Chapter 8 Diagnosis and Surgical Options for Craniosynostosis
• In craniosynostosis, skull growth is arrested in the direction perpendicular to the fused suture and expanded at the sites of unaffected sutures (Virchow’s law), leading to characteristic calvarial deformations. In addition, the skull base and calvarial development are interrelated and changes at one location may affect the growth parameters at the other location.
• Intracranial hypertension can accompany craniosynostosis and is a function of the number of affected sutures, ranging from approximately 14% for single-suture synostosis to roughly 47% in multisuture synostosis. Children suspected of having elevated intracranial pressure may present with irritability, feeding difficulties, failure to thrive, headache, developmental delays, visual changes, calvarial towering, supraorbital recession, or lack of circumferential skull growth. Computed tomography (CT) scan changes may include “beaten copper” appearance of the inner table of the skull and compression of the ventricles and cisterns. Hydrocephalus and Chiari malformation can be associated with children with syndromic craniosynostosis (e.g., Crouzon, Apert, Pfeiffer syndromes).
• An increasing number of growth factor receptors (FGFR, TGF-βR), growth factors (FGF2, TGF-β, BMP), as well as transcription factors (MSX-2 and Twist), have been implicated in the pathogenesis of craniosynostosis and this list will undoubtedly grow in the future.
• The optimal timing of craniosynostosis surgery remains controversial even today, although the majority of craniofacial surgeons operate when patients are between 3 and 12 months of age. Because the normal brain and skull grow most rapidly in the first 2 years of life, early surgery takes advantage of this rapid period of growth and facilitates cranial volume expansion.
• Posterior deformational plagiocephaly, secondary to a supine sleeping position, will generally resolve with positional changes, physiotherapy, or helmet therapy and is only rarely a surgical condition.
Craniosynostosis is defined as the premature closure of a cranial suture which causes abnormal calvarial growth. Skull growth is arrested in the direction perpendicular to the fused suture and expanded at the sites of unaffected sutures, leading to characteristic calvarial deformations (Virchow’s law).1 In addition to the morphological changes accompanying craniosynostosis, functional problems related to brain development and possible intracranial hypertension are major considerations. Although the likelihood of elevated intracranial pressure remains low for patients with single-suture craniosynostosis, children with multiple-suture involvement or delayed presentation of single-suture synostosis are at significantly higher risk.2–4 A broad range of surgical options exist in the armamentarium of contemporary craniofacial surgical reconstruction, all with the primary objective of releasing the affected suture to permit normalization of skull growth in the setting of accelerated cerebral growth. Over time, progressively earlier recognition of craniosynostosis and its subsequent treatment have led to improved surgical results with correspondingly decreased perioperative morbidity. With a greater understanding of technologies relying on dynamic cranial vault alteration, including endoscopic sutural release, spring-assisted cranioplasty, and distraction osteogenesis, new horizons will inevitably unfold.
History
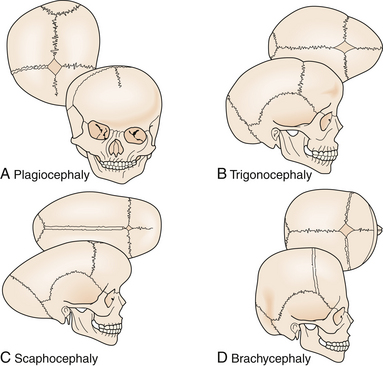
Craniosynostosis has long been recognized as an abnormal process originating at the calvarial suture. Early recognition of the importance of the skull sutures and their relationship to head shape was first made by investigators such as Hippocrates, Galen, and Celsus. In 1791, Sommerring noted that calvarial growth occurred at the suture line and that premature suture closure led to restriction of growth perpendicular to the affected suture.5 In addition to confirming Sommerring’s findings, Virchow was the first to describe the compensatory calvarial growth that occurred at the sites of unaffected sutures and associate a characteristic head shape with its corresponding abnormal suture (Fig. 8.1).1 These observations served as principal tenets directing craniosynostosis surgery over the subsequent century.
Over time, the relationship between calvarial growth and the skull base became better appreciated. In 1959, Moss pointed out the importance of the skull base in the promotion and development of the calvarial vault.6 His contributions included the observation that the cranial base developed prior to the calvarial vault and that characteristic abnormalities in the cranial base were associated with classic sutural abnormalities. Nevertheless, subsequent experimental work in animal models demonstrated that restriction of growth at specific sutures resulted in characteristic skull deformities that mimicked shapes seen in simple (nonsyndromic) craniosynostosis.7–9 As a result, the pathogenesis of craniosynostosis is currently thought to be a combination of skull base and calvarial growth disturbances.
Epidemiology
Craniosynostosis occurs in approximately 1 in 2000 to 1 in 2500 live births.10 This condition can be classified into simple (single-suture) versus complex (multiple sutures) or nonsyndromic versus syndromic (Table 8.1). Single-suture synostosis represents the majority of patients, with multiple-suture synostoses comprising approximately 5% to 15% of cases.10 As reported by large craniofacial centers, syndromic patients account for 15% to 20% of cases, whereas nonsyndromic patients constitute 80% to 85%.11
| Affected Suture | Phenotypic Presentation |
|---|---|
| Sagittal | Dolichocephaly, scaphocephaly |
| Coronal (unilateral) | Anterior plagiocephaly |
| Coronal (bilateral) | Brachycephaly |
| Metopic | Trigonocephaly |
| Lambdoid | Posterior plagiocephaly |
| Multiple sutures | Cloverleaf (Kleeblatschädel), acrocephaly, oxycephaly |
Single-suture synostosis most frequently occurs sporadically, with familial aggregation accounting for 7% to 8% of sagittal and metopic synostosis.11 An equal frequency is found for all ethnic populations; however, gender predilection will vary depending on the type of suture pathology. The most commonly involved location is the sagittal suture, which accounts for 45% to 68% of all individuals12,13 and is marked by a male/female ratio ranging from 3.5:1 to 7:1.14 An autosomal dominant inheritance pattern with 38% penetrance was reported for sagittal synostosis.15 Metopic synostosis is now the second most common form of craniosynostosis (23.7-27.3% of cases), an observation that currently evades definitive etiopathogenesis, and shows a male predominance of 75%.11–13 Unicoronal synostosis, also known as anterior plagiocephaly, accounts for approximately 18% of patients with craniosynostosis,12 with girls outnumbering boys by a 3:2 ratio.16 Lambdoid suture synostosis, referred to as posterior plagiocephaly, is a relatively rare event in children with an observed incidence ranging from 0.9% to 4%.17–19 True lambdoid synostosis must be distinguished from posterior deformational plagiocephaly, also known as positional molding, in which there is occipital flattening on the affected side without associated suture fusion. This epiphenomenon is possibly related to the supine sleeping position in young children, instituted in 1992 to address sudden infant death syndrome (SIDS).20
Although the sporadic nature of simple craniosynostosis makes an accurate prediction of risk difficult to ascertain, it appears that the risk doubles for future siblings if there are no other family members involved. When one parent and child are affected, the subsequent risk rises to 50%. Conversely, if both parents are unaffected and two siblings are affected, the risk for additional sibling involvement approaches 25%.21
More than a hundred syndromes have been associated with craniosynostosis, often marked by an autosomal dominant mode of transmission.22 Among them, Crouzon, Apert, and Pfeiffer (Fig. 8.2) syndromes are the most frequently occurring. Syndromic synostosis is commonly associated with multiple suture closure (coronal, sagittal, etc.) combined with other systemic manifestations (Table 8.2).
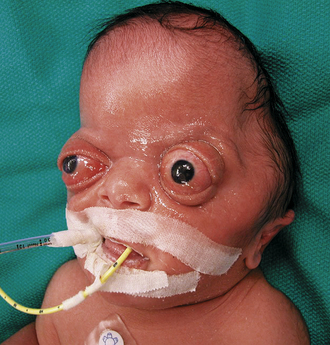
TABLE 8.2 Craniofacial Dysostosis Syndromes
| Syndrome | Involved Suture | Morphological Presentation |
|---|---|---|
| Crouzon | Coronal, sagittal | Midface hypoplasia, shallow orbits, proptosis, hypertelorism |
| Apert | Coronal, sagittal, lambdoid, others | Midface hypoplasia, shallow orbits, proptosis, hypertelorism, symmetrical syndactyly of hands and feet, choanal atresia, ventriculomegaly, genitourinary/cardiovascular anomalies |
| Pfeiffer | Coronal, sagittal | Midface hypoplasia, proptosis, hypertelorism, broad great toe/thumb |
Genetic and Etiological Factors
The etiology of craniosynostosis remains elusive because of its heterogeneous nature. Nevertheless, numerous factors are now known to promote or have been implicated in the development of premature closure of the calvarial sutures. Multiple teratogens, genetic mutations, metabolic disorders, and blood dyscrasias have been associated with craniosynostosis (Table 8.3). Interestingly, maternal smoking has been associated with isolated craniosynostosis23 and advanced paternal age has been found to trend with a higher frequency of metopic synostosis.12
| Hematologic disorders | Thalassemias Sickle cell anemia Polycythemia vera |
| Teratogens | Valproic acid Retinoic acid Aminopterin Diphenylhydantoin |
| Genetic conditions | |
| Metabolic disorders | Rickets Hyperthyroidism |
| Mucopolysaccharidoses | Hurler syndrome Morquio syndrome Mucolipidosis III |
| β-Glucuronidase deficiency | |
| Malformations | Holoprosencephaly Encephalocele Microcephaly Hydrocephalus (shunted) |
With advances in molecular genetics, candidate gene mutations as well as the molecular interactions underlying cranial deformities have been elucidated.24,25 Normal suture growth and morphogenesis is dependent upon a delicate balance between the proliferation of osteoprogenitors within the suture mesenchyme and differentiation to osteoblasts at the osteogenic fronts (Fig. 8.3).26 It is now known that the majority of syndromic craniosynostoses are caused by mutations in genes encoding fibroblast growth factor receptors (FGFR-1, FGFR-2, and FGFR-3) and the transcription factors Twist and MSX-2.27–33 Moreover, these same genes are responsible for approximately 25% of all cases of craniosynostosis.34 In general, gain-of-function mutations are associated with the MSX2 and FGFR genes, while loss of function or haplo-insufficiency abnormalities are found in TWIST gene mutations.35
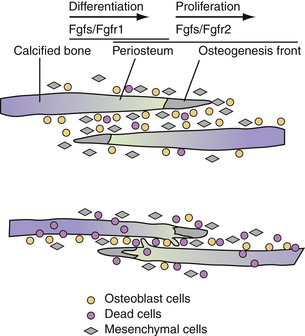
Genetic alterations in FGFR-1 and FGFR-2 have been implicated in Crouzon, Apert, Pfeiffer, and Jackson-Weiss syndromes.30,36–39 FGFR-1 has been found to regulate osteoblast differentiation; therefore, a gain-of-function mutation may precipitate premature suture fusion through promotion of osteoblast differentiation and bone formation.40 Moreover, FGFR-2 has been linked to activation of osteogenic cell apoptosis. As shown by Chen and co-workers, a gain-in-function mutation leads to increased apoptosis and results in decreased cell numbers and distance between two overlapping bones. Ultimately, this develops into physical contact of two opposing bones, eventually leading to premature closure.41
Alterations in growth factor receptors have also been observed in nonsyndromic craniosynostoses. FGFR-3-associated coronal synostosis, also known as Muenke-type craniosynostosis, has been identified in up to 52% of patients with nonsyndromic bicoronal synostosis,42 either as a result of de novo mutations or associated with an autosomal dominant inheritance pattern.43 Gripp and colleagues observed that 10.8% of patients with unilateral coronal synostosis were positive for an FGFR-3 mutation and subsequently recommended testing of all patients with unilateral coronal synostosis to assess the risk of recurrence.44 These guidelines stem from the observation that Muenke-type craniosynostosis has been associated with a reoperation rate of at least 43%.45
Studies have implicated transforming growth factor-beta receptors (TGF-βR) in syndromic and intrauterine head constraint-related craniosynostosis. Loeys and co-workers have discovered a link between mutations of TGF-βR1 and TGF-βR2 and a syndrome of altered cardiovascular, craniofacial, neurocognitive, and skeletal development.46 Hunenko and colleagues demonstrated upregulation of TGF-βR1 and TGF-βR2 in mice undergoing intrauterine constraint leading to coronal suture synostosis.47 Such data points to the ability of mechanical forces to alter growth factor–mediated signaling during craniofacial growth and development.
