Diffuse Lung Disease
Evan J. Zucker, R. Paul Guillerman, Martha P. Fishman, Alicia M. Casey, Craig W. Lillehei and Edward Y. Lee
Childhood interstitial lung diseases (ChILDs), which are associated with significant morbidity and mortality, represent a rare and heterogeneous group of chronic diffuse lung disorders characterized clinically by dyspnea, tachypnea, crackles, and hypoxemia. Although termed “interstitial,” the diseases additionally may involve the alveoli, airways, blood vessels, lymphatic channels, and pleural spaces. With advances in imaging, improved thoracoscopic lung biopsy techniques, and in particular a revised ChILD classification scheme, substantial progress has been made in understanding these previously enigmatic disorders.1–4 In this chapter, we provide an overview of pediatric chronic diffuse lung disease, first discussing the recognized disorders of infancy as defined by the revised ChILD criteria (Box 56-1). We then focus on selected, practically relevant examples of ILDs occurring in older children; even under the new classification system, these diseases remain too numerous to review in their entirety.
Disorders of Infancy
Diffuse Developmental Disorders
Etiology: Diffuse developmental disorders are characterized by marked alveolar gas exchange impairment and are thought to arise early in prenatal lung development. The three main entities within this category are acinar dysplasia, congenital alveolar dysplasia, and alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV). Acinar dysplasia is characterized by arrest of lung development in the pseudoglandular or early canalicular phase. Arrest in the late canalicular/early saccular phase is typical of congenital alveolar dysplasia. ACD/MPV results from an abnormal location of the pulmonary vein branches next to the pulmonary artery branches rather than within the interlobular septae; medial hypertrophy of the pulmonary arterioles, reduced alveolar capillary density, and maldevelopment of pulmonary lobules also occur. A proportion of ACD/MPV cases are caused by FOXF1 gene mutations or 16q24.1 microdeletions.1–3,5–9
Imaging: Generally, only portable chest radiographs are available because of the severity of disease. Initial chest radiographs may be unremarkable, but follow-up examinations typically demonstrate progressive hazy bilateral pulmonary opacities, similar to that seen in children with surfactant deficiency of prematurity or inborn errors of surfactant metabolism. Typically, lung volumes initially are normal to decreased but may be increased with ventilator support. Air leaks such as pneumothorax and pneumomediastinum develop in about half of patients, likely because of barotrauma (Fig. 56-1). Radiographs may show enlargement of the main pulmonary artery in patients with concurrent pulmonary hypertension (PHT). Although imaging findings are nonspecific, this group of disorders should be considered in a full-term neonate with severe respiratory distress similar to PHT of the newborn in the absence of such risk factors as meconium aspiration, asphyxia, prematurity, or sepsis.1–3,10–13
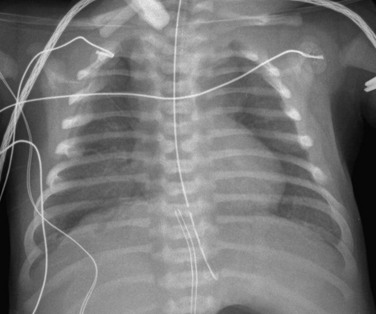
Treatment and Follow-up: Diffuse developmental disorders carry an extremely poor prognosis and are nearly universally fatal, with rapidly progressive respiratory failure typically developing in the first 2 months of life despite such measures as treatment for PHT, intensive ventilation, and extracorporeal membrane oxygenation. Serial chest radiographs can monitor the progression of the disease and help identify acute complications that may be seen with prolonged ventilatory support. Lung transplantation is the only viable treatment. However, patients generally do not survive long enough to receive a transplant, and many families elect to withdraw care upon diagnosis. More than 80% of patients with ACD/MPV have associated extrapulmonary anomalies (e.g., cardiac, gastrointestinal, or genitourinary) for which screening studies may be performed. Because 10% of reported ACD/MPV cases demonstrate a familial association, genetic counseling may be offered to family members.1–3,5–14
Alveolar Growth Disorders
Etiology: The most common form of neonatal ILD, alveolar growth disorders, are characterized by defects in alveolar formation with lobular simplification, lack of alveolar septation, and enlargement of airspaces. Unlike diffuse developmental disorders, in which lung development is preprogrammed to be abnormal, growth disorders are caused by a secondary condition or event affecting lung development. Entities in this category include: (1) pulmonary hypoplasia due to such conditions as oligohydramnios, space-occupying lesions, or neuromuscular disease; (2) postnatal conditions such as prematurity-related chronic lung disease (bronchopulmonary dysplasia [BPD]) and full-term chronic lung disease; (3) structural pulmonary changes seen with such chromosomal abnormalities as trisomy 21; and (4) changes as a result of congenital heart disease in the absence of chromosomal abnormalities.1–3,5,15–20
Imaging: Imaging findings are variable on both plain radiographs and high-resolution computed tomography (HRCT). Chest radiography in infants with classic BPD demonstrates coarse reticular opacities, cystic lucencies, and disordered lung aeration due to alveolar septal fibrosis and hyperinflation. With advances in perinatal medicine allowing delivery as early as 23 weeks’ gestation, when the alveolar ducts and alveoli are just beginning to develop, a shift in the imaging features of BPD has occurred, termed “new” BPD. Findings on chest radiography and CT in infants with new BPD and other alveolar growth abnormalities range from near normal to markedly disordered, variably sized pulmonary lobules, thick perilobular reticular opacities, linear and triangular subpleural opacities, ground-glass opacities, and hyperlucent areas, some of which resemble cysts (Fig. 56-2). These features may be mistaken for “emphysematous changes.” In infants with trisomy 21, small subpleural cysts are particularly common (Fig. 56-3). Chest imaging in patients with abnormal alveolar growth related to X-linked filamin A gene mutations is characterized by central pulmonary artery enlargement, atelectasis, progressive severe pulmonary hyperinflation, hyperlucency, and peripheral pulmonary vascular attenuation similar to congenital lobar or acquired emphysema (e-Fig. 56-4).1–3,5,15–20
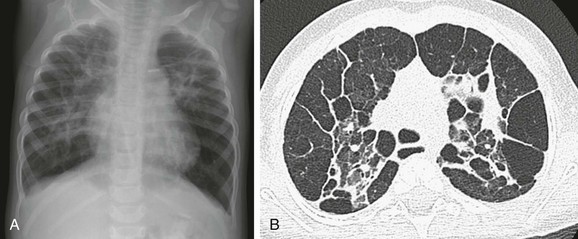
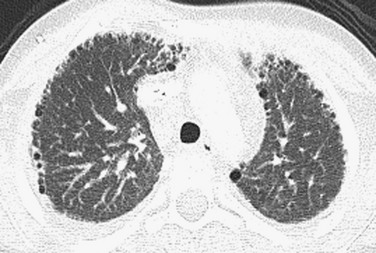
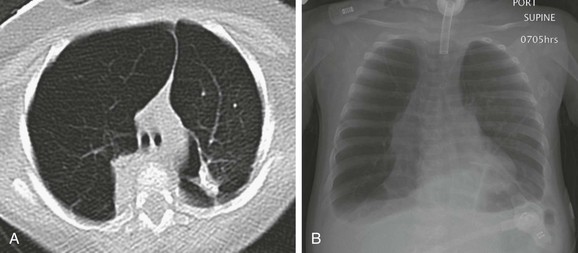
Treatment and Follow-up: Reduction in BPD severity and incidence may be achieved by decreasing respiratory support interventions causing lung injury. Additional treatment strategies including nasal respiratory support, low-dose corticosteroids, fluid restriction, vitamin A, and medical/surgical patent ductus arteriosus closure have shown only lackluster results. The linear and subpleural opacities seen on computed tomography (CT) correspond to interstitial fibroproliferation and are associated with low functional residual capacity, supplemental oxygen, and mechanical ventilation, but they do not correlate with the severity of symptoms. Because a significant proportion of patients with growth abnormalities also have patchy pulmonary interstitial glycogenosis (PIG) (detailed later), which is potentially responsive to steroids, a lung biopsy may be considered to help establish a concurrent diagnosis. Patients with filamin A mutations may experience especially severe respiratory decline requiring lung transplantation for survival.1–3,5,14–22
Surfactant Dysfunction Disorders and Related Abnormalities
