Diagnosis and Management of Liver Failure in the Adult
Decompensation of Chronic Liver Disease
The pathologic basis of acute decompensation in chronic liver disease or acute on chronic liver failure (AoCLF) is incompletely understood but is thought to be precipitated by systemic inflammation, and if looked for hard enough, some signs or symptoms of infection will be seen in most patients.1,2 Patients with well-compensated cirrhosis often decompensate following a defined event. The precipitants can be split into two types: (1) those due to direct liver insult, such as ischemia, a toxic insult such as alcohol, or a superimposed viral infection and (2) those in which the liver is affected as a bystander in a systemic inflammatory process, such as following an episode of sepsis or a gastrointestinal bleed. This can be contrasted to the progressive liver failure of end-stage cirrhosis. Both present with similar clinical pictures but in AoCLF there remains the possibility of improvement and reversibility, and if these patients do recover they end up back on the previous mortality trajectory.1,2
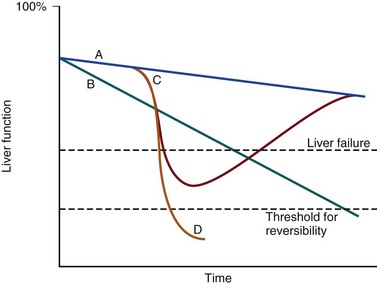
Differentiating between the two is difficult but the presence of a precipitating factor and history can be useful. In patients with end-stage liver cirrhosis there is often a history of gradual deterioration in biochemical parameters, clinical status, and other organ function whereas in AoCLF there is often an acute deterioration. Organ support in the former setting is of dubious utility as it is almost always futile (Fig. 75.1).
One group has proposed a working definition for AoCLF as “acute deterioration in liver function over a period of 2-4 weeks, usually associated with a precipitating event, leading to severe deterioration in clinical status, with jaundice and hepatic encephalopathy and or hepatorenal syndrome (HRS), with a high SOFA/APACHE II score.”1
AoCLF often occurs with multiple organ dysfunction. This may be because of the underlying precipitating event such as pneumonia or bacterial peritonitis or as a result of the severe deterioration in liver function. Often it is difficult to distinguish which came first. Renal failure is the most common organ failure associated with AoCLF and it is the severity of organ dysfunction that dictates the outcome.3
Precipitating Factors of Acute on Chronic Liver Failure
Alcoholic Hepatitis
Alcoholic hepatitis is a common manifestation of alcohol abuse. Between 20% and 30% of heavy drinkers will present with it at some time. The clinical picture is variable from a relatively mild syndrome associated with loss of appetite, nausea, and vomiting with right upper quadrant pain to severe life-threatening liver decompensation, but the most obvious sign is the sudden onset of jaundice, and this can be the first indication of liver disease in a heavy drinker. Overall, following first presentation, 35% to 50% of patients will die within the first month of diagnosis.4 It usually presents following a bout of heavy drinking and often occurs on a background of cirrhosis. Patients frequently deteriorate after stopping alcohol. This may be due to the immunosuppressive effects of alcohol. In patients without cirrhosis abstinence and nutritional support can lead to a marked improvement in liver function over time.
 Features suggestive of malnutrition such as muscle wasting and vitamin deficiency are seen commonly. In severe decompensation other intercurrent illnesses such as pneumonia or urinary tract infection or a gastrointestinal bleed are often present. Systemic inflammation may be the precipitant of decompensation in these patients.5
Features suggestive of malnutrition such as muscle wasting and vitamin deficiency are seen commonly. In severe decompensation other intercurrent illnesses such as pneumonia or urinary tract infection or a gastrointestinal bleed are often present. Systemic inflammation may be the precipitant of decompensation in these patients.5
In alcoholic hepatitis liver enzymes are moderately raised with an aspartate aminotransferase (AST) rarely greater than 300 IU. The ratio of AST to alanine aminotransferase (ALT) is usually raised to greater than 2.6 A high white blood cell count and bilirubin are also typical. The liver is usually enlarged and fatty on ultrasound. Portal hypertension often complicates the clinical picture, presenting as worsening of ascites. Assessment of filling pressures can be difficult if ascites is tense. Further hemodynamic monitoring is recommended if there is any doubt regarding filling status or if cardiac output is thought to be compromised.
The prognosis of patients with alcoholic hepatitis depends on the severity of the disease. Risk stratification is important, both in defining the indications for the use of specific pharmacologic agents aimed at the interruption of progression and in clinical research. Serum markers and liver histologic findings provide the best means for risk stratification, the presence of cirrhosis being a poor marker.7 In many cases, however, biopsy is precluded because of the risks of bleeding. Serum bilirubin, coagulation parameters, and creatinine have been shown to predict outcome and have been combined into several scoring systems; the discriminative function (DF); Child, Turcotte, Pugh (CTP); the Mayo end-stage liver disease (MELD) score; and the Glasgow Alcoholic Hepatitis (GAH) score have been used to evaluate outcome. Of these, the MELD score appears to offer the best prediction of mortality risk because of the inclusion of renal function (creatinine) as well as liver function; renal deterioration in this setting has a particularly poor prognosis.8 Standard critical care prognostic scores have also been used to assess the prognosis of patients with decompensated chronic liver disease of any cause. The sequential organ failure (SOFA) score has been shown to provide useful prognostic information.1,9
In early disease, abstinence and good nutrition, including supplementation with B vitamins, is the mainstay of management. In severe disease, progression to hepatic encephalopathy and organ failure can be rapid. In addition to general supportive care, specific therapies have been used in an attempt to halt and reverse the hepatic inflammation and prevent the fall into multiple organ failure and ultimately death. Of these, and pentoxifylline have been the most extensively studied.10
Portal Hypertensive Bleeding
 Portal hypertension is a significant complication of chronic liver disease leading to the formation of portosystemic collateral vessels. Of these vessels, the most significant clinically are those that occur in the wall of the stomach and esophagus. In patients with portal hypertension as a result of cirrhosis the development of gastrointestinal varices occurs in approximately 60% at the time of diagnosis.11 The incidence of the first acute bleed in an unselected patient is relatively low at about 5% per year but it can be catastrophic when it occurs.12 The mortality rate associated with the event has fallen over the past 20 years from approximately 30% to 50% to about 20% and control of the initial bleeding episode is achieved in about 90% of patients.13 Half of all fatalities occur in the first 5 days following an acute bleed and about half of them are due to uncontrolled bleeding and the rest are due to multiple organ failure. This pattern remains the same when mortality rate in the first 6 weeks following the first bleed is examined.13
Portal hypertension is a significant complication of chronic liver disease leading to the formation of portosystemic collateral vessels. Of these vessels, the most significant clinically are those that occur in the wall of the stomach and esophagus. In patients with portal hypertension as a result of cirrhosis the development of gastrointestinal varices occurs in approximately 60% at the time of diagnosis.11 The incidence of the first acute bleed in an unselected patient is relatively low at about 5% per year but it can be catastrophic when it occurs.12 The mortality rate associated with the event has fallen over the past 20 years from approximately 30% to 50% to about 20% and control of the initial bleeding episode is achieved in about 90% of patients.13 Half of all fatalities occur in the first 5 days following an acute bleed and about half of them are due to uncontrolled bleeding and the rest are due to multiple organ failure. This pattern remains the same when mortality rate in the first 6 weeks following the first bleed is examined.13
Control of bleeding episodes is via direct endoscopic therapy, usually in the form of band ligation in the case of esophageal varices.14 Terlipressin, a synthetic analog of vasopressin, can be used as an adjunct to band ligation. Terlipressin induces splanchnic vasoconstriction and reduces portal pressure and has been shown to reduce mortality rate.15 There is some evidence that somatostatin may reduce the amount of blood transfused but the evidence is weak.16 Uncontrolled bleeding due to either failed endoscopic or pharmacologic therapy or when resources do not allow immediate endoscopic therapy may be managed with balloon tamponade of the stomach or esophageal varices. Primary control of esophageal varices is usually successful but balloons should not be inflated for more than 12 hours because of the risks of mucosal necrosis. Care should be exercised when inserting and inflating a Sensgtaken-Blakemore or Minnesota tube because perforation of the esophagus is usually fatal in this setting. Following control with the insertion of a balloon tamponade device, further endoscopic therapy, surgical shunting, or transjugular intrahepatic portosystemic stent shunts (TIPS) may be attempted. Of the shunt procedures used TIPS is the most commonly attempted, where technical skills are available. The procedure involves decompressing the portal circulation into the hepatic vein via a stent inserted percutaneously through the liver via the internal jugular vein. The procedure is a very successful salvage therapy for controlling bleeding but can induce certain complications, including (a) volume overload as a result of portal shunting into the right atrium, which can induce heart failure; (b) bypassing the portal circulation resulting in ischemic liver injury and, rarely, liver failure; and (c) worsening of encephalopathy grade (the most common complication) resulting in failure to wake from the procedure, the need for extended ventilation, and the risk of hospital-acquired infection. There have not yet been any controlled trials of TIPS as salvage therapy in this setting and it is unclear what role TIPS has in the management of acute bleeding varices as it may just result in prolongation of the dying process. Despite better prevention of rebleeding when compared to endoscopic therapy in patients with bleeding varices, a mortality benefit cannot be shown with TIPS.17 Recently, Garcia-Pagan and colleagues have shown that TIPS, not done as salvage therapy, but used as a primary form of therapy once the initial bleed has been controlled, results in a dramatic reduction in rebleeding rates and mortality rates.18 This study has huge resource implications and needs to be repeated.
Coagulation abnormalities are commonly present in patients with acutely bleeding varices, and abnormalities in clotting factors have been shown to be independent prognostic factors in patients with esophageal varices. Attempts to treat coagulation abnormalities with clotting components should be undertaken during an acute bleed to include fresh frozen plasma (FFP) and platelets targeted to correct abnormal partial thromboplastin time (PTT) and platelet count. Activated factor VII has also been investigated in this setting. The authors showed that in patients with worse cirrhosis, as defined by Child-Pugh grades B and C, recombinant activated factor VII (rFVIIa) increased the chances of controlling the bleeding, although a more recent study by the same group shows no difference between groups.19
Bleeding from varices is associated with infection. Between 35% and 60% of cirrhotic patients with bleeding varices have documented infection over the following 2 weeks.20
The use of antibiotic prophylaxis has been shown to reduce the incidence of infection in this group of patients and to improve short-term mortality rate.20,21 It has been proposed that infection is the precipitant to an acute increase in portal pressure that may trigger bleeding.22
Bacterial Peritonitis
Patients with chronic liver disease are at increased risk of infection. The immunosuppression associated with chronic liver disease is incompletely understood but relates to a range of factors including an impaired innate and adaptive immune response.23,24
In cirrhotic patients with ascites, decompensation to liver failure is often precipitated by infection. Bacterial peritonitis is a common and severe complication. It can be completely asymptomatic but can present with a range of symptoms and signs including local signs of peritonitis, abdominal pain, diarrhea, and signs of systemic inflammation such as fever, rigors, raised white blood cell count, hypotension, and tachycardia.25
Bacterial peritonitis is often spontaneous, without any obvious source, although it can be secondary to other intra-abdominal disease. It is generally caused by aerobic gram-negative bacteria, although gram-positive bacteria including methicillin-resistant Staphylococcus aureus (MRSA) should be suspected if there is treatment failure, especially if the patient has been taking prophylactic antibiotics.26 In any patient with ascites and decompensation of liver disease, spontaneous bacterial peritonitis (SBP) should be suspected. Diagnosis is demonstrated by sampling of the ascitic fluid. An absolute leukocyte count in ascitic fluid of greater than 250/µL is diagnostic. Antibiotics should be started as soon as SBP is suspected, following blood culture and a diagnostic ascitic tap. Empiric broad-spectrum antibiotic coverage for gram-negative and MRSA organisms (depending on local prevalence) should be started until cultures are available.
Renal failure is a significant complication of SBP in patients with cirrhosis and is discussed later. The use of human albumin solution (HAS) in SBP has been shown to reduce the incidence of progression to renal failure and should be considered in this setting.25
Supportive Management in Critical Care
The presentation of AoCLD will depend on the precipitating event or events but the clinical picture and pattern of organ failure will on the whole look very similar. Hyperbilirubinemia and the resulting jaundice are almost universal, as are other biochemical manifestations of poor liver function. Plasma protein production is deranged, leading to a prolongation of prothrombin time and hypoalbuminemia. Thrombocytopenia due to hypersplenism and sepsis is also characteristic. The effects on the coagulation system are complex, however, as there is a concurrent reduction in the production of endogenous anticoagulant proteins in the liver resulting a relative rebalancing. Infection leads to consumption of factors and platelets and so the use of thromboelastography is recommended in bleeding patients. Lower levels of factors can result in difficulties maintaining coagulation during times of stress, particularly during infections, and evidence of a heparin-like effect has been shown.27 It is also interesting to note that the incidence of thromboembolism is not lower in patients with cirrhosis even in the setting of a prolonged international normalization ratio (INR), suggesting that standard measures of coagulation are unreliable.28
The pattern of circulatory changes associated with cirrhosis is distinctive. Hypotension and an increase in cardiac output are typical, resulting in a hyperdynamic circulation. Peripheral vasodilatation, however, is not distributed evenly and occurs mainly in the splanchnic circulation as a result of sinusoidal portal hypertension. Splanchnic vasodilatation results in effective arterial underfilling with the resulting activation of compensatory mechanisms.29 During decompensation circulatory changes become more pronounced with an increase in portal pressure; systemic vasodilatation worsens and blood pressure drops further and becomes less responsive to vasopressor support. Recent work shows that cardiac output may fall in those who develop hepatorenal failure, as do cardiac filling pressures and pulmonary artery pressure. These changes point toward some form of cardiac depressant factor associated with decompensation.29 There is a reduction in intrarenal blood flow, due to the activation of compensatory mechanisms designed to maintain arterial volume, such as the renin-angiotensin system and sympathetic nervous system. This results in a further reduction in glomerular filtration rate (GFR) and urine output. Cardiac function may be further compromised by liver failure–associated cardiomyopathy, which is manifested by a low or normal cardiac output in the setting of reduced afterload.30 There may also be a relative hypovolemia in the setting of a normal or raised central venous pressure because of raised intra-abdominal pressure due to ascites. Echocardiography is very helpful in the delineation of any cardiac dysfunction and can help assess pulmonary artery pressure to assess if the patient has portopulmonary hypertension.31 A pulmonary artery (PA) catheter should be used if there is any doubt.
Acute kidney injury (AKI) is the most common form of organ dysfunction seen in patients with AoCLF and has significant attributable mortality risk.32 There are four main causes: those associated with bacterial infection are the most common, followed by hypovolemia and parenchymal disease, with hepatorenal failure the least common.3 However, hepatorenal failure has a particularly poor outcome. The diagnosis of AKI in the setting of liver disease is problematic, as conventional measures of renal function are less indicative of renal function.
Hepatorenal syndrome is a severe and progressive reduction in renal function in patients with severe liver disease and presents in two clinical patterns, type 1 and type 2. Hepatorenal syndrome represents the renovascular response to the profound circulatory changes associated with cirrhosis. Type 2 HRS is characterized by a less severe and more gradual reduction in renal function associated with diuretic-resistant ascites and hyponatremia. It is usually seen in association with end-stage cirrhosis. Type 1 HRS is characterized by the rapid decline in renal function defined as a 100% increase in serum creatinine to a level greater than 221 mmol/L or a 50% reduction in 24-hour creatinine clearance to a level less than 20 mL/minute in less than 2 weeks.33 Initially HRS is potentially reversible as the kidneys are functionally normal; however, the longer the circulatory changes last without reestablishing the GFR, the less likely are the kidneys to recover, and the distinction between HRS and acute tubular necrosis becomes less clear. In most patients there is a precipitating event, while in others it occurs in close proximity to an event such as the resolution of SBP. The diagnosis is made by the exclusion of other causes and is based on criteria developed by the International Ascites Club.33
Until recently there was no effective treatment for patients with HRS other than liver transplantation, but over the past 15 years observational and interventional studies have shown that countering the compensatory circulatory changes that promote intrarenal vasoconstriction with use of systemic and selective vasoconstrictors plus plasma volume expansion with albumin can lead to significant improvement in renal function.34 The aim of therapy is to focus vasoconstriction on the dilated splanchnic vessels, resulting in a redistribution of the blood volume back into the systemic arterial circulation. Over time this results in suppression of the compensatory mechanisms; a reduction in plasma renin activity, sympathetic activity, and circulating catecholamines; and an associated increase in GFR, sodium excretion, and urine output.35 Many different vasoconstrictors have been tried in this setting. The drug most commonly studied is terlipressin and it is often given with 20% human albumin solution (HAS). This approach has been subjected to a randomized controlled clinical trial in which the addition of albumin was associated with an improvement in outcome.36 Evidence suggests that vasoconstrictor therapy, with or without plasma volume expansion, results in an improvement in renal function in about a third of patients with type I HRS. The best predictors of response to therapy is the baseline serum creatinine, and those who do respond have a significant rise in their arterial pressure.34,37
Ammonia production in the gut is important in the pathogenesis. Ammonia is usually cleared from the portal circulation in the liver. The mainstays of treatment are colonic cleansing with enemas and enteral disaccharides such as lactulose. Both of these procedures have been shown to improve encephalopathy grades. The restriction of protein, once fashionable in patients with liver disease, is contraindicated. The majority of these patients are malnourished and adequate enteral nutrition is essential.38
Large volume paracentesis should be considered in any patient with tense ascites. There is evidence that large volume paracentesis improves lung mechanics and oxygenation in nonventilated patients but data in ventilated patients are lacking.39–41 Hepatic hydrothorax is a persistent pleural effusion, almost always associated with ascites, usually on the right side of the chest. It can be massive in size, containing many liters of fluid, and is thought to be due to communication from the peritoneum. Management can be difficult. Direct drainage with thoracocentesis is usually inadvisable as the fluid accumulation is persistent, leading to continued need for chest tube placement, which carries the risk of infection. Management should be directed at the ascites and includes diuretics, salt restriction, and paracentesis. TIPS can be used to control both ascites and hydrothorax but is associated with worsening encephalopathy grade in some patients.42
Adrenal dysfunction in patients with critical illness has been documented extensively, particularly in patients with septic shock.43 Studies have suggested an improvement in outcome with steroid use but subsequent investigations have been less clear. Currently recommendations suggest steroid treatment should not be based on adrenal stimulation and should be reserved for those with vasopressor-resistant septic shock or early acute respiratory distress syndrome (ARDS) within 14 days of onset.43 Patients with liver failure have been shown to have a high incidence of adrenal suppression as well, although it is not clear if this has a different etiology to other critically ill patients. Patients with adrenal suppression have worse hepatic and renal function, more organ failure, and a higher intensive care unit (ICU) and hospital mortality rate.44 There remains, however, a lack of consensus regarding definitions of adrenal dysfunction and appropriate testing in patients with liver disease and in which patients require treatment.45 However, in patients with cirrhosis and septic shock admitted to the ICU with adrenal insufficiency as defined by a suboptimal response to ACTH stimulation, replacement of hydrocortisone (50 mg every 6 hours) results in a higher incidence of shock resolution and hospital survival when compared to historical control subjects.46
Outcome and Data on ICU Use in Decompensated Cirrhosis
Despite ICU care, the outcome in patients with decompensated cirrhosis is poor. In one study overall cumulative mortality rates were 36% in the critical care unit, 46% in the hospital, and 56% at 6-month follow-up.47
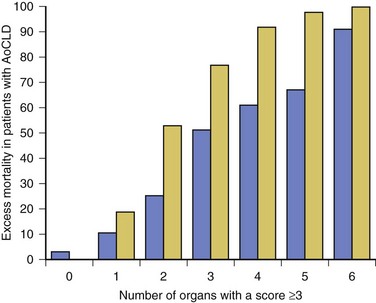
In patients who require organ support within the critical care environment a number of observations can be made. Derangement of acute physiology at admission is a predictor of outcome, as it is for unselected patients admitted to the ICU.48 In addition, the number of organs requiring support also is predictive of outcome, as it is in unselected patients.47,49 If you compare the number of organs failing (defined as an organ-specific SOFA score greater than 3) there is an excess mortality rate associated with cirrhosis when compared to an unselected group (Fig. 75.2).50 Patients with cirrhosis and three organ systems requiring support have a mortality rate in excess of 90% in some studies.49 Severity of liver disease at admission has a significant bearing on outcome irrespective of indication for admission to the ICU.49,51
Acute (Fulminant) Liver Failure
Acute liver failure (ALF) is a syndrome manifested by the rapid cessation of normal function in individuals with previously normal livers. The rate of decline in function dictates the manner in which the syndrome manifests and influences the outcome. The cause is the main influence on the rate of progression and the likelihood of spontaneous recovery.52
The pathologic basis of the massive hepatic necrosis was described in detail by Lucké and Mallory following the Second World War in 1946.53 The presence of the American army in East Asia and Africa resulted in exposure to both epidemic and serum hepatitis, and data regarding the clinical course of the syndrome and its pathologic features were collated via the army medical services.