In addition to the aforementioned growth factor receptors, their corresponding ligands have been found to be an integral component of calvarial osteoblast proliferation and subsequent sutural fusion. FGF2 has been shown to enhance proliferation rates in rat fetal osteoblasts, promote premature fusion of frontal sutures in calvarial organ cultures, and correlate with intrauterine constraint-related coronal suture synostosis.47,48 Inverse patterns of TGF-β isoform expression between fusing and patent sutures have been demonstrated in animal and human models. Opperman and colleagues demonstrated declining levels of TGF-β3 but continued expression of TGF-β1 and TGF-β2 posterior frontal suture fusion.49 Bone morphogenetic proteins (BMPs), members of the TGF-β superfamily, are involved in a broad range of developmental roles, including bone formation, skeletal patterning, and limb development. Several investigators have demonstrated the critical role of BMPs and their antagonists in dictating cranial suture biology. In particular, in situ hybridization of mouse cranial sutures localized expression of BMP-2 and BMP-4 to the osteogenic fronts and BMP-4 to suture mesenchyme and dura mater in the sagittal and posterior frontal sutures.50 Nacamuli and co-workers found BMP-3, a bone morphogenic protein antagonist, to be decreased in normally fusing posterior frontal sutures and increased in normally patent sagittal sutures.51
Mutations of transcription factors have also been implicated in causing syndromic forms of craniosynostosis. Liu and co-workers linked a gain-of-function mutation in the MSX-2 transcription factor with Boston-type craniosynostosis.52 Loss-of-function mutations in Twist proteins, transcription factors activating osteoblast differentiation, have been found to cause Saethre-Chotzen syndrome.28 Woods and colleagues have recommended TWIST1 mutation screening of all patients with either bicoronal or unicoronal synostosis, given that this genetic alteration confers a greater risk of recurrent intracranial hypertension and subsequent reoperation than nonsyndromic synostosis of the same sutures.53
Anatomical and Pathological Considerations
The calvarium grows most rapidly during the first 12 months, with the brain doubling in volume in the first 6 months and again by the second birthday. While calvarial expansion is most pronounced during the first 2 years, growth continues in a linear fashion until the age of 6 to 7 years, at which time the cranium is 90% of the adult size. Most of this cranial growth takes place in the sutures between the bone plates. Within the center of the sutural area, a population of proliferating osteoprogenitor cells is maintained. A portion of these cells enters the pathway of osteogenic differentiation, forming bone-matrix-secreting osteoblasts at the bone edges and contributing to skull expansion.54 Normal cranial suture closure occurs from front to back and from lateral to medial, with the metopic suture usually closing between 9 and 11 months of age55 and the remaining sutures fusing in adulthood.
A disturbance in the balance between proliferation, differentiation, and apoptosis causes premature ossification within the suture and its synostosis.56 Factors disturbing this balance include genetic or acquired changes in growth factor receptor/ligand profiles, loss of direct contact between dural and sutural cells, and increased external mechanical forces. As mentioned previously, many of the syndromic forms of craniosynostosis are attributed to alterations in the FGF/FGFR, TGF-β/TGF-βR, and BMP cascades. Both cerebral hypoplasia and overshunted hydrocephalus have been associated with secondary craniosynostosis, phenomena likely attributed to loss of dural contact.55,57 Both breach positioning and twin pregnancies have been associated with intrauterine constraint-related craniosynostosis, stemming from mechanical force signal transduction.58
Diagnostic Evaluation and Imaging
Preoperative assessment for craniosynostosis includes a detailed medical history, physical examination, and radiographic imaging. Medical history should elicit for asymmetrical calvarial deformities noted by friends or other family members, family history of calvarial deformities, and symptoms of intracranial hypertension (headache/vomiting, developmental changes, irritability, and oculomotor paresis). Physical examination should evaluate for characteristic calvarial shapes and asymmetries, premature closure of the anterior fontanelle (normally open until 12-18 months of age), perisutural ridging (calcification), and signs of intracranial hypertension (papilledema, supraorbital retrusion, severe towering, and severe frontal/occipital bossing). Routine funduscopic examination for papilledema is an accurate predictor of raised pressure in the older child but may not be 100% sensitive for the younger child (<8 years old).59 Craniofacial asymmetries should be documented in the form of head circumferences, cranial indices, and anthropometric measurements. The history combined with the examination is often confirmatory in an experienced primary care physician/nurse or craniofacial surgeon’s initial evaluation.
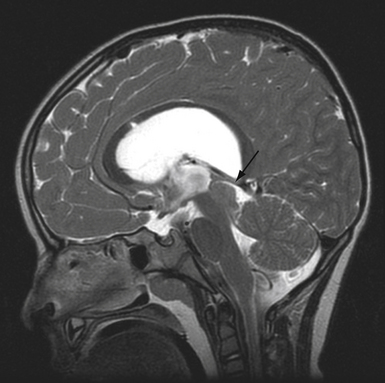
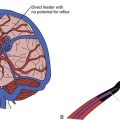
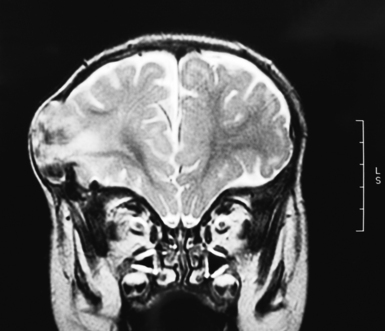
The role of radiological workup for craniosynostosis varies among clinicians. Currently, it is not uncommon for prenatal ultrasound to document craniosynostosis in utero.60–63 In addition to the ultrasound evaluation, fetal magnetic resonance imaging (MRI) (Fig. 8.4) at some centers has offered significant prenatal definition.64 Radiological investigation may be necessary to corroborate the diagnosis and rule out any associated intracranial abnormalities in the postnatal consultation period. Computed tomography (CT) studies remain the most sensitive barometer of bony fusion, as skull plain films suffer from poor sensitivity and a high false positive rate. The recent advent of three-dimensional CT (3D CT) has provided an excellent view of affected suture(s) as well as overall head shape, thereby simplifying the diagnosis and helping with surgical planning.60,65,66 This modality is not mandatory, but rather is reserved for multiple/complicated suture pathology, confirmation of diagnosis, or demonstration of skull base pathology.
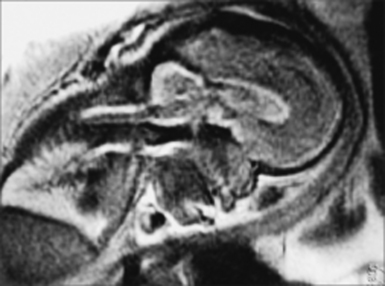
CT scans may also provide radiological evidence for raised intracranial pressure. The presence of intracranial hypertension is dependent on the number of affected sutures, ranging from approximately 14% for single-suture synostosis to approximately 47% in multiple-suture synostosis, as well as on patient age.2,3 Although few children will manifest clinical symptoms of increased intracranial pressure, it is not uncommon to visualize erosion of the inner calvarial table (beaten copper appearance) on CT scan. A diffuse beaten copper appearance has been associated with greater intracranial pressure, as reported by Tuite and co-workers.67 Though it is common to see expanded subarachnoid spaces in all types of craniosynostoses, these spaces usually spontaneously resolve and are not felt to represent an increase in intracranial pressure.68 If there is any evidence for elevated pressure, surgical consideration should be expedited. This is especially vital in patients with syndromic synostosis (e.g., Crouzon, Apert, Pfeiffer syndrome), who are at a greater risk for hydrocephalus and Chiari malformations with associated intracranial hypertension.
CT and MRI studies are also helpful in evaluating the underlying brain for any structural or functional abnormalities. Unrecognized intracranial abnormalities may exist in a small number of patients and may include hydrocephalus (more common in patients with Crouzon syndrome), partial agenesis of the corpus callosum, holoprosencephaly (seen in patients with trigonocephaly), or focal cortical dysplasias. Indeed, as reported by Boop and colleagues, up to 5% of their patients with sagittal synostoses had unappreciated underlying intracranial pathology.69
Therapeutic Considerations
Surgical Indications
Correction of calvarial contour deformities and prevention of psychosocial dysfunction, intracranial hypertension, and mental retardation are the thrusts for surgical intervention in craniosynostosis. In the past, surgical intervention for simple craniosynostosis was undertaken primarily because of cosmetic and psychosocial considerations.70,71 Recently, sutural release in simple craniosynostosis has been advised owing to the concerns regarding raised intracranial pressure as well as mild but significant developmental delay in the aging child with uncorrected single-suture synostosis.2,3,72–74 In contrast to simple craniosynostosis, patients with complex or syndromic synostoses present with increased severity in neurological and cosmetic symptoms;3,75–77 therefore, surgical intervention in these infants is even more imperative.
Timing of Surgery
The optimal timing for reconstructive surgery in craniosynostosis remains controversial, as the age at surgery has different effects on intraoperative hemodynamics, postoperative cranial growth, and subsequent mental development. With regard to intraoperative hemodynamics, Meyer and co-workers demonstrated that older patient age (>6 months) was associated with decreased blood loss.78 In addition to benefiting from decreased blood loss, older infants can tolerate extensive blood loss better than younger infants. From the perspective of long-term skull growth, data are conflicting. In 1987, Whitaker demonstrated that as surgical age increased, the likelihood of secondary surgery also elevated.79 On the other hand, Fearon and colleagues recently found that older patient age (≥12 months) was associated with less diminished cranial growth following correction of all types of single sutural craniosynostosis.80 These findings must be weighed against the need to fully reconstruct any advanced postoperative skull defects in children over 12 months of age, because dura will not regenerate bone as readily. From the standpoint of mental development, Arnaud and co-workers reported that postoperative mental outcome was significantly better when surgery was performed before the patient reached 12 months of age.42
Although the literature is inconclusive regarding the appropriate timing for correction of craniosynostosis, the majority of craniofacial surgeons operate between 3 and 12 months of age. The specific time period is dependent on the type of surgical approach used. In general, endoscopic corrections are done at an earlier age, namely, by 3 to 4 months of age. Open surgical corrections are often done later. Fearon and colleagues perform treatment at 4 months of age for sagittal synostosis and 9 months of age for all other single-suture synostoses (metopic, coronal, and lambdoid).80 Marchac and associates reviewed their craniofacial experience with 983 patients operated on over 20 years, discussing their timing of surgical operations.81 Children with brachycephaly underwent a floating forehead procedure between 2 and 4 months of age, infants with sagittal synostosis underwent parasagittal craniectomies between 2 and 4 months of age or a frontocranial remodeling procedure if presenting between 6 and 9 months of age, and infants with either metopic or unicoronal synostoses had a frontocranial remodeling procedure between 6 and 9 months of age.
Type of Surgery
There is a growing debate in the literature regarding the optimal type of operation for correction of craniosynostosis. An open craniofacial approach was proposed as early as 1890, with Lannelogue advocating early open surgical release of a fused suture to prevent intracranial hypertension.82 Building on the principles of Lannelogue, a number of centers have reported their large-volume experience with open cranial vault remodeling procedures for all types of craniosynostoses.79,80,83–87 In 421 intracranial operations with movement of one or both orbits, Whitaker and co-workers in 1979 reported a 2.2% rate of mortality, 6.2% rate of infection, and 2.2% frequency of CSF leak.85 With increased experience and refinement in technique, Whitaker and colleagues reported a 0% mortality rate, 3.7% infection rate, and 1.2% CSF leak rate in a 1987 report of 164 open craniofacial procedures for craniosynostosis.79 Regarding longevity of the open craniofacial procedure, McCarthy and associates identified a 13.5% rate of reoperation for simple craniosynostoses and 36.8% revision rate for complex craniosynostoses during a 20-year experience.86,87 Reoperation rates have also decreased with experience and refinement of open surgical technique, with Sloan and co-workers reporting a 7.2% overall rate of reoperation in 250 patients and Fearon and associates noting a 2% rate of revision in 248 cases of simple craniosynostosis.80,84
Endoscopic Craniosynostosis Correction
Jimenez and Barone pioneered endoscopic sutural release in the mid-1990s,88–94 and it has been used by increasing numbers of surgeons who treat craniosynostosis. In general, the idea is to perform a minimally invasive strip craniectomy of the fused suture at an early age. The child then wears a cranial molding helmet, which slowly corrects the deformity over several months.
For coronal (Fig. 8.5) or metopic craniosynostosis, the child is positioned supine. One small incision is used to access the fused suture. A burr hole is drilled, and an endoscope is used to separate the dura from the overlying bone. Bone-cutting scissors are then used to cut out the fused suture. Irrigation and Gelfoam are used for hemostasis, and the incision is closed.
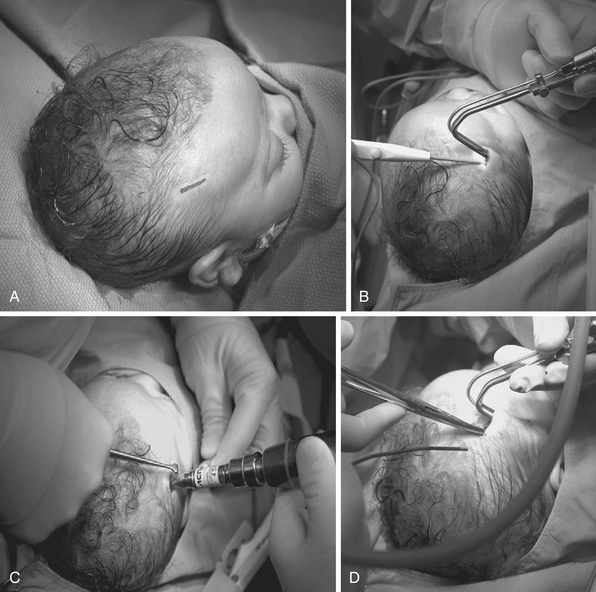
Postoperatively, patients are placed in orthotic helmets for 7 to 12 months to facilitate dynamic cranial vault alteration. Summarizing the data from studies by Jimenez and Barone,88–94 average age at operation is between 3 and 4 months, mean operating time centers around 60 minutes, average estimated blood loss is approximately 30 mL or approximately 5% estimated blood volume (EBV), transfusion rate hovers around 10%, average length of stay is generally 1 day, and complication rates range from 0% to 6%. These numbers are far lower than the blood loss of 25% to 500% of EBV, blood transfusion volume of 25% to 500% of EBV, and 4- to 7-day length of stay associated with open craniofacial techniques.93 MacKinnon and co-workers also found that children treated with endoscopic repair of unicoronal craniosynostosis may have less severe eye findings, such as V-pattern strabismus, than children treated with fronto-orbital advancement at a later age.95
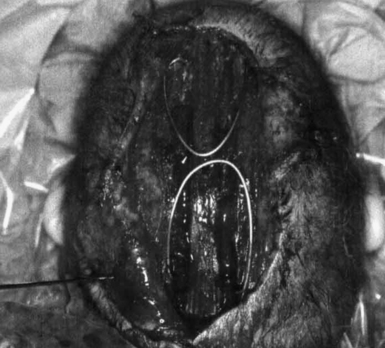
Spring-Assisted Cranioplasty
Spring-assisted cranioplasty was first undertaken on human subjects by Lauritzen in 1997, based upon success in the rabbit model by Persing and co-workers.96,97 This technique relies on a standard open access incision, osteotomies at the sites of stenosed sutures, placement of omega-shaped tension springs across the osteotomy sites, and possible placement of compressive springs along areas of compensatory growth (Fig. 8.6). Implantable springs are typically removed 4 to 7 months postoperatively. In 2008, Lauritzen and co-workers reported their large-volume experience with spring-assisted cranioplasty for all forms of craniofacial surgery.98 Results included an average operative time of 97 to 215 minutes, mean blood loss ranging from 143 to 503 mL, average length of stay between 5 and 6 days, mean cephalic index of 74 in scaphocephalic infants 6 months following cranioplasty, 6% reoperation rate, 3% rate of intracranial hypertension secondary to compressive springs, and a 0% mortality rate. On the heels of this study, David and co-workers reported their experience with the first 75 spring-assisted surgeries for scaphocephaly.99 With a mean follow-up period of 46 months, mean cephalic index was 75.4, which is comparable to patients with open cranial vault reconstruction and was maintained at 3- and 5-year follow-ups. Optimism with this emerging modality must be balanced against the need for a second operation for spring removal as well as the lack of control of spring action.