Etiology: Diseases in surfactant dysfunction disorders and related abnormalities are caused by genetic mutations resulting in surfactant dysfunction. Mutations in surfactant proteins B (SpB) and C (SpC) and the adenosine triphosphate–binding cassette transporter protein A3 (ABCA3) directly impair surfactant metabolism. SpB and ABCA3 mutations demonstrate an autosomal-recessive inheritance pattern, whereas SpC defects are autosomal-dominant loss of function mutations. Other rare genetic disorders such as thyroid transcription factor-1 abnormalities (“brain-lung-thyroid syndrome”), lysinuric protein intolerance, and granulocyte-macrophage colony-stimulating factor–Rα mutations also affect surfactant metabolism and also are included in this category. Additional uncharacterized disorders of surfactant metabolism exist.1–3,21–42
Imaging: Chest radiographs in infants presenting with a surfactant disorder demonstrate diffuse or patchy hazy granular pulmonary opacities. HRCT is characterized by diffuse ground-glass opacity, consolidation, interlobular septal thickening, or a crazy-paving pattern typical of pulmonary alveolar proteinosis (PAP) (detailed separately in a later section) (Fig. 56-5). With increasing age, the ground-glass opacities decrease in extent and thin-walled parenchymal cysts develop, becoming larger and more numerous over time (e-Fig. 56-6). Pectus excavatum is common in patients surviving past infancy, possibly because of the effects of chronic lung disease on the growing chest wall.1–3,21–42
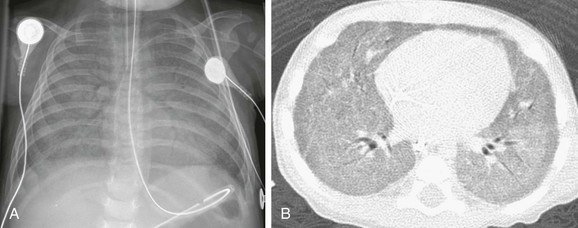
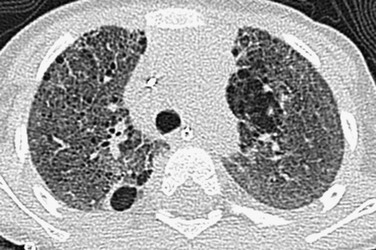
Treatment and Follow-up: Often the diagnosis of inherited surfactant disorder can be established through testing of blood or buccal swab–derived samples for surfactant gene mutations, obviating the need for lung biopsy. The mainstay of treatment is chronic ventilator support in children with respiratory failure. Anecdotal evidence suggests a possible role for pulse corticosteroids, hydroxychloroquine (Plaquenil), and azithromycin. Nutritional supplementation via a gastrostomy tube often is required for growth. Palivizumab (Synagis) may be considered to help prevent lung infections. Patients with rapidly progressive disease may be eligible for lung transplantation. Investigational efforts focus on targeted therapies for the specific genetic mutations involved.1–3,14,21–42
Specific Conditions of Unknown or Poorly Understood Etiology
Neuroendocrine Cell Hyperplasia of Infancy
Etiology: The etiology of neuroendocrine cell hyperplasia of infancy (NEHI) is unknown. Histopathologically, the disease (previously referred to as persistent tachypnea of infancy) is characterized by increased numbers of pulmonary neuroendocrine cells (PNECs) and innervated clusters of PNECs called neuroepithelial bodies in the epithelium of peripheral airways. PNECs function in oxygen sensing and fetal lung development; they usually rapidly decrease in number after the neonatal period. Although increased numbers of PNECs are seen in persons with a variety of pulmonary disorders, the diseases are not likely to be confused with NEHI histopathologically. Some patients with NEHI demonstrate mild inflammation or fibrosis of the airways. Additionally, some cases are familial, suggesting a genetic component.1–3,5,14,43–50
Imaging: Chest radiographs demonstrate hyperinflation and variable increased perihilar opacity resembling bronchiolitis or reactive airways disease (Fig. 56-7). HRCT findings are characteristic with air trapping and a mosaic attenuation pattern affecting at least four lobes and geographic ground-glass opacities most prominent in the right middle lobe, lingula, and paramediastinal lung regions (Fig. 56-8). The sensitivity and specificity of HRCT for the diagnosis is reported to be 78% to 83% and 100% when examinations are interpreted by experienced pediatric thoracic radiologists. Thus in the correct clinical setting, CT may obviate the need for a lung biopsy.1–3,5,14,43–50
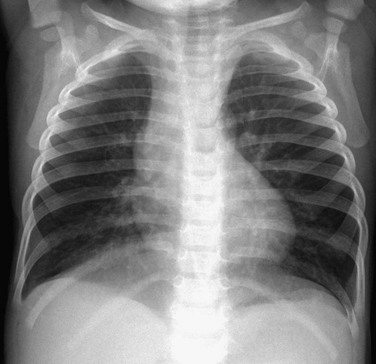
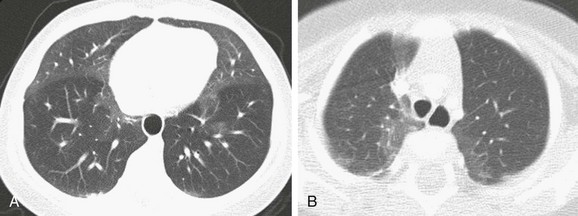
Treatment and Follow-up: Treatment of NEHI at present is supportive and is geared toward preventing hypoxemia and infection and maintaining nutritional support. Corticosteroids are not helpful and are not recommended, except for temporary glucocorticoid pulses if a patient has a concurrent viral infection. Although patients with NEHI have persistent symptoms and require prolonged oxygen therapy, the prognosis is generally favorable with no reported deaths, progression to respiratory failure, or need for lung transplantation attributable to this disorder. However, it should be noted that patients in later life (i.e., adolescence) may experience symptoms such as exercise intolerance related to persistent air trapping and can relapse in the setting of respiratory infection.1–3,5,14,43–50
Pulmonary Interstitial Glycogenosis
Etiology: The etiology of pulmonary interstitial glycogenosis (PIG), previously known as infantile cellular interstitial pneumonitis and histiocytoid pneumonia, remains unknown. PIG is characterized histopathologically by infiltration of the interstitium with immature mesenchymal cells containing copious cytoplasmic glycogen and staining positively for vimentin. Inflammation and fibrosis are not typical. Patchy PIG and alveolar growth abnormalities commonly coexist. The lack of PIG in lung biopsies from children older than 10 months suggests that the disorder may be related to lung growth and development.1–3,5,14–15,51–56
Imaging: Chest radiographs have been reported to demonstrate progressive hyperinflation and evolution from a fine interstitial to a coarse interstitial or alveolar pattern. HRCT findings may include pulmonary architectural distortion, hyperinflated/hyperlucent areas, ground-glass opacities (diffuse, segmental, or subsegmental), interlobular septal thickening, and linear opacities (Fig. 56-9). Because patchy PIG often coexists with alveolar growth abnormalities, the specific imaging features of “pure” PIG are uncertain. In one reported case of PIG, the multiple, small, scattered, air-filled, cystic-appearing changes were most likely attributable to the concomitant alveolar growth abnormality.1–3,5,14–15,51–56
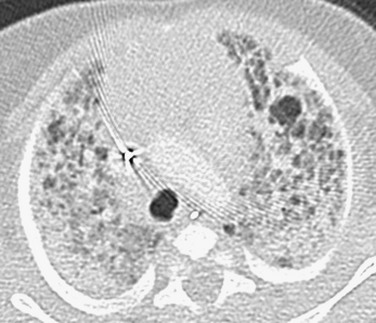
Treatment and Follow-up: Most patients require supplemental oxygen. PIG may respond favorably to pulse glucocorticoids, which are recommended at many ChILD centers. Overall, the prognosis of pure PIG is favorable, with no reported deaths attributable to the disease. However, greater mortality and morbidity rates are expected with concomitant growth abnormalities and PHT. During the first several months of life, marked improvement often occurs clinically, radiologically, and histologically, although hyperinflation may persist for years.1–3,5,14–15,51–56
Disorders of Childhood
Pulmonary Alveolar Proteinosis
Etiology: PAP is characterized by the abnormal accumulation of surfactant, a lipoproteinaceous material, within the alveoli that prevents normal gas exchange. As discussed previously, PAP may arise congenitally as the result of a genetic surfactant deficiency. The acquired form of PAP seen in older children and adults is most commonly an autoimmune process in which autoantibodies to granulocyte macrophage colony-stimulating factor (GM-CSF) are produced. Because GM-CSF normally participates in alveolar macrophage signaling, the disease results in impaired clearance of surfactant-derived intraalveolar lipoproteins. PAP also may be a result of a variety of processes including leukemia, chemotherapy, toxic exposure to fumes and dusts, and other entities that impair alveolar macrophage function.1–3,5,57–63
Imaging: The various etiologies of PAP cannot be differentiated by imaging. Chest radiographs demonstrate symmetric perihilar opacities extending to the peripheral portions of the lungs. These opacities generally are not as consolidative and dense as would be expected for a bacterial pneumonia. HRCT shows bilateral ground-glass opacities with smooth intralobular and interlobular septal thickening in polygonal shapes, an appearance known as “crazy paving”1–3,5,57–63 (Fig. 56-10).
