Chapter 60 Developmental Disorders of the Nervous System
Embryological and Fetal Development of the Nervous System
Mitotic Proliferation of Neuroblasts (Neuronogenesis)
Programmed Cell Death (Apoptosis)
Fissures and Sulci of Cortical Structures
Electrical Polarity of the Cell Membrane
Biosynthesis of Neurotransmitters
Cajal-Retzius Neurons of the Fetal Brain
Suprasegmental Influences on Muscle Maturation
Etiology of Central Nervous System Malformations
Molecular Genetic Classification of Malformations of the Nervous System
Clinical Expression of Selected Malformations of the Nervous System
Embryological and Fetal Development of the Nervous System
Table 60.1 shows known genetic loci and mutations in human central nervous system (CNS) malformations. In most cases, mutations affect the genetic programming of the spatial and temporal sequences of developmental processes. These range from early processes that establish the axes of the neural tube and gradients of genetic expression, to late processes that establish the identity of specific types of neurons, the type of neurotransmitter they synthesize, and the synaptic connections they make. The role of homeobox genes in the differentiation of neural structures is an aspect of development recognized relatively recently. Molecular genetic data are rapidly becoming available because of intense interest in this key to understanding neuroembryology in general and neural induction in particular (Sarnat and Menkes, 2000). Other aspects of current investigative interest include the roles of neurotropic factors, hormones, ion channels, and neurotransmitter systems in fetal brain development. Genetic manipulation in animals has created many genetic models of human cerebral malformations. These contribute greatly to our understanding of human dysgeneses and provide insights into the pathogenesis of epilepsy and other functional results of dysgeneses (Chevassus-au-Louis et al., 1999).
Malformations of the nervous system are unique. No two individual cases are identical, even when categorized as the same anatomical malformation, such as alobar holoprosencephaly, syndromic or isolated agenesis of the corpus callosum, and types 1 and 2 lissencephaly. Functional expression of anatomically similar cases also may vary widely. For example, two cases of holoprosencephaly with nearly identical imaging findings and similar histological patterns of cortical architecture and subcortical heterotopia at autopsy may differ in that one infant may have epilepsy refractory to pharmacological control, whereas the other may have no clinical seizures at all. The difference may be at the level of synaptic organization and the relative maturation of afferent input and neuronal maturation (Sarnat and Born, 1999; Sarnat et al., 2010a). A discussion of the critical sequence of events in neural maturation follows.
Neurulation
Neurulation refers to the formation and closure of the neural tube. The formation of the neural tube from the neural placode starts with the establishment of the axis in the neural plate. The three early axes—longitudinal, horizontal, and vertical—persist during life and correspond to the basic body plan of all vertebrates (Sarnat and Flores-Sarnat, 2001b). At neurulation, grooving of the neural placode occurs in the anteroposterior axis. Subsequently, closure of the lateral margins of the folding neural placode ensues in the dorsal midline to form the neural tube. To accomplish closure, intercellular filaments interdigit cells of the two sides to form a veil at midline closure points and the neuropores. At this time, the neural crest separates bilaterally at the two fusing lips of the closing neural tube, and its cells migrate along predetermined pathways to form the peripheral nervous system, chromaffin tissue, melanocytes, and various other cells. Neural crest cells terminally differentiate only after reaching their final destination. The inhibitory function of versican, a chondroitin sulfate proteoglycan, is an important factor of the extracellular matrix for neural crest cell migration (Dutt et al., 2006).
Disorders of Neurulation (1 to 4 Weeks’ Gestation)
Anencephaly (aprosencephaly with open cranium) is a failure of the anterior neuropore to close at 24 days’ gestation, or perhaps to remain closed. The lamina terminalis and its derivatives fail to form, and most forebrain structures do not develop. Structures derived from the ventral part of the lamina terminalis, the basal telencephalic nuclei, may form imperfectly. Because the deficient forebrain neuroectoderm does not induce development of the overlying mesoderm, the cranium, meninges, and scalp do not close in the sagittal midline, exposing the remaining brain tissue to the surrounding amniotic fluid throughout gestation. The original induction failure, however, is probably that of mesodermal tissue on neuroectoderm and due to a defective rostral end of the notochord. Failure of craniofacial induction by the neural tube, mediated through the prosencephalic and mesencephalic neural crest, is another major pathogenetic factor (Sarnat and Flores-Sarnat, 2005).
Mitotic Proliferation of Neuroblasts (Neuronogenesis)
After formation of the neural tube, proliferation of neuroepithelial cells in the ventricular zone associated with mitoses at the ventricular surface generates neurons and glial cells. The rate of division is greatest during the early first trimester in the spinal cord and brainstem and during the late first and early second trimester in the forebrain. Within the ventricular zone of the human fetal telencephalon, 33 mitotic cycles provide the total number of neurons required for the mature cerebral cortex. Most mitotic activity in the neuroepithelium occurs at the ventricular surface, and the orientation of the mitotic spindle determines the subsequent immediate fate of the daughter cells. If the cleavage plane is perpendicular to the ventricular surface, the two daughter cells become equal neuroepithelial cells preparing for further mitosis. If, however, the cleavage is parallel to the ventricular surface, the two daughter cells are unequal (asymmetrical cleavage). In that case, the one at the ventricular surface becomes another neuroepithelial cell, whereas the one away from the ventricular surface separates from its ventricular attachment and becomes a postmitotic neuroblast ready to migrate to the cortical plate. Furthermore, the products of two genes that determine cell fate, called numb and notch, are on different sides of the neuroepithelial cell. Therefore, with symmetrical cleavages, both daughter cells receive the same amount of each, but with asymmetrical cleavage, the cells receive unequal ratios of each, which also influences their subsequent development (Mione et al., 1997). The orientation of the mitotic spindle requires centractin.
Active mitoses cease well before the time of birth in most parts of the human nervous system, but a few sites retain a potential for postnatal mitoses of neuroblasts. One recognized site is the periventricular region of the cerebral hemispheres (Kendler and Golden, 1996). Another is the external granular layer of the cerebellar cortex, where occasional mitoses persist until 1 year of age. Postnatal regeneration of these neurons after destruction of most by irradiation or cytotoxic drugs occurs in animals and may occur in humans as well. Primary olfactory receptor neurons also retain a potential for regeneration. In fact, if a constant turnover of these neurons did not occur throughout life, the individual would become anosmic after a few upper respiratory infections, which transiently denude the intranasal epithelium. A population of “stem cells” with mitotic potential in the subventricular zone and hippocampal dentate gyrus is reported (Johansson et al., 1999). These have generated considerable interest because of a potential for regeneration of the damaged adult brain and because they may be induced to mature as neurons (Schuldiner et al., 2001).
