[level-membership-for-basic-science-category]
CHAPTER 6 Developmental Genetics
At fertilization the nucleus from a spermatozoon penetrates the cell membrane of an oocyte to form a zygote. This single cell divides to become two, then four, and when the number has doubled some 50 times the resulting organism comprises more than 200 distinct cell types and a total cell number of about 10,000 trillion. This is a fully formed human being with complex biochemistry and physiology, capable of exploring the cosmos and identifying subatomic particles. Not surprisingly, biologists and geneticists are intrigued by the mechanisms of early development and, whilst many mysteries remain, the rate of progress in understanding key events and signaling pathways is rapid.
Prenatal life can be divided into three main stages: pre-embryonic, embryonic, and fetal (Table 6.1). During the pre-embryonic stage, a small collection of cells becomes distinguishable, first as a double-layered or bilaminar disc, and then as a triple-layered or trilaminar disc (Figure 6.1), which is destined to develop into the human infant. During the embryonic stage, craniocaudal, dorsoventral, and proximodistal axes are established, as cellular aggregation and differentiation lead to tissue and organ formation. The final fetal stage is characterized by rapid growth and development as the embryo, now known as a fetus, matures into a viable human infant.
Table 6.1 Main Events in the Development of a Human Infant
| Stage | Time from Conception | Length of Embryo/Fetus |
|---|---|---|
| Pre-embryonic | ||
| First cell division | 30 h | |
| Zygote reaches uterine cavity | 4 d | |
| Implantation | 5–6 d | |
| Formation of bilaminar disc | 12 d | 0.2 mm |
| Lyonization in female | 16 d | |
| Formation of trilaminar disc and primitive streak | 19 d | 1 mm |
| Embryonic stage | ||
| Organogenesis | 4–8 w | |
| Brain and spinal cord are forming, and first signs of heart and limb buds | 4 w | 4 mm |
| Brain, eyes, heart and limbs developing rapidly, and bowel and lungs beginning to develop | 6 w | 17 mm |
| Digits have appeared. Ears, kidneys, liver and muscle are developing | 8 w | 4 cm |
| Palate closes and joints form | 10 w | 6 cm |
| Sexual differentiation almost complete | 12 w | 9 cm |
| Fetal stage | ||
| Fetal movements felt | 16–18 w | 20 cm |
| Eyelids open. Fetus is now viable with specialized care | 24–26 w | 35 cm |
| Rapid weight gain due to growth and accumulation of fat as lungs mature | 28–38 w | 40–50 cm |
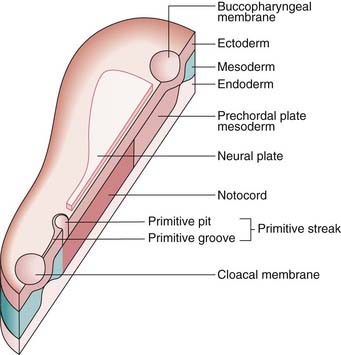
FIGURE 6.1 A schematic trilaminar disc, sectioned along the rostrocaudal axis. Cells from the future ectoderm (top layer) migrate through the primitive streak to form the endoderm (bottom layer) and mesoderm (blue). Formation of the neutral plate in the overlying ectoderm, destined to be the central nervous system, involves sonic hedgehog signaling (p. 86) from the notochord and prechordial plate mesoderm.
(Redrawn with permission from Larsen WJ 1998 Essentials of human embryology. New York: Churchill Livingstone.)
Fertilization and Gastrulation
Fertilization, the process by which the male and female gametes fuse, occurs in the fallopian tube. Of the 100 to 200 million spermatozoa deposited in the female genital tract, only a few hundred reach the site of fertilization. Of these, usually only a single spermatozoon succeeds in penetrating first the corona radiata, then the zona pellucida, and finally the oocyte cell membrane, whereupon the oocyte completes its second meiotic division (see Figure 3.15, p. 40). After the sperm has penetrated the oocyte and the meiotic process has been completed, the two nuclei, known as pronuclei, fuse, thereby restoring the diploid number of 46 chromosomes. This is a potentially chaotic molecular encounter with a high chance of failure, as we know from observations of the early human embryo from in-vitro fertilization programs. It may be likened, somewhat flippantly, to ‘speed dating’, whereby couples test whether they might be compatible on the basis of only a few minutes conversation.
Germ cell and very early embryonic development are two periods characterized by widespread changes in DNA methylation patterns—epigenetic reprogramming (see p. 103). Primordial germ cells are globally demethylated as they mature and are subsequently methylated de novo during gametogenesis, the time when most DNA methylation imprints are established. After fertilization a second wave of change occurs. The oocyte rapidly removes the methyl imprints from the sperm’s DNA, which has the effect of resetting the developmental stopwatch to zero. By contrast, the maternal genome is more passively demethylated in such a way that imprinting marks resist demethylation. A third wave of methylation, de novo, establishes the somatic cell pattern of DNA methylation after implantation. These alternating methylation states help to control which genes are active, or expressed, at a time when two genomes, initially alien to each other, collide.
The fertilized ovum or zygote undergoes a series of mitotic divisions to consist of two cells by 30 hours, four cells by 40 hours, and 12 to 16 cells by 3 days, when it is known as a morula. A key concept in development at all stages is the emergence of polarity within groups of cells—part of the process of differentiation that generates multiple cell types with unique identities. Although precise mechanisms remain elusive, observations suggest that this begins at the very outset; in the fertilized egg of the mouse, the point of entry of the sperm determines the plane through which the first cell cleavage division occurs. This seminal event is the first step in the development of the so-called dorso-ventral, or primary body, axis in the embryo.
Further cell division leads to formation of a blastocyst, which consists of an inner cell mass or embryoblast, destined to form the embryo, and an outer cell mass or trophoblast, which gives rise to the placenta. The process of converting the inner cell mass into first a bilaminar, and then a trilaminar, disc (see Figure 6.1) is known as gastrulation, and takes place between the beginning of the second and the end of the third weeks.
Between 4 and 8 weeks the body form is established, beginning with the formation of the primitive streak at the caudal end of the embryo. The germinal layers of the trilaminar disc give rise to ectodermal, mesodermal, and endodermal structures (Box 6.1). The neural tube is formed and neural crest cells migrate to form sensory ganglia, the sympathetic nervous system, pigment cells, and both bone and cartilage in parts of the face and branchial arches.
Disorders involving cells of neural crest origin, such as neurofibromatosis (p. 298), are sometimes referred to as neurocristopathies. This period between 4 and 8 weeks is described as the period of organogenesis, because during this interval all of the major organs are formed as regional specialization proceeds in a craniocaudal direction down the axis of the embryo.
Developmental Gene Families
Information about the genetic factors that initiate, maintain, and direct embryogenesis is incomplete. However, extensive genetic studies of the fruit fly, Drosophila melanogaster, and vertebrates such as mouse, chick, and zebra-fish have identified several genes and gene families that play important roles in early developmental processes. It has also been possible through painstaking gene expression studies to identify several key developmental pathways, or cascades, to which more detail and complexity is continually being added. The gene families identified in vertebrates usually show strong sequence homology with developmental regulatory genes in Drosophila. Studies in humans have revealed that mutations in various members of these gene families can result in either isolated malformations or multiple congenital anomaly syndromes (see Table 16.5, p. 256). Many developmental genes produce proteins called transcription factors (p. 22), which control RNA transcription from the DNA template by binding to specific regulatory DNA sequences to form complexes that initiate transcription by RNA polymerase.
Early Patterning
The WNT pathway has two main branches: one that is β-catenin–dependent (canonical) and the other independent of β-catenin. In the canonical pathway, Wnt ligand binds to a Frizzled/LRP heterodimer membrane-bound protein complex and the downstream intracellular signaling involves a G protein. The effect of this is to disrupt a large cytoplasmic protein complex that includes Axin, the adenomatous polyposis coli (APC; see p. 221) protein, and the glycogen synthase kinase-3β (GSK-3β) protein. This prevents the phosphorylation of β-catenin, but when β-catenin is not degraded, it accumulates and translocates to the nucleus where it activates the transcription of dorsal-specific regulatory genes. Binding of the ligand to the Fgf receptor results in dimerization of the receptor and transphosphorylation of the receptor’s cytoplasmic domain, with activation of Ras and other kinases, one of which enters the nucleus and activates target transcription factors. Mutated WNT10A in man results in a form of ectodermal dysplasia (odonto-onychodermal dysplasia) but apart from the possibility of WNT4 being implicated in a rare condition called Mayer-Rokitansky-Kuster syndrome, no other members of this gene family are yet implicated in human disease phenotypes.
The TGF-β Superfamily in Development and Disease
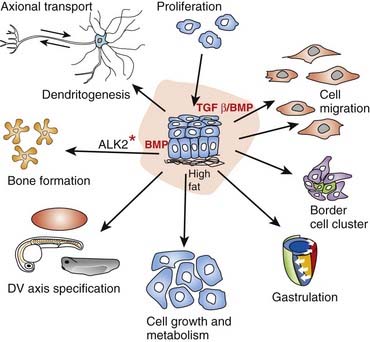
Thus far it recognized that there are 33 members of this cytokine family. Cytokines are a category of signaling molecules—polypeptide regulators—that enable cells to communicate. They differ from hormones in that they are not produced by discrete glands. These extracellular signaling polypeptides are transduced through a cascade to regulate gene expression within the cell nucleus. This is achieved through binding with cell surface receptors that, in a series of reactions, induces phosphorylation and activation of specific receptor kinases. This leads to the translocation of complexes into the nucleus, which execute transcriptional activation or repression of responsive target genes. The TGF-β family can be divided into two groups: (1) the BMPs and (2) the TGF-βs, activins, nodal, and myostatin, acting through various SMAD proteins. Ultimately, this superfamily is actively involved in a very broad range of cellular and developmental processes (Figure 6.2). This includes regulation of the cell cycle, cell migration, cell size, gastrulation and axis specification, and metabolic processes. In relation to health and disease, there are consequences for immunity, cancer, heart disease, diabetes, and Marfan syndrome (p. 300). Hyperactive signalling (overexpression) of BMP4 has been found in the rare bony condition fibrodysplasia ossificans progressiva, where disabling heterotopic bone deposition occurs, which is due to mutated ACVR1, encoding a BMP type 1 receptor. A mutated BMP receptor 2 has been shown to be a cause of familial primary pulmonary hypertension. BMP signalling is also involved in both dendritogenesis and axonal transport.