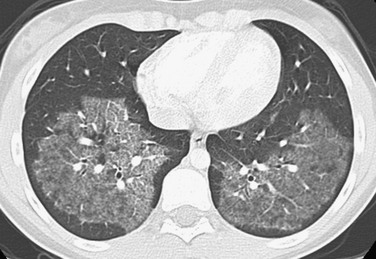
Treatment and Follow-up: After the diagnosis is confirmed, preferably by bronchoscopy with bronchoalveolar lavage and transbronchial biopsy but by a surgical lung biopsy if necessary, serologic testing for GM-CSF antibodies should be performed to distinguish between autoimmune and secondary PAP. For secondary PAP, treatment efforts should focus on identifying the underlying cause. For autoimmune PAP, repeated whole-lung lavage is the current treatment of choice. Aerosolized GM-CSF therapy has shown very promising results in observational studies, although randomized controlled trials are not yet available. Immunosuppression with rituximab or mycophenolate mofetil also has been tried. CT can be used to monitor the response to therapy.1–3,5,57–63
Pulmonary Lymphangiectasia and Lymphangiomatosis
Etiology: Lymphatic disorders are considered “masqueraders” of ILD according to the ChILD classification. However, primary pulmonary lymphangiectasia and lymphangiomatosis can be regarded as true ILDs because they involve the pulmonary interstitium. Pulmonary lymphangiectasia is characterized by dilatation of the lymphatics draining the pulmonary interstitial and subpleural spaces, either on a congenital basis (including some associated genetic syndromes) or an acquired basis (as a result of pulmonary lymphatic or venous obstruction). Pulmonary lymphangiomatosis is characterized by a proliferation of complex lymphatic channels with secondary lymphatic dilatation. In both disorders, the disease may be limited to the lung or may involve additional thoracic and/or extrathoracic manifestations.1–3,15,64–70
Imaging: Chest radiographs in patients who present with the classic sign of severe respiratory distress demonstrate diffuse hazy opacification of the lungs similar to the findings of surfactant deficiency of prematurity or genetic surfactant deficiency. A chest CT scan shows diffuse, smooth thickening of the interlobular septae and peribronchovascular interstitium, patchy ground-glass opacities, and pleural effusions (often chylous) (e-Fig. 56-11). Less diffuse opacity, less severe septal thickening, and greater hyperinflation are characteristic of surviving neonates or patients presenting later in infancy. Magnetic resonance imaging (MRI) demonstrates hyperintensity of the pulmonary interstitium on T2-weighted sequences and pleural effusions. Lung findings are very similar in pulmonary lymphangiectasia and lymphangiomatosis. Unlike lymphangiectasia, lymphangiomatosis usually presents in late childhood and often involves extrapulmonary sites, with lytic bone lesions and mediastinal soft tissue edema occurring frequently.1–3,15,64–70
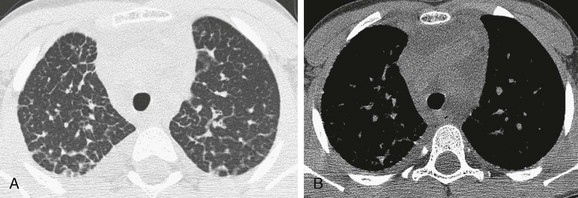
Treatment and Follow-up: Patients with congenital disease may be stillborn or present at birth with severe respiratory distress, which often results in death within the first few hours of life. Mechanical ventilation and pleural drainage invariably are required. Patients with long-term survival have variable degrees of respiratory compromise and are managed with supplemental oxygen at home, symptomatic therapy, fluid restriction, and dietary measures.1–3,15,64–71
Bronchiolitis Obliterans
Etiology: Bronchiolitis obliterans (BO) is characterized by a fibroblastic reparative response to injury of the small airways, resulting in occlusion of the lumen. The inciting injury is usually a respiratory viral infection (often adenovirus or influenza) with marked airway mucosal necrosis. Other preceding conditions include graft-versus-host disease, chronic allograft rejection in patients who have had a lung transplant, and Stevens-Johnson syndrome. Swyer-James-Macleod syndrome is a particular form of BO that predominantly affects one lung and presents several months or years after the initial infection. Terminology for BO in the literature is inconsistent, leading to confusion. The clinical manifestation may be termed “bronchiolitis obliterans syndrome,” whereas the histopathologic correlate is a spectrum termed “constrictive bronchiolitis” or “obliterative bronchiolitis,” depending on the degree of airway lumen occlusion that is present.1–3,5,72–77
Imaging: Findings of chest radiographs are nonspecific and may be normal. The most common abnormality is hyperinflation. A hyperlucent lung on the affected side that is relatively underperfused with normal or decreased volume is characteristic of Swyer-James-Macleod syndrome. CT findings consist of air trapping accentuated on expiration, parenchymal hyperlucency, mosaic attenuation, bronchial wall thickening, bronchiectasis, and pulmonary vascular attenuation. The presence of both hyperlucency and pulmonary vascular attenuation is highly specific for moderate/severe nontransplant BO (e-Fig. 56-12). With a correlating clinical history and a fixed obstructive pattern on pulmonary function testing, CT is diagnostic, bypassing the need for a lung biopsy. In the Swyer-James-Macleod variant, chest radiographs suggest a unilateral abnormality, but in fact, abnormal findings on CT are bilateral in 50% of cases.1–3,5,72–77
Treatment and Follow-up: In the absence of bronchiectasis, BO can be difficult to distinguish from the more common acute viral bronchiolitis with CT. Follow-up imaging thus can be helpful. Imaging findings in persons with acute viral bronchiolitis will normalize on subsequent examinations after symptom resolution (with up to several months lag), whereas persistent or worsening abnormalities will be present in irreversible BO. CT provides valuable prognostic information in postinfectious BO; in children younger than 3 years of age with severe CT abnormalities, lung function is generally poor even after several years. In lung transplant recipients, CT is valuable in screening for posttransplant BO, which is an important contributor to mortality after the first postoperative year. For the BO variant associated with lung transplantation, corticosteroids and the antibiotic azithromycin have shown benefit.1–3,5,72–79
Hypersensitivity Pneumonitis
Etiology: Also known as extrinsic allergic alveolitis, hypersensitivity pneumonitis is characterized by pulmonary inflammation related to inhalational exposure of organic antigens usually from birds, fungi, or dusts carried by family members from the workplace. Other inciting antigens include a variety of highly reactive low molecular weight compounds found in spray paints, glues, epoxy resins, insecticides, and drugs such as methotrexate. Histopathologically, lymphocytic infiltration of the bronchioles and interstitium is seen with giant cells and poorly developed granulomas situated around bronchioles. Three subtypes of hypersensitivity pneumonitis have been described: (1) acute, with symptoms occurring by 4 to 6 hours and lasting up to 22 hours; (2) subacute, characterized by repeated low-level antigen exposure over weeks to months; and (3) chronic, manifested by an insidious, progressive course over months to years, or, alternatively, recurrent acute episodes.1–3,5,64,80–84
Imaging: The acute and subacute forms of hypersensitivity pneumonitis have similar imaging features. Common abnormal findings on chest radiographs are diffuse micronodular interstitial prominence and opacities in the mid to lower lungs, which may resemble pulmonary edema or pneumonia. However, many radiographs will appear normal, with 40% of cases having abnormalities visible only on CT. HRCT demonstrates small (1 to 3 mm) poorly defined centrilobular nodules (reflecting bronchiolitis), ground-glass opacities (reflecting alveolitis), and air trapping, with relative sparing of the upper lungs. On radiography and CT, the chronic form is characterized by volume loss and fibrotic changes predominantly with irregular linear/reticular opacities, architectural distortion, and honeycombing (e-Fig. 56-13).1–3,5,64,80–84