Disorders of Neuronogenesis
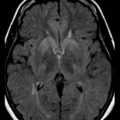
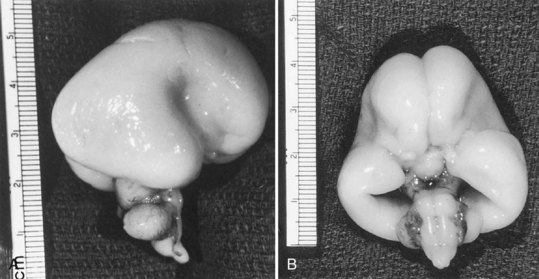
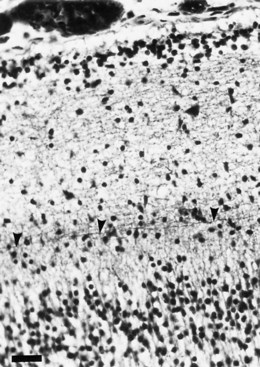
Destructive processes may destroy so many neuroblasts that regeneration of the full complement of cells is impossible. This happens when the insult persists for a long time or is repetitive, destroying each subsequent generation of dividing cells. Inadequate mitotic proliferation of neuroblasts results in hypoplasia of the brain (Fig. 60.1). Such brains are small and grossly malformed, either because of a direct effect on neuroblast migration or by destruction of the glial cells with radial processes that guide migrating nerve cells. The entire brain may be affected, or portions may be selectively involved. Cerebellar hypoplasia often is a selective interference with proliferation of the external granular layer. In some cases, cerebral hypoplasia and microcephaly are the result of precocious development of the ependyma before all mitotic cycles of the neuroepithelium are complete, because ependymal differentiation arrests mitotic activity at the ventricular surface. The mutation of a gene that programs neuronogenesis may be another explanation for generating insufficient neuroepithelial cells, although this pathogenesis remains hypothetical in humans.
Programmed Cell Death (Apoptosis)
Disorders of Programmed Cell Death
Spinal muscular atrophy (see Chapter 74) is an example of a human disease caused by apoptosis not stopping at the proper time. In this disorder, continued loss of spinal motor neurons (SMN) after the normal deletion of surplus embryonic neuroblasts expresses itself as a progressive denervating process. Genetic factors are crucial in determining the arrest of cell death, which accounts for the hereditary character of spinal muscular atrophy. The SMN defective gene at the chromosome 5q13.1 locus has now been isolated and is normally responsible for arresting apoptosis in motor neuroblasts (Roy et al., 1995).
Other neurodegenerative diseases of fetal life and infancy are more widespread within the CNS, rather than limited to one type of neuron such as the motor neuron. The characteristic feature is also progressive neuronal loss that is apoptotic rather than necrotic in character: No inflammatory or glial reaction occurs, and the features of the DNA degradation differ from ischemic necrosis. An example is pontocerebellar hypoplasia, a group of progressive degenerative diseases that begin prenatally and continue postnatally (Barth et al., 1995). Despite the name, they involve much more than the cerebellar system. These diseases are associated with extensive cerebral cortical and basal ganglionic abnormalities even in motor neurons, which cause a clinical presentation at birth resembling spinal muscular atrophy. This autosomal recessive group of diseases, all genetically distinct from olivopontocerebellar atrophy, exemplifies a semantic difficulty. If an atrophic process begins before development is complete, it results in both hypoplasia and superimposed atrophy. In the CNS, glial cells also undergo apoptosis. Glial necrosis intimately links to the interhemispheric passage of commissural fibers in the corpus callosum. In a murine model of callosal agenesis, glial cells that do not degenerate act as a barrier to crossing axons and prevent the corpus callosum from forming.
Neuroblast Migration
No neurons of the mature human brain occupy their site of generation from the neuroepithelium. They migrate to their mature site to establish the proper synaptic connections with appropriate neighboring neurons and send their axons in short or long trajectories to targets. The subependymal germinal matrix (Fig. 60.2) is the subventricular zone of the embryonic concentric layers and consists of postmitotic premigratory neuroblasts and glioblasts. In general, the movement of maturing nerve cells is centrifugal, radiating toward the surface of the brain. The cerebellar cortex is exceptional in that external granule cells first spread over the surface of the cerebellum and then migrate into the folia. Migration of neuroblasts begins at about 6 weeks’ gestation in the human cerebrum and is not completed until at least 34 weeks of fetal life, although the majority of germinal matrix cells after midgestation are glioblasts. Glioblasts continue to migrate until early in the postnatal period. Within the brainstem, neuroblast migration is complete by 2 months’ gestation. Cerebellar external granule cells continue migrating throughout the first year of life.
Neuroblast migration permits a three-dimensional spatial relationship to develop between neurons, which facilitates the formation of complex synaptic circuits. The timing and sequence of successive waves of migrating neuroblasts are precise. In the cerebral cortex, immature nerve cells reach the pial surface and then form deeper layers as more recent arrivals replace their position at the surface. Neurons forming the most superficial layers of neocortex are thus the last to have migrated, although in the three-layered hippocampus, the most superficial neurons represent the earliest migratory wave. Three major groups of molecules control neuroblast migration (Gressens, 2006): (1) molecules of the cytoskeleton that determine the initiation (filamin-A and ADP-ribosylation factor GEF2) and ongoing progression (doublecortin and LIS1) of neuroblast movement; (2) signaling molecules involved in lamination, including reelin and other proteins not yet associated with human diseases; and (3) molecules modulating glycosylation that provide stop signals to migrating neuroblasts (e.g., POMT1 [protein O-mannosyl-transferase], involved in Walker-Warburg syndrome; POMGnT1 [protein O-mannose β-1,2-N-acetylglucosaminyltransferase], involved in muscle-eye-brain disease; and fukutin, involved in Fukuyama muscular dystrophy).