In 1970 Trey and Davidson introduced the term fulminant hepatic failure (FHF) to encompass the current clinicopathologic understanding of the syndrome.54 This definition was an attempt to encapsulate the clinical course and to differentiate it from decompensation of chronic liver disease. They described a syndrome of rapidly progressing liver failure (within 8 weeks) in which the defining point was the onset of hepatic encephalopathy following the onset of symptoms in someone without previous liver disease. They make the point that the syndrome is potentially reversible in some patients. This definition is still used today; however, it has become clear that this definition is too narrow and that subgroups exist. This point is important as these subgroups predict the likely prognosis and potential for survival without a liver transplant.52
The rate of progression from the onset of jaundice or other initial symptoms (such as fatigue or acute viral illness) to development of encephalopathy is used to define subgroups. This in part relates to the cause of liver failure and to the way the pathologic expression of the pattern of organ failure presents. For example, patients with significant acetaminophen-induced hepatotoxicity will generally present with liver failure within 7 days of ingestion, unless ingestion was staggered over a period of time when the timing of liver insult is difficult to define. Patients often present with cardiovascular collapse and renal failure before they become encephalopathic. In contrast, patients presenting with seronegative hepatitis (unknown cause) can have a very variable presentation, some with a prolonged illness over a period of months, resulting in a patient who is deeply jaundiced with evidence of portal hypertension, such as ascites, at the onset of encephalopathy, and others with a relatively short presentation period. These two extreme ends of the syndrome split the group into hyperacute and subacute, with ALF in the middle. Interestingly, it is the hyperacute group that has the best chance for spontaneous recovery, although this group has the highest risk of cerebral edema. The subacute group has the worse prognosis with medical management alone.52
ALF is rare, with about 1 to 6 cases per 1 million of the population in the developed world.55 The incidence in the rest of the world is less clear because of the paucity of data (Box 75.1).
Etiology
Worldwide, and in the developing world in particular, approximately 95% to 100% of patients presenting with ALF will have viral hepatitis.56 Within the United Kingdom and as recently reported in the United States, paracetamol (acetaminophen) hepatotoxicity is the leading cause of ALF. This is followed by liver failure of unknown cause or seronegative hepatitis.57,58
The pattern of ALF within the United Kingdom and United States has been changing over the past 30 years.57,58 Up until the late 1990s, the rate of hospital admission due to paracetamol ingestion had risen year by year. In the United Kingdom paracetamol overdose (POD) is usually due to deliberate self-harm. In contrast, the U.S. data suggest that over half of all patients with ALF due to POD were due to therapeutic misadventure. Some doubts have been expressed regarding this interpretation as misadventure in some cases that appear to be occult suicide attempts.58,59
In 1998 legislation was introduced in the United Kingdom to restrict the over-the-counter sale of paracetamol to 16 tablets from most retail outlets and 32 from pharmacies in the form of blister packs. Interpretation of the effects of the legislation have proved to be complex, in a large part because there were no prospective audits initiated at the time to study it. Early interpretation suggested that admissions to hospital, severe liver toxicity, and transplantation for POD fell.60 The picture is more complex, though. Death rates have fallen; however, much of this reduction can be related to the withdrawal of co-proxamol during the mid-2000s. Co-proxamol, a combination of paracetamol and dextropropoxyphene (a mild opioid), was a prescription-only medication associated with a high degree of fatality if taken in overdose, most of this out of hospital and attributed to the effects of dextropropoxyphene on respiratory depression.61 Admission to hospital because of POD has continued to rise year by year but the number of pills taken on average has fallen and so the case fatality rate is lower. The numbers of patients being referred to transplant centers is also lower than before the legislation, but the number of patients transplanted has remained about the same, indicating the small number of determined overdoses and misadventure cases has remained relatively stable.
In the United Kingdom and the United States, the incidence of acute hepatitis A virus (HAV) and hepatitis B virus (HBV) infection has fallen dramatically since the 1980s57,58 (Figs. 75.3 and 75.4).
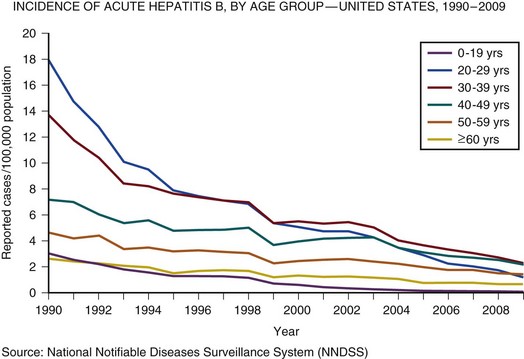
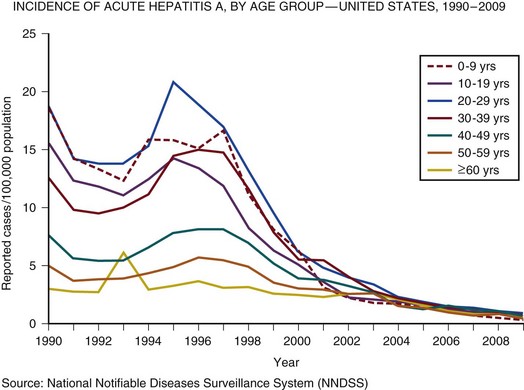
Less than 1% of acute hepatitis A or B progress to ALF. In the United States and United Kingdom, as a proportion of the total, the number of admissions with ALF due to viral hepatitis has fallen steadily and is currently responsible for less than 5% of all admissions in the United Kingdom and 11% in the United States.57,58
Indeterminate hepatitis (non-A to E hepatitis, seronegative ALF, non-A/non-B hepatitis) is often presumed to be viral in origin and is the most common presentation excluding POD in the United Kingdom and United States along with viral hepatitis in the developing world. It is a diagnosis of exclusion and, as diagnostic capabilities improve, is falling in incidence in some centers.57
Acetaminophen (Paracetamol)
Acetaminophen-induced liver failure is the cause of the vast majority of hyperacute liver failure. Acetaminophen poisoning is a common cause of presentation to acute and emergency departments in the United Kingdom and United States; however, the case progression to ALF following paracetamol ingestion is rare at just 0.6% of all presentations in the United Kingdom.62
Following poisoning the half-life of acetaminophen is greatly prolonged because of the saturation of glucuronidation and sulfate conjugation. As a result, there is an increase in the quantity of NAPQI produced. NAPQI is extremely reactive in biologic systems and has a short half-life. Following poisoning, reaction of NAPQI occurs within the centrilobular portions of the liver and leads to necrosis in experimental models. It reacts with cellular constituents in a covalent and noncovalent manner. The exact mechanisms by which NAPQI induces cell death are incompletely understood but include the deactivation of critical cellular proteins, the induction of reactive oxygen species, and the activation of Kupffer cells.63 The loss of regulatory protein function results in abnormal calcium homeostasis and resultant energy failure within the cell and mitochondria.63 Other events such as noncovalent interaction with intracellular signaling and lipid peroxidation also contribute to the toxicity of this molecule. Following this primary toxic phase there is a secondary or extrinsic phase. This extrinsic phase is equated with the recruitment of immune cells to the liver. The liver is one of the major immune organs of the body. Up to 35% of the liver is made up of nonparenchymal cells, including endothelium, Kupffer cells, and resident lymphocytes. These cells, together with macrophages within the liver, perform a major role in immune regulation and in the filtering of antigens from the gut contents via the portal circulation. They are also implicated in the pathologic processes that occur following liver insult. Massive activation of immune cells in response to the intrinsic cellular damage induces the release of cytokines and chemokines both locally and into the systemic circulation.64,65
The minimum dose that can induce hepatic damage appears to be about 125 mg/kg. This represents 15500-mg tablets in a 60-kg individual, although hepatic necrosis has been recorded at much lower doses, especially if associated with hepatic enzyme induction. Doses above 250 mg/kg (30500-mg tablets in a 60-kg individual) will often produce damage, and doses in excess of 350 mg/kg invariably produce significant damage.66
The antidote for acetaminophen poisoning is N-acetylcysteine (NAC). It provides complete protection against hepatotoxicity if given within 12 hours of nonstaggered ingestion.67 Within 12 hours, if the time from ingestion is known with certainty and a plasma acetaminophen level is obtained, reference can be made to the nomogram to see if the potential for hepatotoxicity is present. The nomograms are unreliable if the time from ingestion is uncertain or if there was staggered ingestion over a period of time, as often occurs with therapeutic misadventure or repeated overdose. The use of alcohol often accompanies ingestion, making timing unreliable. Situations that alter normal cytochrome P-450 function such as drug induction, (chronic ethanol use, phenytoin, and isoniazid) again render the information unreliable.68 The use of the nomogram as the only basis for the decision to withhold NAC therapy is to be discouraged because of the uncertainty associated with this timing and the catastrophic potential if NAC is erroneously withheld (Fig. 75.5).
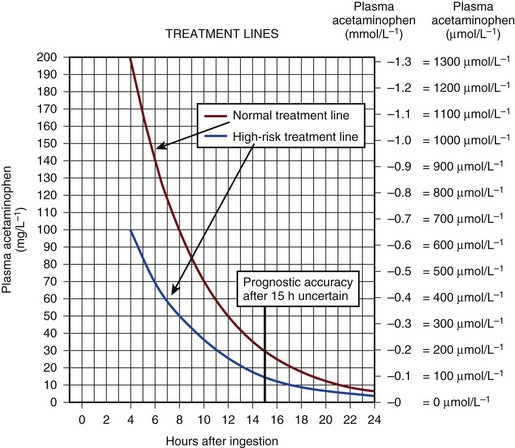
The main effect of NAC is to increase hepatic glutathione production. This promotes the conjugation of NAPQI and its subsequent excretion. In addition, NAC may act as an antioxidant within and outside the liver. It is most effective if given within the first 8 hours following overdose but is still effective following this, although less so. There is some evidence that NAC is effective when administered to the patient up to 72 hours following poisoning, although the mechanism of action is unclear and probably relates to antioxidant effects rather than to any effect on acetaminophen metabolism.69 The role of NAC in established ALF from any cause is more controversial despite widespread use, but recent data have lent support to its use certainly at lower coma grades. However, the mechanism of action is unclear.70,71
Viral Hepatitis
Both epidemic and serum hepatitis were recognized well before the viral form was discovered. In the seminal work of Lucké and Mallory in 1946, they describe 196 patients who died of ALF following both epidemic hepatitis and serum hepatitis, related to the administration of blood products during World War II.53
ALF following acute viral hepatitis is uncommon, with a reported incidence of 0.2% to 4% depending on the underlying cause.72 Liver failure following viral hepatitis tends to run an acute or hyperacute course with the onset of encephalopathy occurring within days or weeks of the first symptoms.73
Hepatitis A is now rare in the United States and Western Europe but is still a common form of acute enterally transmitted hepatitis in the underdeveloped world where it is mainly a mild and self-limiting illness of children.73,74 Infection with hepatitis A carries the lowest risk of conversion to acute hepatic failure of all the hepatotropic viruses. In the West the incidence of ALF following hepatitis A appears to be higher than in the endemic areas. It occurs more commonly in adults and is more severe. Persistent infection with hepatitis A has also been reported75 and even recurring following liver transplantation.76 Diagnosis is made on the basis of IgM antibodies at the time of hospitalization although false-negative results can occur.77
Hepatitis B may lead to ALF is several settings. It occurs most commonly following acute infection but can occur following an acute increase in viral replication following immunosuppressive therapy such as cancer chemotherapy or steroids as well as with coinfection with other viral agents such as delta virus. The host immune response is thought to be responsible for the severity of reaction to the virus, subsequent clearance, and the induction of ALF. This can be seen following the withdrawal of immunosuppressive therapy when there is a very active immune response to the increased viral load. In acute infection surface antigen (HBsAg) is often negative but IgM antibodies to the viral core (HBcAb) will usually be positive. Mutations to the precore stop codon or the core promotor region of the viral genome may be associated with a higher incidence of ALF.78 These particular genes code for HBeAg, and lack of this antigen is associated with a more profound immune response. There have been reports of a very high incidence of ALF associated with outbreaks of acute hepatitis B in the setting of intravenous drug use and chronic hepatitis C infection.79 Lamivudine antiviral therapy for acute HBV-induced hepatitis has been tried, but because of the lack of controlled trials, it is difficult to know if it or other drugs like it help.80
Hepatitis C, as a cause of ALF, is rare in northern Europe and the United States but has been described.81 There is a wide spectrum of clinical presentation associated with acute infection with the more florid presentation associated with a more rapid clearance rate, suggesting that the magnitude of the initial immune response is important.82 Liver failure associated with acute infection appears to be more common in India and the Far East.83 Acute infection may contribute to decompensation in patients with preexisting liver disease, and hepatitis C seropositivity may predispose to liver failure when coinfection with another hepatotropic virus is present.83
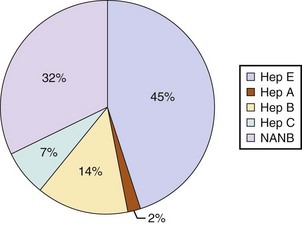
Hepatitis E is likely the most common cause of ALF worldwide and certainly for the Indian subcontinent.83 In the Far East acute HBV infection is the most common cause of ALF due to the high levels of endemicity.84
The existence of hepatitis E was inferred before serologic evidence was available by a process of exclusion. It was long assumed that most if not all epidemic enteric hepatitis was due to the A virus. When serologic markers for hepatitis A became available in the early 1980s it was apparent that the majority of waterborne epidemic hepatitis were due to other agents, producing a syndrome similar clinically to hepatitis A.85 Hepatitis E does not produce a chronic infection and in the vast majority is a self-limiting infection that occurs most commonly in young adults, in contrast to hepatitis A, which is primarily an infection of children. The incidence of hepatitis E associated with ALF is low, with a case-related mortality rate reported at about 0.5% to 4% in the general population but with a much higher mortality rate in pregnancy, as high as 20% in the third trimester. Pregnancy itself appears to be a risk factor for ALF, with a quarter of all infected female patients reported as pregnant in one series. However, this may not be particular to hepatitis E but rather due to the high incidence of epidemic hepatitis E in a relatively immunosuppressed state and pregnant patients do not have a worse prognosis compared to nonpregnant patients with ALF due to hepatitis E.83,86
Five genotypes have been described with 1 to 4 infecting humans and genotype 5 infecting only birds. Genotype 1 is the most common in Asia, with genotype 2 more common in Africa and South America. Genotype 3 can infect both human and animals, whereas genotype 1 and 2 have been described only in humans.87 In the West, travel to endemic areas is a risk factor, but sporadic cases are now being seen more commonly in the developed world. Some of these cases have been associated with contact with animals.87
Seronegative hepatitis is the second most common cause of ALF worldwide in most published series. In northern Europe and the United States it comes in behind paracetamol toxicity, and in the developing world it is second to acute viral hepatitis (Figs. 75.6 and 75.7).
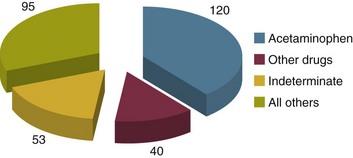
Seronegative hepatitis can be conveniently thought of as a single entity. In reality it is probably an amalgam of various causes that have defied definition or characterization, including acute presentations of autoimmune hepatitis, idiosyncratic drug reactions, and viruses.88,89 Seronegative hepatitis has a variable clinical presentation including a slow insidious onset of general malaise, jaundice, and ascites followed by progressive signs and symptoms of liver failure. At presentation the patient may be deeply jaundiced and may already have ascites and splenomegaly. It can also present with a hyperacute picture. The pattern of signs, symptoms, and organ failure is dictated by the rate of progression. In subacute seronegative hepatitis, the presenting clinical picture can be similar to that of decompensated chronic liver disease, causing occasional diagnostic difficulty. A liver biopsy is sometimes needed to differentiate between the two.