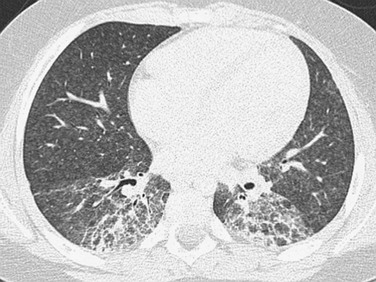
Treatment and Follow-up: The most important aspect of treatment is identifying and eliminating exposure to the inciting antigen. The imaging findings associated with the acute and subacute forms of the disease regress with removal of the antigen. However, the chronic fibrotic changes persist and may even progress. Systemic corticosteroids are the only dependable drug therapy but do not affect the long-term outcome.1–3,5,64,80–84
Diffuse Pulmonary Hemorrhage Disorders
Etiology: Diffuse pulmonary hemorrhage disorders can be subcategorized according to the presence or absence of capillaritis, which is characterized pathologically by inflammatory disruption of the interstitial capillary network. Disorders with capillaritis include idiopathic pulmonary capillaritis, Wegener granulomatosis (recently renamed granulomatosis with polyangiitis), microscopic polyangiitis, Goodpasture syndrome, idiopathic pulmonary-renal syndrome, systemic lupus erythematosus, and drug-induced capillaritis. Disorders without capillaritis include idiopathic pulmonary hemosiderosis, acute idiopathic pulmonary hemorrhage of infancy, Heiner syndrome (pulmonary disease caused by food sensitivity, usually to cow’s milk), coagulation disorders, and cardiovascular disorders such as pulmonary venoocclusive disease and pulmonary arteriovenous malformation.1–3,85–89
Imaging: The classic radiographic appearance of acute diffuse pulmonary hemorrhage consists of bilateral symmetric airspace opacities in a “butterfly” or “batwing” pattern. However, opacities may be asymmetric or unilateral. HRCT is more sensitive and shows patchy ground-glass opacities and consolidation acutely. With organizing or repetitive hemorrhage, findings evolve to interlobular septal thickening, nodular opacities, and potentially a crazy-paving pattern (Fig. 56-14).1–3,85–89
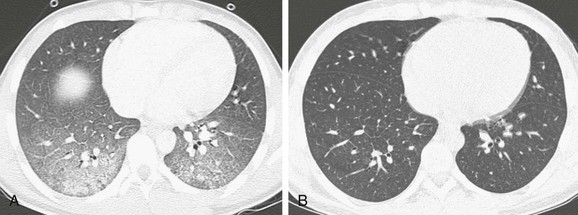
Treatment and Follow-up: Distinguishing between pulmonary hemorrhage with capillaritis versus without capillaritis is critical, because pulmonary capillaritis often requires aggressive immunosuppressive therapy. Because imaging findings are similar in the many conditions causing pulmonary hemorrhage, a lung biopsy often is necessary for a definitive diagnosis. With treatment, CT findings typically improve.1–3,85–89
Nonspecific Interstitial Pneumonia
Etiology: Nonspecific interstitial pneumonia (NSIP) can be idiopathic, familial, or the final common pathway of several other disorders, including autoimmune connective tissue and collagen vascular diseases, genetic surfactant disorders, and hypersensitivity pneumonitis. Histopathologically, there is a characteristic appearance with temporally and spatially uniform interstitial lymphoplasmacytic inflammation and varying degrees of fibrosis. Cellular and fibrotic subtypes have been described.1–3,5,90–93
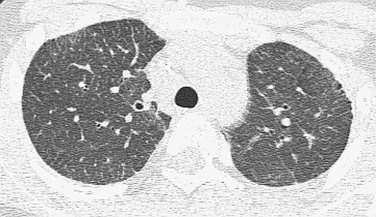
Connective Tissue and Collagen-Vascular Diseases
Etiology: Connective tissue and collagen-vascular diseases are a heterogeneous group of rheumatologic disorders characterized by chronic inflammation and generally thought to have an autoimmune basis. Included entities are systemic lupus erythematosus, rheumatoid arthritis, dermatomyositis, systemic sclerosis, Sjögren syndrome, and mixed connective tissue disease. Most commonly, an NSIP histopathologic pattern (previously detailed) is found. Pulmonary lymphoid hyperplasia, organizing pneumonia (detailed later), vasculopathy, and pleuritis also may occur.1–3,5,90–96
Imaging: Because they share an NSIP pattern, the various entities usually cannot be differentiated by imaging. Occasionally, ancillary findings may suggest the diagnosis, such as a dilated esophagus in a person with systemic sclerosis (e-Fig. 56-16). Childhood-onset systemic lupus erythematosus presents with vasculitis and pulmonary hemorrhage much more commonly than it does with ILD, unlike its adult counterpart.1–3,5,90–96
Organizing Pneumonia
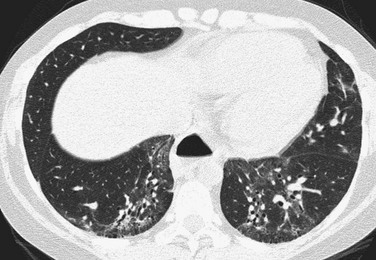
Etiology: Organizing pneumonia is characterized histologically by intraluminal organizing fibrosis in the distal airways and airspaces (bronchioles, alveolar ducts, and alveoli). Organizing pneumonia may be idiopathic and termed “cryptogenic organizing pneumonia,” or it can be a result of a variety of causes including asthma, drug reaction, aspiration pneumonia, autoimmune disease, chemotherapy, bone marrow transplantation, and other conditions stimulating a lung reparative response. The term “bronchiolitis obliterans organizing pneumonia” is no longer favored because of potential confusion with the distinct entity of BO.1–3,97–99
Imaging: Imaging findings are variable, with CT most frequently showing peripheral patchy consolidation with or without surrounding ground-glass opacity. Commonly, air bronchograms and mild bronchial dilatation are found within areas of consolidation. Other recognized findings include the atoll or reverse halo sign (central ground-glass opacity surrounded by consolidation) (Fig. 56-17), small pulmonary nodules along bronchovascular bundles, linear and bandlike subpleural opacities, perilobular thickening, and progressive fibrosis.1–3,97–99
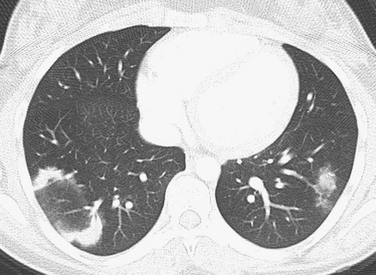
Treatment and Follow-up: Although organizing pneumonia has many underlying causes, overall, corticosteroids are the best treatment option. The prognosis is generally favorable, with up to an 80% cure rate reported. Imaging findings related to inflammation will improve/resolve on follow-up, whereas irreversible fibrotic changes may persist or worsen.1–3,78,97–99
Pulmonary Infiltrate with Eosinophilia
Etiology: The eosinophilic lung diseases are a diverse group of disorders characterized by peripheral or tissue eosinophilia, with interstitial and intraalveolar eosinophils typically present on pathology. Three subcategories are recognized: eosinophilic disease of unknown cause, eosinophilic disease of known cause, and eosinophilic vasculitis. Diseases of unknown cause include simple pulmonary eosinophilia (or Löffler syndrome), acute eosinophilic pneumonia (AEP), chronic eosinophilic pneumonia (CEP), and idiopathic hypereosinophilic syndrome. Diseases of known cause include allergic bronchopulmonary aspergillosis (ABPA), bronchocentric granulomatosis, parasitic infections, and drug reactions. Eosinophilic vasculitis includes allergic angiitis and granulomatosis, also known as Churg-Strauss syndrome.1–3,100–103
Imaging: Imaging findings of interstitial, alveolar, or mixed interstitial-alveolar opacities are in general nonspecific, but certain key features may suggest the underlying diagnosis. CEP and drug-induced pulmonary infiltrate with eosinophilia (PIE) demonstrate a characteristic pattern of peripheral consolidation with sparing of the central lung zones (a “photographic negative” or “reversed” pulmonary edema pattern), allowing a highly specific diagnosis in the setting of peripheral eosinophilia (Fig. 56-18). AEP presents radiographically with bilateral reticular opacities, possibly with consolidation and pleural effusion. On CT, bilateral patchy ground-glass opacity and often interlobular septal thickening, consolidation, or poorly defined nodules are seen. The imaging findings of AEP mimic those of more common entities such as pulmonary edema and acute respiratory distress syndrome, which may result in a delayed diagnosis. ABPA demonstrates central bronchiectasis with or without mucoid impaction; the presence of mucoid impaction of the large airways is referred to as the “finger-in-glove” sign. Simple pulmonary eosinophilia and idiopathic hypereosinophilic syndrome characteristically demonstrate pulmonary nodules with ground-glass halos. Bronchocentric granulomatosis demonstrates focal masses and nodules or lobar consolidation with atelectasis. Findings in persons with Churg-Strauss syndrome include subpleural consolidation, centrilobular nodules, bronchial wall thickening, and interlobular septal thickening.1–3,100–103