Distraction Osteogenesis
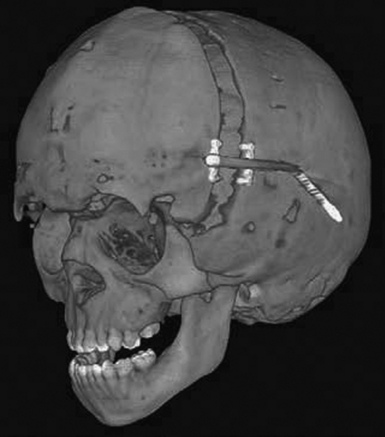
Along the lines of spring-assisted cranioplasty, distraction osteogenesis has been investigated for the treatment of craniosynostosis. This method entails a standard open access incision, osteotomies at the sites of stenosed sutures, and placement of internal versus external distraction devices (Fig. 8.7). After a latency period of 3 to 5 days, the devices are activated for a number of weeks until desired expansion and then followed by a 2- to 3-week consolidation period. Given that appraisal of this technique has been limited to the setting of small case reports,100–112 its safety and efficacy cannot be commented upon at this time.
Clinical Presentation/Therapeutic Considerations
Metopic Synostosis (Trigonocephaly)
Clinical Features
Metopic synostosis is often accompanied with a variable degree of phenotypic severity. Patients may present with mild ridging of the metopic suture, unaccompanied by other manifestations. Premature closure of the metopic suture may also lead to the formation of a triangular head, otherwise known as trigonocephaly. On the severe end of the spectrum, patients may present with a prominent “keel” forehead accompanied by recession of the lateral orbital rims, hypotelorism, and constriction of the anterior frontal fossa (Fig. 8.8).
Among the nonsyndromic craniosynostoses, metopic synostosis is most associated with chromosomal abnormalities, other brain malformations, and cognitive/behavioral dysfunction. Mild variants of metopic synostosis have been recently associated with abnormalities of chromosomes 3, 9, and 11.113–115 In addition, Tubbs and co-workers found a 30% incidence of type I Chiari malformations in the evaluation of patients with simple metopic ridges and postulated that these children were at greater risk secondary to the diminished anterior cranial volume.116 At the other end of the spectrum, severe cases of metopic synostosis have been associated with underlying frontal brain dysmorphology as well as other congenital anomalies.117
Metopic synostosis has also been associated with neurodevelopmental delay, with reported deficits ranging from modest to severe. Historically, metopic synostosis had been considered the form of single-suture synostosis with the highest degree of neuropsychological morbidity.118 Although Becker and co-workers confirmed a high prevalence of speech, cognitive, and behavioral abnormalities in patients with metopic synostosis, reported as 57%, they observed no differences in the degree of neuropsychological morbidity between different forms of single-suture synostoses.119 More recently, however, studies have concluded a modest to no delay in neurodevelopment.120,121 Da Costa and co-workers found that children with nonsyndromic craniosynostoses did not display obvious evidence of intellectual dysfunction, with mean intelligence quotients within the normal range. In 2007, Speltz and colleagues demonstrated a modest but reliable neurodevelopmental delay in children with all forms of single-suture synostoses in comparison to case-matched control subjects.121 Similar to Becker’s study, they found no difference in the extent of neuropsychological morbidity between different types of single-suture synostoses.119,121
Radiological Evaluation
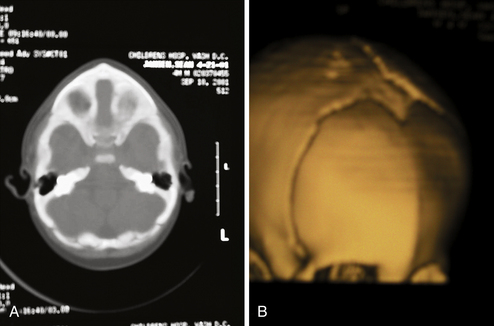
Radiographic imaging by plain skull radiographs may demonstrate a hyperostotic, midline metopic suture in addition to hypoteloric orbits in severe examples of trigonocephaly. Nevertheless, definitive diagnosis is best made by CT, which will offer better bone definition while also evaluating the cerebral parenchyma (Fig. 8.9). Frontal dysmorphology is most commonly seen in this type of craniosynostosis and may consist of corpus callosum dysgenesis, holoprosencephaly, and other frontal dysembryogeneses. Patients suspected of harboring a Chiari malformation are best served by an MRI, although CT scans with low cuts through the posterior fossa may also demonstrate a crowded foramen magnum as a result of tonsillar herniation.
Surgical Therapeutics
Delashaw and colleagues proposed that metopic synostosis and trigonocephaly represent an embryological continuum, directing their surgical approach based on the severity of the frontal calvarial deformities.122 In general, the goals of surgery are the normalization of the forehead with reconstitution of a normal supraorbital rim if necessary.123–129 Individuals presenting solely with a prominent midline keel may be best served by simple contouring of the frontal bone or by removal of the frontal bone flap followed by reconfiguration. These children otherwise demonstrate normal orbital and supraorbital anatomy. Conversely, patients with significant trigonocephaly and hypotelorism will require a fronto-orbital reconstruction, recontouring the frontal bone and laterally expanding the orbits at the same time. Evolution of surgical technique has included more radical treatment of the involved sphenoid bone with simultaneous correction of the hypotelorism.130–132
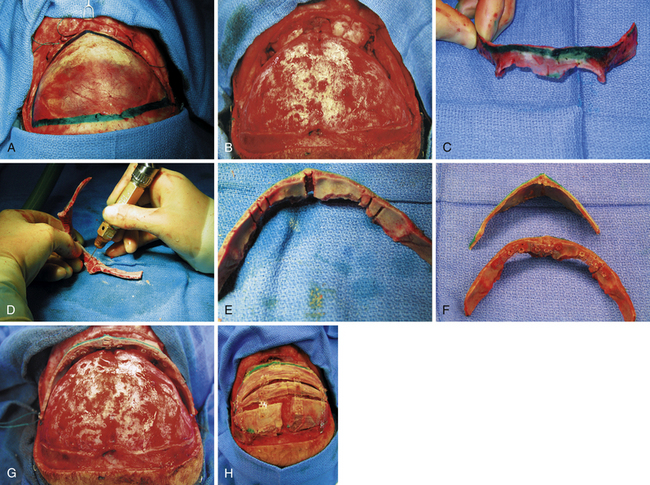
The essentials of fronto-orbital reconstruction involve a standard bicoronal incision, which will provide adequate exposure of the fronto-orbital region while minimizing any postoperative scar. Perioperative antibiotics (cefazolin 10-20 mg/kg loading dose, 8 mg/kg intravenously every 8 hours for 48 hours) as well as steroids (Decadron 0.25 mg/kg intravenous every 6 hours for 48 hours) are given. Prior to the start of surgery, bilateral tarsorrhaphies are undertaken. The incision is infiltrated with 0.5% lidocaine and 1:400,000 parts epinephrine to minimize intraoperative bleeding. The frontal and temporal regions are dissected in the subgaleal plane leaving the periosteum intact on the surface of the bone which also helps to minimize bleeding (Fig. 8.10A). The dissection is taken down to the level of the periorbital tissues, taking care to avoid any injury to the underlying globes. Following exposure of the frontal and orbital regions, the frontal bone is removed, providing access to the intracranial compartment (Fig. 8.10B). The supraorbital rim is then removed in one piece to facilitate reconstruction of the previously triangular supraorbital bar. Care is taken to remove sufficient bone in the region of the sphenoid bone to allow for growth at the midface and orbits (Fig. 8.10C). If the orbits require correction of hypotelorism, it will be necessary to displace the lateral walls of the orbit as well as to split the midline and interpose a calvarial bone graft. Reconfiguration of the supraorbital bar often requires a midline osteotomy to facilitate a flattened forehead with additional partial-thickness bone cuts at the lateral (pterional) angle to promote normalization of the lateral supraorbital angle (Fig. 8.10D and E). The supraorbital reconfiguration is maintained by the utilization of intervening bone grafts as well as absorbable hardware (Fig. 8.10F). Following placement of the supraorbital bar as a foundation (Fig. 8.10G), the frontal bone is reconstructed using the remaining portions of bone. It is often possible to reverse the original frontal bone flap (posterior portion now in an anterior position) to obtain an adequate width and contour with the new frontal bone flap. It is important to provide an adequate enhancement at the pterional region to avoid long-term supratemporal hollowing or recession. Reconstruction is facilitated with absorbable hardware, as permanent hardware has been associated with transcranial migration (Fig. 8.10H).
Outcome and Complications
Following fronto-orbital reconstruction for metopic synostosis, the long-term outcome is generally excellent for more than 90% of patients unless there are manifestations of syndromic overlay.80,83,123,133–137 Recently, Pearson and co-workers reported a 7% rate of reoperation and Fearon and colleagues reported a 2.5% rate of revision in infants treated for metopic synostoses.80,83 Additional surgery may be needed in children who initially do well but subsequently develop new metopic ridging, progressive frontal towering, or continued laterosuperior fronto-orbital restriction.138 It has also been the author’s experience to witness the subsequent development of additional sutural stenosis (sites other than the metopic suture) despite the lack of suture fusion or sclerosis at the time of the initial CT scan.
Surgical morbidity rate remains low for patients undergoing fronto-orbital reconstruction with wound infections, CSF leaks, post-traumatic encephaloceles (Fig. 8.11), and orbital/neural injuries being relatively uncommon events. Intraoperative blood loss and transfusion requirements, leading to hemodynamic instability, constitute greater dangers to the patient and should never be underestimated. Studies have reported blood loss ranging from 19% to 58% of EBV and blood transfusion volumes of approximately 34% of EBV.139–142 It is imperative to accurately gauge the extent of blood loss and match accordingly with packed red blood cells.
Sagittal Synostosis (Scaphocephaly, Dolichocephaly)
Clinical Features
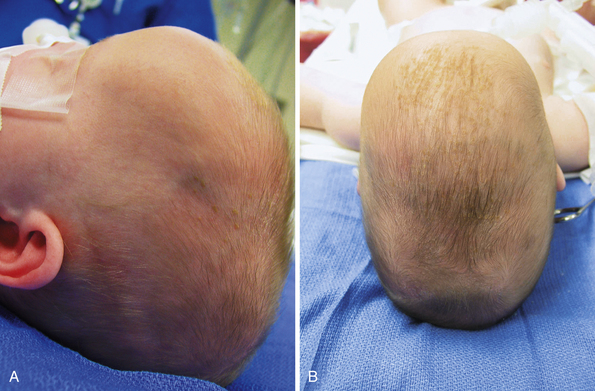
Children with sagittal synostosis will present with a narrow, elongated skull (dolichocephaly—long-headedness, scaphocephaly—boat-shaped). Perisutural ridging is often observed in addition to premature or occasionally delayed closure of the anterior fontanelle. Depending upon the region of greatest premature fusion of the sagittal suture, the child may manifest frontal or occipital bossing, or a combination of both (Fig. 8.12). Some children will also demonstrate a “towering” skull, also known as turricephaly, which may be a harbinger of intracranial hypertension (see earlier section on Diagnostic Evaluation and Imaging).
The presence of neurodevelopmental delay in children with sagittal synostosis remains debated. In a critical review of 17 historical studies, Speltz and co-workers concluded that isolated craniosynostosis was associated with a three- to fivefold increase in risk for cognitive deficits or learning/language disabilities.143 Becker and colleagues reported a 39% rate of speech, cognitive, and behavioral abnormalities in patients with sagittal synostosis, which trended as the lowest rate of neuropsychological morbidity for single-suture synostoses.119 More recently, however, studies have concluded a modest to no delay in neurodevelopment for all forms of single-suture synostoses.120,121
Radiological Evaluation
All patients identified as requiring surgical correction benefit from a preoperative CT scan. A subset of patients may demonstrate scaphocephaly, with an otherwise normal radiographic workup. It has been postulated that these children have “sticky sutures” secondary to microspicules of bone in the involved suture, inhibiting normal growth and development of the calvarium.144 Nevertheless, these children exhibit similar morphological characteristics to those with clear-cut fusion and may benefit from calvarial reconfiguration in severe examples.