The laminated arrangement of the mammalian cerebral cortex requires a large cortical surface area to accommodate increasing numbers of migrating neuroblasts and glioblasts. Initially the cortical plate shows no histological layering, a process beginning at about midgestation, but rather has an immature columnar architecture. The lamination is superimposed upon this columnar pattern, but columnar architecture is still seen postnatally, particularly at the crowns of gyri and the depths of sulci. Even before histological lamination is evident, RNA probes for specific neuronal identities can already detect future organization of the cortical plate (Hevner, 2007). Convolutions provide this large surface area without incurring a concomitant increase in cerebral volume. The formation of gyri and sulci is thus a direct result of migration (Fig. 60.3). Most gyri form in the second half of gestation, which is a period of predominant gliogenesis and glial cell migration. Therefore, the proliferation of glia in the cortex and subcortical white matter may be more important than neuroblast migrations in the formation of convolutions, but the growth of dendrites and synaptogenesis also may influence gyration by contributing mass to the neuropil.
Major Mechanisms of Neuroblast Migration: Radial Glial Fiber Guides and Tangential Migration Along Axons
The majority of neuroblasts arriving at the cortical plate do so by means of radial glial guides from the subventricular zone. A second route, tangential migration, uses axons as the guides for the migratory neuroblasts. The genetically determined programming of neuroblast migration begins when cells are still undifferentiated neuroepithelial cells and even before all their mitotic cycles are complete. Neuroepithelial cells express the gene products of the lissencephaly gene (LIS1), as do ependymal cells and Cajal-Retzius cells of the molecular layer of cerebral cortex. The expression of this gene is defective in type 1 lissencephaly (Miller-Dieker syndrome), a severe disorder of neuroblast migration (Clark et al., 1997). An understanding of its function in migration is incomplete. The guidance of most neurons of the forebrain to their predetermined site from the germinal matrix (embryonic subventricular zone) is by long radiating fibers of specialized fetal astrocytes (Fig. 60.4). The elongated processes of these glial cells span the entire wall of the fetal cerebral hemisphere; their cell bodies are in the periventricular region, and their terminal end-feet are on the limiting pial membrane at the surface of the brain (see Fig. 60.4). Radial glial cells are the first astroglial cells of the human nervous system converted into a mature fibrillary astrocyte of the subcortical white matter; some are still present at birth. Mature astrocytes are present throughout the CNS by 15 weeks’ gestation, and gliogenesis continues throughout fetal and postnatal life. Several types of glial cells are recognizable between 20 and 36 weeks’ gestation.
Facilitating the mechanical process of neuroblasts gliding along a radial glial fiber are several specialized proteins at the radial glial fiber surface membrane or extracellular space. An example is astrotactin, secreted by the neuroblast (Zheng et al., 1996). Glial cells and neural cell adhesion molecules also facilitate gliding (Jouet and Kenwrick, 1995). Fetal ependymal cells have radiating processes that resemble those of the radial glial cell but do not extend beyond the germinal matrix and secrete molecules in the extracellular matrix. Some adhesion molecules are present in the extracellular matrix (Thomas et al., 1996). These molecules serve as lubricants, as adhesion molecules between the membranes of the neuroblast and the radial glial fiber, and as nutritive and growth factors. They stimulate cell movement by a mechanism still poorly understood. Deficient molecules lead to defective migration. For example, the abnormality of the L1 adhesion molecule is the defective genetic program in X-linked hydrocephalus accompanied by polymicrogyria and pachygyria.
In addition to the radial migration to the cerebral cortex, tangential migration also occurs, but the number of neuroblasts is far smaller (Rakic, 1995; Takano et al., 2004). These migrations perpendicular to the radial fibers probably use axons rather than glial processes as guides for migratory neuroblasts. This explains why not all cells in a given region of cortex are from the same clone or vertical column. Most of the tangentially migrating neuroblasts in the cerebral cortical plate are generated in the fetal ganglionic eminence, a deep telencephalic structure of the germinal matrix that gives origin to basal ganglionic neurons and to the γ-aminobutyric acid (GABA)ergic inhibitory interneurons of the cerebral cortex. These neurons in the cortex from tangential migration have some unique metabolic features such as calretinin synthesis (Takano et al., 2004; Ulfig, 2002). Calretinin-reactive inhibitory interneurons in the cerebral cortex comprise about 12% of total neurons and are a subset of total neurons arriving at the cortical plate by tangential migration, which represent about 20% of total cortical neurons.
Tangential migrations occur in the brainstem and olfactory bulb as well as in the cerebrum. The subpial region is another site of neuroblast migration that does not use radial glial cells. Calretinin-reactive neurons are in the cerebellum as well as the cerebral cortex (Yew et al., 1997), particularly Purkinje cells, basket cells, and neurons of the dentate and inferior olivary nuclei of the cerebellar system, but not those of the pontine nuclei, which similarly originated in the rhombic lip of His.
Disorders of Neuroblast Migration
Most disturbances of neuroblast migration involve arrested migration before the journey is complete. These disorders are divisible into three anatomical phases, depending on where the migratory arrest occurred. An example of neuroblasts never having begun migration from the periventricular region is periventricular nodular heterotopia, an X-linked genetic disorder due to defective expression of the gene, filamin-A (FLNA). Subcortical laminar heterotopia results when neuroblasts begin migration but arrest in the subcortical white matter before reaching the cortical plate. This is another X-linked recessive trait but is due to a different gene called doublecortin (DCX). The term double cortex is sometimes used, but this name is incorrect because unlike a true cortex, the subcortical heterotopia lacks lamination. If the neuroblasts reach the cortical plate but lack correct lamination, accompanying this abnormal architecture of the cortical plate are abnormalities of gyration such as lissencephaly or pachygyria. Several different genes, including LIS1 and reelin (RLN), are important in cortical plate organization (Curran and D’Arcangelo, 1998) and mutated in malformations of the terminal phase of neuroblast migration.
