[level-membership-for-pediatrics-category]
Chapter 78 Diabetic Ketoacidosis
Box 78–1 DKA Treatment in Children and Adolescents
Modified from Glaser NS: Pediatric diabetic ketoacidosis and hyperglycemic hyperosmolar state. Pediatr Clin North Am 52:1611-1635, 2005.
Fluids
Insulin
Potassium and other electrolyte replacement
Monitoring
Etiology, Definition, and Presentation
In a child with new onset of type 1 diabetes, declining insulin production results from autoimmune destruction of pancreatic beta cells. The concentration of insulin is decreased relative to glucagon causing excess hepatic glucose production and decreased peripheral glucose uptake in muscle and adipose tissue.1, 2 When the serum glucose concentration rises above approximately 180 to 200 mg/dL, the renal threshold for glucose reabsorption is exceeded causing glycosuria, which leads to osmotic diuresis and compensatory polydipsia. Low insulin concentrations also stimulate the release of free fatty acids (FFA) from adipose tissue to fuel ketogenesis.3 This, in combination with activation of the hepatic β-oxidative enzyme sequence resulting from relative excess of glucagon in relation to insulin, results in markedly increased hepatic ketone production.2–4
Progressive dehydration and increasing acidosis eventually stimulate additional release of the counterregulatory (“stress”) hormones, cortisol, catecholamines, and growth hormone, which accelerate hepatic glucose output and ketone production.5,6 Infection or other illness or injury can likewise contribute to this process by stimulating release of counterregulatory hormones. Elevated cortisol concentrations augment FFA release from adipose tissue and decrease peripheral glucose uptake. Increased epinephrine concentrations directly increase glycogenolysis and stimulate release of gluconeogenic precursors from muscle.7,8 Both epinephrine and norepinephrine also stimulate lipolysis and β-oxidation of FFAs.9,10 Catecholamines may also directly inhibit insulin secretion, thereby accelerating DKA in those with endogenous insulin capacity, such as a new diagnosis of type 1 diabetes or those with type 2 diabetes.11,12 Growth hormone also decreases peripheral glucose uptake, and enhances ketone production by increasing FFA release.13 Elevated concentrations of counterregulatory hormones thus result in increased acidosis, hyperglycemia, and dehydration. This in turn stimulates further counterregulatory hormone release thereby creating a “vicious cycle” resulting in rapid worsening of DKA (Figure 78-1).
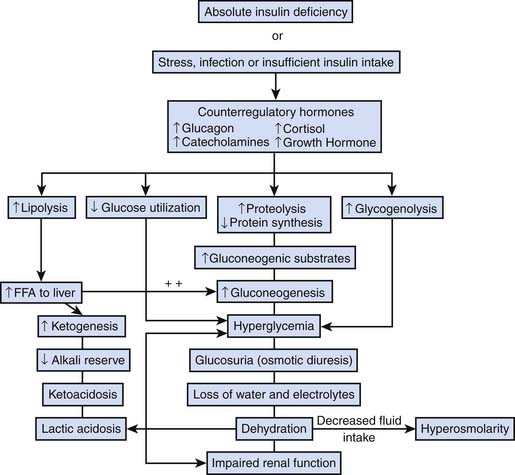
Figure 78–1 Pathophysiology of DKA.
(From Wolfsdorf J, Glaser N, Sperling MA: Diabetic ketoacidosis in infants, children, and adolescents: a consensus statement from the American Diabetes Association, Diabetes Care 29[5]:1150-1159, 2006.)
During DKA, intestinal ileus results from potassium depletion, acidosis, and diminished splanchnic perfusion, causing abdominal pain and vomiting and thereby limiting fluid intake. Progressive dehydration eventually leads to diminished tissue perfusion sufficient to cause accumulation of lactic acid, enhancing acidosis.14 In addition, poor perfusion may result in diminished renal function, limiting the capacity for clearance of glucose and ketones. Ongoing osmotic diuresis and ketonuria in the setting of acidosis also results in urinary losses of electrolytes (potassium, sodium, chloride, calcium, phosphate, and magnesium).
Classical symptoms of DKA include polyuria, polydipsia, polyphagia, weight loss, abdominal pain, nausea, and vomiting. Abdominal tenderness, absence of bowel sounds and guarding are frequent and may even mimic an acute abdomen.15 Tachycardia and signs of hypoperfusion, such as delayed capillary refill time and cool extremities, are also common as well as dry mucous membranes, absence of tears, and poor skin turgor. Despite substantial volume depletion, however, hypotension is unusual in children with DKA and occasional children may present with hypertension. Kussmaul breathing and tachypnea are the result of metabolic acidosis and respiratory compensatory mechanisms. Fruity breath odor (acetone) may be present. Hypothermia has also been described.16
Although hyperglycemia is part of the definition of DKA, in rare cases, the serum glucose concentration may be nearly normal, so called “euglycemic DKA.” This situation has been reported most frequently in pregnant women.17–19 Normal glucose concentrations or even hypoglycemia despite ketosis may also occur in children with known diabetes who administer insulin to treat DKA prior to arrival in the emergency department. In general, however, the persistence and severity of hyperglycemia reflects the severity of dehydration. In the absence of preexisting renal disease or unusually high carbohydrate intake just before presentation, blood glucose concentrations in excess of 500 to 600 mg/dl imply that dehydration is of sufficient severity to diminish the glomerular filtration rate and thereby diminish the capacity for renal clearance of excess glucose.20
Concentrations of ketone bodies (beta-hydroxybutyrate [βOHB] and acetoacetate [AcAc]) are elevated in DKA resulting in acidosis. Hyperchloremic acidosis frequently coexists with increased anion-gap acidosis, and the anion gap reflects the combination of these processes.21 The ratio of βOHB:AcAc (typically 1:1 in the normal state) is increased during DKA and may be as high as 10:1.22 During treatment, this ratio returns to normal. The nitroprusside reaction used to test urine ketone concentrations detects only AcAc and not βOHB. As a result, nitroprusside urine testing cannot be relied on to determine DKA severity or treatment response. Bedside blood ketone meters provide a rapid means for measuring βOHB, and may be useful in place of or in addition to urine testing particularly in patients with anuria or oligoria who produce insufficient amounts of urine for ketone testing.23 Blood ketone measurements are also useful for determining the timing of transition from intravenous to subcutaneous insulin administration. Urine ketones may be present even when blood ketones have normalized as a result of urine stagnating in the bladder.
Hyperglycemia results in fluid shifts from the extravascular to the intravascular space and a decrease in the serum sodium concentration. This decrease can be calculated as an approximately 1.6 mEq/L decrease in sodium concentration for every 100 mg/dl increase in serum glucose higher than 100 mg/dl or Na corrected = Naactual + (glucose in mmol/L – 5.5 mmol/L)}.24,25 Hyperlipidemia may also contribute to a decrease in measured serum sodium concentrations.26 Typically, serum potassium at presentation is in the high-normal range as a result of redistribution of potassium ions from the intracellular to the extracellular space. Several processes are responsible for intracellular potassium depletion including direct effects of low insulin concentrations, intracellular protein and phosphate depletion, and buffering of hydrogen ions in the intracellular compartment.27 Intracellular potassium stores may be profoundly depleted and the serum potassium concentration typically declines rapidly with insulin treatment. Serum phosphate concentrations similarly decrease during treatment.