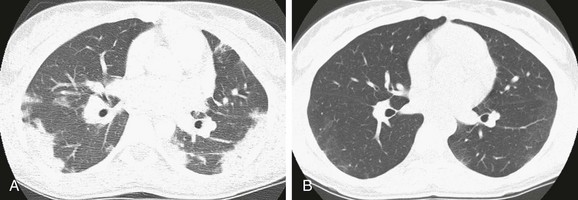
Treatment and Follow-up: Imaging findings, even if nonspecific, can localize potential sites for lung biopsy. Corticosteroids are the treatment of choice, resulting in prompt and complete response. In persons with AEP, relapses do not occur after cessation of corticosteroids, although they can occur in persons with CEP. Therapy may be tailored to the underlying cause; for example, antiparasitic medications may be used in a case of parasite-induced PIE.1–3,100–103
Storage Diseases
Etiology: The lysosomal storage diseases are a group of genetic metabolic disorders resulting in impaired lysosomal function. In the case of Gaucher disease and Niemann-Pick disease, lipid-laden “foamy” macrophages (Gaucher cells or Niemann-Pick cells, respectively) accumulate in tissues. These lipid-laden macrophages may infiltrate the lung, causing pulmonary symptoms. Gaucher disease is the most common lysosomal storage disorder.2–3,104–107
Imaging: Pulmonary findings, if they appear at all, occur late in the course of Gaucher disease, most commonly in the neuronopathic type III form. Chest radiographs may show reticulonodular opacities. CT may reveal a variety of findings, including ground-glass opacities, consolidation, interstitial thickening, bronchial wall thickening, thymic enlargement, and lymphadenopathy. Diffuse interstitial thickening is characteristic of chest radiographs and CT in Niemann-Pick disease type B (Fig. 56-19). A crazy-paving pattern is typical of Niemann-Pick disease type C22–3,104–107 (e-Fig. 56-20).
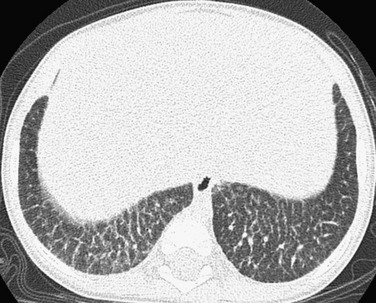
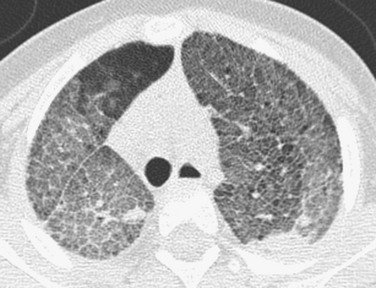
Treatment and Follow-up: Enzyme replacement therapy, approved by the U.S. Food and Drug Administration, is now available for several of the lysosomal storage disorders. In persons who have Gaucher disease with pulmonary involvement, the imaging findings gradually improve with treatment, although complete resolution typically is not achieved. Currently, chest radiographs are recommended every 2 years in persons with Gaucher disease to monitor the lungs.2–3,104–107
Chronic Granulomatous Disease
Etiology: Chronic granulomatous disease (CGD) is a rare inherited immunodeficiency disorder that usually is caused by a mutation in one of the four genes encoding subunits of the phagocyte nicotinamide adenine dinucleotide phosphate. The mutation results in impaired phagocyte nicotinamide adenine dinucleotide phosphate oxidase activity and therefore reduced superoxide production and an impaired oxidative burst. As a result, mechanisms for killing intracellular catalase-positive bacterial organisms and fungal organisms are impaired. The lungs are the most common location of infection. Histologically, granulomatous inflammation is present, often with necrosis, with surrounding chronic inflammation and fibrosis, hence the name chronic granulomatous disease.1–2,108–110
Imaging: A variety of imaging findings may present in patients who have chronic, recurrent infections, including consolidation, ground-glass opacities, tree-in-bud opacities, and centrilobular or random (even miliary) nodules acutely, with bronchiectasis, septal thickening, air trapping, abscess formation, fibrosis, cysts, and honeycomb lung in persons with long-standing disease. Other common thoracic findings include mediastinal and/or hilar lymphadenopathy, pleural thickening, empyema, vertebral or rib osteomyelitis, and chest wall invasion1–2,108–110 (Fig. 56-21).
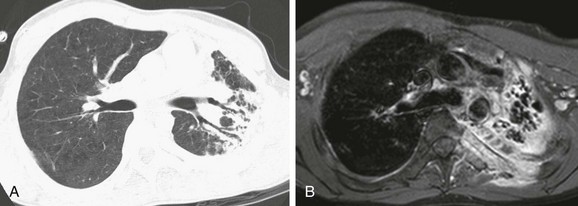
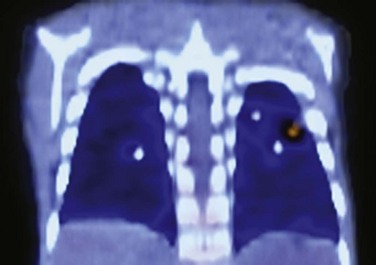
Treatment and Follow-up: CGD is treated with lipophilic antibiotics, antifungal agents, interferon-γ, abscess drainage, surgical resection, and stem cell transplantation. Prophylactic antibiotics are beneficial. Fluorine-18 fluorodeoxyglucose positron emission tomography is more reliable than CT for distinguishing between active and dormant disease activity (e-Fig. 56-22). Improved ability to diagnose and treat CGD has allowed persons with this disease to survive into adulthood.1–2,108–110
Cystic Fibrosis
Etiology: The most common genetic disorder causing chronic pulmonary disease in children, cystic fibrosis (CF) is caused by mutations in the CF transmembrane regulator (CFTR) gene, which is inherited in an autosomal-recessive fashion. Chronic recurrent infections develop, in addition to numerous extrapulmonary manifestations such as meconium ileus in infancy. With progressive disease, chronic inflammatory changes occur that lead to alteration of airway walls with epithelial erosion, partial replacement of the mucosa by granulation tissue, progressive airway dilatation resulting in bronchiectasis, and fibrotic/obliterative changes involving the small airways.1,102,111–113
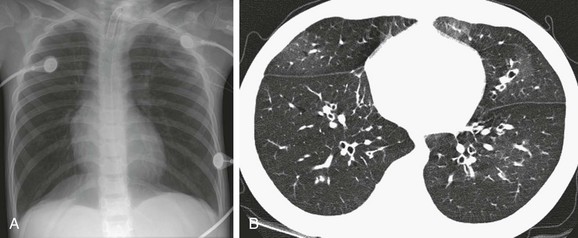
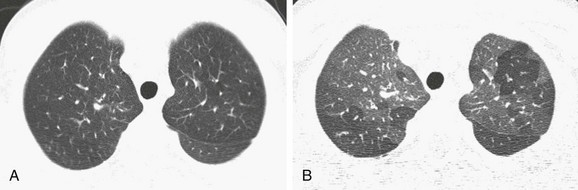
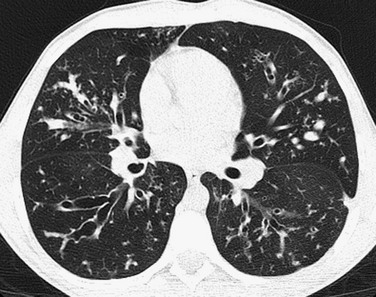
Imaging: Chest imaging in persons with early CF may be normal or show mild to moderate air trapping and/or bronchiectasis (Fig. 56-23). In more advanced disease, bronchiectasis that is predominant in the upper lobe, bronchial wall thickening, centrilobular nodular and tree-in-bud opacities, and mucus plugging with air trapping occur (Fig. 56-24). A finger-in-glove pattern of mucoid impaction similar to ABPA may be observed. Because of chronic/recurrent infections, mediastinal and hilar lymphadenopathy often is present. CT is much more sensitive than pulmonary functions tests for detecting mild or localized lung disease. Although they are not widely used and are of unclear benefit in individualized treatment, CT scoring systems to assess the extent and severity of CF are a valid surrogate endpoint for outcomes in clinical trials. There is a statistically significant correlation of the number of respiratory tract exacerbations and the CT score at baseline and change in score over a 2-year period.111–113
Treatment and Follow-up: Treatment traditionally has focused on the managing/preventing the sequelae of the disease. The standard of care most recently has included oral azithromycin, inhaled tobramycin, hypertonic saline solution, and dornase alfa (Pulmozyme), which functions to break down thick secretions. Additional antibiotics are used depending on the type of infections present and the resistance pattern. Recently, the novel small molecule ivacaftor (Kalydeco) received and Drug Administration approval for CF in patients with at least one G551D mutation. By directly potentiating CFTR with significant improvements in lung function, the medication heralds a new era in drug development for CF, focusing on personalized medicine.114
 What the Clinician Needs to Know
What the Clinician Needs to Know
• The revised ChILD classification scheme has allowed substantial progress in our understanding of pediatric ILD.