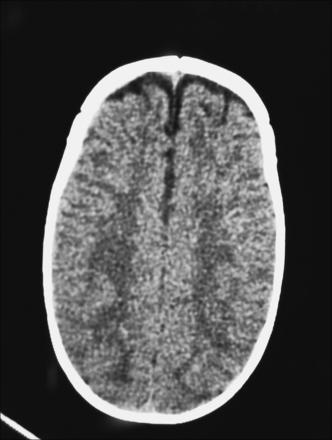
Three-dimensional CT may offer a more accurate depiction of the state of sutural fusion. Unanticipated intracranial pathology is seen in 5% or more of patients with sagittal synostosis,145 including hydrocephalus, agenesis of the corpus callosum, and focal cortical dysplasia. In addition, expanded subarachnoid spaces, often bifrontal, are commonly seen in patients with sagittal synostosis (Fig. 8.13).67,140 Typically, this will resolve spontaneously and does not reflect increased intracranial pressure.
Surgical Therapeutics
The treatment for sagittal synostosis has changed considerably over the past century, with surgical approaches evolving from minimal removal of involved suture and bone82,146 to extensive total calvarectomy.147–151 Despite relative long-term experience and numerous publications, the optimal timing for surgery as well as the extent of bony resection and need for extensive reconstruction remain contested. These issues are especially difficult to answer, given that the extent of calvarial resection and reconfiguration can sometimes be a function of the age at presentation.
A minimalist approach was taken early in the history of reconstructive surgery for sagittal synostosis. In 1892, Lane advocated the simple removal of the pathological sagittal suture, referred to as simple synostectomy or single strip craniectomy.146 Shillito and Matson in 1968 and Hunter and Rudd in 1976 revisited the idea of single strip craniectomy for this deformity.118,152 The general conclusions were that the technique is safe and well tolerated, providing adequate cosmetic results in select patients with mild deformities under the age of 2 to 3 months. Simple synostectomy, however, has several disadvantages stemming from the fact that it strictly addresses the fused suture and not the compensatory changes in skull shape. Specifically, it does not provide immediate or long-term cosmetic improvement in the majority of patients, by failing to shorten the anteroposterior dimension of the skull or address frontal bossing. It also leaves a large unprotected area over the vertex of the skull, an area with a high rate of restenosis and renewed growth restriction.153–156
In an effort to provide more immediate restoration of normal skull contour, extended strip craniectomy procedures were proposed.69,156–160 Venes and Sayers160 and Albright157 described occipitoparietal extension craniectomies as a means of shortening the anteroposterior dimension, expanding the biparietal dimension, and addressing the frontal and occipital prominences.155,156 Immediate cosmetic improvement was secured at the expense of large calvarial defects.
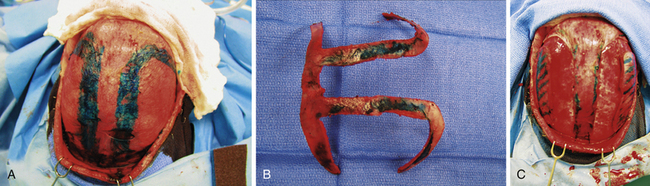
Striving to maintain the advantages of other extended strip craniectomies while simultaneously eliminating the disadvantage of postoperative calvarial defects, Jane and associates reported an extended strip craniectomy in the design of the Greek letter pi (π).158 The pi procedure has become a widely utilized approach for older infants (3-12 months of age) with scaphocephaly. Although multiple modifications with this approach have been proposed, including 12 different variations suggested by Jane depending upon the patient’s clinical presentation,161 the essential steps involve removal of bone along both sides of the sagittal suture as well as over the coronal suture (or lambdoid for the reverse pi) (Fig. 8.14). Cranial bone overlying the sagittal sinus is left intact to minimize bleeding.
To address older infants (>9-12 months of age) as well as those children with significant frontal or occipital prominence, procedures with more aggressive craniectomies and reconstruction have been proposed. In children beyond the period of maximal cerebral growth (>12-18 months of age), correction of the cephalic index will require substantial realignment of the calvarium and skull base. In the setting of significant pathology at the skull base, as proposed by Moss,162,163 many believe that a larger procedure should be undertaken to provide optimal surgical correction as well as avoiding potential recurrence. To accomplish these goals, a number of different approaches have been reported, ranging from variations of the pi procedure with wider or additional craniectomies over the lambdoid or coronal suture to total calvarial reconfiguration.
Total calvarectomy and reconstruction in the setting of severe or late presentation scaphocephaly has been proposed to offer superior cosmetic results with a minimal increase in morbidity.147–151,164,165 This may be accomplished by removal of the frontal, occipital, and both parietal bones. Subsequently, reconfiguration is carried out and aimed at providing a shortened anteroposterior dimension in addition to a widened biparietal diameter (Fig. 8.15). The use of rigid fixation is indispensable in these cases, providing greater three-dimensional conformational stability and decreased intraoperative time, bleeding, and postoperative infection. Although questions regarding transcranial migration of permanent titanium hardware have been raised in the developing infant, there is less controversy in the older patients undergoing total calvarial reconstruction. In addition, the introduction and increasing use of absorbable plates and screws has mitigated concerns regarding transcranial migration and inhibition of calvarial growth.
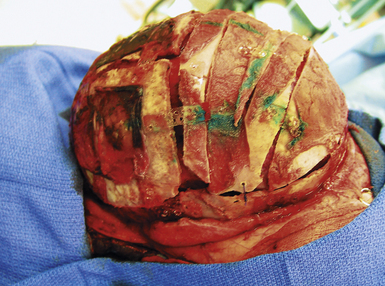
The patient presenting in a delayed fashion (after the age of 18-24 months) merits special consideration. Despite the lack of initial concern over the head shape, the significant calvarial deformity may become readily apparent as the child encounters greater social interaction through preschool, day care, or even later in kindergarten71,73,166 In addition to the myriad number of psychosocial issues, a subset of individuals may also harbor elevated intracranial pressure.3,59,167–170 Older children with significant scaphocephaly, often accompanied by both frontal and occipital bossing in association with midvertex depression, should be evaluated closely for any evidence of intracranial hypertension (see earlier section on Diagnostic Evaluation and Imaging).
Outcome and Complications
Studies have compared outcomes following different types of open craniofacial surgery for sagittal synostosis, with the consensus being improved quantitative and qualitative morphological results with the more extensive procedures.154,164,171,172 Reported cranial indices have been in the range of 71 to 73 for simple strip craniectomy,11,164 71 to 79 for extended strip craniectomy,164,171 and 74 to 78.5 for subtotal/total calvarial remodeling.148,149,164,171 Qualitatively, Kaiser found that extended strip craniectomy had an 83% rate of correction of cranial index, whereas simple strip craniectomy only has a 43% correction rate.171 Maugans and co-workers found the cosmetic outcomes of total calvarial reconstruction to be superior to strip craniectomy, with 79% in the calvarial remodeling group rated as excellent as opposed to 41% in the strip craniectomy group. Additionally, they commented that two patients required a second operation for poor cosmetic results with the strip craniectomy.172
Concerns regarding increased morbidity and mortality rates for more extensive procedures have been alleviated by the favorable outcomes data from several large series. Boop and co-workers reported three simple dural lacerations following 85 cases of the pi procedure,69 and Kanev and Lo described 65 cases without incident.159 Greensmith and co-workers found a 3.3% rate of major complications (one air embolus) and 10% rate of minor complications (one hematoma and two prominent wires) following total cranial vault remodeling.148 Nevertheless, the more extensive procedures do require larger blood transfusions and a longer hospital stay. Mean intraoperative blood transfusion volumes have been reported in the range of 85 to 120 mL for extended strip craniectomies,153,173 159 mL for subtotal calvarial reconfiguration,174 and 460 to 485 mL for total cranial vault remodeling.148,150 Contemporary average lengths of stay have included 3.7 to 5.6 days for extended strip craniectomies,69 2.8 to 8.9 days for subtotal calvarial reconfiguration174,175 and 5 days for total cranial vault remodeling.150
Coronal Synostosis (Anterior Plagiocephaly/Brachycephaly)
Clinical Features
Patients with unicoronal synostosis present with anterior, or frontal, plagiocephaly whereas those with bilateral coronal involvement display brachycephaly. Anterior plagiocephaly shows a predilection for right-sided pathology, with approximately a 3:2 ratio comparing right to left coronal involvement.11 Phenotypic features of anterior plagiocephaly include ipsilateral perisutural ridging, forehead flattening, and orbital recession, coupled with contralateral compensatory frontal bossing (Fig. 8.16). Facial deformities are also common, including nasal root displacement toward the ipsilateral side, anterior displacement of the ipsilateral ear, and chin deviation toward the contralateral side. Pathognomonic for unicoronal synostosis, elevation of the ipsilateral orbit can be seen secondary to superior displacement of the greater wing of the sphenoid, also known as the “harlequin” appearance. The presence of strabismus is common (50-60%), stemming from superior oblique muscle dysfunction as a result of skull base shortening in the anteroposterior direction with concomitant displacement of the orbital roof and trochlea.176,177 Although correction of the plagiocephaly occasionally improves the strabismus, surgery is frequently needed for ultimate correction. Interestingly, left-handedness recently has been shown to be three times more common in patients with anterior plagiocephaly than control subjects and four times more likely with left-sided fusion.178
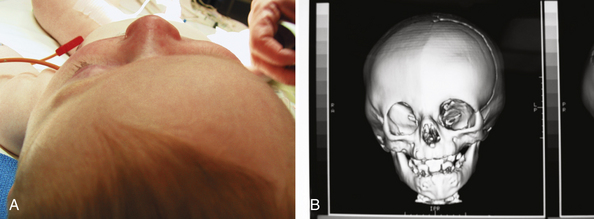
Phenotypic features of brachycephaly include forehead retrusion and flattening, frontal towering, and biparietal widening. The nasal dorsum can be low and hypertelorism may be present. Temporal bulging may be prominent in Muenke-type bicoronal synostosis.179 The presence of a named syndrome can be frequently seen, with an incidence reported as high as 57.1% by Sloan and co-workers.84 Other features that may be associated with bicoronal synostosis include brachydactyly, sensorineural hearing loss, and developmental delay.
Neurodevelopmentally, children with unicoronal and bicoronal synostoses have been associated with varying degrees of functional delay, ranging from minimal to severe.42,119,120,166,180 Becker and co-workers found 52% to 61% of patients with unicoronal and 55% of patients with bicoronal synostosis to demonstrate abnormalities in speech-language, cognitive, or behavioral function.119 In contrast, Speltz and colleagues discovered only a modest neurodevelopmental delay in cognitive and motor skills in patients with unicoronal synostosis.121 Along those lines, Arnaud and co-workers observed a mean preoperative intelligence quotient (IQ) of 95 and postoperative IQ of 97 in children with nonsyndromic brachycephaly, both scores falling within the “normal” range based on normative standards.42
Surgical Therapeutics
Surgical intervention in unicoronal and bicoronal synostoses aims to correct both the frontal and orbital asymmetries. With the current understanding that unilateral coronal synostosis presents with bilateral dysmorphic changes, bilateral correction is now believed to be the optimal approach.87,181–183 For anterior plagiocephaly, a bilateral fronto-orbital advancement procedure is employed for expansion of the affected forehead and orbit with concomitant recession of the contralateral orbit. Bilateral fronto-orbital advancement also serves in the correction of bicoronal synostosis, first proposed by Marchac and Renier in 1979 in the form of the “floating” forehead procedure.184
The timing of surgery remains a debated topic. However, an increasing number of surgeons have favored earlier surgery. Based on experience in 164 patients with craniosynostosis, Whitaker and co-workers proposed that the ideal age for treatment of both asymmetrical and less severe symmetrical synostoses was 4 to 12 months.79 In a 1994 report, Marchac and co-workers recommended performing a frontocranial remodeling procedure between 6 and 9 months for anterior plagiocephaly and between 2 and 4 months for brachycephaly.81 McCarthy and colleagues stated that patients with anterior plagiocephaly should undergo operations at approximately 6 months of age.185 Recently, Whitaker’s group has updated their recommendations and stated that surgery for unicoronal synostosis should be delayed until at least 6 months of age to avoid relapse.186 In contrast to these opinions, Posnick recommended waiting until 10 to 12 months for patients with anterior plagiocephaly.182
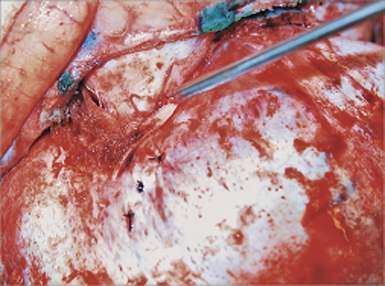
The bilateral fronto-orbital advancement procedure involves the release of both coronal sutures while providing bilateral frontal and orbital correction. After performing bilateral tarsorrhaphies, a standard bicoronal skin incision is made. A bifrontal bone flap is removed typically in one piece, leaving a 1- to 2-cm-wide supraorbital bandeau. Extending the osteotomy posterior to the coronal suture will often provide adequate width and a satisfactory new frontal reconstruction when this bone flap segment is inverted. The sphenoid wing is removed down to the level of the clinoid process and the coronal sutures are opened to the level of the skull base (Fig. 8.17), serving to prevent continued growth restriction and resultant postoperative hollowing of the pterional regions. Care must be taken when performing the osteotomy across the sphenoid wing on the affected side, as its superior displacement may cause technical difficulty upon frontal bone flap removal and lead to dural laceration if caution is not exercised. After removal of the frontal bone flap, the orbital bandeau is freed with osteotomies taken across the lateral orbit at the frontozygomatic suture, the orbital roof, and nasion just above the nasofrontal suture. Care is taken to protect the underlying brain as well as orbital contents with judicious placement of retractors. The bandeau is then reconfigured via partial osteotomies at the midline while utilizing absorbable hardware on the interior surface to maintain the newly contoured shape. The newly configured supraorbital bar is replaced, its lateral aspects and midline fixed to the calvarial foundation with rigid fixation (absorbable hardware) for improved healing and postoperative maintenance of the surgical construct (Fig. 8.18A). It is frequently necessary to overcorrect the expansion of the affected side by 5% to 10%, while also providing a convex shape at the lateral border for a satisfactory reconstruction (Fig. 8.18B). Often, the unaffected orbit is mildly set back to correct for preoperative compensatory overgrowth. The frontal bone flap is then attached to the supraorbital bar, taking care to match it to the previously overbuilt (5-10%) orbital bandeau on the affected side (Fig. 8.18C). Remaining portions of bone are fixed with absorbable plates or suture, and closure is performed in a routine fashion with subgaleal drains.