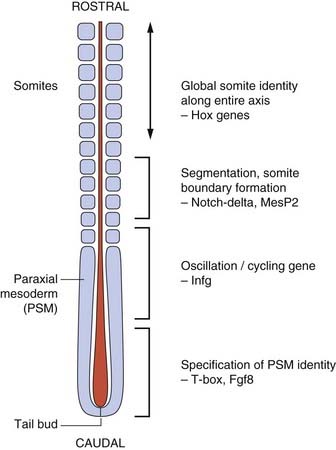
Somatogenesis and the Axial Skeleton
The vertebrate axis is closely linked to the development of the primary body axis during gastrulation, and during this process the presomitic mesoderm (PSM), where somites arise, is laid down in higher vertebrates. Wnt and FGF signals play vital roles in the specification of the PSM. The somites form as blocks of tissue from the PSM in a rostro-caudal direction (Figure 6.3), each being laid down with a precise periodicity that, in the 1970s, gave rise to the concept of the ‘clock and wavefront’ model. Since then, molecular techniques have given substance to this concept, and the key pathway here is notch-delta signaling and the ‘oscillation clock’—a precise, temporally defined wave of cycling gene expression (c-hairy in the chick, lunatic fringe and hes genes in the mouse) that sweeps from the tail-bud region in a rostral direction and has a key role in the process leading to the defining of somite boundaries. Once again, not all of the components are fully understood, but the notch receptor and its ligands, delta-like-1, and delta-like-3, together with presenilin-1 and mesoderm posterior-2, work in concert to establish rostro-caudal polarity within the PSM such that somite blocks are formed. Human phenotypes from mutated genes in this pathway are now well known and include presenile dementia (presenilin-1), which is dominantly inherited, and spondylocostal dysostosis (delta-like-3, mesoderm posterior-2, lunatic fringe, and hairy enhancer of split-7), which is recessively inherited (Figure 6.4). Another component of the pathway is JAGGED1, which, when mutated, results in the dominantly inherited and very variable condition known as Alagille syndrome (arteriohepatic dysplasia) (Figure 6.5). Rarely, mutations in NOTCH2 have been shown to cause some cases of Alagille syndrome, usually with renal malformations.


The Sonic Hedgehog–Patched GLI Pathway
The Sonic hedgehog gene (SHH) is as well known for its quirky name as for its function. SHH induces cell proliferation in a tissue-specific distribution and is expressed in the notochord, the brain, and the zone of polarizing activity of developing limbs. After cleavage and modification by the addition of a cholesterol moiety, the SHH protein binds with its receptor, Patched (Ptch), a transmembrane protein. The normal action of Ptch is to inhibit another transmembrane protein called Smoothened (Smo), but when bound by Shh this inhibition is released and a signaling cascade within the cell is activated. The key intracellular targets are the GLI family of transcription factors (Figure 6.6).
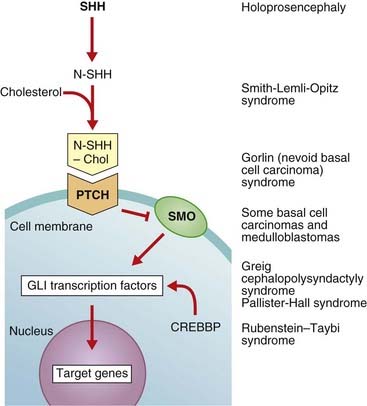
FIGURE 6.6 The Sonic hedgehog (Shh)-Patched (Ptch)-Gli pathway and connection with disease. Different elements in the pathway act as activators (arrows) or inhibitors (bars). The Shh protein is initially cleaved to an active N-terminal form, which is then modified by the addition of cholesterol. The normal action of Ptch is to inhibit Smo, but when Ptch is bound by Shh this inhibition is removed and the downstream signaling proceeds. CREBBP, cAMP response element-binding binding protein.
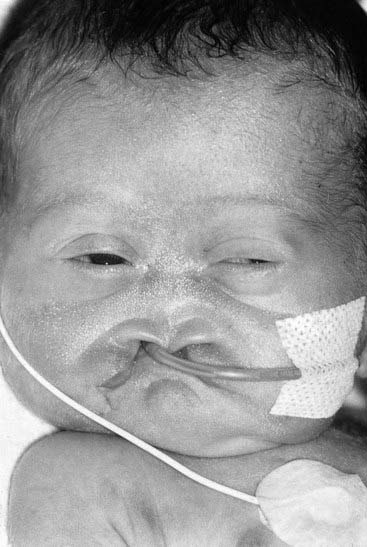
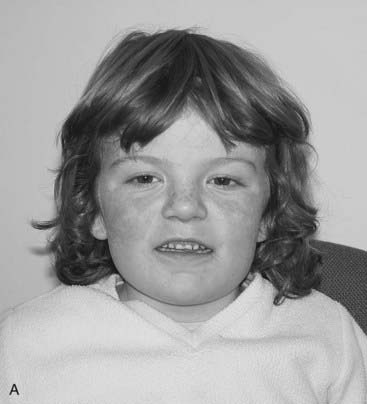
Molecular defects in any part of this pathway lead to a number of apparently diverse malformation syndromes (see Figure 6.6). Mutations in, or deletions of, SHH (chromosome 7q36) cause holoprosencephaly (Figure 6.7), in which the primary defect is incomplete cleavage of the developing brain into separate hemispheres and ventricles. The most severe form of this malformation is cyclopia—the presence of a single central eye. (The complexity of early development can be appreciated by the fact that a dozen or so chromosomal regions have so far been implicated in the pathogenesis of holoprosencephaly [p. 257].) Mutations in PTCH (9q22) result in Gorlin syndrome (nevoid basal cell carcinoma syndrome; Figure 6.8), which comprises multiple basal cell carcinomas, odontogenic keratocysts, bifid ribs, calcification of the falx cerebri, and ovarian fibromata. Mutations in SMO (7q31) are found in some basal cell carcinomas and medulloblastomas. Mutations in GLI3 (7p13) cause Pallister-Hall and Grieg syndromes, which are distinct entities with more or less the same body systems affected. However, there are also links to other conditions, in particular the very variable Smith-Lemli-Opitz syndrome (SLOS), which may include holoprosencephaly as well as some characteristic facial features, genital anomalies and syndactyly. This condition is due to a defect in the final step of cholesterol biosynthesis, which in turn may disrupt the binding of SHH with its receptor Ptch. Some, or all, of the features of SLOS may therefore be due to loss of integrity in this pathway (p. 288). Furthermore, a cofactor for the Gli proteins, CREBBP (16p13) is mutated in Rubenstein-Taybi syndrome (Figure 6.9). Disturbance to different components of the SHH is also clearly implicated in many types of tumor formation.

Homeobox (HOX) Genes
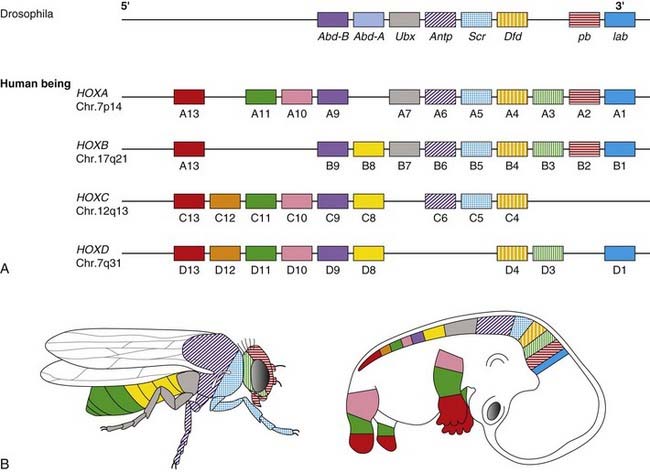
In Drosophila a class of genes known as the homeotic genes has been shown to determine segment identity. Incorrect expression of these genes results in major structural abnormalities; the Antp gene, for example, which is normally expressed in the second thoracic segment, will transform the adult fly’s antennae into legs if incorrectly expressed in the head. Homeotic genes contain a conserved 180-base pair (bp) sequence known as the homeobox, which is believed to be characteristic of genes involved in spatial pattern control and development. This encodes a 60-amino-acid domain that binds to DNA in Hox-response enhancers. Proteins from homeobox-containing (or HOX) genes are therefore important transcription factors that activate and repress batteries of downstream genes. At least 35 downstream targets are known. The Hox proteins regulate other ‘executive’ genes that encode transcription factors or morphogen signals, as well as operating at many other levels, on genes that mediate cell adhesion, cell division rates, cell death, and cell movement. They specify cell fate and help to establish the embryonic pattern along the primary (rostro-caudal) axis as well as the secondary (genital and limb bud) axis. They therefore play a major part in the development of the central nervous system, axial skeleton and limbs, the gastrointestinal and urogenital tracts, and external genitalia.
Drosophila has eight Hox genes arranged in a single cluster, but in humans, as in most vertebrates, there are four homeobox gene clusters containing a total of 39 HOX genes (Figure 6.10). Each cluster contains a series of closely linked genes. In vertebrates such as mice, it has been shown that these genes are expressed in segmental units in the hindbrain and in global patterning of the somites formed from axial presomitic mesoderm. In each HOX cluster, there is a direct linear correlation between the position of the gene and its temporal and spatial expression. These observations indicate that these genes play a crucial role in early morphogenesis. Thus, in the developing limb bud (p. 99) HOXA9 is expressed both anterior to, and before, HOX10, and so on.