• Many pediatric ILDs have a genetic basis with associated anomalies. If ILD is clinically suspected, screening for extrapulmonary abnormalities and a genetic basis should be considered. Additionally, genetic counseling should be offered to family members.
• Alveolar growth abnormalities commonly coexist with a patchy pattern of PIG; because glucocorticoids are recommended for the latter condition, a lung biopsy should be considered for definitive diagnosis. In contrast, the characteristic CT findings of NEHI are reported to have 100% specificity among experienced pediatric radiologists, thus potentially avoiding a lung biopsy.
• BO syndrome is the clinical correlate for the histopathologic entity of constrictive or obliterative bronchiolitis. In distinction, bronchiolitis obliterans organizing pneumonia falls under the category of organizing pneumonia and is termed cryptogenic organizing pneumonia if it is idiopathic.
• Lung transplantation may be the only curative treatment option for infants with respiratory failure from severe alveolar growth abnormalities or genetic surfactant disorders. Specific treatments exist for many etiologies of ILD in children presenting past infancy, including aerosolized GM-CSF therapy, in addition to whole-lung lavage for autoimmune PAP; inciting antigen cessation and corticosteroids for hypersensitivity pneumonitis; immunosuppression for pulmonary hemorrhage with capillaritis; corticosteroids for pulmonary infiltrates with eosinophilia; enzyme replacement therapy for lysosomal storage diseases; antibiotics, interferon-γ, and stem cell transplantation for CGD; and antibiotics, hypertonic saline solution, and dornase alfa for CF, along with the CFTR-potentiator ivacaftor for persons with the G551D mutation.
Deterding, RR. Infants and young children with children’s interstitial lung disease. Pediatr Allergy Immunol Pulmonol. 2010;23:25–31.
Guillerman, RP, Brody, AS. Contemporary perspectives on pediatric diffuse lung disease. Radiol Clin North Am. 2011;49:847–868.
Guillerman, RP. Imaging of childhood interstitial lung disease. Pediatr Allergy Immunol Pulmonol. 2010;23:43–68.
Langston, C, Dishop, MK. Infant lung biopsy: clarifying the pathologic spectrum. Pathol Int. 2004;54:s419–s421.
Lee, EY, Cleveland, RH, Langston, C. Interstitial lung disease in infants and children: new classification system with emphasis on clinical, imaging, and pathologic correlation. In: Cleveland RH, ed. Imaging in pediatric pulmonology. New York: Springer, 2011.
References
1. Lee, EY, Cleveland, RH, Langston, C. Interstitial lung disease in infants and children: new classification system with emphasis on clinical, imaging, and pathologic correlation. In: Cleveland RH, ed. Imaging in pediatric pulmonology. New York: Springer, 2011.
2. Guillerman, RP, Brody, AS. Contemporary perspectives on pediatric diffuse lung disease. Radiol Clin North Am. 2011;49:847–868.
3. Guillerman, RP. Imaging of childhood interstitial lung disease. Pediatr Allergy Immunol Pulmonol. 2010;23:43–68.
4. Langston, C, Dishop, MK. Infant lung biopsy: clarifying the pathologic spectrum. Pathol Int. 2004;54:s419–s421.
5. Dishop, MK. Diagnostic pathology of diffuse lung disease in children. Pediatr Allergy Immunol Pulmonol. 2010;23:69–85.
6. Sen, P, Thakur, N, Stockton, DW, et al. Expanding the phenotype of alveolar capillary dysplasia (ACD). J Pediatr. 2004;145:646–651.
7. Licht, C, Schickendantz, S, Sreeram, N, et al. Prolonged survival in alveolar capillary dysplasia syndrome. Eur J Pediatr. 2004;163:181–182.
8. Stankiewicz, P, Sen, P, Bhatt, SS, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009;84:780–791.
9. Eulmesekian, P, Cutz, E, Parvez, B, et al. Alveolar capillary dysplasia: a six-year single center experience. J Perinat Med. 2005;33:347–352.
10. Michalsky, MP, Arca, MJ, Groenman, F, et al. Alveolar capillary dysplasia: a logical approach to a fatal disease. J Pediatr Surg. 2005;40:1100–1105.
11. Hugosson, CO, Salama, HM, Al-Dayel, F, et al. Primary alveolar capillary dysplasia (acinar dysplasia) and surfactant protein B deficiency: a clinical, radiological and pathological study. Pediatr Radiol. 2005;35:311–316.
12. Gillespie, LM, Fenton, AC, Wright, C. Acinar dysplasia: a rare cause of neonatal respiratory failure. Acta Paediatr. 2004;93:712–713.
13. Newman, B, Yunis, E. Primary alveolar capillary dysplasia. Pediatr Radiol. 1990;21:20–22.
14. Deterding, RR. Infants and young children with children’s interstitial lung disease. Pediatr Allergy Immunol Pulmonol. 2010;23:25–31.
15. Deutsch, GH, Young, LR, Deterding, RR, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1120–1128.
16. Owens, C. Radiology of diffuse interstitial pulmonary disease in children. Eur Radiol. 2004;14:L2–L12.
17. Mahut, B, De Blic, J, Emond, S, et al. Chest computed tomography findings in bronchopulmonary dysplasia and correlation with lung function. Arch Dis Child Fetal Neonatal Ed. 2007;92:F459–F464.
18. Biko, DM, Schwartz, M, Anupindi, SA, et al. Subpleural lung cysts in Down syndrome: prevalence and association with coexisting diagnoses. Pediatr Radiol. 2008;38:280–284.
19. Taylor, PA, Dishop, MK, Lotze, TE, et al. Congenital multilobar emphysema: a characteristic lung growth disorder attributable to Filamin A gene mutations. Pediatr Radiol. 2009;39(suppl 3):S516.
20. Agrons, GA, Courtney, SE, Stocker, JT, et al. From the archives of the AFIP: Lung disease in premature neonates: radiologic-pathologic correlation. Radiographics. 2005;25:1047–1073.
21. Mosca, F, Colnaghi, M, Fumagalli, M. BPD: old and new problems. J Matern Fetal Neonatal Med. 2011;24(suppl 1):80–82.
22. Kugelman, A, Durand, M. A comprehensive approach to the prevention of bronchopulmonary dysplasia. Pediatr Pulmonol. 2011;46(12):1153–1165.
23. Garmany, TH, Wambach, JA, Heins, HB, et al. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res. 2008;63:645–649.
24. Cole, FS, Hamvas, A, Rubinstein, P, et al. Population-based estimates of surfactant protein B deficiency. Pediatrics. 2000;105:538–541.