Acute Presentation of Autoimmune Hepatitis
Autoimmune hepatitis can occur at any time of life from childhood until old age. It is more common in women with a male/female ratio of 1:3. Presentation can be asymptomatic, discovered following routine laboratory testing, or more commonly, the infection may present with jaundice and general malaise. In rare cases autoimmune hepatitis can present as ALF.90 Unfortunately there are no serologic tests with sufficient sensitivity or specificity to make the diagnosis certain. Patterns of markers in the right clinical setting and in the absence of other causes may be useful.91 The diagnosis of autoimmune hepatitis in the setting of ALF is difficult and a degree of uncertainty often lasts. Classically there is a combination of elevated immunoglobulin levels, autoantibodies, and confirmatory histologic evidence of hepatitis in the absence of active viral markers. Elevation of autoantibodies is often seen in patients with ALF due to other causes such as drug-induced liver disease.90 Liver failure should be assessed in the same way as for other causes, and once signs of encephalopathy become apparent, standard prognostic criteria apply. The role of steroids is unproved but should be considered in patients prior to the onset of encephalopathy. Once the patient is listed for transplantation steroid use is controversial because of increased risk of infection.
Drug-Induced Liver Disease
Drug-induced liver disease may be due to a known dose-dependent toxicity, as with paracetamol. Alternatively, unpredictable, rare, idiosyncratic reactions can occur with any drug with a frequency of about 1 in 1000 to 1 in 100,000 patient prescriptions.68 Drug-induced liver failure can mimic all forms of acute and chronic liver disease. However, the predominant clinical presentation consists of either acute hepatitis or cholestatic liver disease. The former has a reported mortality rate of 10% irrespective of the drug.
The patterns of injury associated with these idiosyncratic reactions relate to the mechanism of damage and the cells involved.92 Many patterns have been described but it is massive hepatic necrosis that most often presents as ALF. Liver failure associated with severe cholestasis and veno-occlusive disease is also seen.92
Liver injury usually is seen within 6 months following the initiation of therapy. Even if the diagnosis of drug-induced liver failure is considered, a search for other possible causes should be performed. Although any drug is capable of inducing liver injury, more common causes include herbal remedies and recreationally used drugs such as “ecstasy” and cocaine. If suspected, a comprehensive history with a timetable of drug initiation should be constructed. Management includes stopping the offending drug and supportive care. Transplantation should be considered once liver failure occurs as the outcome from drug-induced liver failure, other than that induced by paracetamol, can be poor.93 Presentation profiles for various drugs causing ALF are shown in Table 75.1.68
Table 75.1
Idiosyncratic Drug Reactions and Effects on Cells
| Type of Reaction | Effect on Cells | Examples of Drugs |
| Hepatocellular | Direct effect or production by enzyme-drug adduct leads to cell dysfunction, membrane dysfunction, cytotoxic T-cell response | Isoniazid, trazodone, diclofenac, nefazodone, venlafaxine, lovastatin |
| Cholestasis | Injury to canalicular membrane and transporters | Chlorpromazine, estrogen, erythromycin and its derivatives |
| Immunoallergic | Enzyme-drug adducts on cell surface induce IgE response | Halothane, phenytoin, sulfamethoxazole |
| Granulomatous | Macrophages, lymphocytes infiltrate hepatic lobule | Diltiazem, sulfa drugs, quinidine |
| Microvesicular fat | Altered mitochondrial respiration, beta-oxidation leads to lactic acidosis and triglyceride accumulation | Didanosine, tetracycline, acetylsalicylic acid, valproic acid |
| Steatohepatitis | Multifactorial | Amiodarone, tamoxifen |
| Autoimmune | Cytotoxic lymphocyte response directed at hepatocyte membrane components | Nitrofurantoin, methyldopa, lovastatin, minocycline |
| Fibrosis | Activation of stellate cells | Methotrexate, excess vitamin A |
| Vascular collapse | Causes ischemic or hypoxic injury | Nicotinic acid, cocaine, methylenedioxymethamphetamine |
| Oncogenesis | Encourages tumor formation | Oral contraceptives, androgens |
| Mixed | Cytoplasmic and canalicular injury, direct damage to bile ducts | Amoxicillin-clavulanate, carbamazepine, herbs, cyclosporine, methimazole, troglitazone |
From Lee WM: Drug-induced hepatotoxicity. N Engl J Med 2003;349(5):474-485.
Pregnancy-Induced Liver Disease
In general, pregnancy-associated liver failure has the best prognosis when compared to all other causes of ALF, and prompt recovery can be expected with delivery of the fetus in most cases, if recognized early enough. Pregnancy-associated liver failure can present in several ways including the syndromes of preeclampsia and the HELLP syndrome (hemolysis, elevated liver function tests, and low platelets), liver rupture, and acute fatty liver of pregnancy.94 Any cause of ALF can occur during pregnancy, and in particular viral hepatitis can be particularly fulminant in its course. This is especially true for hepatitis E and herpes simplex virus infection.
Although originally thought to be a variant of preeclampsia there is evidence that HELLP syndrome may be a separate entity and in fact more related to acute fatty liver of pregnancy.95 Both HELLP and preeclampsia appear to be related to an endothelial injury, possibly immunologically initiated with activation of the coagulation and complement cascades and an imbalance of prostaglandin and thromboxane resulting in increased vascular tone, microangiopathic hemolytic anemia, and vascular thrombosis.96 About 2% to 12% of severe preeclampsia is complicated by HELLP.94 HELLP is thought to occur in approximately 1 in every 1000 live deliveries and patients usually present in the third trimester with nonspecific signs often seen in preeclampsia such as weight gain due to edema and hypertension. In addition, right upper quadrant pain accompanied by nausea and vomiting is commonly seen. Laboratory abnormalities include hyperbilirubinemia due to liver dysfunction and evidence of hemolysis. Transaminases are modestly raised and the platelet count is usually less than 100,000/µL. Liver biopsy, while commonly normal in preeclampsia, shows specific changes of periportal necrosis and fibrin microthrombi. Microvascular steatosis may also be present.96 The liver failure associated with HELLP is manifest as a prolonged prothrombin time and ascites. Renal failure is common. Maternal mortality rate is low but fetal mortality rate has been reported to be between 20% and 60%, although some reports suggest this is lower at 7% and associated with twin pregnancies.96–98 The treatment of choice is delivery of the baby. Conservative therapy is associated with an increase in both maternal and fetal complications.
Spontaneous rupture of the liver can occur in the setting of both preeclampsia and the HELLP syndrome, although it can occur de novo. It often presents with sudden onset of right upper quadrant pain accompanied by signs of hypovolemia or shock and is more common in multiparous women.96 Spontaneous rupture of the liver has high maternal and fetal mortality rates. Its pathogenesis is unclear but it appears that periportal hemorrhage associated with HELLP syndrome may occur close to the capsule, resulting in lifting and bleeding into the potential space. These areas of the capsule then coalesce and rupture. Management of this devastating complication includes prompt delivery of the fetus, local surgical control with packs, and aggressive management of the accompanying coagulopathy. Embolization of any feeding vessels in the liver may be of utility if such skills are available. Hepatectomy followed by liver transplantation can be lifesaving and has been performed.
Acute fatty liver of pregnancy (AFLP) occurs during the third trimester of pregnancy and should be considered in any patient exhibiting signs of liver dysfunction. It is uncommon with an incidence of approximately 1 in 6659 live births.99 If left untreated, maternal and fetal mortality rates are high. The treatment of choice is delivery and prompt recovery can then be expected. There is usually a prodromal illness over a couple of weeks with nonspecific symptoms progressing to jaundice and encephalopathy. Symptoms and signs of preeclampsia or HELLP syndrome are seen in a third of cases and there is some evidence of a common origin due to a fetal fatty acid metabolism disorder.95 Diagnosis is critical and it should be differentiated from viral hepatitis or hepatic failure due to other causes. Liver biopsy can aid in the diagnosis and can be performed via the jugular route if coagulopathy precludes the conventional approach. Characteristic zone 3 microvesicular steatosis is seen. Delivery is the best treatment if diagnosed early. Characteristic features include normoblasts on blood smears and high serum urate. Bleeding can be a major problem during operative delivery. On occasion transplantation may be the only viable option. Triggers for transplantation in pregnancy-induced liver failure are not well defined and the Kings College Criteria perform poorly. Arterial lactate in the setting of encephalopathy appears to be the best predictor.98
Wilson’s Disease
Wilson’s disease is a rare autosomal recessive disorder resulting from copper toxicity with primarily brain and liver manifestation. It usually presents in the second or third decade of life, although it can present from early childhood until late middle age.100 The disease can present with predominantly liver or neurologic symptoms. Neurologic symptoms relate to the distribution of copper to the basal ganglia and result in movement disorders. Patients presenting with liver disease may presents with an active hepatitis, established cirrhosis, or ALF. Other signs such as Kayser-Fleischer rings are associated but not pathognomonic for Wilson’s disease. These greenish brown rings in the cornea result from the deposition of copper.
ALF due to Wilson’s disease can present at any age but more commonly presents in the early 20s. High urinary copper excretion is possibly the most predictable laboratory finding, although the patient may be anuric on presentation. A low serum ceruloplasmin is an additional indicator but again is unreliable in ALF. A high serum bilirubin in combination with modest elevations of transaminases and alkaline phosphatase is often seen, as is intravascular hemolysis, contributing to the raised bilirubin level. Patients with severe liver failure due to Wilson’s disease have an almost 100% mortality rate without liver transplantation, which should be considered as soon as the diagnosis is made.100 There is debate as to whether liver failure secondary to Wilson’s disease is truly ALF, as cirrhosis is invariably present on liver biopsy at the time of presentation. Nevertheless, many patients present acutely and the presentation is often catastrophic. In this sense the timing of the onset and the lack of previous symptoms place fulminant Wilson’s disease in the ALF group. In an uncontrolled series the administration of D-penicillamine before the onset of encephalopathy was associated with survival.101
Neoplastic Infiltration
The syndrome can be considered as part of the hypoxic hepatitis group as zone 3 necrosis is often seen. The metabolic demand of the neoplastic cells and congestion within the liver sinusoids results in infarction of hepatocytes. Hepatosplenomegaly and a raised alkaline phosphatase are often present. Other stigmata including palpable lymphadenopathy and marrow or peripheral blood film changes may be present. Imaging may be diagnostic, especially with massive hepatomegaly, but biopsy may be required.102
Budd-Chiari Syndrome
ALF secondary to Budd-Chiari syndrome is usually fatal without transplantation. The syndrome is defined as outflow obstruction to the hepatic veins and the underlying pathogenesis is thrombosis in the majority but tumor invasion or vascular membrane obstruction may be the cause. This is a rare disorder that occurs predominantly in young adults and affects more women than men. Overall, the 5-year survival rate varies from 50% to 80% in different series.103 The vast majority of patients with the syndrome have at least one predisposing clotting abnormality, either congenital or acquired, such as a malignancy, myeloproliferative disorders, protein C or S deficiency, polycythemia rubra vera, lupus anticoagulant, antithrombin III deficiency, antiphospholipid syndrome, etc.104 The fulminant form of the syndrome in which the patient develops encephalopathy within 8 weeks of the onset of symptoms is rare, and it is much more common for Budd-Chiari syndrome to present in a subacute form over a 3- or 4-month period, characterized by ascites, abdominal pain and hepatomegaly, jaundice, coagulopathy, and raised AST and alkaline phosphatase. Others present with signs of portal hypertension, including refractory ascites and variceal bleeding, and relatively intact hepatocellular function. The diagnosis is made with the combination of clinical presentation and imaging, including Doppler ultrasound studies of the hepatic vessels, plus or minus liver histologic examination, usually via the jugular route because of coagulopathy. Management of the syndrome depends on the manner in which it presents and the underlying cause. Medical management of the syndrome involves the use of anticoagulants and diuretics in an attempt to control ascites. Thrombolysis can be attempted in selected patients with recent onset disease.105 In patients in whom there is progression to signs and symptoms of liver cell failure some sort of portosystemic shunting procedure may reduce symptoms and prevent progression of the disease, allowing time for collateral vessels to develop. Transjugular intrahepatic portosystemic shunt (TIPSS) is most often attempted unless there is evidence of a hypertrophied caudate lobe and inferior vena cava compression, making mesoatrial shunting a better option. In patients with signs of liver failure care must be used when considering a TIPSS procedure, as this can precipitate decompensation and rapid progression to ALF.106 Liver transplantation is ultimately the only option in many patients in which there is a failure of medical and shunt therapy as well as in the fulminant presentation of the syndrome.107 Anticoagulation is usually necessary in the immediate postoperative period.
Veno-occLusive Disease of the Liver
Veno-occlusive disease (VOD) of the liver is a nonthrombotic obstruction of the sinusoids, which may extend to the central veins, in the absence of thrombosis or other underlying disorder of the hepatic veins.108 As a result of a toxic challenge there is acute damage to the endothelial cells, followed by their detachment and their embolization in the central area of the lobule, where they cause a postsinusoidal outflow block.108 The most common cause is myeloablative chemotherapy induction regimens in preparation for bone marrow transplantation (BMT), being seen in up to 54% in some older series.109 Other toxins are known to cause it and it was first described in children following the ingestion of herbal teas in South Africa and it represents a nonspecific response to certain noxious stimuli. Particular induction regimens such as high-dose cyclophosphamide and total body irradiation are implicated in the pathogenesis. The more aggressive induction regimens appear to result in a higher incidence of VOD. Recent reduction in incidence may be due to a reduction in the use of myeloablative regimens and better monitoring of plasma drug concentrations.110 The symptoms of VOD usually occur within 2 weeks of BMT. The development of jaundice, hepatomegaly, abdominal pain, and encephalopathy in the setting of recent BMT strongly suggests the diagnosis. Severe cases are characterized by evidence of hepatocellular necrosis and a high AST concentration. In 25% of cases, which are characterized as severe, the syndrome is progressive, leading to ALF.111 Treatment options in these patients are limited as the outcome is poor with medical management or liver transplantation, although there is hope that newer therapies such as defibrotide (a deoxyribonucleic acid derivative anticoagulant with multiple modes of action), if initiated early before the onset of multiple organ failure, may improve the outlook. In addition, the prophylactic use has been tried in several noncontrolled reports.108
Hypoxic (Ischemic) Hepatitis
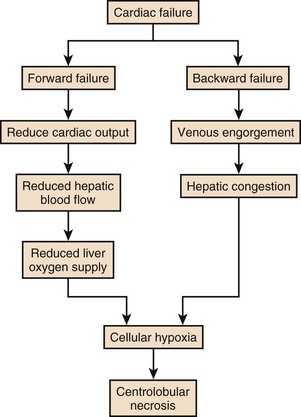
“Shock liver,” “ischemic hepatitis,” or more recently hypoxic hepatitis is the most common cause of significant rise in serum aminotransferase (S-AT) seen in hospitalized patients (Fig. 75.8).
Diagnostically, there are three conditions that should be met: the appropriate clinical setting of circulatory, cardiac, or respiratory failure with a massive rise in S-AT and the exclusion of other causes of massive hepatic necrosis such as viral or drug-induced liver failure.102
In patients with chronic congestive heart failure (CCHF), liver congestion is common. This is usually manifest as mild abnormalities of liver function tests, a prolonged prothrombin time, and mild ascites in some. Hepatic congestion is usually clinically unapparent unless jaundice is present, which can occur following multiple bouts of CCHF. Chronic congestion can lead to fibrosis and ultimately cirrhosis in some patients. Acute rises in serum transaminase and prolongation in prothrombin time, representing acute hepatic necrosis, most commonly develop in patients with CCHF when there is a sudden drop in cardiac output due to an event such as an arrhythmia or myocardial infarction. It is relatively uncommon for the liver to become involved during shock states because of the huge redundancy in blood supply and the ability of the portal system to compensate for any reduction in hepatic arterial flow. If, however, portal flow is already compromised due to passive congestion, then an acute drop in hepatic blood flow can result in ischemia. It is uncommon for hypoxic hepatitis to occur without a recognizable precipitant, but it can occur due to a silent myocardial infarction or arrhythmia, for example. It can occur before the diagnosis of CCHF has been made or be the primary diagnostic event in a younger patient with a cardiomyopathy. Clinically, severe ischemic hepatitis becomes apparent between 24 and 48 hours following an event and is manifest as huge rises in S-AT (up to 10-20 times normal). There is prolongation of the prothrombin time, encephalopathy, hypoglycemia, jaundice, and renal failure. The syndrome is usually self-limiting once the hemodynamic disturbance has receded, and there is a rapid fall in serum transaminases (usually 50% in the first 72 hours).112 Occasionally, progressive liver cellular failure leads rapidly to death. Management should be aimed at investigating and supporting the underlying cause.
Heat Stroke
Liver failure in the setting of heat stroke can be considered to be part of the hypoxic hepatitis group of causes. The argument for its inclusion here is the zone 3 necrosis seen in the liver following autopsy in patients who survive long enough for a reperfusion injury to occur.102 The pathophysiology is complex but is probably related to dehydration and cardiovascular collapse plus increased oxygen demand and acute cardiac failure.102 Exertional heat stroke may occur in new recruits to the army or police force engaging in physical initiation programs. It can also occur in unacclimatized athletes in hot conditions or with drug overdoses such as cocaine and hyperpyrexia syndrome such as those induced by MDMA (methylenedioxy-N-methylamphetamine). It is a potentially devastating syndrome that can lead to multiple organ failure and death. It ranges from mild involvement to ALF and is seen to develop during the first few days following the event. Management is supportive and the majority improves over a period of days to weeks. Liver transplantation has been used in severe cases but the outcome is poor owing to coincident organ failure.113
Mushroom Poisoning
There are many types of poisonous fungi in the world that are responsible for a variety of disorders that can be classified according to the type of poisoning and the timing of onset.114 There are a number of fungi associated with the induction of liver failure following ingestion. Of these, the most common and most deadly are of the genus Amanita. Amanita associated hepatotoxicity follows a triphasic response after ingestion. The first is a self-limiting, nonspecific gastrointestinal upset that occurs within the first 6 to 24 hours. Nausea and vomiting, abdominal cramps, and diarrhea are often seen and it can mimic food poisoning. This is followed by a period of recovery and a few days later by progressive liver failure.