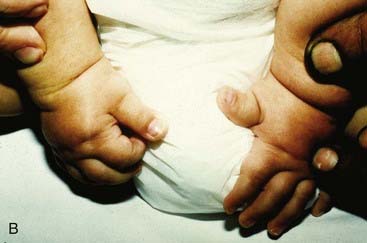
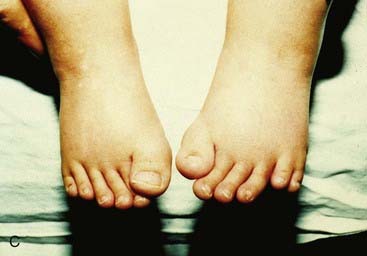
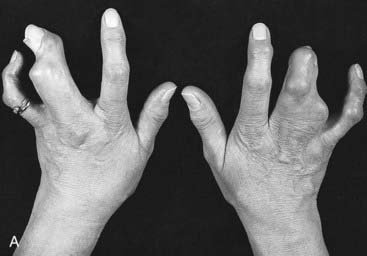
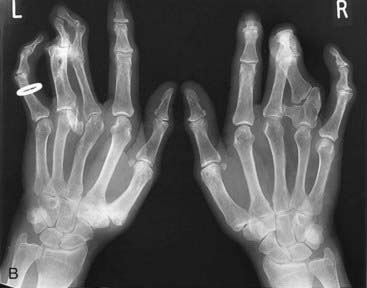
Mutations in HOXA13 cause a rare condition known as the hand-foot-genital syndrome. This shows autosomal dominant inheritance and is characterized by shortening of the first and fifth digits, with hypospadias in males and bicornuate uterus in females. Experiments with mouse Hoxa13 mutants have shown that expression of another gene, EphA7, is severely reduced. Therefore, if this gene is not activated by Hoxa13, there is failure to form the normal chondrogenic condensations in the distal limb primordial. Mutations in HOXD13 result in an equally rare limb developmental abnormality known as synpolydactyly. This also shows autosomal dominant inheritance and is characterized by insertion of an additional digit between the third and fourth fingers and the fourth and fifth toes, which are webbed (Figure 6.11). The phenotype in homozygotes is more severe and reported mutations take the form of an increase in the number of residues in a polyalanine tract. This triplet-repeat expansion probably alters the structure and function of the protein, thereby constituting a gain-of-function mutation (p. 26). Mutated HOXA1 has been found in the rare, recessively inherited Bosley-Saleh-Alorainy syndrome, consisting of central nervous system abnormalities, deafness, and cardiac and laryngotracheal anomalies. A mutation in HOXD10 was found in isolated congenital vertical talus in a large family demonstrating autosomal dominant inheritance, and duplications of HOXD have recently been found in mesomelic limb abnomality syndromes.
Given that there are 39 HOX genes in mammals, it is surprising that so few syndromes or malformations have been attributed to HOX gene mutations. One possible explanation is that most HOX mutations are so devastating that the embryo cannot survive. Alternatively, the high degree of homology between HOX genes in the different clusters could lead to functional redundancy so that one HOX gene could compensate for a loss-of-function mutation in another. In this context HOX genes are said to be paralogous because family members from different clusters, such as HOXA13 and HOXD13, are more similar than adjacent genes in the same cluster.
Paired-Box (PAX) Genes
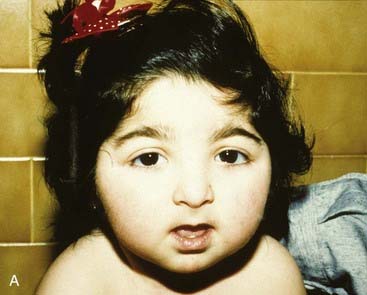
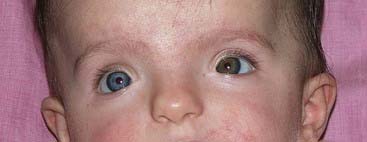
The paired-box is a highly conserved DNA sequence that encodes a 130-amino-acid DNA-binding transcription regulator domain. Nine PAX genes have been identified in mice and humans. In mice these have been shown to play important roles in the developing nervous system and vertebral column. In humans, loss-of-function mutations in five PAX genes have been identified in association with developmental abnormalities (Table 6.2). Waardenburg syndrome type 1 is caused by mutations in PAX3. It shows autosomal dominant inheritance and is characterized by sensorineural hearing loss, areas of depigmentation in hair and skin, abnormal patterns of pigmentation in the iris, and widely spaced inner canthi (Figure 6.12). Waardenburg syndrome shows genetic heterogeneity; the more common type 2 form, in which the inner canthi are not widely separated, is sometimes caused by mutations in the human microphthalmia (MITF) gene on chromosome 3.
Table 6.2 Developmental Abnormalities Associated with PAX Gene Mutations
| Gene | Chromosome Location | Developmental Abnormality |
|---|---|---|
| PAX2 | 10q24 | Renal-coloboma syndrome |
| PAX3 | 2q35 | Waardenburg syndrome type 1 |
| PAX6 | 11p13 | Aniridia |
| PAX8 | 2q12 | Absent or ectopic thyroid gland |
| PAX9 | 14q12 | Oligodontia |
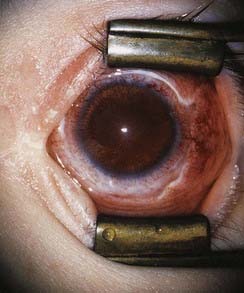
The importance of expression of the PAX gene family in eye development is illustrated by the effects of mutations in PAX2 and PAX6. Mutations in PAX2 cause the renal-coloboma syndrome, in which renal malformations occur in association with structural defects in various parts of the eye, including the retina and optic nerve. Mutations in PAX6 lead to absence of the iris, which is known as aniridia (Figure 6.13). This is a key feature of the WAGR syndrome (p. 282), which results from a contiguous gene deletion involving the PAX6 locus on chromosome 11.
SRY-Type HMG Box (SOX) Genes
SRY is the Y-linked gene that plays a major role in male sex determination (p. 102). A family of genes known as the SOX genes shows homology with SRY by sharing a 79-amino-acid domain known as the HMG (high-mobility group) box. This HMG domain activates transcription by bending DNA in such a way that other regulatory factors can bind with the promoter regions of genes that encode for important structural proteins. These SOX genes are thus transcription regulators and are expressed in specific tissues during embryogenesis. For example, SOX1, SOX2, and SOX3 are expressed in the developing mouse nervous system.
In humans it has been shown that loss-of-function mutations in SOX9 on chromosome 17 cause campomelic dysplasia. This very rare disorder is characterized by bowing of the long bones, sex reversal in chromosomal males, and very poor long-term survival. In-situ hybridization studies in mice have shown that SOX9 is expressed in the developing embryo in skeletal primordial tissue, where it regulates type II collagen expression, as well as in the genital ridges and early gonads. SOX9 is now thought to be one of several genes that are expressed downstream of SRY in the process of male sex determination (p. 102). Mutations in SOX10 on chromosome 22 cause a rare form of Waardenburg syndrome in which affected individuals have a high incidence of Hirschsprung disease. Mutations in SOX2 (3q26) have been shown to cause anophthalmia or microphthalmia, but also a wider syndrome of esophageal atresia and genital hypoplasia in males: the anophthalmia-esophageal-genital syndrome.
Zinc Finger Genes
The term zinc finger refers to a finger-like loop projection consisting of a series of four amino acids that form a complex with a zinc ion. Genes that contain a zinc finger motif act as transcription factors through binding of the zinc finger to DNA. Consequently they are good candidates for single-gene developmental disorders (Table 6.3).
Table 6.3 Developmental Abnormalities Associated with Genes Containing a Zinc Finger Motif
| Gene | Chromosome Location | Developmental Abnormality |
|---|---|---|
| GLI3 | 7p13 | Greig syndrome and Pallister-Hall syndrome |
| WT1 | 11p13 | Denys-Drash syndrome |
| ZIC2 | 13q32 | Holoprosencephaly |
| ZIC3 | Xq26 | Laterality defects |
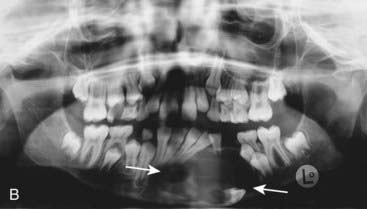
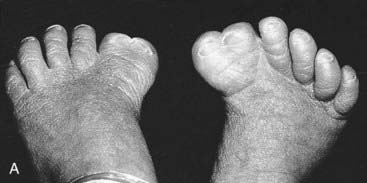
For example, a zinc finger motif–containing gene known as GLI3 on chromosome 7 (as mentioned, a component of the SHH pathway) has been implicated as the cause of two developmental disorders. Large deletions or translocations involving GLI3 cause Greig cephalopolysyndactyly, which is characterized by head, hand and foot abnormalities such as polydactyly and syndactyly (Figure 6.14, A). In contrast, frameshift mutations in GLI3 have been reported in the Pallister-Hall syndrome (Figure 6.14, B), in which the key features are polydactyly, hypothalamic hamartomata and imperforate anus.

Mutations in another zinc finger motif-containing gene known as WT1 on chromosome 11 can cause both Wilms’ tumour and a rare developmental disorder, the Denys-Drash syndrome, in which the external genitalia are ambiguous and there is progressive renal failure as a result of nephritis. Mutations in two other zinc finger motif–containing genes, ZIC2 and ZIC3, have been shown to cause holoprosencephaly and laterality defects, respectively. Just as polarity is a key concept in development, so too is laterality, with implications for the establishment of a normal left-right body axis. In very early development, integrity of many of the same gene families previously mentioned—Nodal, SHH, and Notch—is essential to the establishment of this axis. Clinically, situs solitus is the term given to normal left-right asymmetry and situs inversus to reversal of the normal arrangement. Up to 25% of individuals with situs inversus have an autosomal recessive condition—Kartagener syndrome, or ciliary dyskinesia. Other terms used are isomerism sequence, heterotaxy, asplenia/polyasplenia, and Ivemark syndrome. Laterality defects are characterized by abnormal positioning of unpaired organs such as the heart, liver, and spleen, and more than 20 genes are now implicated from studies in vertebrates, with a number identified in humans by the study of affected families, with all of the main patterns of inheritance represented.
Signal Transduction (’Signaling’) Genes
Signal transduction is the process whereby extracellular growth factors regulate cell division and differentiation by a complex pathway of genetically determined intermediate steps. Mutations in many of the genes involved in signal transduction play a role in causing cancer (p. 213). In some cases they can also cause developmental abnormalities.
FGF Receptors
FGFs play key roles in embryogenesis, including cell division, migration, and differentiation. The transduction of extracellular FGF signals is mediated by a family of four transmembrane tyrosine kinase receptors. These are the fibroblast growth factor receptors (FGFRs), each of which contains three main components: an extracellular region with three immunoglobulin-like domains, a transmembrane segment, and two intracellular tyrosine kinase domains (Figure 6.15).