Leukocytosis is frequent in children with DKA, likely resulting from elevated concentrations of catecholamines and pro-inflammatory cytokines. In children, new onset of type 1 diabetes or insulin omission are far more common causes of DKA than infection. 28 Therefore, an elevated or left-shifted white blood cell count need not prompt a search for an infectious process in the absence of fever or other symptoms or signs of infection. However, in the presence of fever, careful history, physical examination, and laboratory evaluation to assess for infection is prudent.
Epidemiology
Frequency of Diabetic Ketoacidosis at Diagnosis
The frequency of DKA at diagnosis varies widely by geographic region, with an overall estimated frequency of approximately 20% to 67%. In the population-based US study, SEARCH for Diabetes in Youth, data were collected from self-reported health questionnaires and medical record review. In this study, 25.5% of children and adolescents presented at onset of diabetes in DKA.29 A similar frequency was observed in Germany, with 26.3% of children under the age of 15 presenting in DKA at diagnosis of diabetes. In a collaborative study from Germany and Austria, the Diabetes Patienten Verlausdokumentation (an electronic documentation system for children with diabetes), the frequency of DKA at presentation was 21.1%.30 Younger age (<5 years) and female sex were associated with higher likelihood of presenting in DKA.31,32 A delay in diagnosis is associated with a higher likelihood of DKA, and two factors contributing to this are patient age and provider experience. Regions with a higher prevalence of type 1 diabetes generally have a lower frequency of DKA,33 attributed to heightened awareness in providers and thus earlier detection. The non-specific nature of individual symptoms, such as polyuria, tachypnea, and altered mental status, may cause such symptoms to be misconstrued as urinary tract infection, pneumonia, or meningitis, respectively.34 Mallare et al.35 reported a frequency of DKA of 33% in children and adolescents at the initial visit, and almost double (59%) in those in whom the diagnosis of diabetes was missed at the initial visit. The diagnosis of diabetes was more likely to be missed in very young children (34% of children ≤5 years of age compared to 8.5% in those greater than 10 years of age),35 particularly when these very young children are evaluated by family practitioners rather than pediatricians.36
Frequency of Diabetic Ketoacidosis in Children and Adolescents After Diagnosis
Although there are several population-based studies reporting the frequency of DKA at presentation, fewer data are available describing the incidence of DKA in children and adolescents with established diabetes. Reported frequencies range from 1 to 10 per 100 patient years.37 In the Diabetes Control and Complications Trial, the incidence of DKA in adolescents treated with intensive management regimens was 2.8 per 100 patient-years, significantly lower than the incidence in those treated conventionally (4.7 per 100 patient years). Although this is an older study, it was a time and resource-intensive study, and represents a more idealized situation than often encountered in the overall pediatric diabetes population.38 In a more recent study from the Barbara Davis Center for Childhood Diabetes in Denver, Colorado, the overall incidence of DKA was 8 per 100 person-years. In that study, factors associated with higher incidence included older age, higher HbA1C (relative risk [RR] of 1.68 per 1% increase in HbA1C in younger children, RR of 1.43 in older children), higher reported insulin dose, DSM4 psychiatric diagnoses, and “underinsurance” reflecting lower socioeconomic status.39 DKA is also observed more often in children and adolescents on continuous subcutaneous insulin infusion therapy (CSII) than on subcutaneous injections, particularly in the first year of initiation of CSII.40 However, lower rates of DKA are achievable for those on CSII with adequate training and resources.
Over the past two decades T2DM has been occurring with increasing frequency in older children and adolescents. Certain racial/ethnic groups in the United States are disproportionately affected including Native Americans, Hispanics, and African Americans. DKA can be the clinical presentation for T2DM in youth estimated at 5% to 10%.41 Youth with T2DM may also present with hyperglycemic hyperosmolar state (HHS), also referred to hyperosmotic hyperglycemic non-ketotic coma (HHNK, described more fully in the following section).
Morbidity and Mortality Associated with Diabetic Ketoacidosis
Mortality in children presenting with DKA is approximately 0.25% to 0.30%.37 Most of the mortality in DKA occurs in children with cerebral edema, accounting for 57% to 87% of deaths. Neurologic sequelae of DKA are described in the section on DKA-associated complications. Other causes of morbidity and mortality include sepsis and secondary infection, electrolyte abnormalities (e.g., hypokalemia), arrhythmias, rhabdomyolysis, thrombosis, pneumomediastinum, subcutaneous emphysema, and pulmonary edema.
Management Guidelines
Fluids
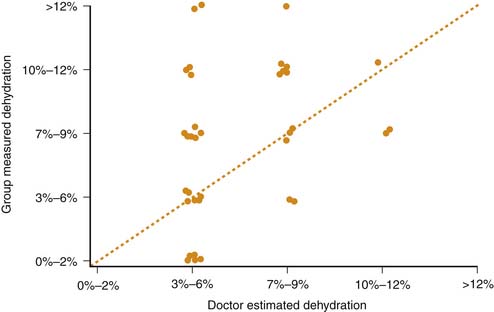
Restoration of adequate peripheral perfusion and hemodynamic stability with bolus administration of intravenous fluids (0.9% saline or other isotonic fluids) should begin as soon as possible. Typical patients require an initial fluid bolus of 10 ml/kg that may be repeated if ongoing hemodynamic instability is present. Studies have shown that clinical assessments of dehydration severity in children with DKA tend to be inaccurate (Figure 78-2).42 The average degree of dehydration for most patients is approximately 7% to 9% of body weight and this figure should be used as a basis for determining the total volume of fluids to be replaced.42,43 The estimated fluid deficit, along with maintenance fluid requirements, should be replaced over 36 to 48 hours using 0.45% to 0.90% saline, generally initially with 0.9% saline, then transitioning to 0.45% saline after several hours assuming serum Na is not falling. Replacement of ongoing urinary fluid losses is usually unnecessary because osmotic diuresis typically resolves rapidly after beginning insulin infusion. However, in circumstances of persistently high urine output, or profuse vomiting or diarrhea, replacement of ongoing losses may be considered.
Insulin
Insulin should be administered intravenously at a rate of 0.1 units/kg/hour. Insulin administration results in resolution of acidosis and hyperglycemia via suppression of ketogenesis, and hepatic glucose output and promotion of peripheral glucose uptake. An initial bolus or loading dose of insulin is not recommended. Maximal suppression of ketogenesis is achieved rapidly with an insulin infusion (0.1 unit/kg/hour).44,45 Even in the absence of insulin administration, the serum glucose concentration usually decreases substantially with initial re-hydration, reflecting improvements in renal perfusion and decreased counter-regulatory hormone concentrations.14 This decline in glucose concentration during the initial period of rehydration should not be interpreted as indicating excessive insulin administration.