25. Gower, WA, Popler, J, Hamvas, A, et al. Clinical improvement in infants with ILD due to mutations in the surfactant protein C gene (SFTPC). Am J Respir Crit Care Med. 2010;181:A6733.
26. Rosen, DM, Waltz, DA. Hydroxychloroquine and surfactant protein C deficiency. N Engl J Med. 2005;352:207–208.
27. Karjalainen, MK, Haataja, R, Hallman, M. Haplotype analysis of ABCA3: association with respiratory distress in very premature infants. Ann Med. 2008;40:56–65.
28. Clement, A, Corvol, H, Epaud, R, et al. Dramatic improvement by macrolides in surfactant deficiency with ABCA3 mutation. Am J Respir Crit Care Med. 2009;179:A3011.
29. Doan, ML, Guillerman, RP, Dishop, MK, et al. Clinical, radiological, and pathological features of ABCA3 mutations in children. Thorax. 2008;63:366–373.
30. Guillot, L, Carre, A, Szinnai, G, et al. NKX2-1 mutations leading to surfactant protein promoter dysregulation cause interstitial lung disease in “Brain-Lung-Thyroid Syndrome. Hum Mutat. 2010;31:E1146–E1162.
31. Iwatani, N, Mabe, H, Devriendt, K, et al. Deletion of NKX2.1 gene encoding thyroid transcription factor-1 in two siblings with hypothyroidism and respiratory failure. J Pediatr. 2000;137:272–276.
32. Deterding, R, Dishop, MK, Uchida, DA, et al. Thyroid transcription factor 1 gene abnormalities; an under recognized cause of children’s interstitial lung disease. Am J Respir Crit Care Med. 2010;181:A6725.
33. Galambos, C, Levy, H, Cannon, CL, et al. Pulmonary pathology in thyroid transcription factor-1 deficiency syndrome. Am J Respir Crit Care Med. 2010;182:549–554.
34. Willemsen, MA, Breedveld, GJ, Wouda, S, et al. Brain-thyroid-lung syndrome: a patient with severe multi-system disorder due to a de novo mutation in the thyroid transcription factor 1 gene. Eur J Pediatr. 2005;164:28–30.
35. Bush, A. Paediatric interstitial lung disease: not just kid’s stuff. Eur Respir J. 2004;24:521–523.
36. Olsen, EØE, Sebire, NJ, Jaffe, A, et al. Chronic pneumonitis of infancy: high-resolution CT findings. Pediatr Radiol. 2004;34:86–88.
37. Hamvas, A. Inherited surfactant protein-B deficiency and surfactant protein-C associated disease: clinical features and evaluation. Semin Perinatol. 2006;30:316–326.
38. Soraisham, AS, Tierney, AJ, Amin, HJ. Neonatal respiratory failure associated with mutation in the surfactant protein C gene. J Perinatol. 2006;26:67–70.
39. Prestridge, A, Woodridge, J, Deutsch, G, et al. Persistent tachypnea and hypoxia in a 3-month-old term infant. J Pediatr. 2006;149:702–706.
40. Stevens, PA, Pettenazzo, A, Brasch, F, et al. Nonspecific interstitial pneumonia, alveolar proteinosis, and abnormal proprotein trafficking resulting from a spontaneous mutation in the surfactant protein C gene. Pediatr Res. 2005;57:89–98.
41. Thouvenin, G, Taam, RA, Flamein, F, et al. Characteristics of disorders associated with genetic mutations of surfactant protein C. Arch Dis Child. 2010;95:449–454.
42. Mechri, M, Epaud, R, Emond, S, et al. Surfactant protein C gene (SFTPC) mutation-associated lung disease: high-resolution computed tomography (HRCT) findings and its relation to histological analysis. Pediatr Pulmonol. 2010;45:1021–1029.
43. Kerby, GS, Wilcox, SL, Hay, TC, et al. Infant pulmonary function testing in children with neuroendocrine cell hyperplasia with and without lung biopsy. Am J Respir Crit Care Med. 2009;179:A3671.
44. Deterding, RR, Pye, C, Fan, LL, et al. Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol. 2005;40:157–165.
45. Popler, J, Young, LR, Deterding, RR. Beyond infancy: persistence of chronic lung disease in neuroendocrine cell hyperplasia of infancy (NEHI). Am J Respir Crit Care Med. 2010;181:A6721.
46. Brody, AS, Guillerman, RP, Hay, TC, et al. Neuroendocrine cell hyperplasia of infancy: diagnosis with high-resolution CT. AJR Am J Roentgenol. 2010;194:238–244.
47. Popler, J, Gower, WA, Mogayzel, PJ, Jr., et al. Familial neuroendocrine cell hyperplasia of infancy. Pediatr Pulmonol. 2010;45:749–755.
48. Bramson, RT, Cleveland, R, Blickman, JG, et al. Radiographic appearance of follicular bronchitis in children. AJR Am J Roentgenol. 1996;166:1447–1450.
49. Young, LR, Brody, AS, Inge, TH, et al. Neuroendocrine cell distribution and frequency distinguish neuroendocrine cell hyperplasia of infancy from other pulmonary disorders. Chest. 2011;139:1060–1071.
50. Deterding, R. Evaluating infants and children with interstitial lung disease. Semin Respir Crit Care Med. 2007;28:333–341.
51. Canakis, AM, Cutz, E, Manson, D, et al. Pulmonary interstitial glycogenosis: a new variant of neonatal interstitial lung disease. Am J Respir Crit Care Med. 2002;165:1557–1565.
52. Onland, W, Molenaar, JJ, Leguit, RJ, et al. Pulmonary interstitial glycogenosis in identical twins. Pediatr Pulmonol. 2005;40:362–366.
53. Castillo, M, Vade, A, Lim-Dunham, JE, et al. Pulmonary interstitial glycogenosis in the setting of lung growth abnormality: radiographs and pathologic correlation. Pediatr Radiol. 2010;40:1562–1565.
54. Smets, K, Dhaene, K, Schelstraete, P, et al. Neonatal pulmonary interstitial glycogen accumulation disorder. Eur J Pediatr. 2004;163:408–409.
55. Deutsch, GH, Young, LR. Pulmonary interstitial glycogenosis: words of caution. Pediatr Radiol. 2010;40:1471–1475.
56. Lanfranchi, M, Allbery, SM, Wheelock, L. Pulmonary interstitial glycogenosis. Pediatr Radiol. 2010;40:361–365.
57. Albafouille, V, Sayegh, N, De Coudenhove, S, et al. CT scan patterns of pulmonary alveolar proteinosis in children. Pediatr Radiol. 1999;29:147–152.
58. Vrielynck, S, Mamou-Mani, T, Emond, S, et al. Diagnostic value of high-resolution CT in the evaluation of chronic infiltrative lung disease in children. AJR Am J Roentgenol. 2008;191:914–920.
59. Copley, SJ, Padley, SP. High-resolution CT of paediatric lung disease. Eur Radiol. 2011;11:2564–2575.
60. Suzuki, T, Sakagami, T, Rubin, BK, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med. 2008;205:2703–2710.
61. Martinez-Moczygemba, M, Doan, ML, Elidemir, O, et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med. 2008;205:2711–2716.
62. Robinson, TE, Trapnell, BC, Goris, ML, et al. Quantitative analysis of longitudinal response to aerosolized granulocyte-macrophage colony-stimulating factor in two adolescents with autoimmune pulmonary alveolar proteinosis. Chest. 2009;135:842–848.
63. Miller, AL, Schissel, S, Levy, BD, et al. Clinical problem-solving. A crazy cause of dyspnea. N Engl J Med. 2011;364:72–77.
64. Clement, A, Nathan, N, Epaud, R, et al. Interstitial lung disease in children. Orphanet J Rare Dis. 2010;5:22.
65. Esther, CR, Jr., Barker, PM. Pulmonary lymphangiectasia: diagnosis and clinical course. Pediatr Pulmonol. 2004;38:308–313.
66. Barker, PM, Esther, CR, Jr., Fordham, LA, et al. Primary pulmonary lymphangiectasia in infancy and childhood. Eur Respir J. 2004;24:413–419.
67. Copley, SJ, Coren, M, Nicholson, AG, et al. Diagnostic accuracy of thin-section CT and chest radiography of pediatric interstitial lung disease. AJR Am J Roentgenol. 2000;174:549–554.