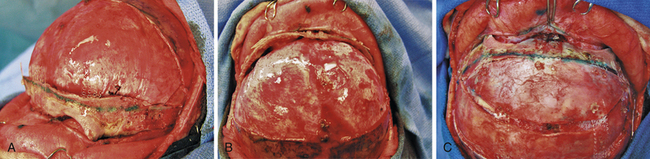
Outcome and Complications
A number of studies have addressed outcomes and complications following correction of unicoronal and bicoronal synostoses.79,80,83,84,87,186–188 Reported reoperation rates have ranged from 3.1% to 29% for unicoronal synostosis79,80,83,84,87,186 and 20% to 50% for bicoronal synostosis.83,84,87,187,188 Interestingly, two recent studies have focused on the impact of surgical age on postoperative outcome in unicoronal synostosis.80,186 Selber and co-workers determined that patients with unicoronal synostosis treated at 6 to 12 months had the lowest rate of secondary surgery (7%), which was statistically significant as compared to patients operated on less than 6 months of age or those receiving surgery at greater than 12 months of age.186 Looking at outcomes from a different perspective, Fearon and colleagues investigated the impact of surgical age on postoperative growth in patients with unicoronal synostosis. Head circumferences in 21 patients with unicoronal synostosis were measured, demonstrating that growth was more than twice as impaired in the early treatment group (mean = 5 months) versus the late treatment group (mean = 14 months).80
A broad range of numbers has been reported for postoperative complications and mortality rate.79,80,84,87,186–188 Complication rates have ranged from 0% to 27% for unicoronal synostosis79,84,87,186 and 20% to 50% for bicoronal synostosis.84,87,187,188 Perioperative morbidity may include wound infection, dural laceration, superficial brain injury, cerebrospinal fluid leak, encephalocele formation, subgaleal hematoma, and ocular injury including corneal abrasion. More serious perioperative complications can involve ischemic brain injury, epidural and subdural hemorrhage, and severe transfusion reactions. Long-term postoperative concerns include recurrent calvarial deformities, cranial bone defects that fail to fill in over time, and hardware-related problems. Defects greater than 2 cm in patients older than 18 to 24 months will often persist and may need eventual correction. Persistent hardware may also be problematic, particularly in patients in whom absorbable hardware was utilized. It is not uncommon for the polylactic/polyglycolic constructs to remain in place for 12 to 18 months before eventual resorption. In rare individuals, sterile abscesses may develop at sites of hardware resorption and subsequently require exploration for débridement. Postoperative mortality rates are near 0% for unicoronal synostosis83,84,186 and range from 0% to 10% for bicoronal synostosis.83,84,87,187
Lambdoid Synostosis and Posterior Deformational Plagiocephaly
Clinical Features
Posterior plagiocephaly due to lambdoid suture synostosis is a rare event today, with the majority of observed posterior plagiocephaly secondary to positional molding. Understanding the phenotypic differences between lambdoid synostosis and posterior deformational plagiocephaly is critical toward making the appropriate diagnosis and designing the proper course of treatment. Children with lambdoid synostosis characteristically have a trapezoid-shaped head in association with posterior displacement of the ipsilateral ear, contralateral occipital bossing, and frequent ridging of the affected lambdoid suture. In contrast, posterior deformational plagiocephaly is marked by a parallelogram-shaped head, anterior displacement of the ipsilateral ear, and ipsilateral frontal bossing in the absence of palpable ridging along the lambdoid sutures (Fig. 8.19).

An increasing number of studies have focused on uncovering the etiological pathogenesis and associated clinical features of positional deformational plagiocephaly. In 2004, Littlefield and co-workers reported an incidence of 1 in 68 infants.189 A prospective cohort study identified male gender, supine position, limited head rotation, lower activity levels, and first-born status as risk factors.190 Indeed, one of the unforeseen consequences of the “back to sleep” campaign was a nearly exponential rise in asymmetrical and symmetrical occipital flattening.20,191–193 Furthermore, about 30% of cases are associated with torticollis, or a tight sternocleidomastoid muscle.11
Radiological Evaluation
Routinely, radiographs are unnecessary in differentiating lambdoid synostosis from posterior deformational plagiocephaly, given that significant morphological differences exist to help separate the two entities on clinical grounds. In situations in which the diagnosis is unclear, skull plain films or facial CT scans can be employed. Lambdoid synostosis is consistent with fusion of the affected lambdoid suture, and posterior deformational plagiocephaly can be associated with sclerosis along a patent lambdoid suture.194
Surgical Therapeutics
Infants with true lambdoid synostosis may benefit from a variety of surgical approaches,195–204 aiming to release the affected suture(s) and normalize the posterior calvarial vault contour. Options include simple synostectomy, unilateral reconfiguration of the affected occipital region, and bilateral occipital reconstruction with or without the use of an occipital bandeau. The majority of lambdoid surgical candidates have significant parietal and frontal compensatory changes in addition to their occipital deformation; therefore, they are best served by a more extended calvarectomy and reconstruction.
For an extended calvarial reconfiguration, the globes are protected with bilateral tarsorrhaphies and patients are placed in a prone position. Perioperative antibiotics and steroids are administered. Intraoperative bleeding is minimized with the use of local anesthetic mixed with epinephrine as well as controlled hypotensive anesthesia. A bicoronal incision is then undertaken followed by subgaleal dissection to expose the occipitoparietal regions. Both parietal bone flaps are subsequently removed with osteotomies taken posterior to the coronal and anterior to the lambdoid sutures. A midline strip of bone is then left to protect the underlying sagittal sinus. After removal of the parietal bone flaps, dissection is carried out at the level of the lambdoid suture under direct visualization, taking great caution at the level of the transverse, sagittal, and sigmoid sinuses. Osteotomies are brought to within 1 cm on either side of the midline, with the final cut made after the underlying dura and sinus have been clearly dissected free under direct vision. Inadvertent entry into the sinus, particularly at the region of the asterion, may lead to significant blood loss over a short period and constitutes the greatest risk encountered for this approach. Nevertheless, with appropriate care and attention, this complication may be avoided in the majority of individuals. The removed calvarial plates are reconfigured to provide adequate normalization of the occipital contour and stabilized with resorbable hardware, deliberately leaving open the region of the prior lambdoid suture (Fig. 8.20). Subgaleal drains are then placed and closure is carried out in routine fashion.
Treatment of posterior deformational plagiocephaly is a function of both the age and severity at presentation. When presenting before the age of 6 months, therapy consists of positional modifications combined with physiotherapy in the case of constrained neck movements or asymmetric neurological development.205 In cases of no improvement or progression of the deformity despite repositioning after 2 to 3 months, cranial orthotic (helmet) therapy (Fig. 8.21) is indicated.11 Additionally, Graham and co-workers recommended helmet therapy if the cephalic index is larger than 90 or if the diagonal diameter difference is more than 1 cm at the age of 6 months.206 Despite conservative management, a small subset of patients (<7%) may require surgical correction for excellent long-term results.195,207

Outcome and Complications
Given the infrequency of unilateral and bilateral lambdoid synostoses, only a handful of studies have large enough patient numbers to glean accurate outcomes data.80,83,84,196,197,199 Reoperation rates range from 0% to 10% and complication rates fall between 5.4% and 13.6%. In addition to improvement of the posterior vault, studies have noted correction of the majority of facial asymmetries with surgeries performed at less than 1 year of age.196,199 Complications can consist of wound infections, transfusion-related problems, hemorrhage, dural injury, and rare intracerebral injury.
Outcome studies have also demonstrated conservative therapy to be beneficial in the reduction of calvarial deformities secondary to positional molding.19,208–215 Postural changes and helmet therapy are most effective between 4 and 12 months of age, during the period of rapid brain growth.205 Approximately 95% of infants can be expected to have satisfactory cosmetic improvement with conservative management. Following a recent systematic review of 16 studies, it was concluded that counterpositioning with or without physiotherapy or helmet therapy may reduce skull deformity; however, it was not possible to draw conclusions regarding the relative effectiveness of these interventions.209 On the issue of duration of therapy, Thompson noted an improvement in head shape after 4 months of helmet therapy, observing better results with increased compliance.211
Future Directions
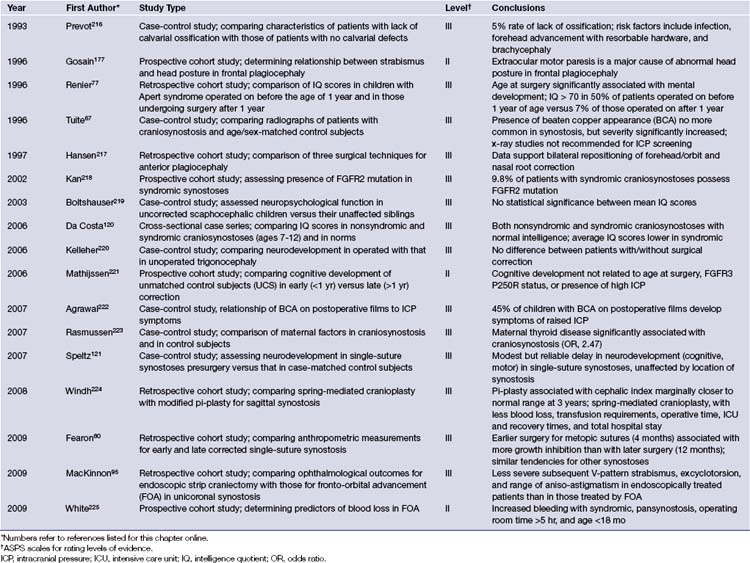
Significant advances over the past decade have contributed to the improvement in diagnosis and treatment of craniosynostosis while concurrently reducing the risks involved in its surgical treatment. A summary of the available evidence-based medicine can be found in Table 8.4.216–225 An understanding of the genetic underpinnings has provided investigators with an in-depth view of the potential mechanisms involved in the pathogenesis of sutural fusion and may eventually lead to new approaches in future treatment. New surgical treatment methods continue to refine the operative arena, with enhanced cosmetic outcomes and decreasing rates of recurrence. Minimally invasive techniques that rely on dynamic cranial vault alteration, including endoscopic sutural release, spring-assisted cranioplasty, and distraction osteogenesis, will undoubtedly play a greater role in the surgical correction of craniosynostosis. In time, answers will be forthcoming with respect to the appropriate surgical approaches for each type of craniosynostosis in addition to the optimal timing for correction. The value of orthotic devices in the setting of positional plagiocephaly will be eventually clarified by objective prospective data. The future will help clarify some of the current issues facing the craniofacial surgeon and no doubt will also bring new challenges to the forefront.
Bellus G.A., Gaudenz K., Zackai E.H., et al. Identical mutations in three different fibroblast growth factor receptor genes in autosomal dominant craniosynostosis syndromes. Nat Genet. 1996;14(2):174-176.
Bialocerkowski A.E., Vladusic S.L., Howell S.M. Conservative interventions for positional plagiocephaly: a systematic review. Dev Med Child Neurol. 2005;47(8):563-570.
Kapp-Simon K.A., Speltz M.L., Cunningham M.L., et al. Neurodevelopment of children with single suture craniosynostosis: a review. Childs Nerv Syst. 2007;23(3):269-281.
Lannelongue M. De la craniectomie dans la microcéphalie. Compt Rend Seances Acad Sci. 1890;50:1382-1385.
Renier D., Sainte-Rose C., Marchac D., et al. Intracranial pressure in craniostenosis. J Neurosurg. 1982;57(3):370-377.
Please go to expertconsult.com to view complete list of references.
1. Virchow R. Uber den Cretinismus, namentlich in Franken und uber pathologische Schadelformen. Verh Phys Med Ges (Wurzburg). 1851;2:230-271.
2. Renier D., Sainte-Rose C., Marchac D., et al. Intracranial pressure in craniostenosis. J Neurosurg. 1982;57(3):370-377.
3. Renier D., Lajeunie E., Arnaud E., et al. Management of craniosynostoses. Childs Nerv Syst. 2000;16(10-11):645-658.
4. Gault D.T., Renier D., Marchac D., et al. Intracranial pressure and intracranial volume in children with craniosynostosis. Plast Reconstr Surg. 1992;90(3):377-381.
5. Sommerring S. Vom Baue des Menschlichen Korpers, 2nd ed. Leipzig: Voss; 1839.
6. Moss M.L. The pathogenesis of premature cranial synostosis in man. Acta Anat (Basel). 1959;37:351-370.
7. Babler W., Persing J.A. Alterations in cranial suture growth associated with premature closure of the sagittal suture in rabbits. Anat Rec. 1985;211:14A.
8. Persing J.A., Babler W.J., Jane J.A., et al. Experimental unilateral coronal synostosis in rabbits. Plast Reconstr Surg. 1986;77(3):369-377.
9. Persson K.M., Roy W.A., Persing J.A., et al. Craniofacial growth following experimental craniosynostosis and craniectomy in rabbits. J Neurosurg. 1979;50(2):187-197.
10. Cohen M.M.Jr., Maclean R.E. Craniosynostosis: Diagnosis, Evaluation, and Management, 2nd ed. New York: Oxford University Press; 2000.