Serum glucose concentrations typically normalize before ketosis and acidosis resolve. To continue insulin administration at dosages sufficient to allow resolution of ketosis, dextrose should be added to the intravenous fluids. Transition to dextrose-containing fluids should occur when the serum glucose concentrations decline below approximately 250 mg/dl. The “two-bag system” for dextrose administration allows a rapid response to changes in serum glucose concentration and is cost-effective.46 Two bags of intravenous fluids with identical electrolyte content, but varying dextrose content (0% and 10%) are administered simultaneously with the relative rates of administration frequently adjusted to increase or decrease the dextrose concentration while maintaining a constant overall rate of administration of fluid and electrolytes.
Electrolytes
Phosphate replacement in children with DKA is controversial. Theoretically, low 2,3-diphosphoglycerate levels in red blood cells may occur in association with hypophosphatemia leading to decreased tissue oxygen delivery.47,48 However, clinical relevancy of this supposition has not been demonstrated. Although the risk of hypocalcemia during DKA treatment is increased with phosphate replacement, symptomatic hypocalcemia is very uncommon when phosphate is administered slowly and in the more modest concentrations recommended in most DKA treatment protocols.49,50 Severe hypophosphatemia during DKA has been shown to be associated with rhabdomyolysis and hemolytic anemia, suggesting that monitoring of serum phosphate concentrations is necessary and treatment of severe hypophosphatemia is essential.51,52
Correction of Acidosis
Routine bicarbonate administration is contraindicated in children with DKA as acidosis generally corrects rapidly with insulin and fluid administration, and hemodynamic instability resulting from acidosis is rare. However bicarbonate administration is associated with several possible adverse effects including an increase in the risk of hypokalemia and a theoretical increase in tissue hypoxia resulting from a leftward shift in the hemoglobin-oxygen dissociation curve.47,53 Paradoxical acidosis of the cerebrospinal fluid has also been documented with bicarbonate administration, likely resulting from diminished respiratory drive and a rise in the partial pressure of CO2, which readily crosses the blood-brain barrier augmenting CSF acidosis.54,55 Bicarbonate administration has also been associated with an increased risk of DKA-related cerebral edema.56 In very rare circumstances (severe hemodynamic instability not responding to standard measures or potentially life threatening hyperkalemia), bicarbonate administration may be considered.
Monitoring
Intensive monitoring is essential for children with DKA and most should be treated in a pediatric intensive care unit (PICU) or other unit with similar capacities.57,58 Blood glucose concentrations are typically measured hourly and electrolyte concentrations every 2 to 4 hours. Determinations of serum pH (every 2 to 4 hours) are helpful, particularly because serum bicarbonate concentrations may not increase during the first several hours. Venous blood gas samples generally are sufficient and arterial samples are rarely needed. Failure of acidosis to improve during treatment should prompt evaluation of the adequacy of insulin infusion, fluid balance, presence of non-anion gap hyperchloremic acidosis and a search for other causes such as renal failure, sepsis, or even appendicitis.
All fluid intake and output should be accurately recorded. Consideration of fluids and other management that may have occurred prior to admission to a PICU is important. Vital signs and mental status should be monitored hourly. One study showed a high frequency of prolonged QT interval corrected for heart rate in children with DKA and arrhythmias have been described in rare cases.59 Therefore, cardiac monitoring is recommended.
Diabetic Ketoacidosis–Associated Complications
Cerebral Edema
Cerebral edema (CE) has been recognized as a complication of diabetes mellitus in children since 1936. It is essentially a clinical diagnosis, based on deterioration of mental state during resuscitation for DKA. Signs and symptoms that should prompt consideration of CE include inappropriate slowing of heart rate, hypertension, severe headache, recurrence of vomiting, irritability, lethargy, or other mental status changes.60 Some patients progress to coma, respiratory arrest, and cerebral herniation. Most episodes of CE occur several hours after initiation of DKA treatment; however, 5% to 20% of cases occur at the time of presentation, before the initiation of therapy. Cerebral edema remains the leading cause of death and morbidity in children with type 1 diabetes mellitus. The frequency of CE associated with DKA remains unchanged despite clinical efforts to the contrary.32
Reported mortality from CE varies widely and is in part dependent on the criteria used to define CE. Rates as high as 50% to 90% have been reported, but more recent studies56,61 report lower rates of 21% to 24%. Overall, the incidence of CE is approximately 0.7% to 0.9% within DKA presentations. In other words, approximately 1 in 400 children with DKA die as a result of CE. Morbidity is significant; in particular, debilitating neurologic sequelae occur in 21% to 26% of children with DKA-related CE.56, 61 Although frank CE is uncommon, there are substantial data to suggest that sub-clinical or asymptomatic CE occurs in many children with DKA, perhaps even in the majority. Limited data suggest that subtle brain injury may also be associated with DKA, even in the absence of clinically apparent CE.62
The pathophysiology of CE remains enigmatic. Several causative theories have been proposed for the occurrence of CE during DKA. Idiogenic, osmotically active substances that regulate cell volume have been thought to play a role in causing DKA-related CE. Taurine (2-aminoethane sulfonic acid), in particular, is thought to mediate a critical role in neuroosmoregulation during DKA.63 Alternatively, cerebral hypoperfusion (caused by volume depletion) before DKA treatment and the effects of reperfusion during DKA therapy have been hypothesized to result in CE and cerebral injury.64,65 Direct effects of ketone bodies and inflammatory cytokines on blood-brain barrier function have also been hypothesized to play a role.66,67 To date, however, the precise pathophysiology of CE remains unresolved and multiple factors may be involved.
Epidemiological studies of risk factors for CE show that children with higher initial blood urea nitrogen concentrations, lower initial PCO2 concentrations and greater acidosis at the time of presentation of DKA seem to be at greatest risk for CE.56,65,68,69 A blunted rise in measured serum sodium concentration during DKA treatment has also been associated with increased risk of CE as has treatment with bicarbonate.56 Early administration of insulin (within the first hour) was also associated with increased CE risk in one study.68 Studies evaluating the impact of variations in fluid administration protocols on risk of CE have yielded conflicting results. As yet, there is no clear association between any aspect of fluid treatment and increased risk of CE.
After the diagnosis of CE is made, treatment is a matter of urgency and should not be delayed while awaiting imaging studies or further testing. Intravenous mannitol (0.25-1 g/kg) should be administered immediately. Recent reports suggest the use of 3% saline in boluses or as a continuous infusion for treatment of CE, but data demonstrating beneficial effects are limited to case reports.70,71 Ongoing intensive care unit monitoring is essential. Pulmonary support by means of endotracheal intubation is likely to be required because of severe alterations in mental status, impaired airway reflexes, and altered respiratory drive. Therapeutic hyperventilation in intubated patients, however, has been associated with poorer outcomes.72 Therefore, decreasing PCO2 below the patient’s own compensation for metabolic acidosis should be avoided in children with DKA except where absolutely necessary to treat impending cerebral herniation. A reasonable approach would be to initially maintain the patient’s current PCO2 level and then gradually allow the PCO2 to increase as acidosis corrects. If the patient’s respiratory drive remains intact, utilization of support rather than mandatory ventilation modes, permits the patient to determine rate and depth of breathing. Central nervous system imaging in patients with suspected CE is recommended to exclude other etiologies of altered mental status such as central nervous system thromboses or infarction.