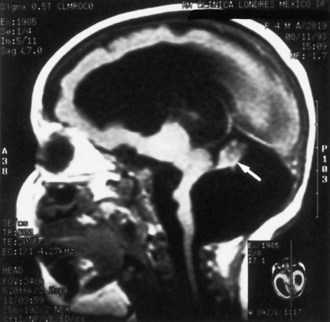
Lissencephaly is a condition of a smooth cerebral cortex without convolutions. Normally at midgestation, the brain is essentially smooth; the interhemispheric, sylvian, and calcarine fissures are the only ones formed. Gyri and sulci develop between 20 and 36 weeks’ gestation, and the mature pattern of gyration is evident at term, although some parts of the cerebral cortex (e.g., frontal lobes) are still relatively small. In lissencephaly type 1 (Miller-Dieker syndrome), the cerebral cortex remains smooth. Lesser degrees of this gross morphological defect exist, with a few excessively wide gyri (pachygyria) or multiple excessively small gyri (polymicrogyria). The histopathological pattern is that of a 4-layer cortex in which the outermost layer (1) is the molecular layer, as in normal 6-layered neocortex. Layer 2 corresponds to layers 2 through 6 of normal neocortex, layer 3 is cell-sparse as a persistent fetal subplate zone, and layer 4 consists of incompletely migrated neurons in the subcortical intermediate zone. In lissencephaly type 2 (Walker-Warburg syndrome), poorly laminated cortex with disorganized and disoriented neurons is seen histologically, and the gross appearance of the cerebrum is one of a smooth brain or a few poorly formed sulci (Fig. 60.5). The term cobblestone refers to the aspect of the surface with multiple shallow furrows not corresponding to normal sulci. The cerebral mantle may be thin, suggesting a disturbance of cell proliferation as well as of neuroblast migration. Malformations of the brainstem and cerebellum often are present as well (see Fig. 60.5). Lissencephaly type 1 and type 2 (Walker-Warburg syndrome, Fukuyama muscular dystrophy, muscle-eye-brain disease of Santavuori) are genetic diseases. Lissencephaly also results from nongenetic disturbances of neuroepithelial proliferation or neuroblast migration, including destructive encephaloclastic processes such as congenital infections during fetal life. More recently it has been recognized that the lissencephalies, including those resulting from mutations in LIS1, DCX, and ARX genes, are disturbances not only in radial migration, but also involve tangentially migrating neuroblasts (Marcorelles et al., 2010).
In sum, either defective genetic programming or acquired lesions in the fetal brain that destroy or interrupt radial glial fibers may cause disorders of neuroblast migration. Cells may not migrate at all and become mature neurons in the periventricular region, as occurs in X-linked periventricular nodular heterotopia (Eksioglu et al., 1996) and in some cases of congenital cytomegalovirus infection. Cells may become arrested along their course as heterotopic neurons in deep subcortical white matter, as occurs in many genetic syndromes of lissencephaly-pachygyria and in many metabolic diseases including cerebrohepatorenal (Zellweger) syndrome and many aminoacidurias and organic acidurias. The same aberration may occur in acquired insults to the radial glial cell during ontogenesis. Cells may overmigrate beyond the limits of the pial membrane into the meninges as ectopic neurons, either singly or in clusters known as marginal glioneuronal heterotopia, or brain warts. Rarely, herniation of the germinal matrix into the lateral ventricle may occur through gaps in the ependyma; those cells mature as neurons, forming a non-neoplastic intraventricular mass that may or may not obstruct cerebrospinal fluid (CSF) flow. Whether disoriented radial glial fibers actually guide neuroblasts to an intraventricular site or neuroblasts are physically pushed in a direction of less resistance is uncertain.
Fissures and Sulci of Cortical Structures
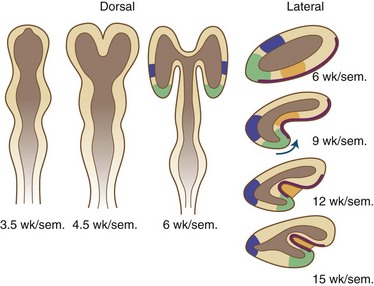
Fissures and sulci are grooves that form in laminated cortices, principally cerebral and cerebellar. Such folding accomplishes a need for an enlarging surface area without a concomitant increase in tissue volume as development proceeds. Without gyration of the cerebral cortex and foliation of the cerebellar cortex, the brain would be so large and voluminous at birth that neither the neonate nor the mother would survive delivery. Fissures and sulci both result from mechanical forces during fetal growth, but they differ in that fissures form from external forces and sulci form from internal forces imposed by the increased volume of neuronal cytoplasm and the formation of neuropil, the processes of neurons and glial cells (Sarnat and Flores-Sarnat 2010a). The ventricular system acts as another external force, surrounded by but outside of the brain parenchyma. Whereas fissures generally form earlier and often are deeper than sulci, these are not the most important differences. Box 60.1 lists the various fissures of the brain, and Fig. 60.6 is a drawing of the development of the human telencephalic flexure, which becomes, after closure of the operculum, the Sylvian fissure. It should be noted that the ventral bending of the primitive oval-shaped telencephalic hemisphere results in the original posterior pole becoming the temporal—not the occipital—lobe, and that the lateral ventricle bends with the brain. The occipital horn of the lateral ventricle is a more recent diverticulum of the original simple ventricle and as such remains the most variable part of the ventricular system, symmetrical in only 25% of normal individuals. Cerebellar folia are the equivalent of cerebral cortical gyri. A temporally and spatially precise sequence of the development of fissures, sulci, and cerebellar folia is genetically programmed and enables the neuroradiologist and neuropathologist to also assess maturational delay of this aspect of ontogenesis. The gestational age of a premature infant may be determined to within a 2-week period or less from the convolutional pattern of the brain.
Disorders of Fissures and Sulci
The telencephalic Sylvian fissures fail to form in holoprosencephaly and form abnormally in many major malformations of the brain, including lissencephalies, schizencephaly, and severe cerebral hypoplasias (Sarnat and Flores-Sarnat, 2010a). Abnormal gyration is a regular feature of many neuroblast migratory disorders, including lissencephaly, pachygyria, and polymicrogyria, enabling an accurate diagnosis by neuroimaging not only postnatally but also by prenatal fetal magnetic resonance imaging (MRI), even though microscopic details of cortical lamination and organization are below the resolution of these techniques.