Management is essentially supportive, although many specific therapies have been tried in the treatment of Amanita toxicity such as high-dose penicillin, silibinin, cimetidine, and NAC, but none has been shown to improve outcome. Liver transplantation should be considered for patients with severe liver failure and standard criteria apply.114
Clinical Course
History and Physical Examination
The history may be self-evident in the case of paracetamol hepatotoxicity, especially if the patient is self-presenting following deliberate self-harm. However, therapeutic misadventure, which is a relatively common cause of hepatotoxicity, especially in the United States, may not be obvious unless considered by the physician.115 Any patient presenting with coma should have a drug screen (including paracetamol).
Subacute liver failure has, by definition, a more insidious onset. Jaundice is often preceded by nonspecific symptoms of fatigue and general malaise. Abnormal liver enzymes will often be revealed during the initial workup. Subsequent investigations will include viral, iron, copper and genetic studies and, in addition, attempt to rule out decompensation of chronic liver disease as a cause the current symptoms and signs. Liver biopsy may be considered in subacute liver failure (Fig. 75.9).
Initial Resuscitation and Emergency Care
The symptoms and signs of hepatic encephalopathy in ALF are subjectively different from those seen in decompensated chronic liver disease or in patients with stable chronic encephalopathy. Agitation and aggressive behavior are more common in ALF. This may be based on an increased turnover of the excitatory neurotransmitter glutamate in ALF associated with an acute increase in cerebral ammonia uptake.116
The West Haven criteria, designed to assess coma grade in patients with cirrhosis, are often applied to patients with ALF and are useful because of familiarity.117 However, once the patient becomes unconscious at grade IV encephalopathy, the scale does not provide any further information and is too crude to provide a clinically useful description of the level of consciousness. The Glasgow Coma Scale score, while not assessed specifically in this setting, is useful in relating clinical information to others.118 Encephalopathy grade can progress very quickly, especially with hyperacute presentation. Intubation and sedation are recommended once grade III encephalopathy is achieved because of the attendant risks to airway and possibilities of raised intracranial pressure, discussed later. Figure 75.10 describes encephalopathy grade and Glasgow Coma Scale score.
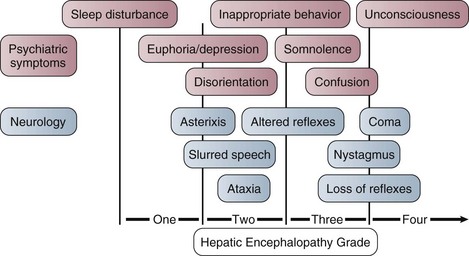
Figure 75.10 Hepatic encephalopathy grade in acute liver failure.
Following the safe and appropriate management of airway and breathing, focus can be directed to the correction of any circulatory dysfunction. Patients with ALF can progress rapidly to circulatory shock. The circulatory changes associated with ALF are predictable, usually associated with systemic vasodilatation and relative hypovolemia. This can be profound and require significant volume resuscitation. It is easy to underestimate the amount of fluid required in the early stages of ALF and the use of prognostic markers is unreliable before adequate circulatory function is restored. Monitoring at this stage should include a central venous catheter and direct arterial access for both blood pressure monitoring and for repeated blood tests. Care must be given to serum electrolytes because they often become deranged during this initial period. In particular, hyponatremia and hypophosphatemia are common.119–121 This may be due to excess quantities of hypotonic fluid administration. A typical flowsheet of investigations of a patient presenting with paracetamol poisoning from a referring hospital is shown in Box 75.2.

For prognostic reasons it is important not to avoid correcting any biochemical coagulopathy at this stage if possible. Despite quite significant prolongation of prothrombin time, bleeding from line sites is uncommon in the initial stages of ALF. In fact, there is evidence that the patients are prothrombotic in vivo.122
Investigation in a Patient with Suspected Acute Liver Failure
Initial investigations provide a baseline from which important diagnostic and prognostic information is taken. Serial blood test measurements should be performed as severity and prognosis are assessed over time from initial presentation (Table 75.2). Depending on the manner and speed of presentation, a complete blood count, clotting tests, liver function tests, arterial blood gas measurements, and ammonia levels should be performed twice daily, if not more often, in the early stages to assess progression of the illness. A liver ultrasound scan to assess liver size and blood supply should be performed.
Table 75.2
Initial Investigation and Common Findings
| Laboratory Test | Finding(s) |
| Complete blood count | Platelets often low |
| Urea and electrolytes | Serum sodium often low, especially in paracetamol hepatotoxicity Urea often low because of reduced production Serum creatinine is useful prognostic marker |
| Liver enzymes | Serum AST/ALT variable, very high in paracetamol toxicity Bilirubin is prognostic marker in nonparacetamol liver failure Also can be high in Wilson’s disease from hemolysis |
| Phosphate | Often low, especially in paracetamol toxicity High level indicative of poor prognosis; suggests lack of regeneration |
| Magnesium | Often low |
| Prothrombin time, international normalized ratio (INR) | Very sensitive indicator of liver function Has prognostic significance in all forms of acute liver failure May improve with vitamin K in deficiency states |
| Viral serology for all known hepatotropic viruses | |
| Urinary copper, plasma ceruloplasmin if Wilson’s disease is suspected | Ceruloplasmin level often very low but can be normal Urine copper usually high |
| Arterial ammonia | Has some prognostic value and may be an indicator of cerebral edema in patients at risk |
| Arterial whole blood lactate | Very sensitive marker for liver function, particularly in paracetamol toxicity High level indicative of poor prognosis |
| Serum glucose | Hypoglycemia common |
| β-Human chorionic gonadotropin in women | Unwanted pregnancy can be a precipitant of deliberate self-harm |
Transfer Criteria Guide
ALF is a sporadic syndrome with a case incidence of about 2000 per year in the United States.123 Management is complex and liver transplantation is the only effective therapy in severe cases. Transfer to a unit with experience in the management of these patients, preferably one with a liver transplant program, will provide optimal care. It is often difficult to decide when or whether to transfer a patient with acute liver dysfunction, and this decision will vary according to cause and presentation. The best described clinical course is that following severe paracetamol poisoning after a deliberate overdose.52 Staggered overdose and therapeutic misadventure are less predictable in their clinical course but often have a poor outcome and can be considered a poor prognostic risk factor.124
The following criteria for transfer are based on expert opinion and have not been subjected to rigorous study and err on the side of caution72 (Fig. 75.11):
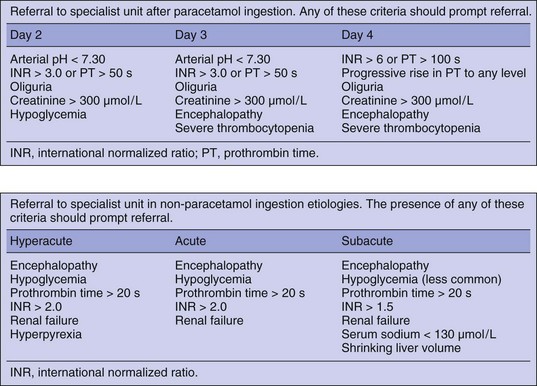
• In patients with a subacute presentation it is better to transfer patients before they have progressed to grade II encephalopathy because of the additional complications associated with transferring ventilated patients.
• Patients with acute or hyperacute liver failure showing signs of early encephalopathy should be intubated and ventilated prior to transfer. This can occasionally result in disagreement between referring hospitals and receiving liver units on how the transfer should be managed. There are many anecdotal stories from liver units around the world describing patients who become unmanageable during transfer in planes, helicopters, and road ambulances resulting in injury to patients and staff.
• Heroic transfers are almost always inappropriate. Patients should not be considered for transfer if they are on rapidly accelerating inotropic support, have severe hypoxemia, or already have fixed dilated pupils. There is little point a patient dying in an ambulance.
Management
Supportive Care
Airway and Ventilation
Controlled ventilation to a normal PaCO2 is recommended at this stage. The use of hyperventilation is not indicated without further monitoring.125,126
Circulation
The circulatory changes associated with ALF can be profound.127 The pathologic basis for this are incompletely understood, but similar in many ways to changes observed in patients with a systemic inflammatory response due to sepsis or trauma. The pattern observed depends on the cause and the rate of onset of the syndrome. In hyperacute liver failure due to paracetamol overdose, patients can develop fulminant peripheral cardiovascular collapse that can be an early mode of death.128 Others with a more subacute onset can develop peripheral vascular changes similar to those with decompensated chronic liver disease and hepatorenal failure.
Fluid resuscitation should be commenced as soon as possible after presentation and should be directed by invasive monitoring. The type of fluid used for resuscitation has not been subjected to controlled trials in this setting but large volumes of hypotonic fluids should be avoided as this may contribute to cerebral swelling seen in this group, and it has been shown that hypertonic saline infusion reduces the incidence of intracranial hypertension in patients with ALF.119
Vasopressin and its longer acting analog terlipressin have been used as vasopressors in septic and cardiogenic shock.129,130 Terlipressin results in profound systemic vasoconstriction and, if used inappropriately, has the potential to reduce cardiac output and hence oxygen delivery. It has been shown to increase intracranial hypertension in ALF due to an increase in cerebral blood volume because of the breakdown in cerebrovascular autoregulation.131 However, the data are mixed and a more recent study suggests that terlipressin may be a useful adjunct to norepinephrine in ALF.132 Because of this uncertainty, monitoring of ICP should be considered if it is used.
The majority of patients presenting with ALF are young adults, and as a result, comorbid cardiac dysfunction is usually absent. Cardiac depression has been reported in association with POD.128 On occasion cardiac disease can present as ALF in the case of ischemic hepatitis.
Relative adrenal insufficiency has been shown to occur in patients with septic shock and in patients with decompensated chronic liver disease and ALF.44,133–135 The use of stress doses of corticosteroids in ALF has been shown to reduce norepinephrine requirements in patients with ALF.136 The effect on outcome is uncertain, though.
Renal Support
Renal failure is common in ALF with a reported incidence of up to 70%.137 Paracetamol causes direct renal tubular dysfunction, possibly by affecting membrane protein function and occasionally renal failure is the predominant organ affected.138,139 As a result, renal failure can be expected in this setting. Although it is most commonly associated with POD, the incidence in other causes, such as Wilson’s disease due to the direct toxic effects of copper, is also high but it is less prominent in ALF due to viral hepatitis or seronegative hepatitis.
There remains debate regarding the cause, pathology, and definition of renal dysfunction in critical illness despite a huge amount of effort.140 The term acute tubular necrosis is inadequate in the setting of critical illness when it fails to describe the full spectrum of renal dysfunction. This is even truer in the setting of ALF. In addition, the use of hepatorenal syndrome in ALF is also inappropriate because this term does not adequately describe the often rapid clinical deterioration. This is not to say that altered systemic and renal hemodynamics do not contribute to the pathophysiology of renal failure as they do in critical illness in general. In the subacute presentation of this syndrome early renal dysfunction can be similar in presentation to that seen in end-stage chronic liver disease with sodium and water retention in the absence of intrinsic renal damage.141
Management of renal failure is essentially the same as in ALF and other forms of multiple organ failure and consists of maintenance of intravascular volume, cardiac output, and mean arterial pressure.123,142
Extracorporeal renal support is necessary in most patients with ALF at some time. Continuous venovenous hemofiltration (CVVH) is the most efficient and safest method for renal support and should be started early.143 Indications for the early use of renal support are not just limited to oliguria. CVVH has been shown to improve hemodynamic stability in patients with critical illness and has been used as salvage therapy in patients on high-dose vasopressor.144 High-volume hemofiltration (4000 mL/hour ultrafiltrate exchange) has been studied in ALF and has been shown to reduce serum lactate, base deficit, and norepinephrine requirements when compared to historical control subjects.145 Evidence for survival benefit for high-dose renal replacement therapy in critical illness is lacking, however, and recent high-quality studies suggest that there is a ceiling of dose, above which there is no additional survival benefit.146,147
Patients with liver failure do not tolerate the lactate load associated with lactate-buffered replacement fluid during CVVH. In fact, the rapid infusion of lactate can induce a systemic acidosis in this setting.148 Also, the infusion of large quantities of lactate-containing fluid reduces the utility of serum lactate as one of the most important prognostic indicators.149 As a consequence, the use of bicarbonate hemofiltration is recommended.150
Intracranial Hypertension
Etiology
The hallmark of liver failure is the development of hepatic encephalopathy, which with severe liver failure progresses to deep coma. In addition to the progressive coma, early reports of patients with signs of raised ICP were noted in the literature.151 What is less clear is whether they possess a common cause. Certainly patients with subacute liver failure can develop a deep coma without signs of cerebral edema, but patients with signs of intracranial hypertension (ICH) always develop high-grade encephalopathy prior to this.
Cerebral edema was noted and commented on in the seminal clinicopathologic review of servicemen presenting with fulminant epidemic hepatitis during the East Asian campaign of the Second World War.53 However, the recognition that cerebral edema was a distinct clinical entity and cause of death associated with ALF did not become clear until much later.151,152
 The recognition that brain swelling is an important component of ALF is now well established. Management strategies place the risk of ICH at the forefront of care, and prophylactic therapy, monitoring, and treatment are widely debated in the literature.153,154 The incidence of cerebral edema in ALF is not completely clear and is dependent on the cause of liver failure, the severity, and the rate of onset. It is important to distinguish between ICH and cerebral edema. The former requires the measurement of pressure within the cranial vault and results in brain ischemia and signs of tentorial herniation. The latter is difficult to define clinically and depends on how hard you look. CT and postmortem studies have suggested an incidence of between 50% and 80% of those with high-grade encephalopathy.151,155 The pathologic changes of cerebral edema and ICH in ALF are not completely understood, but in recent years progress has been made in understanding the processes involved.156
The recognition that brain swelling is an important component of ALF is now well established. Management strategies place the risk of ICH at the forefront of care, and prophylactic therapy, monitoring, and treatment are widely debated in the literature.153,154 The incidence of cerebral edema in ALF is not completely clear and is dependent on the cause of liver failure, the severity, and the rate of onset. It is important to distinguish between ICH and cerebral edema. The former requires the measurement of pressure within the cranial vault and results in brain ischemia and signs of tentorial herniation. The latter is difficult to define clinically and depends on how hard you look. CT and postmortem studies have suggested an incidence of between 50% and 80% of those with high-grade encephalopathy.151,155 The pathologic changes of cerebral edema and ICH in ALF are not completely understood, but in recent years progress has been made in understanding the processes involved.156
Management
In the management of ICH in ALF it is important to target those at risk. The development of ICH sets the management of ALF apart from other forms of critical illness and it remains a leading cause of death despite advances in the understanding of the etiology ALF and management of ALF patients.172,173 Monitoring the pressure within the cranial vault is difficult and invasive. Despite investigation, a reliable noninvasive method for the evaluation of cerebral blood flow, cerebral oxygenation, and ICP remains elusive.174
Predicting Intracranial Hypertension
In ALF the development of cerebral edema is seen in patients with the shortest time between the development of jaundice and the onset of encephalopathy.52 A fulminating presentation, lack of time for cerebral adaptation, and systemic burden of a necrotic liver appear to be the likely reasons. Patients with paracetamol toxicity make up the largest number in this group. Other causes can fall into the hyperacute group including patients with ALF due to hepatotrophic viruses, particularly HBV. In contrast, patients with subacute liver failure have a smaller risk.
It has been suggested that the incidence of ICH following paracetamol toxicity has fallen since the mid-1980s.175 However, it still represents a significant complication in this setting. Recent work from King’s College Hospital in London suggests an incidence of 20% to 30% in all patients with ALF. Data from our own unit suggest that ICH is implicated in the death of 25% of all patients with ALF and 35% of those following paracetamol-induced toxicity.172,173
Young age has consistently been found to be a risk factor.151,173 Arterial ammonia concentration has been shown to correlate with death and cerebral herniation.158,159,173,176 Recent commentary suggests that arterial ammonia should be measured serially in all patients with ALF and that ICP monitoring be instituted if the concentration is greater than or equal to 150 µmol/L.170
Serum sodium is often low in patients with ALF. In a consecutive group of patients with ALF from POD admitted to King’s Hospital liver intensive care unit, 65% were hyponatremic on arrival (Will Bernal, personal communication). Hyponatremia is associated with a poor outcome in ALF,177 and data have shown an inverse relationship between ICP and serum sodium in patients with ALF178) (Box 75.3).
The severity of the inflammatory response and organ failure are positively associated with the risk of developing ICH. Systemic inflammation may exacerbate ICH in a brain primed by ALF.173
Monitoring the Brain
Computed tomography is a standard investigation in any patient with suspected intracranial disease. Cerebral edema can be recognized in CT scans of patients with ALF and the severity correlates crudely to encephalopathy grade, but the correlation between imaging and severity of ICP measurement is poor.155,179 As little additional information is gained, very careful consideration should be undertaken before transporting this very sick group of patients to the CT scanner. Occasionally there are diagnostic difficulties or a
Ammonia has long been implicated as important in hepatic encephalopathy and, as the evidence mounts, increasingly recognized as an important factor in the cause of cerebral edema in ALF.157 Recent work has shown that whole blood ammonia predicts outcome and the chances of cerebral herniation.158,159 Electron microscopic analysis of postmortem brain biopsy together with gravimetric analysis of brain water content, in animal models, have shown that swelling occurs in the gray matter and that astrocytes are the main target.160,161 Ammonia may induce astrocyte swelling by a number of possible mechanisms. It is detoxified in astrocytes by combining with glutamate to produce glutamine. Astrocytes are the only cell type in the brain that contains glutamine synthetase, and this normal pathway maintains the ratio of glutamate to glutamine within astrocytes and neurons. The raised serum ammonia induces a buildup of glutamine within astrocytes, increasing the osmotic potential, absorption of water, and an increase in volume. Evidence of this effect can be inferred by the fact that inhibition of glutamine synthetase, which catalyzes the reaction, prevents brain swelling in experimental models.162 An osmotic effect may not be the only mechanism by which glutamine induces cellular swelling, however, but there is circumstantial evidence that it occurs to some extent. In subacute and chronic liver disease there is time for adaptation to the increase in astrocyte osmotic potential by the excretion of intracellular osmolytes such as myoinositol.163 In ALF there is less time for adaptation because of the rapid increase in intracellular glutamine.