Neuropsychologic Sequelae
Adverse neurodevelopmental outcomes in children with diabetes have in large part been attributed to recurrent hypoglycemia, and this area has been extensively investigated. More recently, hyperglycemia and hyperglycemic extremes such as occur with DKA have been attracting increasing attention and interest for resultant potential neuropsychological sequelae. Short-term effects on neurocognitive performance involving complex skills, such as inhibiting an overlearned response and learning of complex novel information have been described.73 Long-term deficits in memory have been found to be associated with DKA.62 Rapid and significant variability in blood glucose levels may have further impact, particularly on the developing brain and may be more neurotoxic than sustained hyperglycemia.74
Thrombotic Complications
Thrombotic complications are common in children with DKA and central venous catheters are particularly prone to thrombosis.75,76 Cerebral thromboses and pulmonary emboli have also been described. Hyperosmolarity may result in direct osmotic disruption of endothelial cells leading to release of tissue thromboplastins.77 Higher levels of vasopressin stimulated by hypertonicity and decreased vascular volume may also contribute to enhanced coagulation.78 Prophylaxis with low dose heparin should be considered for children with central venous lines.
Other Complications
Rhabdomyolysis79,80 is potentially life threatening. It is characterized by elevated serum creatine kinase, lactate dehydrogenase, and amino alanine transferase concentrations due to muscle injury. Rhabdomyolysis may result in renal failure, compartment syndrome, severe hyperkalemia and other electrolyte disorders leading to arrhythmias. Hyperosmolarity has been thought to be one causative factor and the risk is higher in children who have DKA complicated by features of HHS.79
Acute pancreatitis has been described in case reports of both children and adults with DKA, but occurs rarely. Far more frequent are benign elevations in serum amylase and/or lipase occurring in 24% to 40% of children with DKA. These elevated pancreatic enzyme concentrations typically normalize rapidly with DKA treatment and are not associated with clinical features of pancreatitis.81
Although neurological deterioration in children with DKA is most frequently caused by cerebral edema, cerebral infarctions with and without hemorrhage and cerebral thromboses have also been described.82 Other rare complication of DKA in children include pulmonary edema,83 cardiac arrhythmias,84 renal failure,85 intestinal necrosis,86–88 and rhinocerebral mucormycosis.89,90
Hyperglycemic Hyperosmolar Syndrome
HHS is characterized by extreme elevations in serum glucose (>600 mg/dL) and hyperosmolarity (serum osm >330 mOsm/kg) in the absence of significant ketosis or acidosis (urine ketone concentration <1.5 mmol/L and serum bicarbonate >15 mEq/L). Although HHS is defined as a condition separate from DKA, 30% of cases occur in combination with substantial ketosis and acidosis meeting criteria for both HHS and DKA. Until recently, HHS was thought to occur infrequently in pediatrics. A recent increase in case reports of HHS in children suggest that the frequency may be increasing.91,92 As in adults, HHS in children has a relatively high mortality of 10% to 35%.93,94 The majority of HHS reports in children include acanthosis nigricans, obesity, African-American race, and family history of type 2 diabetes. Most cases of HHS are the initial presentation of diabetes, and most of these youth will subsequently have a clinical diagnosis of type 2 diabetes.
Occurrence of HHS during DKA poses challenges in terms of recognition and treatment. Generally, dehydration is more profound than the clinical assessment would suggest, reflecting difficulties in clinical evaluation due to obesity and relative preservation of intravascular volume because of hyperosmolarity. Electrolyte losses similarly exceed those of DKA as a result of more prolonged osmotic diuresis. Patients who meet criteria for both DKA and HHS require more prolonged and aggressive fluid and electrolyte replacement therapy than typical children with DKA. Replacement of ongoing urinary losses may be necessary. Frequent reassessment of circulatory status and fluid balance is critical. A high frequency of thromboses has been described in children with HHS as well as rhabdomyolysis and a malignant hyperthermia-like syndrome.95,96 Cerebral edema appears to be a rare complication of HHS, with only one case reported.97
Health Care Costs Associated with Diabetic Ketoacidosis
Health care costs for DKA vary by geographic regions in the United States, and comparing costs is often complicated by variations in health care systems, methods of reporting costs (e.g., hospital costs versus payer costs), and contractual arrangements. One strategy to decrease the frequency of DKA is to promote awareness in the general population, in communities, and among providers. A successful campaign to heighten awareness of signs and symptoms of DKA took place in Parma, Italy. In the Parma campaign, simple messages regarding signs and symptoms of diabetes were provided to practitioners and schools, and free access to care arranged. Compared with neighboring regions where the frequency of DKA at the time of diagnosis was quite high at 78%, the Parma region observed a very low frequency of 12.5% during the 8 years of the campaign. Of note, the campaign was relatively inexpensive, the costs $23,000 for the 8 years of the campaign.33
Another more targeted strategy is linked to recognition of diabetes risk. In children enrolled in prevention studies, which are mostly siblings of probands with type 1 diabetes, the frequency of DKA is far less than that of the general population; less than 4% for those participating in the Diabetes and Prevention Trial-1 presented in DKA, and 63.3% were asymptomatic.98 For children and adolescents with diagnosed diabetes, multidisciplinary and intensive team management approaches have been shown to decrease the frequency of DKA. Unfortunately, obtaining sufficient reimbursement for intensive case management in the United States has been challenging, despite demonstrated savings to the health care system. In a relatively small study, the costs for emergency and hospital visits for those not involved in intervention more than exceeded (125%) the costs of intensive case management.99 This included only hospital charges and did not include additional societal costs, such as missed days of work and school, patient and family worry and anxiety, and impact of recurrent DKA, possible cerebral edema, and poor diabetes control on long-term health. In a larger, longer study of multisystemic home-based psychotherapy for adolescents with poorly controlled diabetes, admissions for DKA were reduced by almost half compared with the control group over a 2-year period, resulting in a estimated cost savings of $23,886 to 72,226 (range reflecting hospital costs and third party costs, respectively).100 These examples emphasize the need for preventative rather than crisis-based approaches to the pediatric diabetes population.
References are available online at http://www.expertconsult.com.
1. Gerich J., Lorenzi M., Bier D., et al. Prevention of human diabetic ketoacidosis by somatostatin; evidence for an essential role of glucagon. N Engl J Med. 1975;292:985-989.
2. Schade D., Eaton R. Glucagon regulation of plasma ketone body concentration in human diabetes. J Clin Invest. 1975;56:1340-1344.
3. McGarry J., Foster D. Regulation of ketogenesis and clinical aspects of the ketotic state. Metabolism. 1972;21:471-489.
4. Fukao T., Lopaschuk G., Mitchell G. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry, Prostaglandins. Leukotrienes Essential Fatty Acids. 2004;70:243-251.
5. Burge M., Garcia N., Qualls C.S., DS. Differential effects of fasting and dehydration in the pathogenesis of diabetic ketoacidosis. Metabolism. 2001;50:171-177.
6. Umpierrez G., DiGirolamo M., Tuvlin J. Differences in metabolic and hormonal milieu in diabetic and alcohol-induced ketoacidosis. J Crit Care. 2000;15:52-59.
7. Gustavson S., Chu C., Nishizawa M., et al. Interaction of glucagon and epinephrine in the control of hepatic glucose production in the conscious dog. Am J Physiol Endocrinol Metab. 2002;284:E695-E707.