Synaptogenesis
Synapse formation follows the development of dendritic spines and polarization of the cell membrane. The relation of synaptogenesis to neuroblast migration differs in different parts of the nervous system. In the cerebral cortex, synaptogenesis always follows neuroblast migration. In the cerebellar cortex, however, the external granule cells develop axonal processes that become the long parallel fibers of the molecular layer and make synaptic contact with Purkinje cell dendrites before migrating through the molecular and Purkinje cell layer to their mature internal position within the folium. Synaptophysin immunoreactivity with thermal intensification has proved to be a useful marker for studying normal and abnormal synaptogenesis in the fetus and newborn (Sarnat et al., 2010).
Disorders of Synaptogenesis
Because the formation of dendritic spines and the formation of synapses are so closely related, the same spectrum of diseases already discussed is equally appropriate for consideration in this section. In preterm infants, who are generally unwell even if they do not have specific neurological disease, the rate of maturation of the EEG is often slow, which may reflect an impairment of synapse formation. Chronic hypoxemia particularly delays neurological maturation. Deletions of δ-catenin, a neuron-specific catenin implicated in adhesion and dendritic branching, lead to severe synaptic dysfunction and correlate with the severity of mental retardation in cri du chat syndrome (Israely et al., 2004).
Biosynthesis of Neurotransmitters
In some parts of the brain, transitory fetal transmitters may appear during development and then disappear. Substance P and somatostatin are present in the fetal cerebellum at midgestation, but these neuropeptides are never in the mature cerebellum. In the cerebral cortex of the frontal lobe, the pattern of laminar distribution of cholinergic muscarinic receptors in the mature brain is the inverse of that in the fetus. The functions of these transitory transmitter systems are unknown. Some serve as tropic molecules rather than transmitters in early development. Even amino acid transmitters such as GABA may serve mainly a tropic function at an early stage in development. In situ hybridization and immunocytochemical techniques demonstrate neurotransmitters in neurons of the developing brain of experimental animals and may be applicable to human tissue under some circumstances (Dupuy and Houser, 1997). The ontogeny of neurotransmitter systems depends not only on the mechanisms of synthesis of chemical transmitters but also on the development of highly specific receptors of these chemical signals and their ability to modify excitability of neuronal membranes and trigger action potentials after the recognition of specific molecules (Rho and Storey, 2001; Simeone et al., 2003).
Cajal-Retzius Neurons of the Fetal Brain
Cajal-Retzius cells are large, mature, stellate neurons in the marginal (outermost) zone of the fetal cerebral cortex. They are the first cells to appear at the surface of the embryonic cerebrum, preceding the first wave of radial migration from the subventricular zone and forming a plexus in the marginal (later the molecular) zone. They migrate to the surface either from the ganglionic eminence or from the midbrain neuromere (Sarnat and Flores-Sarnat, 2002). The first afferent processes to enter the marginal layer are dendrites of pyramidal cells of layer VI; synapses between Cajal-Retzius and pyramidal neurons of layer VI form the first intrinsic cortical circuits (Marín-Padilla, 1998). They eventually have synaptic contacts with cortical neurons in all layers.
Cajal-Retzius cells contain acetylcholinesterase and oxidative enzymes and secrete GABA and probably ACh as neurotransmitters. Their long axons extend parallel to the surface of the brain, plunging short branches into layer II (Fig. 60.7). Cajal-Retzius neurons are sparse by term but persist even in the adult, though their function after maturity is uncertain. They strongly express the transcription product of the LIS1 gene, which is defective in X-linked hydrocephalus associated with polymicrogyria and defective neuroblast migration. Cajal-Retzius neurons also strongly express RLN, another gene essential for radial neuroblast migration (Clark et al., 1997; Sarnat and Flores-Sarnat, 2002). This is the only specific disease involving Cajal-Retzius neurons.
Suprasegmental Influences on Muscle Maturation
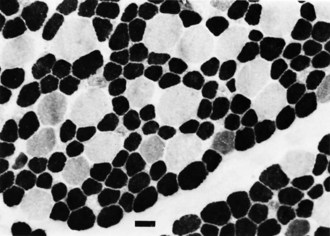
The motor unit is capable of developing normally in the absence of suprasegmental modification, such as occurs in infants with severe hypoplasia of the brain (see Fig. 60.1). Malformations of the brainstem and cerebellar hypoplasia in particular are associated with a variety of aberrations in histochemical differentiation. These aberrations include (1) delayed maturation; (2) more than 80% predominance of type 1 or type 2 myofibers, with or without uniform hypoplasia of one or the other type (Fig. 60.8); and (3) classic congenital muscle fiber–type disproportion. Malformations limited to the cerebral cortex do not cause fiber-type predominance. Muscle histology of children with cerebral palsy from birth asphyxia or other perinatal insults shows only nonspecific type 2 muscle fiber atrophy. The normal ratio of fiber types is preserved. This is similar to the changes that follow disuse or immobilization of muscle. The speculation is that many of the small bulbospinal “subcorticospinal” tracts (i.e., vestibulospinal, reticulospinal, olivospinal, tectospinal, and rubrospinal) are more important than the larger corticospinal tract during the stage of histochemical differentiation of muscle between 20 and 28 weeks’ gestation. These small descending pathways are generally well myelinated and functional at that time, whereas the corticospinal tract does not even begin its myelination cycle or proliferation of axonal terminals until after muscle development is complete.
Etiology of Central Nervous System Malformations
Ischemic Encephalopathy in the Fetus
The existence of fetal watershed zones of the cortical vascular bed is important in the pathogenesis of ulegyria, an atrophy of gyri that grossly resembles polymicrogyria. Focal areas of cortical atrophy and gliotic scarring occur after perinatal ischemic or hypoxic encephalopathy. The four-layered cortex of polymicrogyria is quite a different lesion from ulegyria, resulting from a primary disturbance of neuroblast migration. Some authors question this interpretation, however, and provide evidence of postmigratory laminar necrosis of the cortex. The distribution of polymicrogyria is frequently in vascular territories of fetal brain and often forms a rim surrounding a porencephalic cyst in the territory of the middle cerebral artery. Multicystic encephalomalacia and hydranencephaly are end-stage sequelae of massive cerebral infarction in the developing brain. Watershed zones also exist in the brainstem between the territories supplied by paramedian penetrating short and long circumferential arteries, which originate from the basilar artery. Transitory hypoperfusion in the basilar artery in fetal life may produce watershed infarcts in the tegmentum of the pons and medulla oblongata. This is a probable pathogenesis of Möbius syndrome and probably also of “failure of central respiratory drive” in neonates with hypoventilation not due to pulmonary or neuromuscular disorders (Sarnat, 2004b). The cause is involvement of the tractus solitarius, which receives afferents from chemoreceptors such as the carotid body and provides efferent axons to motor neurons that innervate the diaphragm and intercostal muscles.