11. Van Veelen-Vincent M.L.C., Mathijssen I., Arnaud E., Renier D. Craniosynostosis. In: Lumenta C.B., di Rocco C., editors. European Manual of Medicine. Neurosurgery. Berlin: Springer; 2010:501-528.
12. Selber J., Reid R.R., Chike-Obi C.J., et al. The changing epidemiologic spectrum of single-suture synostoses. Plast Reconstr Surg. 2008;122(2):527-533.
13. Di Rocco F., Arnaud E., Meyer P., et al. Focus session on the changing “epidemiology” of craniosynostosis (comparing two quinquennia: 1985-1989 and 2003-2007) and its impact on the daily clinical practice: a review from Necker Enfants Malades. Childs Nerv Syst. 2009;25(7):807-811.
14. Knoll B.I., Persing J.A. Craniosynostosis. In: Bentz M.L., Bauer B.S., Zuker R.M., editors. Pediatric Plastic Surgery. 2nd ed. St. Louis: Quality Medical Publishing; 2008:541-563.
15. Lajeunie E., Le Merrer M., Bonaiti-Pellie C., et al. Genetic study of scaphocephaly. Am J Med Genet. 1996;62(3):282-285.
16. Persing J.A. MOC-PS(SM) CME article: management considerations in the treatment of craniosynostosis. Plast Reconstr Surg. 2008;121(Suppl 4):1-11.
17. Ellenbogen R.G., Gruss J.S., Cunningham M.L. Update on craniofacial surgery: the differential diagnosis of lambdoid synostosis/posterior plagiocephaly. Clin Neurosurg. 2000;47:303-318.
18. Huang M.H., Gruss J.S., Clarren S.K., et al. The differential diagnosis of posterior plagiocephaly: true lambdoid synostosis versus positional molding. Plast Reconstr Surg. 1996;98(5):765-774. discussion 775–776
19. Mulliken J.B., Vander Woude D.L., Hansen M., et al. Analysis of posterior plagiocephaly: deformational versus synostotic. Plast Reconstr Surg. 1999;103(2):371-380.
20. Turk A.E., McCarthy J.G., Thorne C.H., et al. The “back to sleep campaign” and deformational plagiocephaly: is there cause for concern? J Craniofac Surg. 1996;7(1):12-18.
21. Cohen M.M.Jr. Sutural biology and the correlates of craniosynostosis. Am J Med Genet. 1993;47(5):581-616.
22. Atkinson F.R.B. Hereditary craniofacial dysotosis, or Crouzon’s disease. Med Press Circular. 1937;195:118-124.
23. Kallen K. Maternal smoking and craniosynostosis. Teratology. 1999;60(3):146-150.
24. Muenke M., Schell U. Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 1995;11(8):308-313.
25. Wilkie A.O., Morriss-Kay G.M., Jones E.Y., et al. Functions of fibroblast growth factors and their receptors. Curr Biol. 1995;5(5):500-507.
26. Lenton K.A., Nacamuli R.P., Wan D.C., et al. Cranial suture biology. Curr Top Dev Biol. 2005;66:287-328.
27. Bellus G.A., Gaudenz K., Zackai E.H., et al. Identical mutations in three different fibroblast growth factor receptor genes in autosomal dominant craniosynostosis syndromes. Nat Genet. 1996;14(2):174-176.
28. el Ghouzzi V., Le Merrer M., Perrin-Schmitt F., et al. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15(1):42-46.
29. Howard T.D., Paznekas W.A., Green E.D., et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15(1):36-41.
30. Jabs E.W., Li X., Scott A.F., et al. Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat Genet. 1994;8(3):275-279.
31. Jabs E.W., Muller U., Li X., et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell. 1993;75(3):443-450.
32. Oldridge M., Wilkie A.O., Slaney S.F., et al. Mutations in the third immunoglobulin domain of the fibroblast growth factor receptor-2 gene in Crouzon syndrome. Hum Mol Genet. 1995;4(6):1077-1082.
33. Reardon W., Winter R.M. Saethre-Chotzen syndrome. J Med Genet. 1994;31(5):393-396.
34. Twigg S.R., Kan R., Babbs C., et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci U S A. 2004;101(23):8652-8657.
35. Slater B.J., Lenton K.A., Kwan M.D., et al. Cranial sutures: a brief review. Plast Reconstr Surg. 2008;121(4):170e-178e.
36. Ibrahimi O.A., Eliseenkova A.V., Plotnikov A.N., et al. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci U S A. 2001;98(13):7182-7187.
37. Muenke M., Schell U., Hehr A., et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet. 1994;8(3):269-274.
38. Plomp A.S., Hamel B.C., Cobben J.M., et al. Pfeiffer syndrome type 2: further delineation and review of the literature. Am J Med Genet. 1998;75(3):245-251.
39. Wilkie A.O., Slaney S.F., Oldridge M., et al. Apert syndrome results from localized mutations of FGFR-2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9(2):165-172.
40. Zhou Y.X., Xu X., Chen L., et al. A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Hum Mol Genet. 2000;9(13):2001-2008.
41. Chen L., Li D., Li C., et al. A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone. 2003;33(2):169-178.
42. Arnaud E., Meneses P., Lajeunie E., et al. Postoperative mental and morphological outcome for nonsyndromic brachycephaly. Plast Reconstr Surg. 2002;110(1):6-12. discussion 13
43. Moloney D.M., Wall S.A., Ashworth G.J., et al. Prevalence of Pro250Arg mutation of fibroblast growth factor receptor 3 in coronal craniosynostosis. Lancet. 1997;349(9058):1059-1062.
44. Gripp K.W., McDonald-McGinn D.M., Gaudenz K., et al. Identification of a genetic cause for isolated unilateral coronal synostosis: a unique mutation in the fibroblast growth factor receptor 3. J Pediatr. 1998;132(4):714-716.
45. Honnebier M.B., Cabiling D.S., Hetlinger M., et al. The natural history of patients treated for FGFR3-associated (Muenke-type) craniosynostosis. Plast Reconstr Surg. 2008;121(3):919-931.
46. Loeys B.L., Chen J., Neptune E.R., et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275-281.
47. Hunenko O., Karmacharya J., Ong G., et al. Toward an understanding of nonsyndromic craniosynostosis: altered patterns of TGF-beta receptor and FGF receptor expression induced by intrauterine head constraint. Ann Plast Surg. 2001;46(5):546-553. discussion 553–554
48. Moursi A.M., Winnard P.L., Winnard A.V., et al. Fibroblast growth factor 2 induces increased calvarial osteoblast proliferation and cranial suture fusion. Cleft Palate Craniofac J. 2002;39(5):487-496.
49. Opperman L.A., Nolen A.A., Ogle R.C. TGF-beta 1, TGF-beta 2, and TGF-beta 3 exhibit distinct patterns of expression during cranial suture formation and obliteration in vivo and in vitro. J Bone Miner Res. 1997;12(3):301-310.
50. Kim H.J., Rice D.P., Kettunen P.J., et al. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development. 1998;125(7):1241-1251.
51. Nacamuli R.P., Fong K.D., Lenton K.A., et al. Expression and possible mechanisms of regulation of BMP3 in rat cranial sutures. Plast Reconstr Surg. 2005;116(5):1353-1362.
52. Liu Y.H., Tang Z., Kundu R.K., et al. Msx2 gene dosage influences the number of proliferative osteogenic cells in growth centers of the developing murine skull: a possible mechanism for MSX2-mediated craniosynostosis in humans. Dev Biol. 1999;205(2):260-274.
53. Woods R.H., Ul-Haq E., Wilkie A.O., et al. Reoperation for intracranial hypertension in TWIST1-confirmed Saethre-Chotzen syndrome: a 15-year review. Plast Reconstr Surg. 2009;123(6):1801-1810.
54. Morriss-Kay G.M., Wilkie A.O. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207(5):637-653.
55. Weinzweig J., Kirschner R.E., Farley A., et al. Metopic synostosis: defining the temporal sequence of normal suture fusion and differentiating it from synostosis on the basis of computed tomography images. Plast Reconstr Surg. 2003;112(5):1211-1218.
56. Bourez R.L., Mathijssen I.M., Vaandrager J.M., et al. Apoptotic cell death during normal embryogenesis of the coronal suture: early detection of apoptosis in mice using annexin V. J Craniofac Surg. 1997;8(6):441-445.
57. Dobyns W.B., Truwit C.L. Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics. 1995;26(3):132-147.
58. Kirschner R.E., Gannon F.H., Xu J., et al. Craniosynostosis and altered patterns of fetal TGF-beta expression induced by intrauterine constraint. Plast Reconstr Surg. 2002;109(7):2338-2346. discussion 2347–2354
59. Tuite G.F., Chong W.K., Evanson J., et al. The effectiveness of papilledema as an indicator of raised intracranial pressure in children with craniosynostosis. Neurosurgery. 1996;38(2):272-278.
60. Levi D., Rampa F., Barbieri C., et al. True 3D reconstruction for planning of surgery on malformed skulls. Childs Nerv Syst. 2002;18(12):705-706.
61. Miller C., Losken H.W., Towbin R., et al. Ultrasound diagnosis of craniosynostosis. Cleft Palate Craniofac J. 2002;39(1):73-80.
62. Stelnicki E.J., Mooney M.P., Losken H.W., et al. Ultrasonic prenatal diagnosis of coronal suture synostosis. J Craniofac Surg. 1997;8(4):252-258. discussion 259–261
63. van der Ham L.I., Cohen-Overbeek T.E., Paz y Geuze H.D., et al. The ultrasonic detection of an isolated craniosynostosis. Prenat Diagn. 1995;15(12):1189-1192.
64. Fjortoft M.I., Sevely A., Boetto S., et al. Prenatal diagnosis of craniosynostosis: value of MR imaging. Neuroradiology. 2007;49(6):515-521.
65. Girod S., Teschner M., Schrell U., et al. Computer-aided 3-D simulation and prediction of craniofacial surgery: a new approach. J Craniomaxillofac Surg. 2001;29(3):156-158.
66. Perlyn C.A., Marsh J.L., Vannier M.W., et al. The craniofacial anomalies archive at St. Louis Children’s Hospital: 20 years of craniofacial imaging experience. Plast Reconstr Surg. 2001;108(7):1862-1870.
67. Tuite G.F., Evanson J., Chong W.K., et al. The beaten copper cranium: a correlation between intracranial pressure, cranial radiographs, and computed tomographic scans in children with craniosynostosis. Neurosurgery. 1996;39(4):691-699.
68. Chadduck W.M., Chadduck J.B., Boop F.A. The subarachnoid spaces in craniosynostosis. Neurosurgery. 1992;30(6):867-871.
69. Boop F.A., Chadduck W.M., Shewmake K., et al. Outcome analysis of 85 patients undergoing the pi procedure for correction of sagittal synostosis. J Neurosurg. 1996;85(1):50-55.
70. Pertschuk M.J., Whitaker L.A. Psychosocial adjustment and craniofacial malformations in childhood. Plast Reconstr Surg. 1985;75(2):177-184.
71. Barritt J., Brooksbank M., Simpson D. Scaphocephaly: aesthetic and psychosocial considerations. Dev Med Child Neurol. 1981;23(2):183-191.
72. Cohen S.R., Cho D.C., Nichols S.L., et al. American Society of Maxillofacial Surgeons outcome study: preoperative and postoperative neurodevelopmental findings in single-suture craniosynostosis. Plast Reconstr Surg. 2004;114(4):841-847. discussion 848–849
73. Magge S.N., Westerveld M., Pruzinsky T., et al. Long-term neuropsychological effects of sagittal craniosynostosis on child development. J Craniofac Surg. 2002;13(1):99-104.
74. Shipster C., Hearst D., Somerville A., et al. Speech, language, and cognitive development in children with isolated sagittal synostosis. Dev Med Child Neurol. 2003;45(1):34-43.
75. Noetzel M.J., Marsh J.L., Palkes H., et al. Hydrocephalus and mental retardation in craniosynostosis. J Pediatr. 1985;107(6):885-892.
76. Patton M.A., Goodship J., Hayward R., et al. Intellectual development in Apert’s syndrome: a long term follow up of 29 patients. J Med Genet. 1988;25(3):164-167.
77. Renier D., Arnaud E., Cinalli G., et al. Prognosis for mental function in Apert’s syndrome. J Neurosurg. 1996;85(1):66-72.
78. Meyer P., Renier D., Arnaud E., et al. Blood loss during repair of craniosynostosis. Br J Anaesth. 1993;71(6):854-857.
79. Whitaker L.A., Bartlett S.P., Schut L., et al. Craniosynostosis: an analysis of the timing, treatment, and complications in 164 consecutive patients. Plast Reconstr Surg. 1987;80(2):195-212.
80. Fearon J.A., Ruotolo R.A., Kolar J.C. Single sutural craniosynostoses: surgical outcomes and long-term growth. Plast Reconstr Surg. 2009;123(2):635-642.
81. Marchac D., Renier D., Broumand S. Timing of treatment for craniosynostosis and facio-craniosynostosis: a 20-year experience. Br J Plast Surg. 1994;47(4):211-222.
82. Lannelongue M. De la craniectomie dans la microcéphalie. Compt Rend Seances Acad Sci. 1890;50:1382-1385.
83. Pearson G.D., Havlik R.J., Eppley B., et al. Craniosynostosis: a single institution’s outcome assessment from surgical reconstruction. J Craniofac Surg. 2008;19(1):65-71.
84. Sloan G.M., Wells K.C., Raffel C., et al. Surgical treatment of craniosynostosis: outcome analysis of 250 consecutive patients. Pediatrics. 1997;100(1):E2.
85. Whitaker L.A., Munro I.R., Salyer K.E., et al. Combined report of problems and complications in 793 craniofacial operations. Plast Reconstr Surg. 1979;64(2):198-203.