Other possible mechanisms include a breakdown in cellular energy metabolism induced by an increase in intracellular glutamine and ammonia. Impairment of α-ketoglutarate dehydrogenase and pyruvate dehydrogenase induced by oxidative stress and ammonia results in a breakdown in astrocyte energy production and an increase in glycolysis. This is supported by an increase in extracellular lactate and brain lactate flux in clinical studies.116,125 Mitochondrial dysfunction induced by ammonia may also play a role.157
In addition to the cytotoxic edema seen early in ALF, changes in cerebral blood volume may play an important role in the development of ICH. Cerebral blood flow shows wide variation in patients with ALF, and the normal close relationship between cerebral metabolism and flow appears to be lost.164 In animal models of liver failure there is a gradual increase in cerebral blood flow but this situation is less clear in human studies.156,165 Cerebral metabolism is generally reduced with high-grade encephalopathy in patients with ALF and there is evidence that relative or absolute cerebral hyperemia can contribute to ICH.131,166
An increase in cerebral blood flow may induce ICH by a number of mechanisms and induce further cerebral swelling. This could occur due to an increased flux of potentially toxic metabolites such as ammonia or by an increase in cerebral water content because of an increase in cerebrovascular hydrostatic pressure. The blood-brain barrier has been considered to remain intact in ALF but recent evidence suggests that there is some impairment, although the degree to which this contributes to cerebral swelling is still being debated.161,167
 Cerebrovascular autoregulation is lost in ALF.165,168 The cause is unknown but a gradual cerebral vasoparesis concurs with clinical observation. Autoregulation can be restored by hyperventilation and mild hypothermia but not by the use of indomethacin.169 It has been suggested that the increase in astrocyte glutamine plays a role in this gradual vasoparalysis and loss of autoregulation by the induction of local nitric oxide or carbon monoxide.156 It is relatively common to find a patient with a high ICP and jugular venous oxygen saturation above 80%. This suggests a state of luxury perfusion (unregulated blood supply in excess of demand) in which an increase in cerebral blood volume in an already swollen brain accounts for the associated increase in ICP.170
Cerebrovascular autoregulation is lost in ALF.165,168 The cause is unknown but a gradual cerebral vasoparesis concurs with clinical observation. Autoregulation can be restored by hyperventilation and mild hypothermia but not by the use of indomethacin.169 It has been suggested that the increase in astrocyte glutamine plays a role in this gradual vasoparalysis and loss of autoregulation by the induction of local nitric oxide or carbon monoxide.156 It is relatively common to find a patient with a high ICP and jugular venous oxygen saturation above 80%. This suggests a state of luxury perfusion (unregulated blood supply in excess of demand) in which an increase in cerebral blood volume in an already swollen brain accounts for the associated increase in ICP.170
Patients with ALF often develop a systemic inflammatory response. Depending on the cause and severity of hepatic necrosis, damage-associated molecular pattern molecules and pathogen-associated molecular pattern molecules (DAMPs and PAMPs) are released from the liver. These, in turn, activate the innate and adaptive immune response culminating in the pathophysiologic picture associated with ALF. The severity of the inflammatory response has been shown to correlate with the degree of encephalopathy and the severity of ICH, and there is evidence that neuroinflammation in response to circulating cytokines exacerbates ICH.171
suspected complication of ICP bolt insertion and CT scanning might be considered.
Functional brain imaging, using single positron emission tomography (SPECT), has been used to investigate the distribution of cerebral blood flow in ALF and magnetic resonance imaging (MRI) scanning has been used to investigate the distribution of intracerebral water, but neither has found a place in clinical practice.126,180,181
In patients with suspected ICH the direct monitoring of cerebral oxygenation and blood flow are appealing but current methods have technical and clinical limitations. Tissue PO2 and interstitial metabolites, using intraparenchymal probes, have been investigated in traumatic brain injury and to a limited extent in ALF.116,182 They have the advantage in traumatic injury of providing localized information around the area of injury. The use of cerebral microdialysis (sampling of the cerebral interstitial fluid using a microcoaxial catheter with a semipermeable membrane) remains a research tool in ALF at the present time.
Methods used for the estimation of global cerebral oxygenation include the sampling of jugular venous (JV) blood for oxygen saturation, and products of metabolism such as lactate. A JV saturation of less than 55% suggests an ischemic brain. This can be due to a reduction in blood flow in excess of demand because of brain swelling or cerebral vasoconstriction due to hypocarbia, if the patient is being hyperventilated. An increase in demand due to seizure activity can also manifest as a reduction in JV saturation.183 High jugular saturation (>80%) may represent a hyperemic brain, and steps can be taken to reduce cerebral blood volume if ICP is raised. Very high JV saturation is often seen as a terminal event and may represent a complete loss of oxygen extraction by the brain (Fig. 75.12).
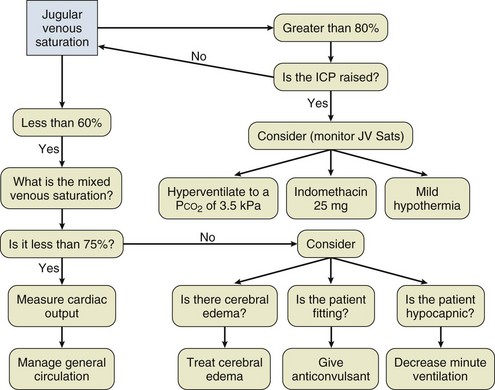
Noninvasive measurement of cerebral blood flow using transcranial Doppler (TCD) has been investigated in ALF. In one study it was found to be predictive of changes in cerebral blood flow induced by hyperventilation.184 More recent investigation has shown changes in TCD waveform as ICP increases, but it is not clear how reproducible these results are.185
The recognition that ICP is raised in a significant proportion of patients with ALF and that this is implicated in significant rates of morbidity and mortality has led to the use of direct measurement of ICP with various forms of monitoring.151,186 These techniques, although fully supported internationally in traumatic brain injury, are controversial in the field of ALF and there remains a dichotomy of opinion in most countries, with some units using them and others not.153,154,187,188
Controversy revolves around the lack of evidence of improved outcome with the monitoring of ICP and the risk of intracranial bleeding complicating insertion. The reported risk of bleeding, from survey data, is between 10% and 20% overall, the majority of which is not clinically significant. Mortality rate has been reported at between 1% and 3% (Alistair Lee, Edinburgh, UK, personal communication).186,188 The risk of bleeding following placement is higher than that seen following traumatic brain injury. There is uncontrolled evidence that activated factor VII can reduce this risk.189
It has not been possible to prove that ICP monitoring improves survival in ALF, as a randomized controlled clinical trial has not been performed to evaluate it. However, it is generally accepted that medical intervention can reduce ICP and prevent cerebral ischemia and brain herniation in patients with ALF.170 Published data suggest that having an ICP monitor increases the intervention rate compared to patients without and increases the length of survival in the critical care unit, if not overall survival.190 The majority of patients with ALF die of multiple organ failure due to sepsis. Intervention to reduce ICP may just prevent early cerebral death.
Prophylactic Measures
Serum sodium is often low in patients with ALF. In a consecutive group of patients with ALF from POD admitted to King’s Hospital liver intensive care unit, 65% were hyponatremic on arrival (Will Bernal, personal communication). Hyponatremia is associated with a poor outcome in ALF.177 Based on retrospective data showing an inverse relationship between ICP and serum sodium in patients with ALF, moderate hypernatremia was investigated as a possible prophylactic intervention.119,178 Maintaining serum sodium between 145 and 155 mmol/L using hypertonic saline was found to reduce ICP from baseline and reduce the incidence of surges in ICP.119 Hypothermia improves outcome following out-of-hospital cardiac arrest and has been investigated in patients with traumatic brain injury. Early reports suggest hypothermia can reduce ICP and ammonia production in patients with ALF. However, prophylactic hypothermia has not been shown to reduce the incidence or improve outcome in this setting.191 Simple measures such as raising the head of the bed to a 30-degree angle and the avoidance of excessive stimulation are also prudent.
Cerebral Perfusion Pressure
In traumatic brain injury there is a consensus of opinion supporting the use of cerebral perfusion pressure (CPP) as a treatment goal.187 In ALF the concept of CPP-directed therapy is less useful. To assume a correlation with cerebral blood flow there has to be a consistent cerebrovascular resistance, which is not the case in ALF.170 This is due to the loss of cerebrovascular autoregulation, and attempts to increase cerebral perfusion are often unsuccessful as the use of a vasopressor results in an increase in ICP as brain blood volume increases.131,192 However, this is not to say that CPP should be ignored entirely but the safe lower limit of CPP has yet to be defined, as there are many reports of patients surviving with normal cerebral function despite a low CCP.193 The normal lower limit of cerebral autoregulation is reached at a mean arterial blood pressure of about 50 mm Hg, below which flow becomes pressure dependent. In patients with absent autoregulation, such as in ALF, CCP should probably be maintained above 40 mm Hg (the normal lower limit of autoregulation with an ICP of 10 mm Hg or less), but no data exist to back this statement up. Maintenance of CPP in ALF is best achieved by decreasing ICP and aiming for a mean arterial pressure with fluid and a vasopressor that does not increase ICP above 25 mm Hg. Attempting to improve cerebral oxygen balance is also attractive in this setting. This may be attempted with intravenous indomethacin (has been shown to improve CPP without compromising cerebral oxygenation), hypothermia, increased sedation, and hyperventilation.169,194–196 Monitoring cerebral oxygenation is very useful during such a maneuver.
General Management of Patients with Raised Intracranial Pressure
ICP is normally less than 15 mm Hg in an adult. The definition of ICH is not precise and will vary among patients. Available data are derived from patients with traumatic brain injury (TBI) where observational studies suggest that intervention to reduce pressure should be instituted between 20 and 25 mm Hg, although pupillary abnormalities and brain herniation can occur at lower pressures.187 There have not been any studies investigating treatment threshold in ALF and so similar thresholds to traumatic brain injury are used.
The management of ICH is usually escalated along standard algorithms (Figs. 75.13 and 75.14). Elevate the patient to an angle of 30 degrees and avoid tight straps around the neck to encourage venous drainage. ICP tends to increase during nursing intervention. If the ICP rises following nursing intervention and does not resolve after several minutes it implies poor intracranial compliance. Treatment is usually instituted for a sustained rise in ICP (more than 10 minutes over the treatment threshold) or clinical signs suggesting cerebral ischemia or impending herniation.
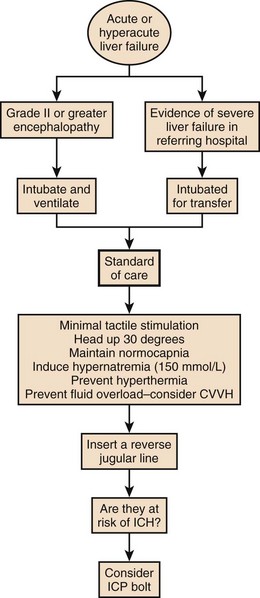
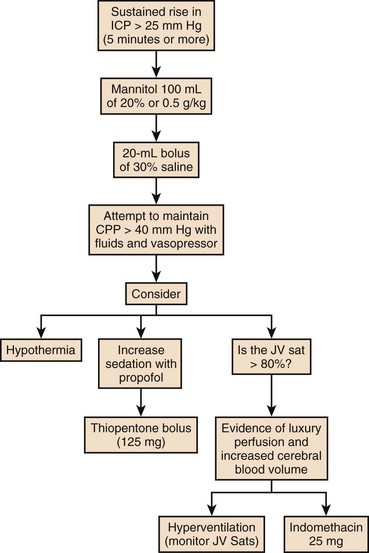
• Sedation should be increased. Propofol is probably the agent of choice.194
• Osmotherapy is the mainstay of treatment following these simple measures.
• Mannitol as a rapid infusion (0.5-1.0 g/kg) has been shown to reduce ICP reliably in ALF.197 The dose can be repeated but care must be used in renal failure due to accumulation, and multiple administrations can result in a hyperosmolar syndrome. Plasma osmolality should be monitored if multiple doses are used. Current practice is to remove 500 mL of ultrafiltrate via CVVH following each bolus dose of mannitol.
• Bolus doses of 20 mL hypertonic saline (30%) has a similar effect to mannitol in this setting in the author’s experience. Hypertonic saline has a higher reflectance coefficient at the blood-brain barrier compared to mannitol and there appears to be less tachyphylaxis to multiple administration.119
In patients with a raised ICP and cerebral hyperemia, suggested by a JV oxygen saturation of 80% or greater (luxury perfusion), short-term hyperventilation will induce cerebral vasoconstriction and reduce blood volume. This maneuver has been shown not to impair cerebral oxygenation but close monitoring of cerebral oxygenation should be employed if it is attempted.195 Short-term hyperventilation has not been shown to improve outcome in ALF but does prolong survival in the ICU.198 Hyperventilation may be lifesaving and may buy time for definitive treatment (transplantation). Indomethacin induces cerebral vasoconstriction and reduces ICP in patients with both traumatic brain injury and ALF without impairing cerebral oxygenation, although confirmatory studies are needed.169
Seizures
Ammonia toxicity and cerebral edema are associated with seizure activity and it has been recognized that subclinical seizures occur more commonly than previously thought.183 The use of mechanical ventilation facilitated by sedatives and muscular paralysis can mask clinical signs. Seizures adversely affect cerebral oxygen consumption and may contribute to cerebral edema and are a cause of low jugular oxygen saturation. It has been suggested that prophylactic phenytoin be used in all patients with ALF and high-grade encephalopathy. Others have questioned this approach because of the significant side effects and apparent lack of effect on outcome.199 If confirmed, seizure activity should be managed along standard management guidelines.
Infection and Immunosuppression
Patients with ALF have multiple immune defects and are susceptible to infections.200 Infection is a common cause of progression and complication in ALF.201 Antibiotic prophylaxis has been shown to reduce the incidence of infection and enable transplantation to proceed but has not been shown to improve survival. Selective decontamination of the digestive tract has not been shown to be superior to intravenous antibiotics alone.200,202 More recent work has shown how the microbiologic flora of a liver unit has changed over the past 20 years—for example, bacteremia appearing later in the ICU stay and being predominantly gram-negative in nature.203
It is current practice to prescribe broad-spectrum antibacterial and antifungal medication to patients with ALF depending on local sensitivities. There is a general trend to the timing of infection with gram-positive infections occurring earlier than gram-negative. Early gram-negative infections tend to be less resistant “endogenous” bacteria followed later by resistant hospital-acquired organisms.204,205
Nutrition
 Within the general intensive care literature there is a consensus toward enteral nutrition (EN) as the route of choice.206 There are few additional data on which to base decisions in patients with ALF. There is, however, wide regional variation in the prescribing of parenteral nutrition (PN) compared to EN. The reason for this is unclear as EN is associated with a reduction in infectious complications.206 However, it is clear that some centers prefer PN.207 Nutritional requirements in ALF are not well understood.
Within the general intensive care literature there is a consensus toward enteral nutrition (EN) as the route of choice.206 There are few additional data on which to base decisions in patients with ALF. There is, however, wide regional variation in the prescribing of parenteral nutrition (PN) compared to EN. The reason for this is unclear as EN is associated with a reduction in infectious complications.206 However, it is clear that some centers prefer PN.207 Nutritional requirements in ALF are not well understood.
Energy expenditure is raised during ALF. This is surprising considering the normal contribution of the liver, a large metabolically active organ, on the overall energy expenditure, illustrated by the effects of hepatectomy on energy expenditure during elective liver transplantation.208 There is organ-specific lactate production in both the liver and the lungs and evidence of a systemic inflammatory response in many patients.201,209,210 The cause of systemic inflammation in ALF is activation of inflammatory cells (leukocytes and endothelium) and the release of systemic cytokines. This may be initiated by infection.201 Patients in the early stages of ALF are markedly catabolic with insulin resistance despite the frequent presence of a hypoglycemic state.211 The Harris-Benedict equation is inaccurate in ALF and indirect calorimetry should be used if energy requirements are sought.
Serum amino acids are consistently deranged during liver failure. There is a low or normal concentration of glutamine, branched chain amino acids, and tryptophan and an increase in others.212 High brain concentration of ammonia is thought to contribute to cerebral edema and encephalopathy in ALF. Manipulation of ingested amino acids has been investigated in an attempt to reduce ammonia concentration. L-Ornithine and L-arginine (LOLA) infusion encourages alternate pathway metabolism and reduces hepatic encephalopathy in chronic liver disease.213 LOLA has been investigated in animal models of ALF and resulted in normalization of plasma ammonia concentration, significant delay in the onset of encephalopathy, and a reduction in brain water concentration.214 LOLA has been investigated in 2009 with a randomised controlled clinical trial in ALF. Despite theoretical promise, the investigators were unable to show any reduction in mortality rate or ammonia between the groups.215
N-Acetylcysteine (NAC) is primarily used as a glutathione precursor and antidote in paracetamol poisoning but is used widely in patients with ALF induced by other causes and later on in its subsequent course. Results of a recent randomized controlled clinical trial have shown efficacy with NAC in patients with low-grade encephalopathy and nonacetaminophen-induced liver failure. The mechanism of action is unknown.70
Serum electrolytes are often deranged in the early stages of ALF. Hypophosphatemia is common in patients with ALF and is a good prognostic sign. High or normal phosphate may indicate a lack of hepatic regeneration and renal impairment.120 Hypomagnesemia is also common and should be corrected.
Artificial Liver Support
The liver is a complex organ, and to be an ideal liver replacement, any system has to support a wide range of biosynthetic and metabolic functions. Any working system will also have to counter the systemic burden of the necrotic, dying liver.216
Artificial liver support can be split into two main approaches. In the first there is an attempt to simulate or replace all or most of the functions of the liver. These systems include hepatocytes either from human or animal sources. Another view of liver failure suggests that toxins either excreted by the dying liver or not metabolized because of an acute reduction in function are responsible for the majority of the signs and symptoms. In this view extracorporeal blood purification with dialysis or adsorption techniques are employed and serum proteins not produced are replaced with plasma (Box 75.4).