Molecular Genetic Classification of Malformations of the Nervous System
Classification is a fundamental human thought process, allowing us to organize data in a systematic manner and understand relations. The traditional basis for classification of CNS malformations is descriptive morphogenesis. New insights into the molecular genetic programming of neural development require the integrations of this information with the anatomical criteria (Sarnat and Flores-Sarnat, 2001b, 2004; Simeone, 2002). For example, lissencephaly and holoprosencephaly are two important malformations, each formerly thought to be distinctive. It is now recognized that many different genetic defects cause each; hence they are end stages of ontogenetic errors with diverse causes (see following discussion). A pure genetic classification to replace anatomical criteria, by contrast, would not be useful to clinicians, radiologists, or pathologists and would be incomplete because many genetic mutations remain unknown. A compromise that addresses the deficiencies of both pure anatomical and pure genetic schemes of classification is one based on patterns of genetic expression in which the precise genetic mutation may or may not be known but is stated while preserving anatomical criteria (Sarnat, 2000; Sarnat and Flores-Sarnat, 2001b). The up-regulation or down-regulation of a dorsalizing or ventralizing gene may be recognizable by its anatomical effect on neural tube development, even if the precise gene is unknown.
The traditional categories of CNS development that allow categories of ontogenetic processes, such as neuronogenesis, neuroblast migration, and synaptogenesis, and their disturbances in malformations may be preserved in the proposed new scheme of classification. They are supplemented by new categories such as “disturbances of cellular lineage” (e.g., tuberous sclerosis; hemimegalencephaly) and disorders of embryonic neuromeric segmentation (e.g., absence of the midbrain and upper pons; absence of the basal ganglia; Chiari malformations). Some genes specify particular types of cellular differentiation and may change the cell type at different stages of development (Marquardt and Pfaff, 2001). One of the most important concepts in the integrated morphological-molecular-genetic scheme is the gradients of genetic expression (Sarnat and Flores-Sarnat, 2001b). The gradients are those of the axes of the neural tube: dorsoventral and ventrodorsal, rostrocaudal and sometimes caudorostral, and mediolateral. Nearly all genes have gradients of expression, with stronger expression in some regions and gradually lesser influence more distally. For example, if the rostrocaudal gradient in holoprosencephaly extends as far as the midbrain, mesencephalic neural crest migration is impaired, and midfacial hypoplasia results regardless of the severity of the forebrain malformation (see following discussion). Some authors attempt to develop schemes of regional classification for malformations (e.g., limited to the cerebral cortex for use in genetic epilepsies). All classifications should consider the entire CNS, however, because the rostrocaudal gradients of genetic expression may cause important subcortical defects, and indeed some seizure disorders may even originate in subcortical structures. The up-regulation and down-regulation of genes also is sometimes easier to understand in the anatomically simpler structures of the brainstem and spinal cord, allowing extrapolation to more complex forebrain structures.
Clinical Expression of Selected Malformations of the Nervous System
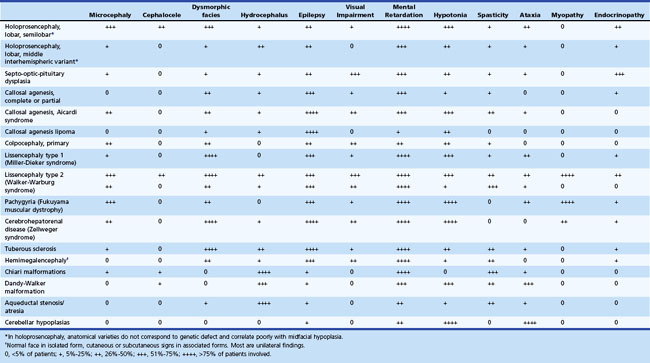
Table 60.2 summarizes the clinical features of major malformations of the brain.
Disorders of Symmetry and Cellular Lineage
Hemimegalencephaly
Hemimegalencephaly is one of the most enigmatic cerebral malformations, because it is a severe dysgenesis limited to one cerebral hemisphere or, less commonly, includes the ipsilateral cerebellar hemisphere and brainstem (total hemimegalencephaly). Though traditionally regarded as another disorder of neuroblast migration, this feature is probably only secondary to involvement of radial glial cells and perhaps the neuroblasts themselves, and the primary process is a disturbance of cellular lineage and also involvement of genes of symmetry expressed as early as gastrulation (Flores-Sarnat, 2002a, 2008; Flores-Sarnat et al., 2003). Individual neural cells exhibit both glial and neuronal proteins and have abnormal growth and morphology. Some cases of hemimegalencephaly are isolated, but others are particularly associated with neurocutaneous syndromes: epidermal nevus syndrome, Proteus syndrome, and Klippel-Trenaunay syndrome (Flores-Sarnat, 2006). Neurological clinical features and neuropathological findings are virtually identical in isolated and associated forms. Partial epilepsy is the principal clinical feature in severe and moderate forms, often refractory to medical treatment and abolished only by hemispherectomy or other surgical resections. Other less constant features include variable mental retardation and contralateral motor deficit. Mild as well as severe forms occur. Associated forms additionally include the features of the particular syndrome, such as lipoma formation in the ipsilateral face in epidermal nevus syndrome and Proteus syndrome.
Disorders of Neurulation (1 to 4 Weeks’ Gestation)
Anencephaly (Aprosencephaly with Open Cranium)
Anencephaly is a failure of the closing of the anterior neuropore at 24 days’ gestation. Death in utero occurs in approximately 7% of anencephalic pregnancies, 34% of such babies are premature, and 53% at term. Stillbirth, presumably resulting from intrapartum death, occurs in 20% of these deliveries. In one study of 211 pregnancies, 72% (153) of anencephalic offspring were liveborn; of those, 67% (103) died within 24 hours, but 6 survived 6 or more days (maximum 28 days) (Jaquier et al., 2006). The prenatal diagnosis of anencephaly is by examination of amniotic fluid for elevation of α-fetoprotein, and confirmation is by sonographic imaging as early as 12 weeks’ gestation. The face may show a midline hypoplasia, similar to holoprosencephaly (see following section on Holoprosencephaly), probably because the rostrocaudal gradient of a defective genetic expression extends to the midbrain and interferes with mesencephalic neural crest migration (Sarnat and Flores-Sarnat, 2001a).