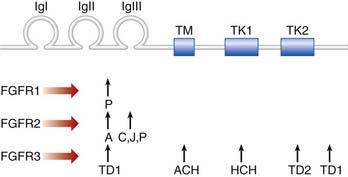
Mutations in the genes that code for FGFRs have been identified in two groups of developmental disorders (Table 6.4). These are the craniosynostosis syndromes and the achondroplasia family of skeletal dysplasias. The craniosynostosis syndromes, of which Apert syndrome (Figure 6.16) is the best known, are characterized by premature fusion of the cranial sutures, often in association with hand and foot abnormalities such as syndactyly (fusion of the digits). Apert syndrome is caused by a mutation in one of the adjacent FGFR2 residues in the peptides that link the second and third immunoglobulin loops (see Figure 6.15). In contrast, mutations in the third immunoglobulin loop can cause either Crouzon syndrome, in which the limbs are normal, or Pfeiffer syndrome, in which the thumbs and big toes are broad. Achondroplasia is the most commonly encountered form of genetic short stature (Figure 6.17). The limbs show proximal (‘rhizomelic’) shortening and the head is enlarged with frontal bossing. Intelligence and life expectancy are entirely normal. Achondroplasia is almost always caused by a mutation in, or close to, the transmembrane domain of FGFR3. The common transmembrane domain mutation leads to the replacement of a glycine amino-acid residue by an arginine—an amino acid that is never normally found in cell membranes. This in turn appears to enhance dimerization of the protein that catalyzes downstream signaling. Hypochondroplasia, a milder form of skeletal dysplasia with similar trunk and limb changes but normal head shape and size, is caused by mutations in the proximal tyrosine kinase domain (intracellular) of FGFR3. Finally, thanatophoric dysplasia, a much more severe and invariably lethal form of skeletal dysplasia, is caused by mutations in either the peptides linking the second and third immunoglobulin domains (extracellular) of FGFR3, or the distal FGFR3 tyrosine kinase domain.
Table 6.4 Developmental Disorders Caused by Mutations in Fibroblast Growth Factor Receptors
| Gene | Chromosome | Syndrome |
|---|---|---|
| Craniosynostosis syndromes | ||
| FGFR1 | 8p11 | Pfeiffer |
| FGFR2 | 10q25 | Apert Crouzon Jackson-Weiss Pfeiffer |
| FGFR3 | 4p16 | Crouzon (with acanthosis nigricans) |
| Skeletal dysplasias | ||
| FGFR3 | 4p16 | Achondroplasia Hypochondroplasia Thanatophoric dysplasia |

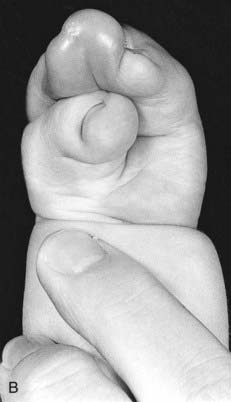
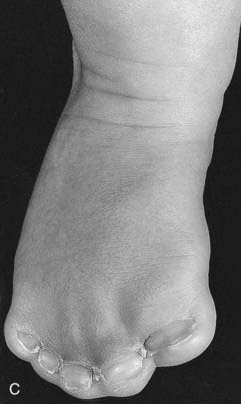
The mechanism by which these mutations cause skeletal shortening is not understood at present. The mutations cannot have loss-of-function effects as children with the Wolf-Hirschhorn syndrome (pp. 280–281), which is due to chromosome microdeletions that include FGFR3, do not show similar skeletal abnormalities. Instead, the mutations probably involve a gain of function mediated by increased ligand binding or receptor activation.
The Pharyngeal Arches
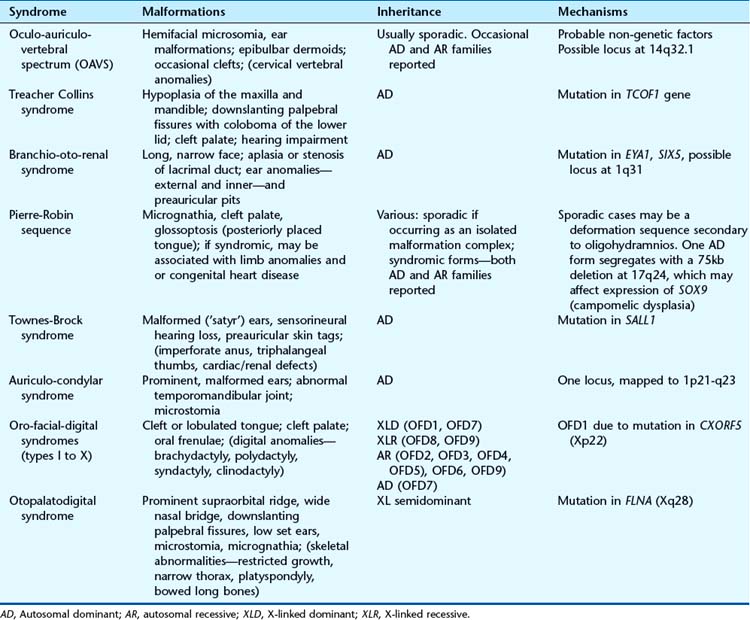
The pharyngeal (or branchial) arches correspond to the gill system of lower vertebrates and appear in the fourth and fifth weeks of development. Five (segmented) pharyngeal arches in humans arise lateral to the structures of the head (Figure 6.18) and each comprises cells from the three germ layers and the neural crest. The lining of the pharynx, thyroid, and parathyroids arises from the endoderm, and the outer epidermal layer arises from the ectoderm. The musculature arises from the mesoderm, and bony structures from neural crest cells. Separating the arches are the pharyngeal clefts externally and the pharyngeal pouches internally; these have important destinies. Numbered from the rostral end, the first arch forms the jaw and muscles of mastication, the first cleft is destined to be the external auditory meatus, and the first pouch the middle ear apparatus. The second arch forms the hyoid apparatus and muscles of facial expression, whereas the third pouch develops into the thymus, and the third and fourth pouches become the parathyroids. The arteries within the arches have important destinies too and, after remodeling, give rise to the aortic and pulmonary arterial systems. Some of the syndromes, malformations, inheritance patterns and genetic mechanisms associated with the first and second pharyngeal arches are listed in Table 6.5.
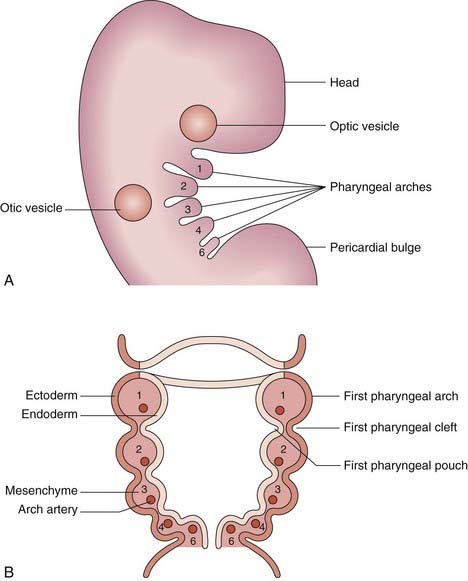
However, the most well known, and probably most common, condition due to disturbed development of pharyngeal structures—the third and fourth pouches—is DiGeorge syndrome (DGS), also known as velocardiofacial syndrome (VCFS), and well described even earlier by Sedláčková of Prague in 1955. This is described in more detail in Chapter 18 (pp. 282–283); it results from a 3Mb submicroscopic chromosome deletion of band 22q11 with the loss of some 30 genes. Studies in mice (the equivalent, or syntenic, region is on mouse chromosome 16) suggest that the most significant gene loss is that of Tbx1, strongly expressed throughout the pharyngeal apparatus. Heterozygous Tbx1 knock-out mice show hypoplastic or absent fourth pharyngeal arch arteries, suggesting that TBX1 in humans is the key. Indeed, mutations in this gene have now been found in some congenital heart abnormalities and it is possible that TBX1 is the key gene for other elements of the phenotype. However, there are still unanswered questions in the whole DGS/VCFS/Sedláčková story.
Role of Cilia in Developmental Abnormalities
Cilia are the equivalent of flagella in wider biology and they share structural identity. They are hairlike protrusions from the cell surface (Figure 6.19), up to 20 µm long and, present in large numbers on the apical cell surface, beat in coordinated waves. In cross-section they consist of a scaffold of nine microtubule doublets surrounding a central pair. The central and outer doublets are connected by radial spokes, which produce the force necessary to bend the cilium; dynein arms facilitate this movement. They clear mucous from the respiratory epithelium, drive sperm along the Fallopian tube, and move cerebrospinal fluid in the cavities of the central nervous system. In development the cilia at the organizational node of the vertebrate embryo conduct a circular motion, wafting molecules unidirectionally and helping to establish left-right asymmetry.
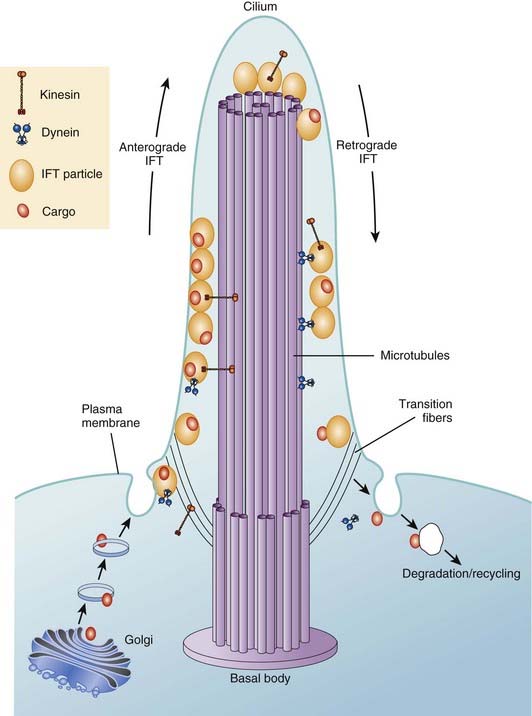
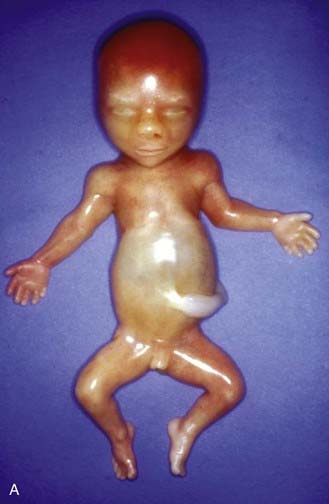
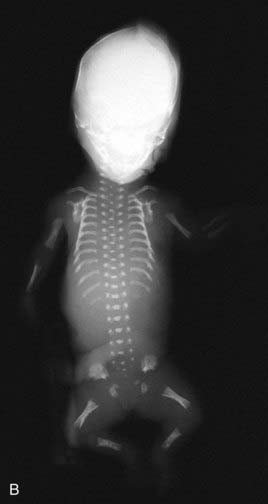
Apart from their obvious mechanical function, which conceptually is straightforward, it appears increasingly likely that cilia behave like molecular antennae that sense extracellular signaling molecules. The sonic hedgehog and Wnt signaling pathways depend to an extent on cilial functional integrity for optimal signaling. Defective cilial function can therefore impact on a broad range of developmental processes and pathways. Defects in the cilial proteins themselves lead to wide-ranging phenotypic effects that include retinal degeneration, anosmia, renal, hepatic and pancreatic cyst formation, postaxial polydactyly, and situs inversus. The growing list of recognizable syndromes is now referred to as ‘ciliopathies’ (Table 6.6). One of these, short-rib polydactyly syndrome, which follows autosomal recessive inheritance and is due to mutated DYNC2H1, is shown in Figure 6.20. The features of the listed syndromes in Table 6.6 overlap with many other multiple congenital abnormality syndromes, and we shall certainly see the relevance of cilia to developmental genetics increase in the years ahead.