8. Gustavson S., Chu C., Nishizawa M., et al. Glucagon’s actions are modified by the combination of epinephrine and gluconeogenic precursor infusion. Am J Physiol Endocrinol Metab. 2003;285:E534-E544.
9. Avogaro A., Valerio A., Gnudi L., et al. The effects of different plasma insulin concentrations on lipolytic and ketogenic responses to epinephrine in normal and Type 1 (insulin-dependent) diabetic humans. Diabetologia. 1992;35:129-138.
10. Krentz A., Freedman D., Greene R. Differential effects of physiological versus pathophysiological plasma concentrations of epinephrine and norepinephrine on ketone body metabolism and hepatic portal blood flow in man. Metabolism. 1996;45:1214-1220.
11. Nonogaki K. New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia. 2000;43:533-549.
12. Porte D. Sympathetic regulation of insulin secretion. Arch Intern Med. 1969;123:252-260.
13. Butler P., Kryshak E., Rizza R. Mechanism of growth hormone-induced postprandial carbohydrate intolerance in humans. Am J Physiol. 1991;260:E513-E520.
14. Waldhausl W., Kleinberger G., Korn A. Severe hyperglycemia: effects of rehydration on endocrine derangements and blood glucose concentration. Diabetes. 1979;28:577-584.
15. Valerio D. Acute diabetic abdomen in children. Lancet. 1976;1(7950):66-68.
16. Matz R. Hypothermia in diabetic acidosis. Hormones. 1972;3:36-41.
17. Burge M., Hardy K., Schade D. Short term fasting is a mechanism for the development of euglycemic ketoacidosis during periods of insulin deficiency. J Clin Endocrinol Metab. 1993;76:1192-1198.
18. Cullen M., Reece E., Homko C., Sivan E. The changing presentations of diabetic ketoacidosis during pregnancy. Am J Perinatol. 1996;13:449-451.
19. Jenkins D., Close C., Krentz A. Euglycemic diabetic ketoacidosis: does it exist? Acta Diabetol. 1993;30:251-253.
20. Halperin M., Goldstein M., Richardson R., Robson L. Quantitative aspects of hyperglycemia in the diabetic: a theoretical approach. Clin Invest Med. 1980;2(4):127-130.
21. Adrogue H., Wilson H., Boyd A. Plasma acid-base patterns in diabetic ketoacidosis. New Engl J Med. 1982;307:1603-1610.
22. Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diab Metab Res Rev. 1999;15:412-426.
23. Ham M., Okada P., White P. Bedside ketone determination in diabetic children with hyperglycemia and ketosis in the acute care setting. Pediatr Diab. 2004;5:39-43.
24. Katz M. Hyperglycemia-induced hyponatremia — calculation of expected serum sodium depression. N Engl J Med. 1973;289:843-844.
25. Oh G., Anderson S., Tancredi D. Hyponatremia in pediatric diabetic ketoacidosis: reevaluating the correction factor for hyperglycemia. Arch Pediatr Adolesc Med. 2009;163:771-772.
26. Kaminska E., Pourmotabbed G. Spurious laboratory values in diabetic ketoacidosis and hyperlipidemia. Am J Emerg Med. 1993;11:77-80.
27. Halperin M., Bear R., Goldstein M. Interpretation of the serum potassium concentration in metabolic acidosis. Clin Invest Med. 1979;2(1):55-57.
28. Flood R., Chiang V. Rate and prediction of infection in children with diabetic ketoacidosis. Am J Emerg Med. 2001;19:270-273.
29. Rewers A., Klingensmith G., Davis C., et al. Presence of diabetic ketoacidosis at diagnosis of diabetes mellitus in youth: the Search for Diabetes in Youth Study. Pediatrics. 2008;121:e1258-e1266.
30. Neu A., Hofer S., Karges B., et al. Ketoacidosis at diabetes onset is still frequent in children and adolescents: a multicenter analysis of 14,664 patients from 106 institutions. Diab Care. 2009;32:1647-1648.
31. Neu A., Willasch A., Ehehalt S. Ketoacidosis at onset of type 1 diabetes mellitus in children–frequency and clinical presentation. Pediatr Diab. 2003;4:77-81.
32. Bui T., Werther G., Cameron F. Trends in diabetic ketoacidosis in childhood and adolescence: a 15-yr experience. Pediatr Diab. 2002;3:82-88.
33. Vanelli M., Chiari G., Ghizzoni L. Effectiveness of a prevention program for diabetic ketoacidosis in children. Diab Care. 1999;22:7-9.
34. Vanelli M.C.F. Treatment of diabetic ketoacidosis in children and adolescents. Acta Bio Medica. 2003;74:59-68.
35. Mallare J., Cordice C., Ryan B. Identifying risk factors for the development of diabetic ketoacidosis in new onset type 1 diabetes mellitus. Clin Pediatr. 2003;42:591-597.
36. Pawlowicz M.B.D., Niedzwiecki M., Balcerska A. Difficulties or mistakes in diagnosing type 1 diabetes in children? demographic factors influencing delayed diagnosis. Pediatr Diab. 2009;10(8):542-549.
37. Dunger D., Sperling M., Acerini C., et al. ESPE / LWPES Consensus statement on diabetic ketoacidosis in children and adolescents. Arch Dis Child. 2003;89:188-194.
38. Diabetes Control and Complications Trial Research Group. Effect of intensive diabetes treatment on the development and progression of long-term complications in adolescents with insulin-dependent diabetes mellitus: Diabetes Control and Complications Trial. J Pediatr. 1994;125:177-188.
39. Rewers A., Chase H., Mackenzie T., et al. Predictors of acute complications in children with type 1 diabetes. JAMA. 2002;287:2511-2518.
40. Hanas R.L.F., Lindblad B.A. 2-yr national population study of pediatric ketoacidosis in Sweden: predisposing conditions and insulin pump use. Pediatr Diab. 2009;10(1):33-37.
41. Wolfsdorf J.C.M., Daneman D., Dunger D. Diabetic ketoacidosis in children and adolescents with diabetes. Pediatr Diab. 2009;10(Suppl 12):118-133.
42. Koves I., Neutze J., Donath S., et al. The accuracy of clinical assessment of dehydration during diabetic ketoacidosis in childhood. Diab Care. 2004;27:2485-2487.
43. Smith L., Rotta A. Accuracy of clinical estimates of dehydration in pediatric patients with diabetic ketoacidosis. Pediatr Emerg Care. 2002;18:395-396.
44. Luzi L., Barrett E., Groop L. Metabolic effects of low-dose insulin therapy on glucose metabolism in diabetic ketoacidosis. Diabetes. 1988;37:1470-1477.
45. Schade D., Eaton R. Dose response to insulin in man: differential effects on glucose and ketone body regulation. J Clin Endocrinol Metab. 1977;44:1038-1053.
46. Grimberg A., Cerri R., Satin-Smith M., Cohen P. The “two bag system” for variable intravenous dextrose and fluid administration: benefits in diabetic ketoacidosis management. J Pediatr. 1999;134:376-378.
47. Alberti G., Darley J., Emerson P., Hockaday T. 2,3-Diphosphoglycerate and tissue oxygenation in uncontrolled diabetes mellitus. Lancet. 1972;2(7774):391-395.