Biologic Systems
 Biologic systems consist of a bioreactor within which the cellular biomass is contained, and a mechanism of containing and separating the biomass from the circulation of the patient. They require an extracorporeal system to deliver blood or plasma to the bioreactor and may also contain an adsorption or dialysis component. Data from liver resection suggest that approximately 250 mL of liver by volume is required to prevent death from liver failure. This typically represents 20% to 30% of liver mass.217
Biologic systems consist of a bioreactor within which the cellular biomass is contained, and a mechanism of containing and separating the biomass from the circulation of the patient. They require an extracorporeal system to deliver blood or plasma to the bioreactor and may also contain an adsorption or dialysis component. Data from liver resection suggest that approximately 250 mL of liver by volume is required to prevent death from liver failure. This typically represents 20% to 30% of liver mass.217
Immortalized cell lines that proliferate in culture, while retaining some liver-specific functionality, can be used in an attempt to overcome the limitations of primary hepatocytes. Many lines have been created by retroviral transfection with regulatory genes that stimulate cell division. The insertion of “terminator” genes that give the cells a limited life or enable switching the immortalizing gene off have also been developed to improve safety.218 Other sources of immortal and readily cultured cells are tumor derived such as the ubiquitous Hep G2/C3A hepatoblastoma line. Finally, stem cell sources appear to offer the most hope in terms of a readily available and functional supply of differentiated hepatocytes.219
Clinical trials in the use of bioartificial liver support have been relatively disappointing. The bioartificial liver (BAL) uses porcine hepatocytes and a charcoal column in series. The largest trial published so far in the field was powered for survival advantage in ALF and primary nonfunction following liver transplantation.220 The study was terminated early by the data and safety-monitoring board because the trial was likely to be futile based on the results at interim analysis.220 Post hoc analysis suggested some effect in the ALF group alone. The extracorporeal liver assist device (ELAD) system uses Hep G2/C3A hepatoblastoma line and has been investigated in a number of phase 1 and phase 2 studies designed to report safety and activity. In a randomized controlled study, not powered for mortality, showed a trend for improved survival in the treatment group.221,222 Since then, two further studies of the ELAD machine have been conducted in patients with AoCLD, both published as abstracts, both suggesting an improvement in outcome.223,224
Nonbiologic Systems
 Many of the molecules that accumulate within the blood during ALF are small or middle-sized.216 These molecules can be targeted by a variety of extracorporeal purification techniques, including dialysis through various types of membranes and adsorption onto carriers such as charcoal, resins, or albumin.
Many of the molecules that accumulate within the blood during ALF are small or middle-sized.216 These molecules can be targeted by a variety of extracorporeal purification techniques, including dialysis through various types of membranes and adsorption onto carriers such as charcoal, resins, or albumin.
Early work with hemodialysis was unsuccessful and was largely abandoned with the conclusion that the toxemia of ALF was not due solely to small water-soluble molecules. The advent of synthetic membranes in the 1970s rekindled the interest in convective therapy for ALF. These allowed larger molecules to pass through compared to the cuprophane alternatives. Opolon reported clinical improvement with the use of high-permeability membrane hemodialysis and hemofiltration in patients with ALF.225 Hemofiltration, as a form of renal support, is used in most liver critical care units as part of general supportive care and there is some evidence that increased convective exchange, with high-volume (>35 mL/kg/hour) hemofiltration, is associated with improved hemodynamic stability and improved encephalopathy scores.145,225
Charcoal hemoperfusion has been extensively investigated. Initial trials were encouraging, but later larger randomized controlled trials were unable to show an improvement in outcome.137 Large volume plasma exchange showed some improvement in hemodynamics and other parameters in initial studies.226 The therapy is logistically difficult to perform. However, in 2010 a large randomized controlled clinical trial of high-volume plasma exchange (10 L/day for 3 days) was presented and published in abstract form, showing improved survival, and it may become a standard form of therapy.227
In an attempt to improve the efficacy of dialysis techniques, adsorbents have been added to the dialysis fluid to widen the range of molecules removed. There are a number of approaches to doing this. One is dialysis of blood or plasma against albumin solution. This is done via a recycled circulation in the MARS machine or via single pass albumin dialysis. Another is the use of plasma separation, adsorption, and filtration and then recombining the “cleaned” plasma with the patient’s blood.223
Nonbiologic systems have been shown to improve blood pressure, increase vascular resistance, and improve short-term encephalopathy scores in patients with AoCLF. Three large studies have been reported, two in the management of AoCLF (the RELIEF study and the HELIOS study) and the FULMAR study in patients with ALF. Unfortunately, none of the studies has shown an improvement in outcome.223
Liver Transplantation
When and Whom to Transplant
Timing is important, and in those with severe liver injury there is a window of opportunity beyond which transplantation often becomes futile because of deteriorating organ function.228 It was recognized early in the history of transplantation for ALF that the challenge was to develop robust prognostic indicators. These have to be sensitive, early enough to provide maximum advantage to the patient, and specific enough not to result in unnecessary transplants.
A “super-urgent” designation exists in the national transplant sharing scheme in the United Kingdom with a similar “category 1A” designation in the United States (see http://www.unos.org/ for details). These categories recognize the role of early transplantation in ALF and the detrimental effect of delay in this setting.
In the United Kingdom, the super-urgent designation (Box 75.5) is closely linked to the prognostic score developed in King’s College Hospital in the late 1980s using retrospective multivariate analysis with prospective validation.229 The prognostic criteria have been subsequently validated in other centers and shown to be robust.230 Other criteria have been developed.231
The criteria are not perfect. First, despite their good specificity (i.e., if the patient meets criteria the patient is likely to die), the sensitivity and negative predictive value are not as good, and there is a substantial proportion of patients who will die without ever reaching transplant criteria. In addition, awaiting positive criteria can lead to delay in listing and worsening of organ failure that often then preclude listing. This contributes to the fact that published rates of transplantation in those who reach criteria are only 50% following POD.232 Clinical practice has changed since this designation was first defined. For example, it is rare to see a patient following POD with a pH below 7.3 or a creatinine above 300 mmol/L because of improved resuscitation and early renal support at the referring hospital. Because of these factors, ongoing efforts to establish markers that increase sensitivity and occur even earlier in the course of the syndrome, while maintaining good specificity and not reducing the positive predictive value to unacceptable levels leading to unnecessary transplants, continue.
Serum phosphate levels are higher in nonsurvivors following both POD and in other causes of ALF.120,233 However, there appears to be an unacceptable overlap and others have suggested that the use of serum phosphate does not provide any additional benefit to existing markers.121,234,235 Other factors investigated include α-fetoprotein levels and nuclear magnetic resonance (NMR) analysis of peripheral blood.120,236 Acute physiologic scoring as a basis for prediction has also been utilized.237
The liver plays a central role in lactate metabolism. In fact, in patients with severe liver necrosis the liver changes from being a consumer of lactate to being a net producer.209 Arterial blood lactate levels have been shown to improve the sensitivity and maintain the specificity if added to the original King’s College Hospital criteria and are achieved earlier in the course of the syndrome.149 However, the additional advantage of lactate over the original criteria has been questioned by some.238
On the practical issue of actually managing patients with fulminant hepatic failure some room for clinical interpretation has been included in the super-urgent listing rules. For example, there is a group of patients who do not achieve King’s College Hospital criteria but subsequently die, usually of cerebral edema or multiple organ failure secondary to sepsis.172,175 These patients often have worse acute physiologic scores compared to survivors.232,237 As a result the U.K. super-urgent criteria allow an assessment of deteriorating acute physiology based on cardiovascular, respiratory, or cerebral disease. Similarly, the UNOS 1A criteria allow for patients “not expected to survive a further 7 days.”
Outcome from Transplantation
In Europe the 1-year survival rate following transplantation for ALF is worse than that seen in chronic liver disease. The excess mortality rate is seen in the first month or so after transplantation. This represents the severity of organ dysfunction seen prior to transplantation in ALF. Following this initial period, the curve flattens and the survival rate is actually better than that of patients with chronic liver disease (Fig. 75.15). This probably represents a younger age group and less disease recurrence. There is a huge degree of heterogeneity in ALF, and those transplanted with seronegative hepatitis display a better survival profile than patients transplanted for other causes, although they exhibit a similar early mortality rate while in the ICU.89
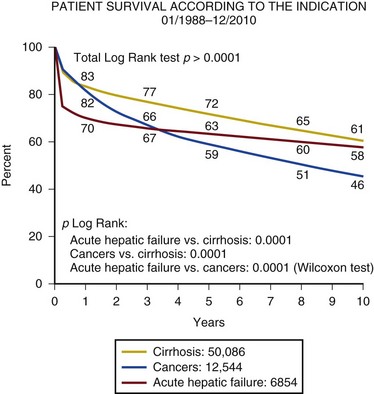
Figure 75.15 Patient survival according to the first indication for liver transplant. (From European Liver and Intestine Transplant Association, accessed at http:www.eltr.org.)
There appear to be both recipient and donor factors that help predict the outcome from transplantation in ALF.89,232,239 In theory, the severity of illness and organ dysfunction prior to transplantation should predict outcome. However, because unstable patients tend to be either not listed or withdrawn from the waiting list, there is inherent bias in retrospective analysis. Age of the recipient is a significant factor, certainly for seronegative ALF, but also in POD, in which age is often used to exclude listing.232 In non-paracetamol ALF, serum creatinine at the time of transplantation is a predictor of 2-month survival. Following POD, time from ingestion to transplantation has been shown to be a good predictor of 2-month survival; all patients transplanted later than 6 days from ingestion died. APACHE III score at transplantation and the severity of metabolic acidosis are also predictive.239
Donor factors found to be important are the use of reduced size grafts in paracetamol-induced ALF and evidence of early graft dysfunction as defined by a high AST or INR in the early postoperative period. In addition a high donor body mass index (BMI) is a risk factor for death in seronegative hepatitis.89,232
In 2012 a review of the European liver transplant database for patients transplanted following ALF was done in which 4903 patients were evaluated. The 1-, 5-, and 10-year patient and graft survival rates were 74%, 68%, and 63%, respectively. Death and graft loss were independently associated with male recipients, recipients older than 50 years, incompatible ABO matching, donors older than 60 years, and reduced size graft. Recipients over 50 receiving a graft >60 years old had a 57% 1-year mortality rate or graft loss.240
Auxiliary Transplantation
It provides the potential to support the patient during the acute phase of liver failure, enabling regeneration of the native liver. This is attractive because in a number of patients immunosuppressive drugs can be withdrawn, allowing the graft to atrophy or be removed, and eliminating the risks associated with lifelong immunosuppression. Data on this procedure have been accumulating since the mid-1990s. Initial reports suggested that the procedure was associated with a high incidence of technical problems, primary dysfunction, and retransplantation. Later reports, however, suggest that many of these issues are resolving with greater experience in patient and graft selection. The best outcome has been seen in patients aged younger than 40 years with either acute viral hepatitis or paracetamol hepatotoxicity in which 1-year graft and patient survival are similar to those for standard transplantation for ALF. Withdrawal of immunosuppression can be achieved in 30% to 70% of patients transplanted.241–243
Living Related Lobe Donation
In many countries the only chance of transplantation for ALF is in a living related donation of a liver lobe. This is most often performed in children when an adult left lobe can be used. In adults, a right lobe is usually required, increasing the risk to the donor. With the worldwide shortage of donor organs, living related transplantation for ALF is widely accepted in many countries but not all. There are significant issues related to living related transplantation in ALF including a donor mortality rate of 1% and major morbidity rate of 40% to 60%. There are also ethical implications of adequately preparing the donor, medically and psychologically, in a time of acute crisis.244
References
1. Sen, S, Williams, R, Jalan, R. The pathophysiological basis of acute-on-chronic liver failure. Liver. 2002; 22(Suppl 2):5–13.
2. Katoonizadeh, A, Laleman, W, Verslype, C, et al. Early features of acute-on-chronic alcoholic liver failure: A prospective cohort study. Gut. 2010; 59(11):1561–1569.
3. Martin-Llahi, M, Guevara, M, Torre, A, et al. Prognostic importance of the cause of renal failure in patients with cirrhosis. Gastroenterology. 2011; 140(2):488–496.
4. Mathurin, P. Is alcoholic hepatitis an indication for transplantation? Current management and outcomes. Liver Transpl. 2005; 11(Suppl 2):S21–S24.
5. Hines, IN, Wheeler, MD. Recent advances in alcoholic liver disease III. Role of the innate immune response in alcoholic hepatitis. Am J Physiol Gastrointest Liver Physiol. 2004; 287(2):G310–G314.
6. Sorbi, D, Boynton, J, Lindor, KD. The ratio of aspartate aminotransferase to alanine aminotransferase: Potential value in differentiating nonalcoholic steatohepatitis from alcoholic liver disease. Am J Gastroenterol. 1999; 94(4):1018–1022.
7. Desmet, VJ, Roskams, T. Cirrhosis reversal: A duel between dogma and myth. J Hepatol. 2004; 40(5):860–867.
8. Dunn, W, Jamil, LH, Brown, LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology. 2005; 41(2):353–358.
9. Warrillow, SJ. Predictions and outcomes for the critically ill patient with cirrhosis: Is it time to settle on the SOFA and let jaundiced views on outcome MELD away? Crit Care Med. 2010; 38(11):2259–2260.
10. Mathurin, P. Corticosteroids for alcoholic hepatitis—What’s next? J Hepatol. 2005; 43(3):526–533.
11. D’Amico, G, Pagliaro, L, Bosch, J. The treatment of portal hypertension: A meta-analytic review. Hepatology. 1995; 22(1):332–354.
12. Zhang, C, Thabut, D, Kamath, PS, Shah, VH. Oesophageal varices in cirrhotic patients: From variceal screening to primary prophylaxis of the first oesophageal variceal bleeding. Liver Int. 2011; 31(1):108–119.
13. D’Amico, G, De Franchis, R. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology. 2003; 38(3):599–612.
14. Garcia-Tsao, G, Bosch, J. Management of varices and variceal hemorrhage in cirrhosis. N Engl J Med. 2010; 362(9):823–832.
15. Ioannou, G, Doust, J, Rockey, DC. Terlipressin for acute esophageal variceal hemorrhage. Cochrane Database Syst Rev. (1):2003.
16. Gotzsche, PC, Hrobjartsson, A. Somatostatin analogues for acute bleeding oesophageal varices. Cochrane Database Syst Rev. (1):2005.
17. Khan, S, Tudur Smith, C, Williamson, P, Sutton, R. Portosystemic shunts versus endoscopic therapy for variceal rebleeding in patients with cirrhosis. Cochrane Database Syst Rev. (4):2006.
18. Garcia-Pagan, JC, Caca, K, Bureau, C, et al. Early use of TIPS in patients with cirrhosis and variceal bleeding. N Engl J Med. 2010; 362(25):2370–2379.
19. Bosch, J, Thabut, D, Albillos, A, et al. Recombinant factor VIIa for variceal bleeding in patients with advanced cirrhosis: A randomized, controlled trial. Hepatology. 2008; 47(5):1604–1614.
20. Bernard, B, Grange, JD, Khac, EN, et al. Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: A meta-analysis. Hepatology. 1999; 29(6):1655–1661.
21. Chavez-Tapia, NC, Barrientos-Gutierrez, T, Tellez-Avila, F, et al. Meta-analysis: Antibiotic prophylaxis for cirrhotic patients with upper gastrointestinal bleeding—An updated Cochrane review. Aliment Pharmacol Ther. 2011; 34(5):509–518.
22. Goulis, J, Patch, D, Burroughs, AK. Bacterial infection in the pathogenesis of variceal bleeding. Lancet. 1999; 353(9147):139–142.
23. Gustot, T, Durand, F, Lebrec, D, et al. Severe sepsis in cirrhosis. Hepatology. 2009; 50(6):2022–2033.
24. Wasmuth, HE, Kunz, D, Yagmur, E, et al. Patients with acute on chronic liver failure display “sepsis-like” immune paralysis. J Hepatol. 2005; 42(2):195–201.
25. European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010; 53(3):397–417.
26. Fernandez, J, Navasa, M, Gomez, J, et al. Bacterial infections in cirrhosis: Epidemiological changes with invasive procedures and norfloxacin prophylaxis. Hepatology. 2002; 35(1):140–148.
27. Pluta, A, Gutkowski, K, Hartleb, M. Coagulopathy in liver diseases. Adv Med Sci. 2010; 55(1):16–21.
28. Dabbagh, O, Oza, A, Prakash, S, et al. Coagulopathy does not protect against venous thromboembolism in hospitalized patients with chronic liver disease. Chest. 2010; 137(5):1145–1149.
29. Ruiz-del-Arbol, L, Monescillo, A, Arocena, C, et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005; 42(2):439–447.
30. Olson, JC, Kamath, PS. Acute-on-chronic liver failure: Concept, natural history, and prognosis. Curr Opin Crit Care. 2011; 17(2):165–169.
31. Kawut, SM, Krowka, MJ, Trotter, JF, et al. Clinical risk factors for portopulmonary hypertension. Hepatology. 2008; 48(1):196–203.
32. Cholongitas, E, Senzolo, M, Patch, D, et al. Cirrhotics admitted to intensive care unit: The impact of acute renal failure on mortality. Eur J Gastroenterol Hepatol. 2009; 21(7):744–750.
33. Salerno, F, Gerbes, A, Gines, P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007; 56(9):1310–1318.
34. Boyer, TD, Sanyal, AJ, Garcia-Tsao, G, et al. Predictors of response to terlipressin plus albumin in hepatorenal syndrome (HRS) type 1: Relationship of serum creatinine to hemodynamics. J Hepatol. 2011; 55(2):315–321.
35. Moreau, R, Lebrec, D. The use of vasoconstrictors in patients with cirrhosis: Type 1 HRS and beyond. Hepatology. 2006; 43(3):385–394.
36. Ortega, R, Gines, P, Uriz, J, et al. Terlipressin therapy with and without albumin for patients with hepatorenal syndrome: Results of a prospective, nonrandomized study. Hepatology. 2002; 36(4 Pt 1):941–948.
37. Sanyal, AJ, Boyer, T, Garcia-Tsao, G, et al. A randomized, prospective, double-blind, placebo-controlled trial of terlipressin for type 1 hepatorenal syndrome. Gastroenterology. 2008; 134(5):1360–1368.
38. Mizock, BA. Nutritional support in hepatic encephalopathy. Nutrition. 1999; 15(3):220–228.
39. Gupta, D, Aggarwal, AN, Dhiman, RK, et al. Pulmonary function changes after large volume paracentesis. Trop Gastroenterol. 2000; 21(2):68–70.
40. Byrd, RP, Jr., Roy, TM, Simons, M. Improvement in oxygenation after large volume paracentesis. South Med J. 1996; 89(7):689–692.
41. Duranti, R, Laffi, G, Misuri, G, et al. Respiratory mechanics in patients with tense cirrhotic ascites. Eur Respir J. 1997; 10(7):1622–1630.
42. Gur, C, Ilan, Y, Shibolet, O. Hepatic hydrothorax—Pathophysiology, diagnosis and treatment—Review of the literature. Liver Int. 2004; 24(4):281–284.
43. Marik, PE, Pastores, SM, Annane, D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: Consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008; 36(6):1937–1949.