The Limb as a Developmental Model
Tissue Differentiation and Growth
Once limb formation has been initiated, a localized area of thickened ectoderm at the limb tip, known as the apical ectodermal ridge (AER), produces growth signals such as FGF4 and FGF8, which maintain further growth and establish the proximo-distal axis (Figure 6.21). Expression of the gene TP63 is crucial for sustaining the AER and, when this gene is mutated, split hand-foot (ectrodactyly) malformations result, often together with oral clefting and other anomalies—ectrodactyly-ectodermal dysplasia-clefting syndrome. Signals from another localized area on the posterior margin of the developing bud, known as the zone of polarizing activity, determine the anteroposterior axis. One of these signals is SHH (pp. 86–88), which acts in concert with other FGF genes, GLI3, and another gene family, which produces BMPs. Another morphogen, retinoic acid, is believed to play a major role at this stage in determining development at the anterior margin of the limb bud.
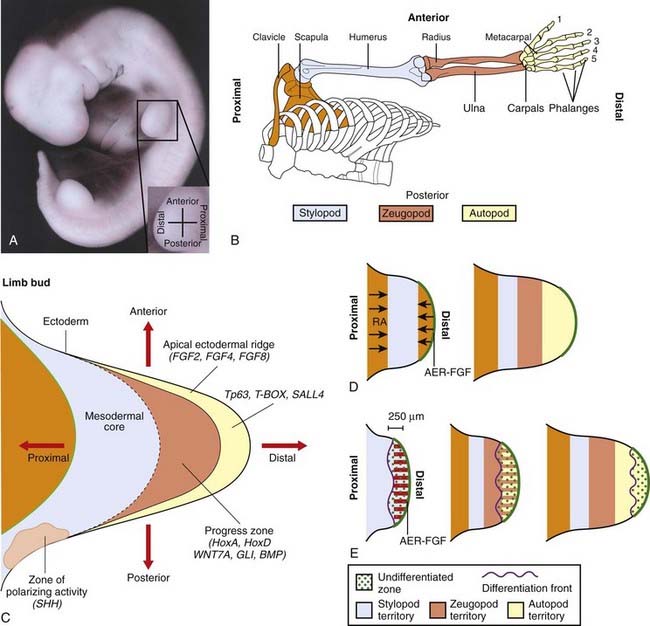
Subsequent development involves the activation of genes from the HOXA and HOXD clusters in the undifferentiated proliferating mesenchymal cells beneath the AER. This area is known as the progress zone. Cells in different regions express different combinations of HOX genes that determine local cell proliferation, adhesion, and differentiation. Downstream targets of the HOX gene clusters remain to be identified. Other genes that clearly have a key role are those of the T-box family, already discussed, and SALL4, which is mutated in Okihiro syndrome (radial ray defects with abnormal eye movements resulting from congenital palsy affecting the sixth cranial nerve).
FGFs continue to be important during the later stages of limb development. In this context it becomes easy to understand why limb abnormalities are a feature of disorders such as Apert syndrome (see Figure 6.16), in which mutations have been identified in the extracellular domains of FGFR2.
Developmental Genes and Cancer
Several genes that play important roles in embryogenesis have also been shown to play a role in causing cancer (Table 6.7). This is not surprising, given that many developmental genes are expressed throughout life in processes such as signal transduction and signal transcription (p. 213). It has been shown that several different mechanisms can account for the phenotypic diversity demonstrated by these so-called teratogenes.

Gain-of-Function versus Loss-of-Function Mutations
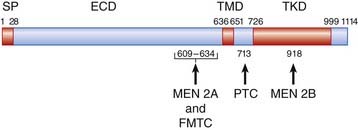
Mention has already been made of the causal role of the RET proto-oncogene in familial Hirschsprung disease, as well as in both inherited and sporadic thyroid cancer (p. 93). The protein product encoded by RET consists of three main domains: an extracellular domain that binds to a glial cell line–derived neurotrophil factor, a transmembrane domain, and an intracellular tyrosine kinase domain that activates signal transduction (Figure 6.22). Mutations causing loss of function result in Hirschsprung disease. These include whole gene deletions, small intragenic deletions, nonsense mutations, and splicing mutations leading to synthesis of a truncated protein.
Positional Effects and Developmental Genes
The discovery of a chromosomal abnormality, such as a translocation or inversion, in a person with a single-gene developmental syndrome provides a strong indication of the probable position of the disease locus, because it is likely that one of the breakpoints involved in the rearrangement will have disrupted the relevant gene. However, in a few instances, it has emerged that the chromosome breakpoint actually lies approximately 10 to 1000 kb upstream or downstream of the gene that is subsequently shown to be mutated in other affected individuals (Table 6.8). The probable explanation is that the breakpoint has separated the coding part of the gene from contiguous regulatory elements (p. 21). These observations have created obvious difficulties for those carrying out the original research when the putative disease gene in translocation families has been found not to contain an intragenic mutation.
| Gene | Chromosome | Developmental Anomaly |
|---|---|---|
| GLI3 | 7p13 | Greig cephalopolysyndactyly |
| SHH | 7q36 | Holoprosencephaly |
| PAX6 | 11p13 | Aniridia |
| SOX9 | 17q24 | Campomelic dysplasia |
Hydatidiform Moles
Occasionally conception results in an abnormal pregnancy in which the placenta consists of a proliferating disorganized mass known as a hydatidiform mole. These changes can be either partial or complete (Table 6.9).
Table 6.9 Characteristics of Partial and Complete Hydatidiform Moles
| Partial Mole | Complete Mole | |
|---|---|---|
| No. of chromosomes | 69 | 46 |
| Parental origin of chromosomes | 23 maternal 46 paternal |
All 46 paternal |
| Fetus present | Yes, but not viable | No |
| Malignant potential | Very low | High |
Partial Hydatidiform Mole
Chromosome analysis of tissue from partial moles reveals the presence of 69 chromosomes—i.e., triploidy (p. 276). Using DNA polymorphisms, it has been shown that 46 of these chromosomes are always derived from the father, with the remaining 23 being maternal in origin. This doubling of the normal haploid paternal contribution of 23 chromosomes can be due to either fertilization by two sperm, which is known as dispermy, or to duplication of a haploid sperm chromosome set by a process known as endoreduplication.
Different Parental Expression in Trophoblast and Embryoblast
Studies in mice have shown that when all nuclear genes in a zygote are derived from the father, the embryo fails to develop, whereas trophoblast development proceeds relatively unimpaired. In contrast, if all of the nuclear genes are maternal in origin, the embryo develops normally, but extra-embryonic development is poor. The observations outlined previously on partial and complete moles indicate that a comparable situation exists in humans, with paternally derived genes being essential for trophoblast development and maternally derived genes being necessary for early embryonic development. These phenomena are relevant to the concept of epigenetics (see the following section, p. 103) and genomic imprinting (p. 121).
Sexual Differentiation and Determination
The sex of an individual is determined by the X and Y chromosomes (p. 33). The presence of an intact Y chromosome leads to maleness regardless of the number of X chromosomes present. Absence of a Y chromosome results in female development.
Although the sex chromosomes are present from conception, differentiation into a phenotypic male or female does not commence until approximately 6 weeks. Up to this point both the müllerian and Wolffian duct systems are present and the embryonic gonads, although consisting of cortex and medulla, are still undifferentiated. From 6 weeks onwards, the embryo develops into a female unless the testis-determining factor initiates a sequence of events that prompt the undifferentiated gonads to develop into testes.
The Testis-Determining Factor—SRY
In 1990 it was shown that the testis-determining factor or gene is located on the short arm of the Y chromosome close to the pseudoautosomal region (p. 118). This gene is now referred to as being located in the sex-determining region of the Y chromosome (SRY). It consists of a single exon that encodes a protein of 204 amino acids that include a 79-amino-acid HMG box (p. 92), indicating that it is likely to be a transcription regulator.
From a biological viewpoint (i.e., the maintenance of the species), it would clearly be impossible for the SRY gene to be involved in crossing over with the X chromosome during meiosis I. Hence SRY has to lie outside the pseudoautosomal region. However, there has to be pairing of X and Y chromosomes, as otherwise they would segregate together into the same gamete during, on average, 50% of meioses. Nature’s compromise has been to ensure that only a small portion of the X and Y chromosomes are homologous, and therefore pair during meiosis I. Unfortunately, the close proximity of SRY to the pseudoautosomal region means that, occasionally, it can get caught up in a recombination event. This almost certainly accounts for the majority of XX males, in whom molecular and fluorescent in-situ hybridization studies show evidence of Y-chromosome sequences at the distal end of one X-chromosome short arm (see Figure 18.22, p. 288).
Expression of SRY triggers off a series of events that involves other genes such as SOX9 (17q24), leading to the medulla of the undifferentiated gonad developing into a testis, in which the Leydig cells begin to produce testosterone (Figure 6.23). This leads to stimulation of the Wolffian ducts, which form the male internal genitalia, and also to masculinization of the external genitalia. This latter step is mediated by dihydrotestosterone, which is produced from testosterone by the action of 5α-reductase (pp. 174, 287). The Sertoli cells in the testes produce a hormone known as müllerian inhibitory factor, which causes the müllerian duct system to regress. In campomelic dysplasia, resulting from mutated SOX9, ambiguous genitalia is common in cases with a 46,XY karyotype. Ambiguous genitalia, or sex reversal, is also frequent in cases of deletion 9p24.3 syndrome. This is probably due to haploinsufficiency for the DMRT1 gene, a transcriptional regulator expressed in Sertoli cells, spermatogonia, and spermatocytes.