48. Fisher J., Kitabchi A. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab. 1983;57:177-180.
49. Becker D., Brown D., Steranka B., Drash A. Phosphate replacement during treatment of diabetic ketoacidosis. Am J Dis Child. 1983;137:241-246.
50. Gibby O., Veale K., Hayes T. Oxygen availability from the blood and the effect of phosphate replacement on erythrocyte 2,3-diposphoglycerate and haemoglobin-oxygen affinity in diabetic ketoacidosis. Diabetologia. 1978;15:381-385.
51. Lord G., Scott J., Pusey C., et al. Diabetes and rhabdomyolysis. A rare complication of a common disease. BMJ. 1993;307:1126-1128.
52. Shilo S., Werner D., Hershko C. Acute hemolytic anemia caused by severe hypophosphatemia in diabetic ketoacidosis. Acta Haemat. 1985;73:55-57.
53. Soler N., Bennet M., Dixon K. Potassium balance during treatment of diabetic ketoacidosis with special reference to the use of bicarbonate. Lancet. 1972;30:665-667.
54. Assal J., Aoki T., Manzano F., Kozak G. Metabolic effects of sodium bicarbonate in management of diabetic ketoacidosis. Diabetes. 1973;23:405-411.
55. Bureau M., Begin R., Berthiaume Y. Cerebral hypoxia from bicarbonate infusion in diabetic acidosis. J Pediatr. 1980;96:968-973.
56. Glaser N., Barnett P., McCaslin I., et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. N Engl J Med. 2001;344:264-269.
57. Sperling M., Dunger D., Acerini C., et al. ESPE / LWPES consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics. 2003;113:e133-e140.
58. Wolfsdorf J., Glaser N., Sperling M. Diabetic ketoacidosis in infants, children and adolescents: A consensus statement from the American Diabetes Association. Diab Care. 2006;29:1150-1159.
59. Kuppermann N., Park J., Glatter K., Prolonged QT interval corrected for heart rate during diabetic ketoacidosis in children, Arch Pediatr Adolesc Med2008:162(6);544-549,
60. Muir A., Rosenbloom A. Cerebral edema in childhood diabetic ketoacidosis: natural history, radiographic findings and early identification. Diab Care. 2004;27:1541-1546.
61. Edge J., Hawkins M., Winter D., Dunger D. The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch Dis Child. 2001;85:16-22.
62. Ghetti S., Lee J., Holtpatrick C. Diabetic ketoacidosis and memory impairment in children with type 1 diabetes. J Pediatr. 2010;156(1):109-114.
63. Cameron F., Kean M., Wellard R. Insights into the acute cerebral metabolic changes associated with childhood diabetes. Diabet Med. 2005;22:648-653.
64. Glaser N. Cerebral injury and cerebral edema in children with diabetic ketoacidosis: could cerebral ischemia and reperfusion injury be involved? Pediatr Diab. 2009;10:534-541.
65. Mahoney C., Vlcek B., Del Aguila M. Risk factors for developing brain herniation during diabetic ketoacidosis. Pediatr Neurol. 1999;21:721-727.
66. Hoffman W., Burek C., Waller J. Cytokine response to diabetic ketoacidosis and its treatment. Clin Immunol. 2003;108:175-181.
67. Isales C., Min L., Hoffman W. Acetoacetate and B-hydroxybutyrate differentially regulate endothelin-1 and vascular endothelial growth factor in mouse brain microvascular endothelial cells. J Diab Comp. 1999;13(2):91-97.
68. Edge J., Jakes R., Roy Y., et al. The UK case-control study of cerebral oedema complicating diabetic ketoacidosis in children. Diabetologia. 2006;49:2002-2009.
69. Lawrence S., Cummings E., Gaboury I., Daneman D. Population-based study of incidence and risk factors for cerebral edema in pediatric diabetic ketoacidosis. J Pediatr. 2005;146:688-692.
70. Curtis J., Bohn D., Daneman D. Use of hypertonic saline in the treatment of cerebral edema in diabetic ketoacidosis (DKA). Pediatr Diab. 2001;2:191-194.
71. Kamat P., Vats A., Gross M., Checchia P. Use of hypertonic saline for the treatment of altered mental status associated with diabetic ketoacidosis. Pediatr Crit Care Med. 2003;4:239-242.
72. Marcin J., Glaser N., Barnett P., et al. Clinical and therapeutic factors associated with adverse outcomes in children with DKA-related cerebral edema. J Pediatr. 2003;141:793-797.
73. Spencer-Smith M., Northam E., Koves I. Neurocognitive changes over a 6 month period in children newly diagnosed type 1 diabetes mellitus and diabetic ketoacidosis. ISPAD conference abstract. 2007.
74. Northam E.A., Rankins D., Lin A. Central nervous system function in youth with type 1 diabetes 12 years after disease onset. Diab Care. 2009;32(3):445-450.
75. Davis J., Surendran T., Thompson S., Corkey C., Dka C.V.L., DVT. increased risk of deep venous thrombosis in children with diabetic ketoacidosis and femoral central venous lines. Ir Med J. 2007;100:344.
76. Gutierrez J., Bagatell R., Sampson M. Femoral central venous catheter-associated deep venous thrombosis in children with diabetic ketoacidosis. Crit Care Med. 2003;31:80-83.
77. McDonnell C., Pedreira C., Vadamalayan B. Diabetic ketoacidosis, hyperosmolarity and hypernatremia: are high-carbohydrate drinks worsening initial presentation? Pediatr Diab. 2005;6:90-94.
78. Grant P., Wiles P., Dean H. The physiological effects of vasopressin on haemostasis are not mediated by catecholamine release. Thromb Res. 1987;45:839-843.
79. Buckingham B., Roe T., Yoon J. Rhabdomyolysis in diabetic ketoacidosis. Am J Dis Child. 1981;135:352-354.
80. Casteels K., Beckers D., Wouters C., Van Geet C. Rhabdomyolysis in diabetic ketoacidosis. Pediatr Diab. 2003;4:29-31.
81. Quiros J., Marcin J., Kuppermann N., et al. Elevated serum amylase and lipase in pediatric diabetic ketoacidosis. Pediatr Crit Care Med. 2008;9:418-422.
82. Rosenbloom A. Intracerebral crises during treatment of diabetic ketoacidosis. Diabetes Care. 1990;13:22-33.
83. Hoffman W., Locksmith J., Burton E., et al. Interstitial pulmonary edema in children and adolescents with diabetic ketoacidosis. J Diab Comp. 1998;12:314-320.
84. Malone J., Brodsky S. The value of electrocardiogram monitoring in diabetic ketoacidosis. Diab Care. 1980;3:543-547.
85. Murdoch I., Pryor D., Haycock G., Cameron S. Acute renal failure complicating diabetic ketoacidosis. Acta Paediatr. 1993;82:498-500.
86. Chan-Cua S., Jones K., Lynch F., Friedenberg G. Necrosis of the ileum in a diabetic adolescent. J Pediatr Surg. 1992;27:1236-1238.
87. Dimeglio L., Chaet M., Quigley C., Grosfled J. Massive ischemic intestinal necrosis at the onset of diabetes mellitus with ketoacidosis in a three-year old girl. J Pediatr Surg. 2003;38:1537-1539.