Cephalocele (Encephalocele; Exencephaly)
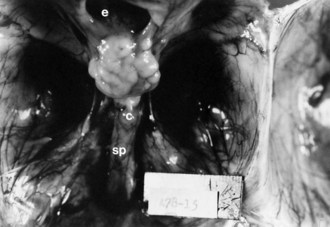
Most encephaloceles are parietal or occipital (Fig. 60.9) and contain supratentorial tissue, cerebellar tissue, or both. Frontal encephaloceles are less common in North America and Europe but are the most frequent variety in Thailand, Vietnam, and surrounding countries. They usually include olfactory tissue. Cases of encephaloceles related to Agent Orange (containing the herbicides, 2,4-dichlorophenoxyacetic acid [2,4-D] and 2,4,5-trichlorophenoxyacetic acid [2,4,5-T]), which was used in the Vietnam War, are still reportedly observed in Cambodia.
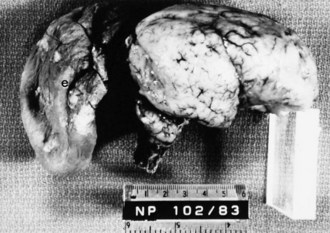
Skin may completely cover the encephalocele, or thin, distorted meningeal membranes may be exposed. When the ventricular system also is herniated into the encephalocele sac, hydrocephalus develops. Leaking CSF rapidly leads to infection. Some encephaloceles, particularly those of the occipital midline, may become so large that they exceed the size of the infant’s head. Nasopharyngeal encephaloceles are rare but may be a source of meningitis from CSF leak through the nose. Malformations of the visceral organs often coexist with encephaloceles, and other congenital anomalies of the eyes and face, cleft palate, and polydactyly are also common. The entire brain may be severely hypoplastic (see Fig. 60.1).
Congenital Aqueductal Stenosis
Another aspect for consideration in the category of disorders of neurulation is the down-regulation of genes in the vertical axis of the neural tube. In the case of the ventrodorsal gradient due to defective SHH expression, sacral agenesis with dysplastic spinal cord at the levels of the deficient vertebrae (and notochord) is the best example. Down-regulation in the dorsoventral gradient of several genes or gene families, including ZIC2, SHH in the forebrain, BMP, and PAX, may result in holoprosencephaly (see following section on Holoprosencephaly) or may cause defective development of the dorsomedial septum of the midbrain with aqueductal stenosis (Sarnat and Flores-Sarnat, 2010b). Box 60.2 lists the various causes of congenital aqueductal stenosis (Sarnat and Flores-Sarnat, 2010b).
Box 60.2
Causes of Congenital Aqueductal Stenosis
Genetic or Presumed Genetic Causes
Acquired Causes in Utero
Intraventricular hemorrhage with thrombus in aqueduct
Congenital infections (e.g., cytomegalovirus infection, mumps)
Ependymitis/ventriculitis with gliosis around and within aqueduct
Aqueductal membrane across lumen
Aneurysms, venous angiomas, and other vascular malformations
Cystic dilatation of perivascular Virchow-Robin spaces in midbrain
Tumors of aqueduct (e.g., ependymoma, astrocytoma, glioneuronal hamartoma, neurepithelial tumor of subcommissural organ)
Tumors that compress the midbrain tectum from above (e.g. pineal tumors and cysts, arachnoidal cysts, lipomas)
From Sarnat, H.B., Flores-Sarnat, L., 2010. Congenital aqueductal stenosis associated with aplasia or hypoplasia of the dorsal median septum of the midbrain. A disorder of genetic gradients along the axes of the neural tube. [To be submitted.]
Midline Malformations of the Forebrain (4 to 8 Weeks’ Gestation)
Holoprosencephaly
Traditionally, HPE was a single malformation with three variants: alobar, semilobar, lobar. A fourth was added later: the middle interhemispheric variant (Hahn and Pinter, 2002; Simon et al., 2002). Another variant recently described is septopreoptic holoprosencephaly, demonstrated as noncleavage restricted to the preoptic and septal region by MRI in 7 patients (Hahn et al., 2010); we have now recognized two additional cases (unpublished). Recent molecular genetic data redefine HPE as a common end-stage malformation with six known different genetic mutations demonstrated in various cases (Golden, 1998) (Table 60.3). Other chromosomal defects (in loci 3p26, 4,5, 6, 14q13, 14q21.1-q21.2, 20, 21q22.3) are known in which the specific genetic mutation is not yet identified. All six known defective genes together account for only about 20% of cases, so many more gene defects remain undiscovered. Furthermore, each of the traditional anatomical variants of HPE is demonstrable in each of the six known genetic forms, signifying that these merely represent degrees of severity without etiological implication. A defect in the ZIC2 gene is associated with chromosome 13q deletions, and HPE is frequent in infants with trisomy 13 (Brown et al., 1998). One of the most studied of the genetic mutations is the strong ventralizing gene, sonic hedgehog (SHH); lack of expression of this gene in the prechordal mesoderm ventral to the rostral end of the neural tube results in no neural induction (Roessler et al., 1996). Abnormal SHH expression also may be altered in metabolic diseases with impaired cholesterol synthesis and high serum levels of the cholesterol precursor molecule, 7-dehydrocholesterol, as in the Smith-Lemli-Opitz syndrome associated with HPE (Kelley et al., 1996).
Table 60.3 Best Documented Genetic Mutations in Holoprosencephaly
| Chromosomal Locus | Defective Gene | Vertical Gradient Effect |
|---|---|---|
| 2p21 | SIX3 | Dorsoventral |
| 7q36 | SHH | Ventrodorsal (spinal cord, hindbrain), dorsoventral (midbrain, forebrain)* |
| 13q32 | ZIC2 | Dorsoventral |
| 18q11.3 | TGIF | Ventrodorsal |
| 9q22.3 | PTCH | Ventrodorsal |
| 10q11.2 | DKK | Ventrodorsal |
* Although SHH is a powerful ventralizing gene in the embryonic spinal cord and hindbrain, recent evidence indicates that at the level of the midbrain and most rostral regions of the neural tube, it changes its gradient and becomes dorsalizing in the vertical axis.