86. McCarthy J.G., Glasberg S.B., Cutting C.B., et al. Twenty-year experience with early surgery for craniosynostosis: II. The craniofacial synostosis syndromes and pansynostosis—results and unsolved problems. Plast Reconstr Surg. 1995;96(2):284-295. discussion 296–298
87. McCarthy J.G., Glasberg S.B., Cutting C.B., et al. Twenty-year experience with early surgery for craniosynostosis: I. Isolated craniofacial synostosis—results and unsolved problems. Plast Reconstr Surg. 1995;96(2):272-283.
88. Barone C.M., Jimenez D.F. Endoscopic craniectomy for early correction of craniosynostosis. Plast Reconstr Surg. 1999;104(7):1965-1973. discussion 1974–1975
89. Barone C.M., Jimenez D.F. Endoscopic approach to coronal craniosynostosis. Clin Plast Surg. 2004;31(3):415-422. vi
90. Jimenez D.F., Barone C.M. Endoscopic craniectomy for early surgical correction of sagittal craniosynostosis. J Neurosurg. 1998;88(1):77-81.
91. Jimenez D.F., Barone C.M. Endoscopy-assisted wide-vertex craniectomy, “barrel-stave” osteotomies, and postoperative helmet molding therapy in the early management of sagittal suture craniosynostosis. Neurosurg Focus. 2000;9(3):e2.
92. Jimenez D.F., Barone C.M. Early treatment of anterior calvarial craniosynostosis using endoscopic-assisted minimally invasive techniques. Childs Nerv Syst. 2007;23(12):1411-1419.
93. Jimenez D.F., Barone C.M., Cartwright C.C., et al. Early management of craniosynostosis using endoscopic-assisted strip craniectomies and cranial orthotic molding therapy. Pediatrics. 2002;110(1 Pt 1):97-104.
94. Jimenez D.F., Barone C.M., McGee M.E., et al. Endoscopy-assisted wide-vertex craniectomy, barrel stave osteotomies, and postoperative helmet molding therapy in the management of sagittal suture craniosynostosis. J Neurosurg. 2004;100(Suppl 5):407-417.
95. MacKinnon S., Rogers G.F., Gregas M., et al. Treatment of unilateral coronal synostosis by endoscopic strip craniectomy or fronto-orbital advancement: ophthalmologic findings. J AAPOS. 2009;13(2):155-160.
96. Lauritzen C., Sugawara Y., Kocabalkan O., et al. Spring mediated dynamic craniofacial reshaping. Case report. Scand J Plast Reconstr Surg Hand Surg. 1998;32(3):331-338.
97. Persing J.A., Babler W.J., Nagorsky M.J., et al. Skull expansion in experimental craniosynostosis. Plast Reconstr Surg. 1986;78(5):594-603.
98. Lauritzen C.G., Davis C., Ivarsson A., et al. The evolving role of springs in craniofacial surgery: the first 100 clinical cases. Plast Reconstr Surg. 2008;121(2):545-554.
99. David L.R., Plikaitis C.M., Couture D., et al. Outcome analysis of our first 75 spring-assisted surgeries for scaphocephaly. J Craniofac Surg. 2010;21(1):3-9.
100. Akai T., Iizuka H., Kawakami S. Treatment of craniosynostosis by distraction osteogenesis. Pediatr Neurosurg. 2006;42(5):288-292.
101. Akizuki T., Komuro Y., Ohmori K. Distraction osteogenesis for craniosynostosis. Neurosurg Focus. 2000;9(3):e1.
102. Anderson P.J., Tan E., David D.J. Simultaneous multiple vector distraction for craniosynostosis syndromes. Br J Plast Surg. 2005;58(5):626-631.
103. Choi J.W., Koh K.S., Hong J.P., et al. One-piece frontoorbital advancement with distraction but without a supraorbital bar for coronal craniosynostosis. J Plast Reconstr Aesthet Surg. 2009;62(9):1166-1173.
104. do Amaral C.M., Di Domizio G., Tiziani V., et al. Gradual bone distraction in craniosynostosis. Preliminary results in seven cases. Scand J Plast Reconstr Surg Hand Surg. 1997;31(1):25-37.
105. Imai K., Komune H., Toda C., et al. Cranial remodeling to treat craniosynostosis by gradual distraction using a new device. J Neurosurg. 2002;96(4):654-659.
106. Kim S.W., Shim K.W., Plesnila N., et al. Distraction vs. remodeling surgery for craniosynostosis. Childs Nerv Syst. 2007;23(2):201-206.
107. Kim Y.O., Choi J.W., Kim D.S., et al. Cranial growth after distraction osteogenesis of the craniosynostosis. J Craniofac Surg. 2008;19(1):45-55.
108. Lee J.A., Park D.H., Yoon S.H., et al. Distractor breakage in cranial distraction osteogenesis for children with craniosynostosis. Pediatr Neurosurg. 2008;44(3):216-220.
109. Nishimoto S., Oyama T., Nagashima T., et al. Gradual distraction fronto-orbital advancement with “floating forehead” for patients with syndromic craniosynostosis. J Craniofac Surg. 2006;17(3):497-505.
110. Nonaka Y., Oi S., Miyawaki T., et al. Indication for and surgical outcomes of the distraction method in various types of craniosynostosis. Advantages, disadvantages, and current concepts for surgical strategy in the treatment of craniosynostosis. Childs Nerv Syst. 2004;20(10):702-709.
111. Park D.H., Chung J., Yoon S.H. The role of distraction osteogenesis in children with secondary craniosynostosis after shunt operation in early infancy. Pediatr Neurosurg. 2010;45(6):437-445.
112. Tellado M.G., Lema A. Coronal suturectomy through minimal incisions and distraction osteogenesis are enough without other craniotomies for the treatment of plagiocephaly due to coronal synostosis. J Craniofac Surg. 2009;20(6):1975-1977.
113. Teebi A.S., Gibson L., McGrath J., et al. Molecular and cytogenetic characterization of 9p-abnormalities. Am J Med Genet. 1993;46(3):288-292.
114. Preus M., Vekemans M., Kaplan P. Diagnosis of chromosome 3 duplication q23®qter, deletion p25®pter in a patient with the C (trigonocephaly) syndrome. Am J Med Genet. 1986;23(4):935-943.
115. Obregon M.G., Mingarelli R., Digilio M.C., et al. Deletion 11q23®qter (Jacobsen syndrome). Report of three new patients. Ann Genet. 1992;35(4):208-212.
116. Tubbs R.S., Elton S., Blount J.P., et al. Preliminary observations on the association between simple metopic ridging in children without trigonocephaly and the Chiari I malformation. Pediatr Neurosurg. 2001;35(3):136-139.
117. Schneider E.N., Bogdanow A., Goodrich J.T., et al. Fronto-ocular syndrome: newly recognized trigonocephaly syndrome. Am J Med Genet. 2000;93(2):89-93.
118. Shillito J.Jr., Matson D.D. Craniosynostosis: a review of 519 surgical patients. Pediatrics. 1968;41(4):829-853.
119. Becker D.B., Petersen J.D., Kane A.A., et al. Speech, cognitive, and behavioral outcomes in nonsyndromic craniosynostosis. Plast Reconstr Surg. 2005;116(2):400-407.
120. Da Costa A.C., Walters I., Savarirayan R., et al. Intellectual outcomes in children and adolescents with syndromic and nonsyndromic craniosynostosis. Plast Reconstr Surg. 2006;118(1):175-181. discussion 182–183
121. Speltz M.L., Kapp-Simon K., Collett B., et al. Neurodevelopment of infants with single-suture craniosynostosis: presurgery comparisons with case-matched controls. Plast Reconstr Surg. 2007;119(6):1874-1881.
122. Delashaw J.B., Persing J.A., Park T.S., et al. Surgical approaches for the correction of metopic synostosis. Neurosurgery. 1986;19(2):228-234.
123. Albin R.E., Hendee R.W.Jr., O’Donnell R.S., et al. Trigonocephaly: refinements in reconstruction. Experience with 33 patients. Plast Reconstr Surg. 1985;76(2):202-211.
124. Anderson F.M. Treatment of coronal and metopic synostosis: 107 cases. Neurosurgery. 1981;8(2):143-149.
125. Fearon J.A., Kolar J.C., Munro I.R. Trigonocephaly-associated hypotelorism: is treatment necessary? Plast Reconstr Surg. 1996;97(3):503-509. discussion 510–511
126. Marsh J.L., Schwartz H.G. The surgical correction of coronal and metopic craniosynostoses. J Neurosurg. 1983;59(2):245-251.
127. Oi S., Matsumoto S. Trigonocephaly (metopic synostosis). Clinical, surgical and anatomical concepts. Childs Nerv Syst. 1987;3(5):259-265.
128. Salkind G., Sutton L.N., Bruce D.A., et al. Management of trigonocephaly. Surg Neurol. 1986;25(2):159-162.
129. Shaffrey M.E., Persing J.A., Delashaw J.B., et al. Surgical treatment of metopic synostosis. Neurosurg Clin North Am. 1991;2(3):621-627.
130. Goodrich J.T., Hall C.D. Metopic synostosis and trigonocephaly: a spectrum of dysmorphology. In: Goodrich J.T., Hall C.D., editors. Craniofacial Anomalies: Growth and Development from a Surgical Perspective. New York: Thieme; 1995:15-22.
131. McCarthy J.G., Bradley J.P., Longaker M.T. Step expansion of the frontal bar: correction of trigonocephaly. J Craniofac Surg. 1996;7(5):333-335.
132. Sadove A.M., Kalsbeck J.E., Eppley B.L., et al. Modifications in the surgical correction of trigonocephaly. Plast Reconstr Surg. 1990;85(6):853-858.
133. Cohen S.R., Maher H., Wagner J.D., et al. Metopic synostosis: evaluation of aesthetic results. Plast Reconstr Surg. 1994;94(6):759-767.
134. Dhellemmes P., Pellerin P., Lejeune J.P., et al. Surgical treatment of trigonocephaly. Experience with 30 cases. Childs Nerv Syst. 1986;2(5):228-232.
135. Di Rocco C., Velardi F., Ferrario A., et al. Metopic synostosis: in favor of a “simplified” surgical treatment. Childs Nerv Syst. 1996;12(11):654-663.
136. Posnick J.C., Lin K.Y., Chen P., et al. Metopic synostosis: quantitative assessment of presenting deformity and surgical results based on CT scans. Plast Reconstr Surg. 1994;93(1):16-24.
137. Wall S.A., Goldin J.H., Hockley A.D., et al. Fronto-orbital re-operation in craniosynostosis. Br J Plast Surg. 1994;47(3):180-184.
138. Persing J.A., Mayer P.L., Spinelli H.M., et al. Prevention of “temporal hollowing” after fronto-orbital advancement for craniosynostosis. J Craniofac Surg. 1994;5(4):271-274.
139. Faberowski L.W., Black S., Mickle J.P. Blood loss and transfusion practice in the perioperative management of craniosynostosis repair. J Neurosurg Anesthesiol. 1999;11(3):167-172.
140. Kang J.K., Lee S.W., Baik M.W., et al. Perioperative specific management of blood volume loss in craniosynostosis surgery. Childs Nerv Syst. 1998;14(7):297-301.
141. Kearney R.A., Rosales J.K., Howes W.J. Craniosynostosis: an assessment of blood loss and transfusion practices. Can J Anaesth. 1989;36(4):473-477.
142. Steinbok P., Heran N., Hicdonmez T., et al. Minimizing blood transfusions in the surgical correction of coronal and metopic craniosynostosis. Childs Nerv Syst. 2004;20(7):445-452.
143. Speltz M.L., Kapp-Simon K.A., Cunningham M., et al. Single-suture craniosynostosis: a review of neurobehavioral research and theory. J Pediatr Psychol. 2004;29(8):651-668.
144. Burke M.J., Winston K.R., Williams S. Normal sutural fusion and the etiology of single sutural craniosynostosis: the microspicule hypothesis. Pediatr Neurosurg. 1995;22(5):241-246. discussion 247
145. Cerovac S., Neil-Dwyer J.G., Rich P., et al. Are routine preoperative CT scans necessary in the management of single suture craniosynostosis? Br J Neurosurg. 2002;16(4):348-354.
146. Lane L.C. Pioneer craniectomy for the relief of mental imbecility due to premature sutural closure and microcephalus. JAMA. 1892;18:49.
147. Burstein F.D., Hudgins R.J., Cohen S.R., et al. Surgical correction of severe scaphocephalic deformities. J Craniofac Surg. 1994;5(4):228-235. discussion 236
148. Greensmith A.L., Holmes A.D., Lo P., et al. Complete correction of severe scaphocephaly: the Melbourne method of total vault remodeling. Plast Reconstr Surg. 2008;121(4):1300-1310.
149. Heller J.B., Heller M.M., Knoll B., et al. Intracranial volume and cephalic index outcomes for total calvarial reconstruction among nonsyndromic sagittal synostosis patients. Plast Reconstr Surg. 2008;121(1):187-195.
150. Hudgins R.J., Burstein F.D., Boydston W.R. Total calvarial reconstruction for sagittal synostosis in older infants and children. J Neurosurg. 1993;78(2):199-204.
151. Weinzweig J., Baker S.B., Whitaker L.A., et al. Delayed cranial vault reconstruction for sagittal synostosis in older children: an algorithm for tailoring the reconstructive approach to the craniofacial deformity. Plast Reconstr Surg. 2002;110(2):397-408.
152. Hunter A.G., Rudd N.L. Craniosynostosis. I. Sagittal synostosis: its genetics and associated clinical findings in 214 patients who lacked involvement of the coronal suture(s). Teratology. 1976;14(2):185-193.
153. Greene C.S.Jr., Winston K.R. Treatment of scaphocephaly with sagittal craniectomy and biparietal morcellation. Neurosurgery. 1988;23(2):196-202.
