipids. Studies with cell lines and isolated enterocytes that overexpress FATP4 demonstrated that FATP4 is both necessary and sufficient for efficient uptake of long-chain and very-long-chain fatty acids.90 Detailed substrate studies based on 14C-labeled fatty acids have been presented for FATP1 and FATP4.87,90 Both studies showed that uptake of fatty acids shorter than 10 carbon atoms, such as butyric acid and octanoic acid, was unaffected by FATP expression, whereas uptake of common long-chain fatty acids, such as palmitate and oleate, was robustly enhanced.90 More recently, a wrinkle-free phenotype has been associated with the spontaneous autosomal recessive mutation of the gene for FATP4, resulting in a very tight and thick-skinned phenotype.91
Nutrients, hormones, and cytokines have been reported to regulate FATP expression. Rats fed a high-fat diet showed increased FATP expression in the heart, but not the liver. Several reports have shown a positive regulation of mouse FATP by ligands that activate either PPAR-γ, PPAR-α, or PPAR-γ/RXR heterodimers in hepatoma cell lines, the liver, and the intestine. Further, a PPAR binding site was identified in the murine FATP1 promoter. TNF-α is a negative regulator of FATP expression and down-regulates FATP mRNA in liver and FATP1 and FATP4 proteins in adipocytes.88