44. Marik, PE. Adrenal-exhaustion syndrome in patients with liver disease. Intensive Care Med. 2006; 32(2):275–280.
45. Fede, G, Spadaro, L, Tomaselli, T, et al. Adrenocortical dysfunction in liver disease: A systematic review. Hepatology. 2012; 55(4):1282–1291.
46. Fernandez, J, Escorsell, A, Zabalza, M, et al. Adrenal insufficiency in patients with cirrhosis and septic shock: Effect of treatment with hydrocortisone on survival. Hepatology. 2006; 44(5):1288–1295.
47. Wehler, M, Kokoska, J, Reulbach, U, et al. Short-term prognosis in critically ill patients with cirrhosis assessed by prognostic scoring systems. Hepatology. 2001; 34(2):255–261.
48. Aggarwal, A, Ong, JP, Younossi, ZM, et al. Predictors of mortality and resource utilization in cirrhotic patients admitted to the medical ICU. Chest. 2001; 119(5):1489–1497.
49. Cholongitas, E, Senzolo, M, Patch, D, et al. Risk factors, sequential organ failure assessment and model for end-stage liver disease scores for predicting short term mortality in cirrhotic patients admitted to intensive care unit. Aliment Pharmacol Ther. 2006; 23(7):883–893.
50. Moreno, R, Vincent, JL, Matos, R, et al. The use of maximum SOFA score to quantify organ dysfunction/failure in intensive care. Results of a prospective, multicentre study. Working Group on sepsis related problems of the ESICM. Intensive Care Med. 1999; 25(7):686–696.
51. Rabe, C, Schmitz, V, Paashaus, M, et al. Does intubation really equal death in cirrhotic patients? Factors influencing outcome in patients with liver cirrhosis requiring mechanical ventilation. Intensive Care Med. 2004; 30(8):1564–1571.
52. O’Grady, JG, Schalm, SW, Williams, R. Acute liver failure: Redefining the syndromes. Lancet. 1993; 342(8866):273–275.
53. Lucke, B, Mallory, T. The fulminant form of epidemic hepatitis. Am J Pathol. 1946; 22(5):867–945.
54. Trey, C, Davidson, CS. The management of fulminant hepatic failure. Prog Liver Dis. 1970; 3:282–298.
55. Bower, WA, Johns, M, Margolis, HS, et al. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007; 102(11):2459–2463.
56. Dhiman, RK, Seth, AK, Jain, S, et al. Prognostic evaluation of early indicators in fulminant hepatic failure by multivariate analysis. Dig Dis Sci. 1998; 43(6):1311–1316.
57. Bernal, W. Changing patterns of causation and the use of transplantation in the United Kingdom. Semin Liver Dis. 2003; 23(3):227–237.
58. Lee, WM. Acute liver failure in the United States. Semin Liver Dis. 2003; 23(3):217–226.
59. Ostapowicz, G, Fontana, RJ, Schiodt, FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002; 137(12):947–954.
60. Hawton, K, Simkin, S, Deeks, J, et al. UK legislation on analgesic packs: Before and after study of long term effect on poisonings. BMJ. 2004; 329(7474):1076.
61. Sandilands, EA, Bateman, DN. Co-proxamol withdrawal has reduced suicide from drugs in Scotland. Br J Clin Pharmacol. 2008; 66(2):290–293.
62. Jones, A. Over-the-counter analgesics: A toxicology perspective. Am J Ther. 2002; 9(3):245–257.
63. James, LP, Mayeux, PR, Hinson, JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003; 31(12):1499–1506.
64. Bernal, W, Donaldson, P, Underhill, J, et al. Tumor necrosis factor genomic polymorphism and outcome of acetaminophen (paracetamol)-induced acute liver failure. J Hepatol. 1998; 29(1):53–59.
65. Bernal, W, Langley, P, Wendon, J. Circulating markers of endothelial activation in acetaminophen induced acute liver failure. Crit Care. 1998; 2(Suppl 1):P009.
66. Markin, A, Williams, R. Acetaminophen-induced acute liver liver failure. In: Lee W, Williams R, eds. Acute Liver Failure. Cambridge, England: Cambridge University Press; 1997:32–42.
67. Prescott, LF, Illingworth, RN, Critchley, JA, et al. Intravenous N-acetylcystine: The treatment of choice for paracetamol poisoning. Br Med J. 1979; 2(6198):1097–1100.
68. Lee, WM. Drug-induced hepatotoxicity. N Engl J Med. 2003; 349(5):474–485.
69. Keays, R, Harrison, PM, Wendon, JA, et al. Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: A prospective controlled trial. BMJ. 1991; 303(6809):1026–1029.
70. Lee, WM, Hynan, LS, Rossaro, L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009; 137(3):856–864.
71. Sklar, GE, Subramaniam, M. Acetylcysteine treatment for non-acetaminophen-induced acute liver failure. Ann Pharmacother. 2004; 38(3):498–500.
72. O’Grady, JG. Acute liver failure. Postgrad Med J. 2005; 81(953):148–154.
73. Schiodt, FV, Davern, TJ, Shakil, AO, et al. Viral hepatitis-related acute liver failure. Am J Gastroenterol. 2003; 98(2):448–453.
74. Chadha, MS, Walimbe, AM, Chobe, LP, Arankalle, VA. Comparison of etiology of sporadic acute and fulminant viral hepatitis in hospitalized patients in Pune, India during 1978-81 and 1994-97. Indian J Gastroenterol. 2003; 22(1):11–15.
75. Schiff, ER. Atypical clinical manifestations of hepatitis A. Vaccine. 1992; 10(Suppl 1):S18–S20.
76. Fagan, E, Yousef, G, Brahm, J, et al. Persistence of hepatitis A virus in fulminant hepatitis and after liver transplantation. J Med Virol. 1990; 30(2):131–136.
77. Liaw, YF, Yang, CY, Chu, CM, Huang, MJ. Appearance and persistence of hepatitis A IgM antibody in acute clinical hepatitis A observed in an outbreak. Infection. 1986; 14(4):156–158.
78. Sato, S, Suzuki, K, Akahane, Y, et al. Hepatitis B virus strains with mutations in the core promoter in patients with fulminant hepatitis. Ann Intern Med. 1995; 122(4):241–248.
79. Garfein, RS, Bower, WA, Loney, CM, et al. Factors associated with fulminant liver failure during an outbreak among injection drug users with acute hepatitis B. Hepatology. 2004; 40(4):865–873.
80. Tillmann, HL, Zachou, K, Dalekos, GN. Management of severe acute to fulminant hepatitis B: To treat or not to treat or when to treat? Liver Int. 2012; 32(4):544–553.
81. Farci, P, Alter, HJ, Shimoda, A, et al. Hepatitis C virus-associated fulminant hepatic failure. N Engl J Med. 1996; 335(9):631–634.
82. Vogt, M, Lang, T, Frosner, G, et al. Prevalence and clinical outcome of hepatitis C infection in children who underwent cardiac surgery before the implementation of blood-donor screening. N Engl J Med. 1999; 341(12):866–870.
83. Acharya, SK, Panda, SK, Saxena, A, Gupta, SD. Acute hepatic failure in India: A perspective from the East. J Gastroenterol Hepatol. 2000; 15(5):473–479.
84. Cheng, VC, Lo, CM, Lau, GK. Current issues and treatment of fulminant hepatic failure including transplantation in Hong Kong and the Far East. Semin Liver Dis. 2003; 23(3):239–250.
85. Emerson, SU, Purcell, RH. Running like water—The omnipresence of hepatitis E. N Engl J Med. 2004; 351(23):2367–2368.
86. Bhatia, V, Singhal, A, Panda, SK, Acharya, SK. A 20-year single-center experience with acute liver failure during pregnancy: Is the prognosis really worse? Hepatology. 2008; 48(5):1577–1585.
87. Wedemeyer, H, Pischke, S, Manns, MP. Pathogenesis and treatment of hepatitis E virus infection. Gastroenterology. 2012; 142(6):1388–1397.
88. Gow, P, Hathaway, M, Gunson, B, et al. Association of fulminant non-A non-B hepatitis with homozygosity for HLA A1-B8-DR3. J Gastroenterol Hepatol. 2005; 20(4):555–561.
89. Wigg, AJ, Gunson, BK, Mutimer, DJ. Outcomes following liver transplantation for seronegative acute liver failure: Experience during a 12-year period with more than 100 patients. Liver Transpl. 2005; 11(1):27–34.
90. Manns, MP, Vogel, A. Autoimmune hepatitis, from mechanisms to therapy. Hepatology. 2006; 43(2 Suppl 1):S132–S144.
91. Vergani, D, Alvarez, F, Bianchi, FB, et al. Liver autoimmune serology: A consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J Hepatol. 2004; 41(4):677–683.
92. Bissell, DM, Gores, GJ, Laskin, DL, Hoofnagle, JH. Drug-induced liver injury: Mechanisms and test systems. Hepatology. 2001; 33(4):1009–1013.
93. Devlin, J, O’Grady, J. Indications for referral and assessment in adult liver transplantation: A clinical guideline. British Society of Gastroenterology. Gut. 1999; 45(Suppl 6):VI1–VI22.
94. Hay, JE. Liver disease in pregnancy. Hepatology. 2008; 47(3):1067–1076.
95. Ibdah, JA, Bennett, MJ, Rinaldo, P, et al. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med. 1999; 340(22):1723–1731.
96. Guntupalli, SR, Steingrub, J. Hepatic disease and pregnancy: An overview of diagnosis and management. Crit Care Med. 2005; 33(10 Suppl):S332–S339.
97. Hamid, SS, Jafri, SM, Khan, H, et al. Fulminant hepatic failure in pregnant women: Acute fatty liver or acute viral hepatitis? J Hepatol. 1996; 25(1):20–27.
98. Westbrook, RH, Yeoman, AD, Joshi, D, et al. Outcomes of severe pregnancy-related liver disease: Refining the role of transplantation. Am J Transplant. 2010; 10(11):2520–2526.
99. Castro, MA, Fassett, MJ, Reynolds, TB, et al. Reversible peripartum liver failure: A new perspective on the diagnosis, treatment, and cause of acute fatty liver of pregnancy, based on 28 consecutive cases. Am J Obstet Gynecol. 1999; 181(2):389–395.
100. Brewer, GJ, Askari, FK. Wilson’s disease: Clinical management and therapy. J Hepatol. 2005; 42(Suppl 1):S13–S21.
101. Durand, F, Bernuau, J, Giostra, E, et al. Wilson’s disease with severe hepatic insufficiency: Beneficial effects of early administration of D-penicillamine. Gut. 2001; 48(6):849–852.
102. Henrion, J. Hypoxic hepatitis. Liver Int. 2012; 32(7):1039–1052.
103. Darwish Murad, S, Valla, DC, de Groen, PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology. 2004; 39(2):500–508.
104. Menon, KV, Shah, V, Kamath, PS. The Budd-Chiari syndrome. N Engl J Med. 2004; 350(6):578–585.
105. Slakey, DP, Klein, AS, Venbrux, AC, Cameron, JL. Budd-Chiari syndrome: Current management options. Ann Surg. 2001; 233(4):522–527.
106. Mancuso, A, Fung, K, Mela, M, et al. TIPS for acute and chronic Budd-Chiari syndrome: A single-centre experience. J Hepatol. 2003; 38(6):751–754.
107. Mentha, G, Giostra, E, Majno, PE, et al. Liver transplantation for Budd-Chiari syndrome: A European study on 248 patients from 51 centres. J Hepatol. 2006; 44(3):520–528.
108. Plessier, A, Rautou, PE, Valla, DC. Management of hepatic vascular diseases. J Hepatol. 2012; 56(Suppl 1):S25–S38.
109. McDonald, GB, Hinds, MS, Fisher, LD, et al. Veno-occlusive disease of the liver and multiorgan failure after bone marrow transplantation: A cohort study of 355 patients. Ann Intern Med. 1993; 118(4):255–267.
110. MacQuillan, GC, Mutimer, D. Fulminant liver failure due to severe veno-occlusive disease after haematopoietic cell transplantation: A depressing experience. Q J Med. 2004; 97(9):581–589.
111. Baron, F, Deprez, M, Beguin, Y. The veno-occlusive disease of the liver. Haematologica. 1997; 82(6):718–725.
112. Naschitz, JE, Slobodin, G, Lewis, RJ, et al. Heart diseases affecting the liver and liver diseases affecting the heart. Am Heart J. 2000; 140(1):111–120.
113. Hadad, E, Ben-Ari, Z, Heled, Y, et al. Liver transplantation in exertional heat stroke: A medical dilemma. Intensive Care Med. 2004; 30(7):1474–1478.
114. Diaz, JH. Syndromic diagnosis and management of confirmed mushroom poisonings. Crit Care Med. 2005; 33(2):427–436.
115. Larson, AM, Polson, J, Fontana, RJ, et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology. 2005; 42(6):1364–1372.
116. Tofteng, F, Jorgensen, L, Hansen, BA, et al. Cerebral microdialysis in patients with fulminant hepatic failure. Hepatology. 2002; 36(6):1333–1340.
117. Atterbury, CE, Maddrey, WC, Conn, HO. Neomycin-sorbitol and lactulose in the treatment of acute portal-systemic encephalopathy. A controlled, double-blind clinical trial. Am J Dig Dis. 1978; 23(5):398–406.
118. Vaquero, J, Chung, C, Cahill, ME, Blei, AT. Pathogenesis of hepatic encephalopathy in acute liver failure. Semin Liver Dis. 2003; 23(3):259–269.
119. Murphy, N, Auzinger, G, Bernel, W, Wendon, J. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology. 2004; 39(2):464–470.
120. Schmidt, LE, Dalhoff, K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology. 2002; 36(3):659–665.
121. Macquillan, GC, Seyam, MS, Nightingale, P, et al. Blood lactate but not serum phosphate levels can predict patient outcome in fulminant hepatic failure. Liver Transpl. 2005; 11(9):1073–1079.
122. Yamaguchi, M, Gabazza, EC, Taguchi, O, et al. Decreased protein C activation in patients with fulminant hepatic failure. Scand J Gastroenterol. 2006; 41(3):331–337.
123. Polson, J, Lee, WM. AASLD position paper: The management of acute liver failure. Hepatology. 2005; 41(5):1179–1197.
124. Craig, DG, Bates, CM, Davidson, JS, et al. Staggered overdose pattern and delay to hospital presentation are associated with adverse outcomes following paracetamol-induced hepatotoxicity. Br J Clin Pharmacol. 2012; 73(2):285–294.
125. Wendon, JA, Harrison, PM, Keays, R, Williams, R. Cerebral blood flow and metabolism in fulminant liver failure. Hepatology. 1994; 19(6):1407–1413.
126. Strauss, GI, Hogh, P, Moller, K, et al. Regional cerebral blood flow during mechanical hyperventilation in patients with fulminant hepatic failure. Hepatology. 1999; 30(6):1368–1373.
127. Ellis, A, Wendon, J. Circulatory, respiratory, cerebral, and renal derangements in acute liver failure: Pathophysiology and management. Semin Liver Dis. 1996; 16(4):379–388.
128. McCormick, PA, Treanor, D, McCormack, G, Farrell, M. Early death from paracetamol (acetaminophen) induced fulminant hepatic failure without cerebral oedema. J Hepatol. 2003; 39(4):547–551.
129. Landry, DW, Levin, HR, Gallant, EM, et al. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation. 1997; 95(5):1122–1125.
130. Dunser, MW, Mayr, AJ, Ulmer, H, et al. Arginine vasopressin in advanced vasodilatory shock: A prospective, randomized, controlled study. Circulation. 2003; 107(18):2313–2319.
131. Shawcross, DL, Davies, NA, Mookerjee, RP, et al. Worsening of cerebral hyperemia by the administration of terlipressin in acute liver failure with severe encephalopathy. Hepatology. 2004; 39(2):471–475.
132. Eefsen, M, Dethloff, T, Frederiksen, HJ, et al. Comparison of terlipressin and noradrenalin on cerebral perfusion, intracranial pressure and cerebral extracellular concentrations of lactate and pyruvate in patients with acute liver failure in need of inotropic support. J Hepatol. 2007; 47(3):381–386.
133. Tsai, MH, Chen, YC, Lien, JM, et al. Hemodynamics and metabolic studies on septic shock in patients with acute liver failure. J Crit Care. 2008; 23(4):468–472.
134. Harry, R, Auzinger, G, Wendon, J. The clinical importance of adrenal insufficiency in acute hepatic dysfunction. Hepatology. 2002; 36(2):395–402.
135. Sprung, CL, Annane, D, Keh, D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008; 358(2):111–124.
136. Harry, R, Auzinger, G, Wendon, J. The effects of supraphysiological doses of corticosteroids in hypotensive liver failure. Liver Int. 2003; 23(2):71–77.
137. O’Grady, JG, Gimson, AE, O’Brien, CJ, et al. Controlled trials of charcoal hemoperfusion and prognostic factors in fulminant hepatic failure. Gastroenterology. 1988; 94(5 Pt 1):1186–1192.
138. Trumper, L, Coux, G, Monasterolo, LA, et al. Effect of acetaminophen on the membrane anchoring of Na+, K+ATPase of rat renal cortical cells. Biochem Biophys Acta. 2005; 1740(3):332–339.
139. Cobden, I, Record, CO, Ward, MK, Kerr, DN. Paracetamol-induced acute renal failure in the absence of fulminant liver damage. Br Med J. 1982; 284(6308):21–22.
140. Brochard, L, Abroug, F, Brenner, M, et al. An Official ATS/ERS/ESICM/SCCM/SRLF Statement: Prevention and management of acute renal failure in the ICU patient: An international consensus conference in intensive care medicine. Am J Respir Crit Care Med. 2010; 181(10):1128–1155.
141. Bal, C, Longkumer, T, Patel, C, et al. Renal function and structure in subacute hepatic failure. J Gastroenterol Hepatol. 2000; 15(11):1318–1324.
142. Lee, WM, Stravitz, RT, Larson, AM. Introduction to the revised American Association for the Study of Liver Diseases Position Paper on acute liver failure 2011. Hepatology. 2012; 55(3):965–967.
143. Davenport, A. The management of renal failure in patients at risk of cerebral edema/hypoxia. New Horiz. 1995; 3(4):717–724.
144. Rimmele, T, Kellum, JA. High-volume hemofiltration in the intensive care unit: A blood purification therapy. Anesthesiology. 2012; 116(6):1377–1387.
145. Bernal, W, Wong, T, Wendon, J. High-volume continuous veno-venous haemofiltration in hyper-acute liver failure: A pilot study. Crit Care. 1999; 3(Suppl 1):106.