88. Todani T., Sato Y., Watanabe Y. Ischemic jejunal stricture developing after diabetic coma in a girl: a case report. Europ J Pediatr Surg. 1993;3(2):115-117.
89. Dokmetas H., Canbay E., Yilmaz S., et al. Diabetic ketoacidosis and rhino-orbital mucormycosis. Diab Res Clin Pract. 2002;57:139-142.
90. Khanna S., Soumekh B., Bradley J., et al. A case of fatal rhinocerebral mucormycosis with new onset diabetic ketoacidosis. J Diab Comp. 1998;12:224-227.
91. Cochran J., Walters S., Losek J. Pediatric hyperglycemic hyperosmolar syndrome: diagnostic difficulties and high mortality rate. Am J Emerg Med. 2006;24:297-301.
92. Fourtner S, Weinzimer D, Murphy K, Katz L. Hyperglycemic hyperosmolar nonketotic (HHNK) syndrome in children, Proc Endocr Soc 85th Ann Mtg 2003, 42–6.
93. Rosenbloom A. Hyperglycemic crises and their complications in children. J Pediatr Endocrinol Metab. 2007;20:5-18.
94. Canarie M., Bogue C., Banasiak K. Decompensated hyperglycemic hyperosmolarity without significant ketoacidosis in the adolescent and young adult population. J Pediatr Endocrinol Metab. 2007;20:1115-1124.
95. Gangopadhyay K.K., Ryder R.E. Nontraumatic rhabdomyolysis: an unusual complication of diabetic hyperosmolar nonketotic (HONK) state. J R Soc Med. 2006;99(4):200.
96. Kilbane B.J., Mehta S., Backeljauw P.F. Approach to management of malignant hyperthermia-like syndrome in pediatric diabetes mellitus. Pediatr Crit Care Med. 2006;7(2):169-173.
97. Carchman R.M.D.-Z.M., Calikoglu A.S., Harris B.D. A new challenge in pediatric obesity: pediatric hyperglycemic hyperosmolar syndrome. Pediatr Crit Care Med. 2005;6(1):20-24.
98. Triolo T., Chase H., Barker J. DPT-1 study group. Diabetic subjects diagnosed through the Diabetes Prevention Trial-Type 1 (DPT-1) are often asymptomatic with normal A1C at diabetes onset. Diab Care. 2009;32(5):769-773.
99. Beck J.K., Logan K.J., Hamm R.M. Reimbursement for pediatric diabetes intensive case management: A model for chronic disease? Pediatrics. 2004;113:e47-e50.
100. Ellis D., Naar-King S., Templin T. Multisystemic therapy for adolescents with poorly controlled type 1 diabetes: reduced diabetic ketoacidosis admissions and related costs over 24 months. Diabetes. 2008;31(9):1746-1747.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 78 Diabetic Ketoacidosis
Box 78–1 DKA Treatment in Children and Adolescents
Modified from Glaser NS: Pediatric diabetic ketoacidosis and hyperglycemic hyperosmolar state. Pediatr Clin North Am 52:1611-1635, 2005.
Fluids
Insulin
Potassium and other electrolyte replacement
Monitoring
Etiology, Definition, and Presentation
In a child with new onset of type 1 diabetes, declining insulin production results from autoimmune destruction of pancreatic beta cells. The concentration of insulin is decreased relative to glucagon causing excess hepatic glucose production and decreased peripheral glucose uptake in muscle and adipose tissue.1, 2 When the serum glucose concentration rises above approximately 180 to 200 mg/dL, the renal threshold for glucose reabsorption is exceeded causing glycosuria, which leads to osmotic diuresis and compensatory polydipsia. Low insulin concentrations also stimulate the release of free fatty acids (FFA) from adipose tissue to fuel ketogenesis.3 This, in combination with activation of the hepatic β-oxidative enzyme sequence resulting from relative excess of glucagon in relation to insulin, results in markedly increased hepatic ketone production.2–4
Progressive dehydration and increasing acidosis eventually stimulate additional release of the counterregulatory (“stress”) hormones, cortisol, catecholamines, and growth hormone, which accelerate hepatic glucose output and ketone production.5,6 Infection or other illness or injury can likewise contribute to this process by stimulating release of counterregulatory hormones. Elevated cortisol concentrations augment FFA release from adipose tissue and decrease peripheral glucose uptake. Increased epinephrine concentrations directly increase glycogenolysis and stimulate release of gluconeogenic precursors from muscle.7,8 Both epinephrine and norepinephrine also stimulate lipolysis and β-oxidation of FFAs.9,10 Catecholamines may also directly inhibit insulin secretion, thereby accelerating DKA in those with endogenous insulin capacity, such as a new diagnosis of type 1 diabetes or those with type 2 diabetes.11,12 Growth hormone also decreases peripheral glucose uptake, and enhances ketone production by increasing FFA release.13 Elevated concentrations of counterregulatory hormones thus result in increased acidosis, hyperglycemia, and dehydration. This in turn stimulates further counterregulatory hormone release thereby creating a “vicious cycle” resulting in rapid worsening of DKA (Figure 78-1).
Figure 78–1 Pathophysiology of DKA.
(From Wolfsdorf J, Glaser N, Sperling MA: Diabetic ketoacidosis in infants, children, and adolescents: a consensus statement from the American Diabetes Association, Diabetes Care 29[5]:1150-1159, 2006.)
During DKA, intestinal ileus results from potassium depletion, acidosis, and diminished splanchnic perfusion, causing abdominal pain and vomiting and thereby limiting fluid intake. Progressive dehydration eventually leads to diminished tissue perfusion sufficient to cause accumulation of lactic acid, enhancing acidosis.14 In addition, poor perfusion may result in diminished renal function, limiting the capacity for clearance of glucose and ketones. Ongoing osmotic diuresis and ketonuria in the setting of acidosis also results in urinary losses of electrolytes (potassium, sodium, chloride, calcium, phosphate, and magnesium).
