Chapter 552 Diabetes Insipidus
Diabetes insipidus (DI) manifests clinically with polyuria and polydipsia and can result from either vasopressin deficiency (central DI) or vasopressin insensitivity at the level of the kidney (nephrogenic DI). Both central DI and nephrogenic DI can arise from inherited defects of congenital or neonatal onset or can be secondary to a variety of causes (Table 552-1).
Physiology of Water Balance
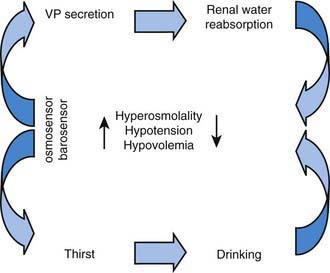
The control of extracellular tonicity (osmolality) and volume within a narrow range is critical for normal cellular structure and function (Chapter 52). Extracellular fluid tonicity is regulated almost exclusively by water intake and excretion, whereas extracellular volume is regulated by sodium intake and excretion. The control of plasma tonicity and intravascular volume involves a complex integration of endocrine, neural, behavioral, and paracrine systems (Fig. 552-1). Vasopressin, secreted from the posterior pituitary, is the principal regulator of tonicity, with its release largely stimulated by increases in plasma tonicity. Volume homeostasis is largely regulated by the renin-angiotensin-aldosterone system, with contributions from both vasopressin and the natriuretic peptide family.
Causes of Hypernatremia
Hypernatremia is discussed in Chapter 52.3.
Central Diabetes Insipidus
Central diabetes insipidus can result from multiple etiologies, including genetic mutations in the vasopressin gene; trauma (accidental or surgical) to vasopressin neurons; congenital malformations of the hypothalamus or pituitary; neoplasms; infiltrative, autoimmune, and infectious diseases affecting vasopressin neurons or fiber tracts; and increased metabolism of vasopressin. In approximately 10% of children with central DI, the etiology is idiopathic. Other pituitary hormone deficiencies may be present (Chapter 551).
Autosomal dominant central DI usually occurs within the first 5 yr of life and results from mutations in the vasopressin gene. A number of mutations can cause gene-processing defects in a subset of vasopressin-expressing neurons, which have been postulated to result in endoplasmic reticulum (ER) stress and cell death. Wolfram syndrome, which includes DI, diabetes mellitus, optic atrophy, and deafness, also results in vasopressin deficiency. Mutations in two genes, which give rise to ER proteins, have been associated with this condition. Congenital brain abnormalities (Chapter 585) such as septo-optic dysplasia with agenesis of the corpus callosum, the Niikawa-Kuroki syndrome, holoprosencephaly, and familial pituitary hypoplasia with absent stalk may be associated with central DI and defects in thirst perception. Empty sella syndrome, possibly resulting from unrecognized pituitary infarction, can be associated with DI in children.
Given the anatomic distribution of vasopressin neurons over a large area within the hypothalamus, tumors that cause DI must either be very large and infiltrative or be strategically located near the base of the hypothalamus, where vasopressin axons converge before their entry into the posterior pituitary. Germinomas and pinealomas typically arise in this region and are among the most common primary brain tumors associated with DI. Germinomas can be very small and undetectable by MRI for several yr following the onset of polyuria. Quantitative measurement of the β-subunit of human chorionic gonadotropin, often secreted by germinomas and pinealomas, should be performed in children with idiopathic or unexplained DI in addition to serial MRI scans. Craniopharyngiomas and optic gliomas can also cause central DI when they are very large, although this is more often a postoperative complication of the treatment for these tumors (Chapter 491). Hematologic malignancies, as with acute myelocytic leukemia, can cause DI via infiltration of the pituitary stalk and sella.
Blanco EJ, Lane AH, Aijaz N, et al. Use of subcutaneous DDAVP in infants with central diabetes insipidus. J Pediatr Endocrinol Metab. 2006;19:919-925.
Hoorn EJ, Zietse R. Water balance disorders after neurosurgery: the triphasic response revisited. NDT Plus. 2010;3:42-44.
Knoers N, Monnens LH. Nephrogenic diabetes insipidus: clinical symptoms, pathogenesis, genetics and treatment. Pediatr Nephrol. 1992;6:476-482.
Maghnie M, Cosi G, Genovese E, et al. Central diabetes insipidus in children and young adults. N Engl J Med. 2000;343:988-1007.
Muglia LJ, Majzoub JA. Disorders of the posterior pituitary. In Sperling MA, editor: Pediatric endocrinology, ed 2, Philadelphia: WB Saunders, 2002.
Ranadive SA, Rosenthal SM. Pediatric disorders of water balance. Endocrinol Metab Clin North Am. 2009;38:663-672.