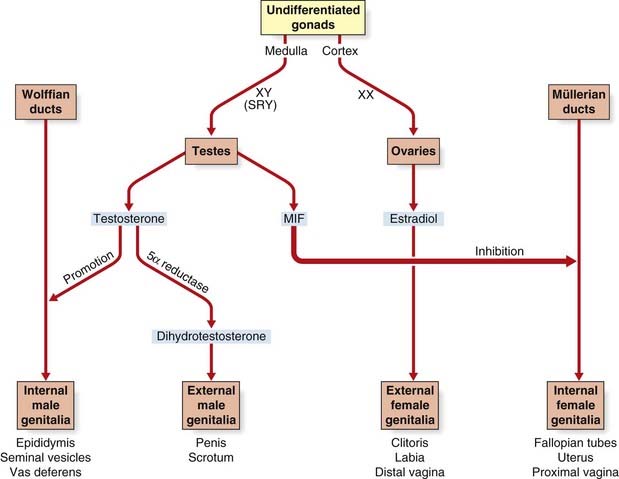
Normally sexual differentiation is complete by 12 to 14 weeks’ gestation, although the testes do not migrate into the scrotum until late pregnancy. Abnormalities of sexual differentiation are uncommon but they are important causes of infertility and sexual ambiguity. They are considered further in Chapter 18.
Epigenetics and Development
The most common form of DNA modification—the biochemical mechanism for epigenesis—is direct covalent methylation of nucleotides. This appears to lead to a series of steps that alters local chromatin structure. In human genetics the best recognized epigenetic phenomena are X-chromosome inactivation, described in the following section, and parent-of-origin specific gene expression (parental imprinting), which is realized in Prader-Willi and Angelman syndromes (pp. 122–123), and Beckwith-Wiedemann and Russell-Silver syndromes (pp. 124–125)—i.e., when errors occur. There is much interest, however, in the possibility that epigenetic states can be influenced by environmental factors. In animal studies there is evidence that the nutritional and behavioral environment may lead to different ‘epialleles’, and in human populations epidemiological studies have shown convincing correlations of maternal (and in some cases grandparental) nutritional status with late-onset cardiovascular and metabolic-endocrine disease.
X-Chromosome Inactivation
The process of XCI occurs early in development at around 15 to 16 days’ gestation, when the embryo consists of approximately 5000 cells. Normally either of the two X chromosomes can be inactivated in any particular cell. Thereafter the same X chromosome is inactivated in all daughter cells (Figure 6.24). This differs from the case in marsupials, in which the paternally derived X chromosome is consistently inactivated.
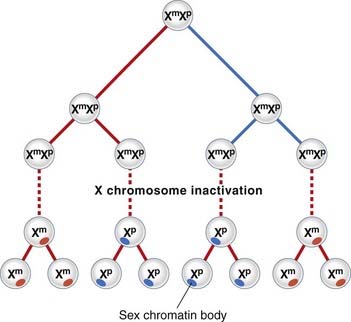
The inactive X chromosome exists in a condensed form during interphase when it appears as a darkly staining mass of chromatin known as the sex chromatin, or Barr body. During mitosis the inactive X chromosome is late replicating. Laboratory techniques have been developed for distinguishing which of the X chromosomes is late replicating in each cell. This can be useful for confirming that one of the X chromosomes is structurally abnormal, as usually an abnormal X chromosome will be preferentially inactivated; or, more correctly, only those hematopoietic stem cells in which the normal X chromosome is active will have survived. Apparent non-random inactivation also occurs when one of the X chromosomes is involved in a translocation with an autosome (p. 116).
The epigenetic process of XCI is achieved by differential methylation (a form of imprinting; see p. 121) and is initiated by a gene, XIST (‘X inactivation specific transcript’), which maps within the X-inactivation centre at Xq13.3. XIST is expressed only from the inactive X chromosome and produces RNA that spreads an inactivation methylation signal up and down the X chromosome on which it is located. This differential methylation of the X chromosomes has been utilized in carrier detection studies for X-linked immunodeficiency diseases (e.g., Wiskott-Aldrich syndrome) using methylation-sensitive restriction enzymes (p. 204). Not all of the X chromosome is inactivated. Genes in the pseudoautosomal region at the tip of the short arm remain active, as do other loci elsewhere on the short and long arms, such as XIST. There are more genes that escape XCI in Xp compared with Xq. This probably explains why more severe phenotypic effects are seen in women with small Xp chromosome deletions compared with those in women with small deletions in Xq. If all loci on the X chromosome were inactivated, then all women would have the clinical features of Turner syndrome and the presence of more than one X chromosome in a male (e.g., 47,XXY) or two in a female (e.g., 47,XXX) would have no phenotypic effects. There are, in fact, quite characteristic clinical features in these disorders (p. 278).
XCI provides a satisfactory explanation for several observations, described below.
Barr Bodies
In men and women with more than one X chromosome, the number of Barr bodies (p. 39) visible at interphase is always one less than the total number of X chromosomes. For example, men with a 47,XXY karyotype have a single Barr body, whereas women with a 47,XXX karyotype have two Barr bodies.
Mosaicism
Mice that are heterozygous for X-linked genes affecting coat color show mosaicism with alternating patches of different color rather than a homogeneous pattern. This is consistent with patches of skin being clonal in origin in that they are derived from a single stem cell in which one or other of the X chromosomes is expressed, but not both. Thus, each patch reflects which of the X chromosomes was active in the original stem cell. Similar effects are seen in tissues of clonal origin in women who are heterozygous for X-linked mutations such as ocular albinism (Figure 6.25).
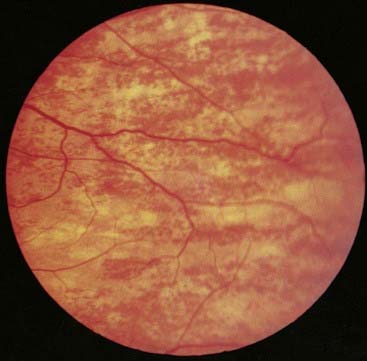
FIGURE 6.25 The fundus of a carrier of X-linked ocular albinism showing a mosaic pattern of retinal pigmentation.
(Courtesy Mr. S.J. Charles, The Royal Eye Hospital, Manchester, UK.)
Other evidence confirming that X-inactivation leads to mosaicism in females comes from studies of the expression of the enzyme glucose-6-phosphate dehydrogenase (p. 187) in clones of cultured fibroblasts from women heterozygous for variants of this gene. Each clone is derived from a single cell and expresses one of the variants, but never both. The clonal origin of tumors can be confirmed in women who are heterozygous for such variants by demonstration of the expression of only one of the variants in the tumor.
Problems of Carrier Detection
Carrier detection for X-linked recessive disorders based only on examination of clinical features or on indirect assay of gene function is notoriously difficult and unreliable. Cells in which the X chromosome with the normal gene is active can have a selective advantage, or they can correct the defect in closely adjacent cells in which the X chromosome with the mutant gene is active. For example, only a proportion of carriers of Duchenne muscular dystrophy (DMD) show evidence of muscle damage as indicated by measurement of creatine kinase in serum (p. 314). Similarly, distorted ratios of very long chain fatty acids are seen in many, but not all, carriers of X-linked adrenoleukodystrophy.
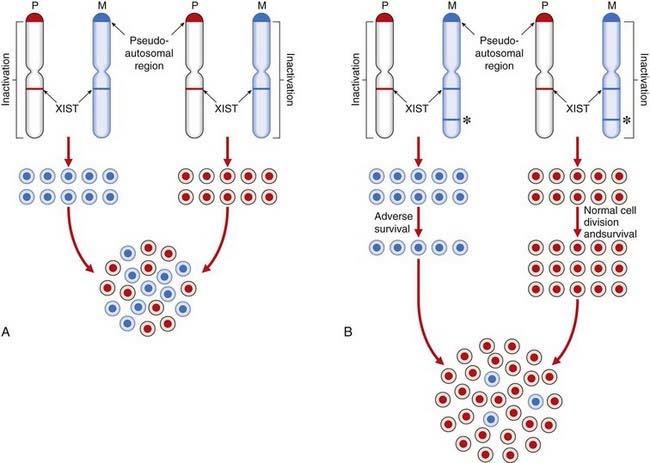
Fortunately, the development of molecular methods for carrier detection in X-linked disorders can bypass these problems by use of PCR primers that distinguish the products of methylated and unmethylated DNA—if there has been selection against the cell line in which the mutant-bearing X chromosome is active (Figure 6.26).
Manifesting Heterozygotes
Occasionally a woman is encountered who shows mild or even full expression of an X-linked recessive disorder, such as DMD or hemophilia A. One possible explanation is that she is a manifesting heterozygote in whom, by chance, the X chromosome bearing the normal gene has been inactivated in significantly more than 50% of relevant cells. This is referred to as skewed X-inactivation (p. 116). There is some evidence that X-chromosome inactivation can itself be under genetic control, because families with several manifesting carriers of disorders such as DMD and Fabry disease have been reported. In a few families, marked skewing of X-inactivation in several females has been shown to be associated with an underlying mutation in XIST.
Recent Research
Research has suggested that, just as XCI is not an all-or-none phenomenon for the whole chromosome, it is probably not all-or-none for every gene. In a study of skin fibroblasts, which express more than 600 of the 1098 genes identified on the X chromosome, about 20% were found to be inactivated in some but not all samples. About 15% escaped XCI completely, whereas only 65% were fully silenced and thus expressed in one dose. In addition to non-random XCI, the variable dosage of genes that escape XCI may account for variation among normal females as well as those who are heterozygous for X-linked disease genes.
Twinning
Twins can be identical or non-identical—i.e., monozygotic (MZ) (uniovular) or dizygotic (DZ) (biovular) —depending on whether they originate from a single conception or from two separate conceptions (Table 6.10). Comparison of the incidence of disease in MZ and DZ twins reared apart and together can provide information about the relative contributions of genetics and the environment to the cause of many of the common diseases of adult life (see Chapter 15), especially the study of mental health and behavior.
Table 6.10 Summary of Differences between Monozygotic and Dizygotic Twins
| Monozygotic | Dizygotic | |
|---|---|---|
| Origin | Single egg fertilized | Two eggs, each fertilized by a single sperm |
| Incidence | 1 in 300 pregnancies | Varies from 1 in 100 to 1 in 500 pregnancies |
| Proportion of genes in common | 100% | 50% (on average) |
| Fetal membranes | 70% monochorionic and diamniotic; 30% dichorionic and diamniotic; rarely monochorionic and monoamniotic | Always dichorionic and diamniotic |
Monozygotic Twins
The sex ratio for conjoined twins is markedly distorted, with about 75% being female. The later the twinning event, the more distorted the sex ratio in favor of females, and X-inactivation studies suggest that MZ twinning occurs around the time of X-inactivation, a phenomenon limited to female zygotes, of course.