68. Faul, JL, Berry, GJ, Colby, TV, et al. Thoracic lymphangiomas, lymphangiectasias, lymphangiomatosis, and lymphatic dysplasia syndrome. Am J Respir Crit Care Med. 2000;161:1037–1046.
69. Swenson, SJ, Hartman, TE, Mayor, JR, et al. Diffuse pulmonary lymphangiomatosis: CT findings. J Comput Assist Tomogr. 1995;19:348–352.
70. Chung, CJ, Fordham, LA, Barker, P, et al. Children with congenital pulmonary lymphangiectasia: after infancy. AJR Am J Roentgenol. 1999;173:1583–1588.
71. Bellini, C, Boccardo, F, Campisi, C, et al. Pulmonary lymphangiectasia. Lymphology. 2005;38:111–121.
72. Yalçin, E, Dog, D, Halilog, M, et al. Postinfectious bronchiolitis obliterans in children: clinical and radiological profile and prognostic factors. Respiration. 2003;70:371–375.
73. Zhang, L, Irion, K, da Silva Porto, N, et al. High-resolution computed tomography in pediatric patients with postinfectious bronchiolitis obliterans. J Thorac Imaging. 1999;14:85–89.
74. Smith, KJ, Dishop, MK, Fan, LL, et al. Diagnosis of bronchiolitis obliterans with computed tomography in children. Pediatr Allergy Immunol Pulmonol. 2011;23:253–259.
75. Moonnumakal, SP, Fan, LL. Bronchiolitis obliterans in children. Curr Opin Pediatr. 2008;20:272–278.
76. Mattiello, R, Sarria, EE, Mallol, J, et al. Post-infectious bronchiolitis obliterans: can CT scan findings in early age anticipate lung function? Pediatr Pulmonol. 2010;45:315–319.
77. Lucaya, J, Gartner, S, García-Peña, P, et al. Spectrum of manifestations of Swyer-James-Macleod syndrome. J Comput Assist Tomogr. 1998;22:592–597.
78. Kim, TO, Oh, IJ, Kang, HW, et al. Temozolomide-associated bronchiolitis obliterans organizing pneumonia successfully treated with high-dose corticosteroid. J Korean Med Sci. 2012;27:450–453.
79. Vos, R, Vanaudenaerde, BM, Verleden, SE, et al. Antiinflammatory and immunomodulatory properties of azithromycin involved in treatment and prevention of chronic lung allograft rejection. Transplantation. 2012;94(2):101–109.
80. MacDonald, S, Müller, NL. Insights from HRCT: how they affect the management of diffuse parenchymal lung disease. Semin Respir Crit Care Med. 2003;24:357–364.
81. Fan, LL. Hypersensitivity pneumonitis in children. Curr Opin Pediatr. 2002;14:323–326.
82. Hartman, TE. The HRCT features of extrinsic allergic alveolitis. Semin Respir Crit Care Med. 2003;24:419–426.
83. Vece, TJ, Fan, LL. Interstitial lung disease in children older than 2 years. Pediatr Allergy Immunol Pulmonol. 2010;23:33–41.
84. Zacharisen, MC, Fink, JN. Hypersensitivity pneumonitis and related conditions in the work environment. Immunol Allergy Clin North Am. 2011;31:769–786.
85. Fullmer, JJ, Langston, C, Dishop, MK, et al. Pulmonary capillaritis in children: a review of eight cases with comparison to other alveolar hemorrhage syndromes. J Pediatr. 2005;146:376–381.
86. Susarla, SC, Fan, LL. Diffuse alveolar hemorrhage syndromes in children. Curr Opin Pediatr. 2007;19:314–320.
87. Nuesslein, TG, Teig, N, Rieger, CH. Pulmonary haemosiderosis in infants and children. Paediatr Respir Rev. 2006;7:45–48.
88. Ravenel, JG, McAdams, HP. Pulmonary vasculitis: CT features. Semin Respir Crit Care. 2003;24:427–436.
89. Connolly, B, Manson, D, Eberhard, A, et al. CT appearance of pulmonary vasculitis in children. AJR Am J Roentgenol. 1996;167:901–904.
90. Brody, AS. Imaging considerations: interstitial lung disease in children. Radiol Clin North Am. 2005;43:391–403.
91. Gordon, IO, Cipriani, N, Arif, Q, et al. Update in nonneoplastic lung diseases. Arch Pathol Lab Med. 2009;63:366–373.
92. Kligerman, SJ, Groshong, S, Brown, KK, et al. Nonspecific interstitial pneumonia: radiologic, clinical, and pathologic considerations. Radiographics. 2009;29:73–87.
93. Flaherty, KR, Martinez, FJ. Nonspecific interstitial pneumonia. Semin Respir Crit Care Med. 2006;27:652–658.
94. Lilleby, C, Aalokeen, TM, Johansen, B, et al. Pulmonary involvement with childhood-onset systemic lupus erythematosus. Clin Exp Rheumatol. 2006;24:203–208.
95. Schirmer, M, Dejaco, C, Duftner, C. Advances in the evaluation and classification of chronic inflammatory rheumatic diseases. Discov Med. 2012;13:299–304.
96. Leask, A. Emerging targets for the treatment of scleroderma. Expert Opin Emerg Drugs. 2012;17(2):173–179.
97. Fan, LL, Deterding, RR, Langston, C. Pediatric interstitial lung diseases revisited. Pediatr Pulmonol. 2004;38:369–378.
98. Polverosi, R, Maffesanti, M, Dalpiaz, G. Organizing pneumonia: typical and atypical HRCT patterns. Radiol Med. 2006;111:202–212.
99. Epler, GR. Bronchiolitis obliterans organizing pneumonia, 25 years: a variety of causes, but what are the treatment options? Expert Rev Respir Med. 2011;5:353–361.
100. Jeong, YJ, Kim, KI, Seo, IJ, et al. Eosinophilic lung diseases: a clinical, radiologic, and pathologic overview. Radiographics. 2007;27:617–637.
101. Oermann, CM, Panesar, KS, Langston, C, et al. Pulmonary infiltrates with eosinophilia syndromes in children. J Pediatr. 2000;136:351–358.
102. Martinez, S, Heyneman, LE, McAdams, HP, et al. Mucoid impactions: finger-in-glove sign and other CT and radiographic features. Radiographics. 2008;28:1369–1382.
103. Fernández Pérez, ER, Olson, AL, Frankel, SK. Eosinophilic lung diseases. Med Clin North Am. 2011;95:1163–1187.
104. McHugh, K, Olsen, EØE, Vellodi, A. Gaucher disease in children: radiology of non-central nervous system manifestations. Clin Radiol. 2004;59:117–123.
105. Goiten, O, Elstein, D, Abrahamov, A, et al. Lung involvement and enzyme replacement therapy in Gaucher’s disease. QJM. 2001;94:407–415.
106. Guillemot, N, Troadec, C, de Villemeur, TB, et al. Lung disease in Niemann-Pick disease. Pediatr Pulmonol. 2007;42:1207–1214.
107. Griese, M, Brasch, F, Aldana, VR, et al. Respiratory disease in Niemann-Pick type C2 is caused by pulmonary alveolar proteinosis. Clin Genet. 2010;77:119–130.
108. Towbin, AJ, Chaves, I. Chronic granulomatous disease. Pediatr Radiol. 2010;40:657–668.
109. Khanna, G, Kao, SC, Kirby, P, et al. Imaging of chronic granulomatous disease in children. Radiographics. 2005;25:1183–1195.
110. Gungor, T, Engel-Bicik, I, Eich, G, et al. Diagnostic and therapeutic impact of whole body positron emission tomography using fluorine-18-fluoro-2-deoxy-D-glucose in children with chronic granulomatous disease. Arch Dis Child. 2001;85:341–345.
111. Cleveland, RH, Neish, AS, Zurakowski, D, et al. Cystic fibrosis: predictors of accelerated decline and distribution of disease in 230 patients. AJR Am J Roentgenol. 1998;171:1311–1315.
112. Ramsey, BW. Use of lung imaging studies as outcome measures for development of new therapies in cystic fibrosis. Proc Am Thorac Soc. 2007;4:359–363.
113. Brody, AS, Sucharew, H, Campbell, JD, et al. Computed tomography correlates with pulmonary exacerbations in children with cystic fibrosis. Am J Respir Crit Care Med. 2005;172:1128–1132.
114. Ramsey, BW, Davies, J, McElvaney, NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672.