Classical symptoms of DKA include polyuria, polydipsia, polyphagia, weight loss, abdominal pain, nausea, and vomiting. Abdominal tenderness, absence of bowel sounds and guarding are frequent and may even mimic an acute abdomen.15 Tachycardia and signs of hypoperfusion, such as delayed capillary refill time and cool extremities, are also common as well as dry mucous membranes, absence of tears, and poor skin turgor. Despite substantial volume depletion, however, hypotension is unusual in children with DKA and occasional children may present with hypertension. Kussmaul breathing and tachypnea are the result of metabolic acidosis and respiratory compensatory mechanisms. Fruity breath odor (acetone) may be present. Hypothermia has also been described.16
Although hyperglycemia is part of the definition of DKA, in rare cases, the serum glucose concentration may be nearly normal, so called “euglycemic DKA.” This situation has been reported most frequently in pregnant women.17–19 Normal glucose concentrations or even hypoglycemia despite ketosis may also occur in children with known diabetes who administer insulin to treat DKA prior to arrival in the emergency department. In general, however, the persistence and severity of hyperglycemia reflects the severity of dehydration. In the absence of preexisting renal disease or unusually high carbohydrate intake just before presentation, blood glucose concentrations in excess of 500 to 600 mg/dl imply that dehydration is of sufficient severity to diminish the glomerular filtration rate and thereby diminish the capacity for renal clearance of excess glucose.20
Concentrations of ketone bodies (beta-hydroxybutyrate [βOHB] and acetoacetate [AcAc]) are elevated in DKA resulting in acidosis. Hyperchloremic acidosis frequently coexists with increased anion-gap acidosis, and the anion gap reflects the combination of these processes.21 The ratio of βOHB:AcAc (typically 1:1 in the normal state) is increased during DKA and may be as high as 10:1.22 During treatment, this ratio returns to normal. The nitroprusside reaction used to test urine ketone concentrations detects only AcAc and not βOHB. As a result, nitroprusside urine testing cannot be relied on to determine DKA severity or treatment response. Bedside blood ketone meters provide a rapid means for measuring βOHB, and may be useful in place of or in addition to urine testing particularly in patients with anuria or oligoria who produce insufficient amounts of urine for ketone testing.23 Blood ketone measurements are also useful for determining the timing of transition from intravenous to subcutaneous insulin administration. Urine ketones may be present even when blood ketones have normalized as a result of urine stagnating in the bladder.
Hyperglycemia results in fluid shifts from the extravascular to the intravascular space and a decrease in the serum sodium concentration. This decrease can be calculated as an approximately 1.6 mEq/L decrease in sodium concentration for every 100 mg/dl increase in serum glucose higher than 100 mg/dl or Na corrected = Naactual + (glucose in mmol/L – 5.5 mmol/L)}.24,25 Hyperlipidemia may also contribute to a decrease in measured serum sodium concentrations.26 Typically, serum potassium at presentation is in the high-normal range as a result of redistribution of potassium ions from the intracellular to the extracellular space. Several processes are responsible for intracellular potassium depletion including direct effects of low insulin concentrations, intracellular protein and phosphate depletion, and buffering of hydrogen ions in the intracellular compartment.27 Intracellular potassium stores may be profoundly depleted and the serum potassium concentration typically declines rapidly with insulin treatment. Serum phosphate concentrations similarly decrease during treatment.
Leukocytosis is frequent in children with DKA, likely resulting from elevated concentrations of catecholamines and pro-inflammatory cytokines. In children, new onset of type 1 diabetes or insulin omission are far more common causes of DKA than infection. 28 Therefore, an elevated or left-shifted white blood cell count need not prompt a search for an infectious process in the absence of fever or other symptoms or signs of infection. However, in the presence of fever, careful history, physical examination, and laboratory evaluation to assess for infection is prudent.
Epidemiology
Frequency of Diabetic Ketoacidosis at Diagnosis
The frequency of DKA at diagnosis varies widely by geographic region, with an overall estimated frequency of approximately 20% to 67%. In the population-based US study, SEARCH for Diabetes in Youth, data were collected from self-reported health questionnaires and medical record review. In this study, 25.5% of children and adolescents presented at onset of diabetes in DKA.29 A similar frequency was observed in Germany, with 26.3% of children under the age of 15 presenting in DKA at diagnosis of diabetes. In a collaborative study from Germany and Austria, the Diabetes Patienten Verlausdokumentation (an electronic documentation system for children with diabetes), the frequency of DKA at presentation was 21.1%.30 Younger age (<5 years) and female sex were associated with higher likelihood of presenting in DKA.31,32 A delay in diagnosis is associated with a higher likelihood of DKA, and two factors contributing to this are patient age and provider experience. Regions with a higher prevalence of type 1 diabetes generally have a lower frequency of DKA,33 attributed to heightened awareness in providers and thus earlier detection. The non-specific nature of individual symptoms, such as polyuria, tachypnea, and altered mental status, may cause such symptoms to be misconstrued as urinary tract infection, pneumonia, or meningitis, respectively.34 Mallare et al.35 reported a frequency of DKA of 33% in children and adolescents at the initial visit, and almost double (59%) in those in whom the diagnosis of diabetes was missed at the initial visit. The diagnosis of diabetes was more likely to be missed in very young children (34% of children ≤5 years of age compared to 8.5% in those greater than 10 years of age),35 particularly when these very young children are evaluated by family practitioners rather than pediatricians.36
Frequency of Diabetic Ketoacidosis in Children and Adolescents After Diagnosis
Although there are several population-based studies reporting the frequency of DKA at presentation, fewer data are available describing the incidence of DKA in children and adolescents with established diabetes. Reported frequencies range from 1 to 10 per 100 patient years.37 In the Diabetes Control and Complications Trial, the incidence of DKA in adolescents treated with intensive management regimens was 2.8 per 100 patient-years, significantly lower than the incidence in those treated conventionally (4.7 per 100 patient years). Although this is an older study, it was a time and resource-intensive study, and represents a more idealized situation than often encountered in the overall pediatric diabetes population.38 In a more recent study from the Barbara Davis Center for Childhood Diabetes in Denver, Colorado, the overall incidence of DKA was 8 per 100 person-years. In that study, factors associated with higher incidence included older age, higher HbA1C (relative risk [RR] of 1.68 per 1% increase in HbA1C in younger children, RR of 1.43 in older children), higher reported insulin dose, DSM4 psychiatric diagnoses, and “underinsurance” reflecting lower socioeconomic status.39 DKA is also observed more often in children and adolescents on continuous subcutaneous insulin infusion therapy (CSII) than on subcutaneous injections, particularly in the first year of initiation of CSII.40 However, lower rates of DKA are achievable for those on CSII with adequate training and resources.
Over the past two decades T2DM has been occurring with increasing frequency in older children and adolescents. Certain racial/ethnic groups in the United States are disproportionately affected including Native Americans, Hispanics, and African Americans. DKA can be the clinical presentation for T2DM in youth estimated at 5% to 10%.41 Youth with T2DM may also present with hyperglycemic hyperosmolar state (HHS), also referred to hyperosmotic hyperglycemic non-ketotic coma (HHNK, described more fully in the following section).
Morbidity and Mortality Associated with Diabetic Ketoacidosis
Mortality in children presenting with DKA is approximately 0.25% to 0.30%.37 Most of the mortality in DKA occurs in children with cerebral edema, accounting for 57% to 87% of deaths. Neurologic sequelae of DKA are described in the section on DKA-associated complications. Other causes of morbidity and mortality include sepsis and secondary infection, electrolyte abnormalities (e.g., hypokalemia), arrhythmias, rhabdomyolysis, thrombosis, pneumomediastinum, subcutaneous emphysema, and pulmonary edema.
Management Guidelines
Fluids
Restoration of adequate peripheral perfusion and hemodynamic stability with bolus administration of intravenous fluids (0.9% saline or other isotonic fluids) should begin as soon as possible. Typical patients require an initial fluid bolus of 10 ml/kg that may be repeated if ongoing hemodynamic instability is present. Studies have shown that clinical assessments of dehydration severity in children with DKA tend to be inaccurate (Figure 78-2).42 The average degree of dehydration for most patients is approximately 7% to 9% of body weight and this figure should be used as a basis for determining the total volume of fluids to be replaced.42,43
[/not-level-membership-for-pediatrics-category]