146. Prowle, JR, Schneider, A, Bellomo, R. Clinical review: Optimal dose of continuous renal replacement therapy in acute kidney injury. Crit Care. 2011; 38(5):207.
147. Van Wert, R, Friedrich, JO, Scales, DC, et al. High-dose renal replacement therapy for acute kidney injury: Systematic review and meta-analysis. Crit Care Med. 2010; 38(5):1360–1369.
148. Davenport, A, Will, EJ, Davison, AM. Hyperlactataemia and metabolic acidosis during haemofiltration using lactate-buffered fluids. Nephron. 1991; 59(3):461–465.
149. Bernal, W, Donaldson, N, Wyncoll, D, Wendon, J. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: A cohort study. Lancet. 2002; 359(9306):558–563.
150. Davenport, A. Dialysate and substitution fluids for patients treated by continuous forms of renal replacement therapy. Contrib Nephrol. 2001; 132:313–322.
151. Ware, AJ, D’Agostino, AN, Combes, B. Cerebral edema: A major complication of massive hepatic necrosis. Gastroenterology. 1971; 61(6):877–884.
152. Hanid, MA, Mackenzie, RL, Jenner, RE, et al. Intracranial pressure in pigs with surgically induced acute liver failure. Gastroenterology. 1979; 76(1):123–131.
153. Bernuau, J, Durand, F. Intracranial pressure monitoring in patients with acute liver failure: A questionable invasive surveillance. Hepatology. 2006; 44(2):502–504.
154. Wendon, JA, Larsen, FS. Intracranial pressure monitoring in acute liver failure. A procedure with clear indications. Hepatology. 2006; 44(2):504–506.
155. Wijdicks, EF, Plevak, DJ, Rakela, J, Wiesner, RH. Clinical and radiologic features of cerebral edema in fulminant hepatic failure. Mayo Clin Proc. 1995; 70(2):119–124.
156. Blei, AT. The pathophysiology of brain edema in acute liver failure. Neurochem Int. 2005; 47(1-2):71–77.
157. Norenberg, MD, Rao, KV, Jayakumar, AR. Mechanisms of ammonia-induced astrocyte swelling. Metab Brain Dis. 2005; 20(4):303–318.
158. Clemmesen, JO, Larsen, FS, Kondrup, J, et al. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999; 29(3):648–653.
159. Bhatia, V, Singh, R, Acharya, SK. Predictive value of arterial ammonia for complications and outcome in acute liver failure. Gut. 2006; 55(1):98–104.
160. Traber, PG, Dal Canto, M, Ganger, DR, Blei, AT. Electron microscopic evaluation of brain edema in rabbits with galactosamine-induced fulminant hepatic failure: Ultrastructure and integrity of the blood-brain barrier. Hepatology. 1987; 7(6):1272–1277.
161. Kato, M, Hughes, RD, Keays, RT, Williams, R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology. 1992; 15(6):1060–1066.
162. Takahashi, H, Koehler, RC, Brusilow, SW, Traystman, RJ. Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol. 1991; 261(3 Pt 2):H825–H829.
163. Cordoba, J, Gottstein, J, Blei, AT. Glutamine, myo-inositol, and organic brain osmolytes after portocaval anastomosis in the rat: Implications for ammonia-induced brain edema. Hepatology. 1996; 24(4):919–923.
164. Blei, AT, Larsen, FS. Pathophysiology of cerebral edema in fulminant hepatic failure. J Hepatol. 1999; 31(4):771–776.
165. Larsen, FS, Pott, F, Ejlersen, E, et al. Dissociated cerebral vasoparalysis in acute liver failure. A hypothesis of gradual cerebral hyperaemia. J Hepatol. 1996; 25(2):145–151.
166. Aggarwal, S, Obrist, W, Yonas, H, et al. Cerebral hemodynamic and metabolic profiles in fulminant hepatic failure: Relationship to outcome. Liver Transpl. 2005; 11(11):1353–1360.
167. Nguyen, JH. Blood-brain barrier in acute liver failure. Neurochem Int. 2012; 60(7):676–683.
168. Strauss, G, Hansen, BA, Kirkegaard, P, et al. Liver function, cerebral blood flow autoregulation, and hepatic encephalopathy in fulminant hepatic failure. Hepatology. 1997; 25(4):837–839.
169. Tofteng, F, Larsen, FS. The effect of indomethacin on intracranial pressure, cerebral perfusion and extracellular lactate and glutamate concentrations in patients with fulminant hepatic failure. J Cereb Blood Flow Metab. 2004; 24(7):798–804.
170. Toftengi, F, Larsen, FS. Management of patients with fulminant hepatic failure and brain edema. Metab Brain Dis. 2004; 19(3-4):207–214.
171. Butterworth, RF. Neuroinflammation in acute liver failure: Mechanisms and novel therapeutic targets. Neurochem Int. 2011; 59(6):830–836.
172. Boeckx, NK, Haydon, G, Rusli, F, Murphy, N. Multiorgan failure is the commonest cause of death in fulminant hepatic failure: A single centre experience. Liver Int. 2004; 24(6):702–703.
173. Bernal, W, Hall, C, Karvellas, CJ, et al. Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007; 46(6):1844–1852.
174. Jalan, R. Intracranial hypertension in acute liver failure: Pathophysiological basis of rational management. Semin Liver Dis. 2003; 23(3):271–282.
175. Makin, AJ, Wendon, J, Williams, R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology. 1995; 109(6):1907–1916.
176. Tofteng, F, Hauerberg, J, Hansen, BA, et al. Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure. J Cereb Blood Flow Metab. 2006; 26(1):21–27.
177. Tandon, BN, Joshi, YK, Tandon, M. Acute liver failure. Experience with 145 patients. J Clin Gastroenterol. 1986; 8(6):664–668.
178. Murphy, N, Wendon, J. Serum sodium is inversely proportional to intracranial pressure in acute liver failure. Crit Care. 1999; 3(Suppl 1):P220.
179. Munoz, SJ, Robinson, M, Northrup, B, et al. Elevated intracranial pressure and computed tomography of the brain in fulminant hepatocellular failure. Hepatology. 1991; 13(2):209–212.
180. Ranjan, P, Mishra, AM, Kale, R, et al. Cytotoxic edema is responsible for raised intracranial pressure in fulminant hepatic failure: In vivo demonstration using diffusion-weighted MRI in human subjects. Metab Brain Dis. 2005; 20(3):181–192.
181. Chavarria, L, Alonso, J, Rovira, A, Cordoba, J. Neuroimaging in acute liver failure. Neurochem Int. 2011; 59(8):1175–1180.
182. Nortje, J, Gupta, AK. The role of tissue oxygen monitoring in patients with acute brain injury. Br J Anaesth. 2006; 97(1):95–106.
183. Ellis, AJ, Wendon, JA, Williams, R. Subclinical seizure activity and prophylactic phenytoin infusion in acute liver failure: A controlled clinical trial. Hepatology. 2000; 32(3):536–541.
184. Strauss, GI, Moller, K, Holm, S, et al. Transcranial Doppler sonography and internal jugular bulb saturation during hyperventilation in patients with fulminant hepatic failure. Liver Transpl. 2001; 7(4):352–358.
185. Aggarwal, S, Brooks, DM, Kang, Y, et al. Noninvasive monitoring of cerebral perfusion pressure in patients with acute liver failure using transcranial Doppler ultrasonography. Liver Transpl. 2008; 14(7):1048–1057.
186. Blei, AT, Olafsson, S, Webster, S, Levy, R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet. 1993; 341(8838):157–158.
187. The Brain Trauma Foundation. The American Association of Neurological Surgeons. The Joint Section on Neurotrauma and Critical Care. Indications for intracranial pressure monitoring. J Neurotrauma. 2000; 17(6-7):479–491.
188. Vaquero, J, Fontana, RJ, Larson, AM, et al. Complications and use of intracranial pressure monitoring in patients with acute liver failure and severe encephalopathy. Liver Transpl. 2005; 11(12):1581–1589.
189. Shami, VM, Caldwell, SH, Hespenheide, EE, et al. Recombinant activated factor VII for coagulopathy in fulminant hepatic failure compared with conventional therapy. Liver Transpl. 2003; 9(2):138–143.
190. Keays, RT, Alexander, GJ, Williams, R. The safety and value of extradural intracranial pressure monitors in fulminant hepatic failure. J Hepatol. 1993; 18(2):205–209.
191. Larsen, FS, Murphy, N, Bernal, W, et al. The prophylactic effect of mild hypothermia to prevent brain edema in patients with acute liver failure: Results of a multicenter randomized, controlled trial. J Hepatol. 2011; 54(Suppl 1):S26.
192. Jalan, R, Olde Damink, SW, Deutz, NE, et al. Restoration of cerebral blood flow autoregulation and reactivity to carbon dioxide in acute liver failure by moderate hypothermia. Hepatology. 2001; 34(1):50–54.
193. Davies, MH, Mutimer, D, Lowes, J, et al. Recovery despite impaired cerebral perfusion in fulminant hepatic failure. Lancet. 1994; 343(8909):1329–1330.
194. Wijdicks, EF, Nyberg, SL. Propofol to control intracranial pressure in fulminant hepatic failure. Transpl Proc. 2002; 34(4):1220–1222.
195. Strauss, GI, Moller, K, Larsen, FS, et al. Cerebral glucose and oxygen metabolism in patients with fulminant hepatic failure. Liver Transpl. 2003; 9(12):1244–1252.
196. Jalan, R, Olde Damink, SW, Deutz, NE, et al. Moderate hypothermia in patients with acute liver failure and uncontrolled intracranial hypertension. Gastroenterology. 2004; 127(5):1338–1346.
197. Canalese, J, Gimson, AE, Davis, C, et al. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut. 1982; 23(7):625–629.
198. Ede, RJ, Gimson, AE, Bihari, D, Williams, R. Controlled hyperventilation in the prevention of cerebral oedema in fulminant hepatic failure. J Hepatol. 1986; 2(1):43–51.
199. Bhatia, V, Batra, Y, Acharya, SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—A controlled clinical trial. J Hepatol. 2004; 41(1):89–96.
200. Rolando, N, Philpott-Howard, J, Williams, R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis. 1996; 16(4):389–402.
201. Rolando, N, Wade, J, Davalos, M, et al. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000; 32(4 Pt 1):734–739.
202. Rolando, N, Gimson, A, Wade, J, et al. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology. 1993; 17(2):196–201.
203. Karvellas, CJ, Pink, F, McPhail, M, et al. Predictors of bacteraemia and mortality in patients with acute liver failure. Intensive Care Med. 2009; 35(8):1390–1396.
204. Wade, J, Rolando, N, Philpott-Howard, J, Wendon, J. Timing and aetiology of bacterial infections in a liver intensive care unit. J Hosp Infect. 2003; 53(2):144–146.
205. Vaquero, J, Polson, J, Chung, C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003; 125(3):755–764.
206. Gramlich, L, Kichian, K, Pinilla, J, et al. Does enteral nutrition compared to parenteral nutrition result in better outcomes in critically ill adult patients? A systematic review of the literature. Nutrition. 2004; 20(10):843–848.
207. Schutz, T, Bechstein, WO, Neuhaus, P, et al. Clinical practice of nutrition in acute liver failure—A European survey. Clin Nutr. 2004; 23(5):975–982.
208. Walsh, TS, Wigmore, SJ, Hopton, P, et al. Energy expenditure in acetaminophen-induced fulminant hepatic failure. Crit Care Med. 2000; 28(3):649–654.
209. Murphy, ND, Kodakat, SK, Wendon, JA, et al. Liver and intestinal lactate metabolism in patients with acute hepatic failure undergoing liver transplantation. Crit Care Med. 2001; 29(11):2111–2118.
210. Walsh, TS, McLellan, S, Mackenzie, SJ, Lee, A. Hyperlactatemia and pulmonary lactate production in patients with fulminant hepatic failure. Chest. 1999; 116(2):471–476.
211. Clark, SJ, Shojaee-Moradie, F, Croos, P, et al. Temporal changes in insulin sensitivity following the development of acute liver failure secondary to acetaminophen. Hepatology. 2001; 34(1):109–115.
212. Strauss, GI, Knudsen, GM, Kondrup, J, et al. Cerebral metabolism of ammonia and amino acids in patients with fulminant hepatic failure. Gastroenterology. 2001; 121(5):1109–1119.
213. Kircheis, G, Wettstein, M, Dahl, S, Haussinger, D. Clinical efficacy of L-ornithine-L-aspartate in the management of hepatic encephalopathy. Metab Brain Dis. 2002; 17(4):453–462.
214. Rose, C, Michalak, A, Rao, KV, et al. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology. 1999; 30(3):636–640.
215. Acharya, SK, Bhatia, V, Sreenivas, V, et al. Efficacy of L-ornithine L-aspartate in acute liver failure: A double-blind, randomized, placebo-controlled study. Gastroenterology. 2009; 136(7):2159–2168.
216. Atillasoy, E, Berk, P. Extracorporeal liver support. In: Lee W, Williams R, eds. Acute Liver Failure. Cambridge, England: Cambridge University Press; 1997:223–244.
217. Shirabe, K, Shimada, M, Gion, T, et al. Postoperative liver failure after major hepatic resection for hepatocellular carcinoma in the modern era with special reference to remnant liver volume. J Am Coll Surg. 1999; 188(3):304–309.
218. Kobayashi, N, Noguchi, H, Totsugawa, T, et al. Insertion of a suicide gene into an immortalized human hepatocyte cell line. Cell Transplant. 2001; 10(4-5):373–376.
219. Selden, C, Hodgson, H. Cellular therapies for liver replacement. Transplant Immunol. 2004; 12(3-4):273–288.
220. Demetriou, AA, Brown, RSJ, Busuttil, RW, et al. Prospective, randomized, multicenter, controlled trial of a bioartificial liver in treating acute liver failure. Ann Surg. 2004; 239(5):660–667.
221. Ellis, AJ, Hughes, RD, Wendon, JA, et al. Pilot-controlled trial of the extracorporeal liver assist device in acute liver failure. Hepatology. 1996; 24(6):1446–1451.
222. Millis, JM, Cronin, DC, Johnson, R, et al. Initial experience with the modified extracorporeal liver-assist device for patients with fulminant hepatic failure: System modifications and clinical impact. Transplantation. 2002; 74(12):1735–1746.
223. Tritto, G, Davies, NA, Jalan, R. Liver replacement therapy. Semin Respir Crit Care Med. 2012; 33(1):70–79.
224. Teperman, LW. A phase 2b study of safety & efficacy of a human cell-based biological liver support system (ELAD®) in subjects with acute-on-chronic hepatitis (AOCH) due either to acute alcoholic hepatitis or acute decompensation of cirrhosis. Liver Transpl. 2012; 18(Suppl 1):S82.
225. Opolon, P. High-permeability membrane hemodialysis and hemofiltration in acute hepatic coma: Experimental and clinical results. Artif Organs. 1979; 3(4):354–360.
226. Larsen, FS, Hansen, BA, Jorgensen, LG, et al. High-volume plasmapheresis and acute liver transplantation in fulminant hepatic failure. Transpl Proc. 1994; 26(3):1788.
227. Larsen, F, Schmid, L, Wendon, J, et al. Liver assisting with high volume plasma exchange in patients with acute liver failure. Hepatology. 2010; 52(4):376A.
228. O’Grady, JG, Wendon, J, Tan, KC, et al. Liver transplantation after paracetamol overdose. BMJ. 1991; 303(6796):221–223.
229. O’Grady, JG, Alexander, GJ, Hayllar, KM, Williams, R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989; 97(2):439–445.
230. Anand, AC, Nightingale, P, Neuberger, JM. Early indicators of prognosis in fulminant hepatic failure: An assessment of the King’s criteria. J Hepatol. 1997; 26(1):62–68.
231. Bernuau, J, Goudeau, A, Poynard, T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology. 1986; 6(4):648–651.
232. Bernal, W, Wendon, J, Rela, M, et al. Use and outcome of liver transplantation in acetaminophen-induced acute liver failure. Hepatology. 1998; 27(4):1050–1055.
233. Baquerizo, A, Anselmo, D, Shackleton, C, et al. Phosphorus as an early predictive factor in patients with acute liver failure. Transplantation. 2003; 75(12):2007–2014.
234. Bernal, W, Wendon, J. More on serum phosphate and prognosis of acute liver failure. Hepatology. 2003; 38(2):533–534.
235. Ng, KL, Davidson, JS, Bathgate, AJ. Serum phosphate is not a reliable early predictor of outcome in paracetamol induced hepatotoxicity. Liver Transpl. 2004; 10(1):158–159.
236. Dabos, KJ, Newsome, PN, Parkinson, JA, et al. Biochemical prognostic markers of outcome in non-paracetamol-induced fulminant hepatic failure. Transplantation. 2004; 77(2):200–205.
237. Mitchell, I, Bihari, D, Chang, R, et al. Earlier identification of patients at risk from acetaminophen-induced acute liver failure. Crit Care Med. 1998; 26(2):279–284.
238. Schmidt, LE, Larsen, FS. Is lactate concentration of major value in determining the prognosis in patients with acute liver failure? Hardly. J Hepatol. 2010; 53(1):211–212.
239. Devlin, J, Wendon, J, Heaton, N, et al. Pretransplantation clinical status and outcome of emergency transplantation for acute liver failure. Hepatology. 1995; 21(4):1018–1024.
240. Germani, G, Theocharidou, E, Adam, R, et al. Liver transplantation for acute liver failure in Europe: Outcomes over 20 years from the ELTR database. J Hepatol. 2012; 57(2):288–296.
241. Chenard-Neu, MP, Boudjema, K, Bernuau, J, et al. Auxiliary liver transplantation: Regeneration of the native liver and outcome in 30 patients with fulminant hepatic failure—A multicenter European study. Hepatology. 1996; 23(5):1119–1127.
242. Girlanda, R, Vilca-Melendez, H, Srinivasan, P, et al. Immunosuppression withdrawal after auxiliary liver transplantation for acute liver failure. Transpl Proc. 2005; 37(4):1720–1721.
243. van Hoek, B, de Boer, J, Boudjema, K, et al. Auxiliary versus orthotopic liver transplantation for acute liver failure. EURALT Study Group. European Auxiliary Liver Transplant Registry. J Hepatol. 1999; 30(4):699–705.
244. Neuberger, J, Price, D. Role of living liver donation in the United Kingdom. BMJ. 2003; 327(7416):676–679.