Determination of Zygosity
Zygosity used to be established by study of the placenta and membranes and also by analysis of polymorphic systems such as the blood groups, the human leukocyte antigens and other biochemical markers. Now it is determined most reliably by the use of highly polymorphic molecular (DNA) markers (pp. 69–70) and single nucleotide polymorphisms (SNPs).
Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet. 2009;151C:281-295.
Dreyer SD, Zhou G, Lee B. The long and the short of it: developmental genetics of the skeletal dysplasias. Clin Genet. 1998;54:464-473.
A short review of developmental genes known to cause abnormal skeletal development.
Hall JG. 2003 Twinning. Lancet. 2003;362:735-743.
Hammerschmidt M, Brook A, McMahon AP. The world according to hedgehog. Trends Genet. 1997;13:14-20.
A comprehensive account of the role of the hedgehog gene family in early vertebrate development.
Kleinjan DJ, van Heyningen V. Position effect in human genetic disease. Hum Mol Genet. 1998;7:1611-1618.
Kornak U, Mundlos S. Genetic disorders of the skeleton: a developmental approach. Am J Hum Genet. 2003;73:447-474.
An up-to-date summary of current knowledge.
Lacombe D. Transcription factors in dysmorphology. Clin Genet. 1999;55:137-143.
Lindor NM, Ney JA, Gaffey TA, et al. A genetic review of complete and partial hydatidiform moles and nonmolar triploidy. Mayo Clin Proc. 1992;67:791-799.
A detailed review of the mechanisms that can lead to the formation of hydatidiform moles.
Lyon MF. Gene action in the X chromosome of the mouse (Mus musculus L). Nature. 1961;190:372-373.
The original proposal of X-inactivation—very short and easily understood.
Manouvrier-Hanu S, Holder-Espinasse M, Lyonnet S. Genetics of limb anomalies in humans. Trends Genet. 1999;15:409-417.
A detailed and well-illustrated account of vertebrate limb development.
Muenke M, Schell U. Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 1995;11:308-313.
A concise review of the functions of the fibroblast growth factors and their receptors.
Muragaki Y, Mundlos S, Upton J, Olsen BR. Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science. 1996;272:548-551.
The long-awaited first report of a human malformation caused by a mutation in a HOX gene.
Passos-Bueno MR, Ornelas CC, Fanganiello RD. Syndromes of the first and second pharyngeal arches: a review. Am J Med Genet Part A. 2009;149A:1853-1859.
An excellent short overview of pharyngeal arches and associated syndromes.
Saga Y, Takeda H. The making of the somite: molecular events in vertebrate segmentation. Nature Rev Genet. 2001;2:835-844.
An excellent review of somite development.
Tickle C, editor. Patterning in vertebrate development. Oxford: Oxford University Press, 2003.
Villavicencio EH, Walterhouse DO, Iannaccone PM. The Sonic hedgehog-patched-Gli pathway in human development and disease. Am J Hum Genet. 2000;67:1047-1054.
An excellent short overview of the Sonic hedgehog pathway.
Elements
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 6 Developmental Genetics
At fertilization the nucleus from a spermatozoon penetrates the cell membrane of an oocyte to form a zygote. This single cell divides to become two, then four, and when the number has doubled some 50 times the resulting organism comprises more than 200 distinct cell types and a total cell number of about 10,000 trillion. This is a fully formed human being with complex biochemistry and physiology, capable of exploring the cosmos and identifying subatomic particles. Not surprisingly, biologists and geneticists are intrigued by the mechanisms of early development and, whilst many mysteries remain, the rate of progress in understanding key events and signaling pathways is rapid.
Prenatal life can be divided into three main stages: pre-embryonic, embryonic, and fetal (Table 6.1). During the pre-embryonic stage, a small collection of cells becomes distinguishable, first as a double-layered or bilaminar disc, and then as a triple-layered or trilaminar disc (Figure 6.1), which is destined to develop into the human infant. During the embryonic stage, craniocaudal, dorsoventral, and proximodistal axes are established, as cellular aggregation and differentiation lead to tissue and organ formation. The final fetal stage is characterized by rapid growth and development as the embryo, now known as a fetus, matures into a viable human infant.
Table 6.1 Main Events in the Development of a Human Infant
| Stage | Time from Conception | Length of Embryo/Fetus |
|---|---|---|
| Pre-embryonic | ||
| First cell division | 30 h | |
| Zygote reaches uterine cavity | 4 d | |
| Implantation | 5–6 d | |
| Formation of bilaminar disc | 12 d | 0.2 mm |
| Lyonization in female | 16 d | |
| Formation of trilaminar disc and primitive streak | 19 d | 1 mm |
| Embryonic stage | ||
| Organogenesis | 4–8 w | |
| Brain and spinal cord are forming, and first signs of heart and limb buds | 4 w | 4 mm |
| Brain, eyes, heart and limbs developing rapidly, and bowel and lungs beginning to develop | 6 w | 17 mm |
| Digits have appeared. Ears, kidneys, liver and muscle are developing | 8 w | 4 cm |
| Palate closes and joints form | 10 w | 6 cm |
| Sexual differentiation almost complete | 12 w | 9 cm |
| Fetal stage | ||
| Fetal movements felt | 16–18 w | 20 cm |
| Eyelids open. Fetus is now viable with specialized care | 24–26 w | 35 cm |
| Rapid weight gain due to growth and accumulation of fat as lungs mature | 28–38 w | 40–50 cm |
FIGURE 6.1 A schematic trilaminar disc, sectioned along the rostrocaudal axis. Cells from the future ectoderm (top layer) migrate through the primitive streak to form the endoderm (bottom layer) and mesoderm (blue). Formation of the neutral plate in the overlying ectoderm, destined to be the central nervous system, involves sonic hedgehog signaling (p. 86) from the notochord and prechordial plate mesoderm.
(Redrawn with permission from Larsen WJ 1998 Essentials of human embryology. New York: Churchill Livingstone.)
Fertilization and Gastrulation
Fertilization, the process by which the male and female gametes fuse, occurs in the fallopian tube. Of the 100 to 200 million spermatozoa deposited in the female genital tract, only a few hundred reach the site of fertilization. Of these, usually only a single spermatozoon succeeds in penetrating first the corona radiata, then the zona pellucida, and finally the oocyte cell membrane, whereupon the oocyte completes its second meiotic division (see Figure 3.15, p. 40). After the sperm has penetrated the oocyte and the meiotic process has been completed, the two nuclei, known as pronuclei, fuse, thereby restoring the diploid number of 46 chromosomes. This is a potentially chaotic molecular encounter with a high chance of failure, as we know from observations of the early human embryo from in-vitro fertilization programs. It may be likened, somewhat flippantly, to ‘speed dating’, whereby couples test whether they might be compatible on the basis of only a few minutes conversation.
Germ cell and very early embryonic development are two periods characterized by widespread changes in DNA methylation patterns—epigenetic reprogramming (see p. 103). Primordial germ cells are globally demethylated as they mature and are subsequently methylated de novo during gametogenesis, the time when most DNA methylation imprints are established. After fertilization a second wave of change occurs. The oocyte rapidly removes the methyl imprints from the sperm’s DNA, which has the effect of resetting the developmental stopwatch to zero. By contrast, the maternal genome is more passively demethylated in such a way that imprinting marks resist demethylation. A third wave of methylation, de novo, establishes the somatic cell pattern of DNA methylation after implantation. These alternating methylation states help to control which genes are active, or expressed, at a time when two genomes, initially alien to each other, collide.
The fertilized ovum or zygote undergoes a series of mitotic divisions to consist of two cells by 30 hours, four cells by 40 hours, and 12 to 16 cells by 3 days, when it is known as a morula. A key concept in development at all stages is the emergence of polarity within groups of cells—part of the process of differentiation that generates multiple cell types with unique identities. Although precise mechanisms remain elusive, observations suggest that this begins at the very outset; in the fertilized egg of the mouse, the point of entry of the sperm determines the plane through which the first cell cleavage division occurs. This seminal event is the first step in the development of the so-called dorso-ventral, or primary body, axis in the embryo.
Further cell division leads to formation of a blastocyst, which consists of an inner cell mass or embryoblast, destined to form the embryo, and an outer cell mass or trophoblast, which gives rise to the placenta. The process of converting the inner cell mass into first a bilaminar, and then a trilaminar, disc (see Figure 6.1) is known as gastrulation, and takes place between the beginning of the second and the end of the third weeks.
Between 4 and 8 weeks the body form is established, beginning with the formation of the primitive streak at the caudal end of the embryo. The germinal layers of the trilaminar disc give rise to ectodermal, mesodermal, and endodermal structures (Box 6.1). The neural tube is formed and neural crest cells migrate to form sensory ganglia, the sympathetic nervous system, pigment cells, and both bone and cartilage in parts of the face and branchial arches.
Disorders involving cells of neural crest origin, such as neurofibromatosis (p. 298), are sometimes referred to as neurocristopathies. This period between 4 and 8 weeks is described as the period of organogenesis, because during this interval all of the major organs are formed as regional specialization proceeds in a craniocaudal direction down the axis of the embryo.
Developmental Gene Families
Information about the genetic factors that initiate, maintain, and direct embryogenesis is incomplete. However, extensive genetic studies of the fruit fly, Drosophila melanogaster, and vertebrates such as mouse, chick, and zebra-fish have identified several genes and gene families that play important roles in early developmental processes. It has also been possible through painstaking gene expression studies to identify several key developmental pathways, or cascades, to which more detail and complexity is continually being added. The gene families identified in vertebrates usually show strong sequence homology with developmental regulatory genes in Drosophila. Studies in humans have revealed that mutations in various members of these gene families can result in either isolated malformations or multiple congenital anomaly syndromes (see Table 16.5, p. 256). Many developmental genes produce proteins called transcription factors (p. 22), which control RNA transcription from the DNA template by binding to specific regulatory DNA sequences to form complexes that initiate transcription by RNA polymerase.