154. Marsh J.L., Jenny A., Galic M., et al. Surgical management of sagittal synostosis. A quantitative evaluation of two techniques. Neurosurg Clin North Am. 1991;2(3):629-640.
155. McComb J.G. Occipital reduction-biparietal widening technique for correction of sagittal synostosis. Pediatr Neurosurg. 1994;20(1):99-106.
156. Vollmer D.G., Jane J.A., Park T.S., et al. Variants of sagittal synostosis: strategies for surgical correction. J Neurosurg. 1984;61(3):557-562.
157. Albright A.L. Operative normalization of skull shape in sagittal synostosis. Neurosurgery. 1985;17(2):329-331.
158. Jane J.A., Edgerton M.T., Futrell J.W., et al. Immediate correction of sagittal synostosis. J Neurosurg. 1978;49(5):705-710.
159. Kanev P.M., Lo A.K. Surgical correction of sagittal craniosynostosis: complications of the pi procedure. J Craniofac Surg. 1995;6(2):98-102.
160. Venes J.L., Sayers M.P. Sagittal synostectomy. Technical note. J Neurosurg. 1976;44(3):390-392.
161. Jane J.A., Francel P.C. The evolution of treatment for sagittal synostosis: a personal record. In: Goodrich J.T., Hall C.D., editors. Craniofacial Anomalies: Growth and Development from a Surgical Perspective. New York: Thieme; 1995:15-22.
162. Moss M.L. Functional anatomy of cranial synostosis. Childs Brain. 1975;1(1):22-33.
163. Moss M.L., Salentijn L. The primary role of functional matrices in facial growth. Am J Orthod. 1969;55(6):566-577.
164. Panchal J., Marsh J.L., Park T.S., et al. Sagittal craniosynostosis outcome assessment for two methods and timings of intervention. Plast Reconstr Surg. 1999;103(6):1574-1584.
165. Pensler J.M., Ciletti S.J., Tomita T. Late correction of sagittal synostosis in children. Plast Reconstr Surg. 1996;97(7):1362-1367. discussion 1368–1370
166. Kapp-Simon K.A. Mental development and learning disorders in children with single suture craniosynostosis. Cleft Palate Craniofac J. 1998;35(3):197-203.
167. Cohen S.R., Persing J.A. Intracranial pressure in single-suture craniosynostosis. Cleft Palate Craniofac J. 1998;35(3):194-196.
168. Martinez-Lage J.F., Alamo L., Poza M. Raised intracranial pressure in minimal forms of craniosynostosis. Childs Nerv Syst. 1999;15(1):11-15. discussion 16
169. Stavrou P., Sgouros S., Willshaw H.E., et al. Visual failure caused by raised intracranial pressure in craniosynostosis. Childs Nerv Syst. 1997;13(2):64-67.
170. Thompson D.N., Malcolm G.P., Jones B.M., et al. Intracranial pressure in single-suture craniosynostosis. Pediatr Neurosurg. 1995;22(5):235-240.
171. Kaiser G. Sagittal synostosis—its clinical significance and the results of three different methods of craniectomy. Childs Nerv Syst. 1988;4(4):223-230.
172. Maugans T.A., McComb J.G., Levy M.L. Surgical management of sagittal synostosis: a comparative analysis of strip craniectomy and calvarial vault remodeling. Pediatr Neurosurg. 1997;27(3):137-148.
173. Olds M.V., Storrs B., Walker M.L. Surgical treatment of sagittal synostosis. Neurosurgery. 1986;18(3):345-347.
174. Fearon J.A., McLaughlin E.B., Kolar J.C. Sagittal craniosynostosis: surgical outcomes and long-term growth. Plast Reconstr Surg. 2006;117(2):532-541.
175. Guimaraes-Ferreira J., Gewalli F., David L., et al. Clinical outcome of the modified pi-plasty procedure for sagittal synostosis. J Craniofac Surg. 2001;12(3):218-224. discussion 225–226
176. Bagolini B., Campos E.C., Chiesi C. Plagiocephaly causing superior oblique deficiency and ocular torticollis. A new clinical entity. Arch Ophthalmol. 1982;100(7):1093-1096.
177. Gosain A.K., Steele M.A., McCarthy J.G., et al. A prospective study of the relationship between strabismus and head posture in patients with frontal plagiocephaly. Plast Reconstr Surg. 1996;97(5):881-891.
178. Oh A.K., Mulliken J.B., LaBrie R.A., et al. Increased frequency of left-handedness in patients with unilateral coronal synostosis. Cleft Palate Craniofac J. 2009;46(3):237-244.
179. Lajeunie E., El Ghouzzi V., Le Merrer M., et al. Sex related expressivity of the phenotype in coronal craniosynostosis caused by the recurrent P250R FGFR3 mutation. J Med Genet. 1999;36(1):9-13.
180. Kapp-Simon K.A., Speltz M.L., Cunningham M.L., et al. Neurodevelopment of children with single suture craniosynostosis: a review. Childs Nerv Syst. 2007;23(3):269-281.
181. Bartlett S.P., Whitaker L.A., Marchac D. The operative treatment of isolated craniofacial dysostosis (plagiocephaly): a comparison of the unilateral and bilateral techniques. Plast Reconstr Surg. 1990;85(5):677-683.
182. Posnick J.C. Unilateral coronal synostosis (anterior plagiocephaly): current clinical perspectives. Ann Plast Surg. 1996;36(4):430-447.
183. Sgouros S., Goldin J.H., Hockley A.D., et al. Surgery for unilateral coronal synostosis (plagiocephaly): unilateral or bilateral correction? J Craniofac Surg. 1996;7(4):284-289.
184. Marchac D., Renier D. [The “floating forehead.” Early treatment of craniofacial stenosis. Ann Chir Plast. 1979;24(2):121-126.
185. McCarthy J.G., Epstein F., Sadove M., et al. Early surgery for craniofacial synostosis: an 8-year experience. Plast Reconstr Surg. 1984;73(4):521-533.
186. Selber J.C., Brooks C., Kurichi J.E., et al. Long-term results following fronto-orbital reconstruction in nonsyndromic unicoronal synostosis. Plast Reconstr Surg. 2008;121(5):251e-260e.
187. Marchac D., Renier D., Jones B.M. Experience with the “floating forehead.”. Br J Plast Surg. 1988;41(1):1-15.
188. Wagner J.D., Cohen S.R., Maher H., et al. Critical analysis of results of craniofacial surgery for nonsyndromic bicoronal synostosis. J Craniofac Surg. 1995;6(1):32-37. discussion 38–39
189. Littlefield T.R., Saba N.M., Kelly K.M. On the current incidence of deformational plagiocephaly: an estimation based on prospective registration at a single center. Semin Pediatr Neurol. 2004;11(4):301-304.
190. Hutchison B.L., Thompson J.M., Mitchell E.A. Determinants of nonsynostotic plagiocephaly: a case-control study. Pediatrics. 2003;112(4):e316.
191. Argenta L.C., David L.R., Wilson J.A., et al. An increase in infant cranial deformity with supine sleeping position. J Craniofac Surg. 1996;7(1):5-11.
192. Kane A.A., Mitchell L.E., Craven K.P., et al. Observations on a recent increase in plagiocephaly without synostosis. Pediatrics. 1996;97(6 Pt 1):877-885.
193. Peitsch W.K., Keefer C.H., LaBrie R.A., et al. Incidence of cranial asymmetry in healthy newborns. Pediatrics. 2002;110(6):e72.
194. Dias M.S., Klein D.M. Occipital plagiocephaly: deformation or lambdoid synostosis? II. A unifying theory regarding pathogenesis. Pediatr Neurosurg. 1996;24(2):69-73.
195. David D.J., Menard R.M. Occipital plagiocephaly. Br J Plast Surg. 2000;53(5):367-377.
196. Goodrich J.T., Argamaso R. Lambdoid stenosis (posterior plagiocephaly) and craniofacial asymmetry: long-term outcomes. Childs Nerv Syst. 1996;12(11):720-726.
197. Jimenez D.F., Barone C.M. The Sunrise Technique: the correction of occipital plagiocephaly using bandeau occipital plate and radial osteotomies. Pediatr Neurosurg. 1995;22(3):162-165. discussion 166
198. Jimenez D.F., Barone C.M., Argamaso R.V., et al. Asterion region synostosis. Cleft Palate Craniofac J. 1994;31(2):136-141.
199. McComb J.G. Treatment of functional lambdoid synostosis. Neurosurg Clin North Am. 1991;2(3):665-672.
200. Kaiser G. The clinical significance of bilateral synostosis of the lambdoid suture and the usefulness of its treatment. Childs Brain. 1984;11(2):87-98.
201. Muakkassa K.F., Hoffman H.J., Hinton D.R., et al. Lambdoid synostosis. Part 2: review of cases managed at the Hospital for Sick Children, 1972-1982. J Neurosurg. 1984;61(2):340-347.
202. Persing J.A., Delashaw J.B., Jane J.A., et al. Lambdoid synostosis: surgical considerations. Plast Reconstr Surg. 1988;81(6):852-860.
203. Thaller S.R., Hoyt J., Boggan J. Surgical correction of unilateral lambdoid synostosis: occipital rotation flap. J Craniofac Surg. 1992;3(1):12-17. discussion 18–19
204. Zoller J.E., Mischkowski R.A., Speder B. Preliminary results of standardized occipital advancement in the treatment of lambdoid synostosis. J Craniomaxillofac Surg. 2002;30(6):343-348.
205. Persing J., James H., Swanson J., et al. Prevention and management of positional skull deformities in infants. American Academy of Pediatrics Committee on Practice and Ambulatory Medicine, Section on Plastic Surgery and Section on Neurological Surgery. Pediatrics. 2003;112(1 Pt 1):199-202.
206. Graham J.M.Jr., Gomez M., Halberg A., et al. Management of deformational plagiocephaly: repositioning versus orthotic therapy. J Pediatr. 2005;146(2):258-262.
207. Keating R.F. Management of posterior plagiocephaly and positional deformation. Goodrich J.T., editor, Techniques in Neurosurgery: Craniofacial Disorders. 1997:198-206.
208. Lee R.P., Teichgraeber J.F., Baumgartner J.E., et al. Long-term treatment effectiveness of molding helmet therapy in the correction of posterior deformational plagiocephaly: a five-year follow-up. Cleft Palate Craniofac J. 2008;45(3):240-245.
209. Bialocerkowski A.E., Vladusic S.L., Howell S.M. Conservative interventions for positional plagiocephaly: a systematic review. Dev Med Child Neurol. 2005;47(8):563-570.
210. Hutchison B.L., Hutchison L.A., Thompson J.M., et al. Plagiocephaly and brachycephaly in the first two years of life: a prospective cohort study. Pediatrics. 2004;114(4):970-980.
211. Thompson J.T., David L.R., Wood B., et al. Outcome analysis of helmet therapy for positional plagiocephaly using a three-dimensional surface scanning laser. J Craniofac Surg. 2009;20(2):362-365.
212. Vles J.S., Colla C., Weber J.W., et al. Helmet versus nonhelmet treatment in nonsynostotic positional posterior plagiocephaly. J Craniofac Surg. 2000;11(6):572-574.
213. Loveday B.P., de Chalain T.B. Active counterpositioning or orthotic device to treat positional plagiocephaly? J Craniofac Surg. 2001;12(4):308-313.
214. Rogers G.F., Miller J., Mulliken J.B. Comparison of a modifiable cranial cup versus repositioning and cervical stretching for the early correction of deformational posterior plagiocephaly. Plast Reconstr Surg. 2008;121(3):941-947.
215. Littlefield T.R., Beals S.P., Manwaring K.H., et al. Treatment of craniofacial asymmetry with dynamic orthotic cranioplasty. J Craniofac Surg. 1998;9(1):11-17. discussion 18–19
216. Prevot M., Renier D., Marchac D. Lack of ossification after cranioplasty for craniosynostosis: a review of relevant factors in 592 consecutive patients. J Craniofac Surg. 1993;4(4):247-254. discussion 255–256
217. Hansen M., Padwa B.L., Scott R.M., et al. Synostotic frontal plagiocephaly: anthropometric comparison of three techniques for surgical correction. Plast Reconstr Surg. 1997;100(6):1387-1395.
218. Kan S.H., Elanko N., Johnson D., et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002;70(2):472-486.
219. Boltshauser E., Ludwig S., Dietrich F., et al. Sagittal craniosynostosis: cognitive development, behavior, and quality of life in unoperated children. Neuropediatrics. 2003;34(6):293-300.
220. Kelleher M.O., Murray D.J., McGillivary A., et al. Behavioral, developmental, and educational problems in children with nonsyndromic trigonocephaly. J Neurosurg. 2006;105(Suppl 5):382-384.
221. Mathijssen I., Arnaud E., Lajeunie E., et al. Postoperative cognitive outcome for synostotic frontal plagiocephaly. J Neurosurg. 2006;105(Suppl 1):16-20.
222. Agrawal D., Steinbok P., Cochrane D.D. Significance of beaten copper appearance on skull radiographs in children with isolated sagittal synostosis. Childs Nerv Syst. 2007;23(12):1467-1470.
223. Rasmussen S.A., Yazdy M.M., Carmichael S.L., et al. Maternal thyroid disease as a risk factor for craniosynostosis. Obstet Gynecol. 2007;110(2 Pt 1):369-377.
224. Windh P., Davis C., Sanger C., et al. Spring-assisted cranioplasty vs. pi-plasty for sagittal synostosis—a long-term follow-up study. J Craniofac Surg. 2008;19(1):59-64.
225. White N., Marcus R., Dover S., et al. Predictors of blood loss in fronto-orbital advancement and remodeling. J Craniofac Surg. 2009;20(2):378-381.