Early Patterning
The WNT pathway has two main branches: one that is β-catenin–dependent (canonical) and the other independent of β-catenin. In the canonical pathway, Wnt ligand binds to a Frizzled/LRP heterodimer membrane-bound protein complex and the downstream intracellular signaling involves a G protein. The effect of this is to disrupt a large cytoplasmic protein complex that includes Axin, the adenomatous polyposis coli (APC; see p. 221) protein, and the glycogen synthase kinase-3β (GSK-3β) protein. This prevents the phosphorylation of β-catenin, but when β-catenin is not degraded, it accumulates and translocates to the nucleus where it activates the transcription of dorsal-specific regulatory genes. Binding of the ligand to the Fgf receptor results in dimerization of the receptor and transphosphorylation of the receptor’s cytoplasmic domain, with activation of Ras and other kinases, one of which enters the nucleus and activates target transcription factors. Mutated WNT10A in man results in a form of ectodermal dysplasia (odonto-onychodermal dysplasia) but apart from the possibility of WNT4 being implicated in a rare condition called Mayer-Rokitansky-Kuster syndrome, no other members of this gene family are yet implicated in human disease phenotypes.
The TGF-β Superfamily in Development and Disease
Thus far it recognized that there are 33 members of this cytokine family. Cytokines are a category of signaling molecules—polypeptide regulators—that enable cells to communicate. They differ from hormones in that they are not produced by discrete glands. These extracellular signaling polypeptides are transduced through a cascade to regulate gene expression within the cell nucleus. This is achieved through binding with cell surface receptors that, in a series of reactions, induces phosphorylation and activation of specific receptor kinases. This leads to the translocation of complexes into the nucleus, which execute transcriptional activation or repression of responsive target genes. The TGF-β family can be divided into two groups: (1) the BMPs and (2) the TGF-βs, activins, nodal, and myostatin, acting through various SMAD proteins. Ultimately, this superfamily is actively involved in a very broad range of cellular and developmental processes (Figure 6.2). This includes regulation of the cell cycle, cell migration, cell size, gastrulation and axis specification, and metabolic processes. In relation to health and disease, there are consequences for immunity, cancer, heart disease, diabetes, and Marfan syndrome (p. 300). Hyperactive signalling (overexpression) of BMP4 has been found in the rare bony condition fibrodysplasia ossificans progressiva, where disabling heterotopic bone deposition occurs, which is due to mutated ACVR1, encoding a BMP type 1 receptor. A mutated BMP receptor 2 has been shown to be a cause of familial primary pulmonary hypertension. BMP signalling is also involved in both dendritogenesis and axonal transport.
Somatogenesis and the Axial Skeleton
The vertebrate axis is closely linked to the development of the primary body axis during gastrulation, and during this process the presomitic mesoderm (PSM), where somites arise, is laid down in higher vertebrates. Wnt and FGF signals play vital roles in the specification of the PSM. The somites form as blocks of tissue from the PSM in a rostro-caudal direction (Figure 6.3), each being laid down with a precise periodicity that, in the 1970s, gave rise to the concept of the ‘clock and wavefront’ model. Since then, molecular techniques have given substance to this concept, and the key pathway here is notch-delta signaling and the ‘oscillation clock’—a precise, temporally defined wave of cycling gene expression (c-hairy in the chick, lunatic fringe and hes genes in the mouse) that sweeps from the tail-bud region in a rostral direction and has a key role in the process leading to the defining of somite boundaries. Once again, not all of the components are fully understood, but the notch receptor and its ligands, delta-like-1, and delta-like-3, together with presenilin-1 and mesoderm posterior-2, work in concert to establish rostro-caudal polarity within the PSM such that somite blocks are formed. Human phenotypes from mutated genes in this pathway are now well known and include presenile dementia (presenilin-1), which is dominantly inherited, and spondylocostal dysostosis (delta-like-3, mesoderm posterior-2, lunatic fringe, and hairy enhancer of split-7), which is recessively inherited (Figure 6.4). Another component of the pathway is JAGGED1, which, when mutated, results in the dominantly inherited and very variable condition known as Alagille syndrome (arteriohepatic dysplasia) (Figure 6.5). Rarely, mutations in NOTCH2 have been shown to cause some cases of Alagille syndrome, usually with renal malformations.
The Sonic Hedgehog–Patched GLI Pathway
The Sonic hedgehog gene (SHH) is as well known for its quirky name as for its function. SHH induces cell proliferation in a tissue-specific distribution and is expressed in the notochord, the brain, and the zone of polarizing activity of developing limbs. After cleavage and modification by the addition of a cholesterol moiety, the SHH protein binds with its receptor, Patched (Ptch), a transmembrane protein. The normal action of Ptch is to inhibit another transmembrane protein called Smoothened (Smo), but when bound by Shh this inhibition is released and a signaling cascade within the cell is activated. The key intracellular targets are the GLI family of transcription factors (Figure 6.6).
FIGURE 6.6 The Sonic hedgehog (Shh)-Patched (Ptch)-Gli pathway and connection with disease. Different elements in the pathway act as activators (arrows) or inhibitors (bars). The Shh protein is initially cleaved to an active N-terminal form, which is then modified by the addition of cholesterol. The normal action of Ptch is to inhibit Smo, but when Ptch is bound by Shh this inhibition is removed and the downstream signaling proceeds. CREBBP, cAMP response element-binding binding protein.
Molecular defects in any part of this pathway lead to a number of apparently diverse malformation syndromes (see Figure 6.6). Mutations in, or deletions of, SHH (chromosome 7q36) cause holoprosencephaly (Figure 6.7), in which the primary defect is incomplete cleavage of the developing brain into separate hemispheres and ventricles. The most severe form of this malformation is cyclopia—the presence of a single central eye. (The complexity of early development can be appreciated by the fact that a dozen or so chromosomal regions have so far been implicated in the pathogenesis of holoprosencephaly [p. 257].) Mutations in PTCH (9q22) result in Gorlin syndrome (nevoid basal cell carcinoma syndrome; Figure 6.8), which comprises multiple basal cell carcinomas, odontogenic keratocysts, bifid ribs, calcification of the falx cerebri, and ovarian fibromata. Mutations in SMO (7q31) are found in some basal cell carcinomas and medulloblastomas. Mutations in GLI3 (7p13) cause Pallister-Hall and Grieg syndromes, which are distinct entities with more or less the same body systems affected. However, there are also links to other conditions, in particular the very variable Smith-Lemli-Opitz syndrome (SLOS), which may include holoprosencephaly as well as some characteristic facial features, genital anomalies and syndactyly. This condition is due to a defect in the final step of cholesterol biosynthesis, which in turn may disrupt the binding of SHH with its receptor Ptch. Some, or all, of the features of SLOS may therefore be due to loss of integrity in this pathway (p. 288). Furthermore, a cofactor for the Gli proteins, CREBBP (16p13) is mutated in Rubenstein-Taybi syndrome (Figure 6.9). Disturbance to different components of the SHH is also clearly implicated in many types of tumor formation.
Homeobox (HOX) Genes
In Drosophila a class of genes known as the homeotic genes has been shown to determine segment identity. Incorrect expression of these genes results in major structural abnormalities; the Antp gene, for example, which is normally expressed in the second thoracic segment, will transform the adult fly’s antennae into legs if incorrectly expressed in the head. Homeotic genes contain a conserved 180-base pair (bp) sequence known as the homeobox, which is believed to be characteristic of genes involved in spatial pattern control and development. This encodes a 60-amino-acid domain that binds to DNA in Hox-response enhancers. Proteins from homeobox-containing (or HOX) genes are therefore important transcription factors that activate and repress batteries of downstream genes. At least 35 downstream targets are known. The Hox proteins regulate other ‘executive’ genes that encode transcription factors or morphogen signals, as well as operating at many other levels, on genes that mediate cell adhesion, cell division rates, cell death, and cell movement. They specify cell fate and help to establish the embryonic pattern along the primary (rostro-caudal) axis as well as the secondary (genital and limb bud) axis. They therefore play a major part in the development of the central nervous system, axial skeleton and limbs, the gastrointestinal and urogenital tracts, and external genitalia.
Drosophila has eight Hox genes arranged in a single cluster, but in humans, as in most vertebrates, there are four homeobox gene clusters containing a total of 39 HOX genes (Figure 6.10). Each cluster contains a series of closely linked genes. In vertebrates such as mice, it has been shown that these genes are expressed in segmental units in the hindbrain and in global patterning of the somites formed from axial presomitic mesoderm. In each HOX cluster, there is a direct linear correlation between the position of the gene and its temporal and spatial expression. These observations indicate that these genes play a crucial role in early morphogenesis. Thus, in the developing limb bud (p. 99) HOXA9 is expressed both anterior to, and before, HOX10, and so on.
Mutations in HOXA13 cause a rare condition known as the hand-foot-genital syndrome. This shows autosomal dominant inheritance and is characterized by shortening of the first and fifth digits, with hypospadias in males and bicornuate uterus in females. Experiments with mouse Hoxa13 mutants have shown that expression of another gene, EphA7, is severely reduced. Therefore, if this gene is not activated by Hoxa13, there is failure to form the normal chondrogenic condensations in the distal limb primordial. Mutations in HOXD13 result in an equally rare limb developmental abnormality known as synpolydactyly. This also shows autosomal dominant inheritance and is characterized by insertion of an additional digit between the third and fourth fingers and the fourth and fifth toes, which are webbed (Figure 6.11). The phenotype in homozygotes is more severe and reported mutations take the form of an increase in the number of residues in a polyalanine tract. This triplet-repeat expansion probably alters the structure and function of the protein, thereby constituting a gain-of-function mutation (p. 26). Mutated HOXA1 has been found in the rare, recessively inherited Bosley-Saleh-Alorainy syndrome, consisting of central nervous system abnormalities, deafness, and cardiac and laryngotracheal anomalies. A mutation in HOXD10 was found in isolated congenital vertical talus in a large family demonstrating autosomal dominant inheritance, and duplications of HOXD have recently been found in mesomelic limb abnomality syndromes.
Given that there are 39 HOX genes in mammals, it is surprising that so few syndromes or malformations have been attributed to HOX gene mutations. One possible explanation is that most HOX mutations are so devastating that the embryo cannot survive. Alternatively, the high degree of homology between HOX genes in the different clusters could lead to functional redundancy so that one HOX gene could compensate for a loss-of-function mutation in another. In this context HOX genes are said to be paralogous because family members from different clusters, such as HOXA13 and HOXD13, are more similar than adjacent genes in the same cluster.
Paired-Box (PAX) Genes
The paired-box is a highly conserved DNA sequence that encodes a 130-amino-acid DNA-binding transcription regulator domain. Nine PAX genes have been identified in mice and humans. In mice these have been shown to play important roles in the developing nervous system and vertebral column. In humans, loss-of-function mutations in five PAX genes have been identified in association with developmental abnormalities (Table 6.2). Waardenburg syndrome type 1 is caused by mutations in PAX3
[/not-level-membership-for-basic-science-category]