Data from Blaess, S., Corrales, J.D., Joyner, A.L., 2006. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development 133, 1799-1809.
After chromosomal defects, the most common association of HPE is maternal diabetes mellitus; sacral agenesis is another common malformation in infants of diabetic mothers. Both involve down-regulation of SHH. A defect at the same chromosome 7p36.2 locus associated with an autosomal dominant form of HPE also affects SHH at the posterior, rather than the anterior, end of the neural tube and results in sacral agenesis (Lynch et al., 1995). Disturbed insulin metabolism may affect SHH in programming the neural tube.
The diagnosis of HPE often occurs at the time of delivery, because 93% of patients exhibit midline facial dysplasias. Midfacial hypoplasia is present in most patients with HPE, but others have a normal face. The facial dysmorphism ranges from mild hypotelorism and vomer bones to severe forms including cebocephaly with a single naris, severe hypotelorism and absence of the premaxilla and vomer bones to produce a midline cleft lip and palate, or cyclopia with a midline proboscis dorsal to the single median eye. The severity of the facial dysmorphism does not correlate as well with the anatomical variant as originally expressed in the often cited statement “the face predicts the brain.” Midfacial hypoplasia does correlate, however, with the rostrocaudal extent of the defective genetic expression. If the gradient extends to the embryonic mesencephalic neuromere and causes hypoplasia of the midbrain, neural crest formation and migration are affected (Sarnat and Flores-Sarnat, 2001a). The mesencephalic neural crest is the most rostral origin of neural crest, and this tissue forms not only peripheral neural structures such as the ciliary ganglion but also most of the membranous bones of the face, globe of the eye (except the retina and choroid), and much of the facial connective tissue.
The various forms of HPE are well demonstrated by most imaging techniques (Fig. 60.10), including prenatal ultrasound. The imaging features of each anatomical variant are distinctive (Hahn and Pinter, 2002) and correspond well to the gross neuropathological findings (Golden, 1998). The anterior cerebral artery is usually a single azygous vessel coursing just beneath the inner table of the skull, a pathognomonic finding. The sagittal sinuses, deformed or replaced by a network of large abnormal veins, resemble the early embryonic pattern of venous drainage.
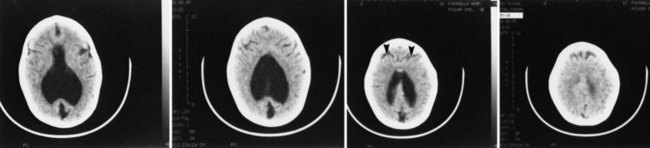
The EEG in HPE shows multifocal spikes that often evolve into hypsarrhythmia. In the neonatal period, the characteristic feature of the waking EEG is almost continuous high-voltage alpha-theta monorhythmic activity, becoming discontinuous in sleep. Visual evoked potentials also are abnormal or altogether absent. The characteristic clinical course of HPE is severe developmental delay and a mixed pattern of seizures that often are refractory to antiepileptic drugs. The presence or absence of seizures does not correlate with the anatomical severity or variant of the defective forebrain and correlates poorly with the genetic mutation (Hahn and Pinter, 2002). A better correlation may be with the degree of mediolateral extension of genetic expression in disrupting the histological architecture of the cortex, or it may relate to an abnormal sequence of maturation of axosomatic (inhibitory) and axodendritic (excitatory) synapses in relation to the maturation of the neuron innervated by these axonal terminations (Sarnat and Flores-Sarnat, 2001a). Some patients develop hydrocephalus that requires a ventriculoperitoneal shunt. This condition is paradoxically more common in the less severe anatomical forms of the malformation. In the severe alobar form, a “dorsal cyst” occupies the entire posterior one-half to two-thirds of the intracranial space and occasionally even protrudes through the anterior fontanelle as a unique encephalocele that may be larger than the rest of the head. No other type of encephalocele occurs at the anterior fontanelle. The dorsal cyst seems to originate from a dilated suprapineal recess of the third ventricle and later is a dorsal membrane that includes the roof of the forebrain, extending from the hippocampi (Sarnat and Flores-Sarnat, 2001a).
Endocrine dysfunction may be present, associated with hypothalamic or pituitary involvement, and vasopressin-sensitive diabetes insipidus occurs in about 86% of cases, other hypothalamic-pituitary dysfunction being much less frequent (Plawner et al., 2002). The basis of this specific involvement of the paraventricular and supraoptic hypothalamic nuclei may be hypoplasia in some cases in which the midline hypoplasia involves the diencephalon as well as the forebrain (most patients), but it also occurs in some children without hypothalamic noncleavage. One hypothesis is that the primary gene defect suppresses expression of the gene, orthopedia (OTP). OTP and downstream genes such as SIM1 and BRN2 are essential for terminal differentiation of neuroendocrine cells of these hypothalamic nuclei (Sarnat and Flores-Sarnat, 2001a).
Isolated Arhinencephaly and Kallmann Syndrome
Absence of olfactory bulbs, tracts, and tubercles commonly accompanies extensive malformations such as HPE and septo-optic dysplasia but may occur with callosal agenesis or as an isolated cerebral anomaly. Kallmann syndrome is an X-linked autosomal dominant condition limited to males, in which anosmia secondary to arhinencephaly without other forebrain malformations is associated with lack of secretion of gonadotropic hormones. The defective gene is KAL1 at the chromosome Xp22.3 locus. Also implicated is the EMX2 gene, though schizencephaly does not occur with Kallmann syndrome (Taylor et al., 1999). Olfactory reflexes may be elicited in the neonate consistently after 32 weeks’ gestation and provide a useful supplement to the neurological examination of newborns suspected of cerebral dysgenesis.
Septo-Optic–Pituitary Dysplasia
Midline cerebellar defects and hydrocephalus occur inconsistently in septo-optic dysplasia. One cerebellar lesion, called rhombencephalosynapsis is aplasia of the vermis and midline fusion of the cerebellar hemispheres and of the dentate nuclei, probably the down-regulation of a dorsalizing gene at the level of rhombomere 1 (Sarnat, 2000).
Clinical manifestations relate mainly to the endocrine deficiencies and vision impairment. Ataxia may be compensable if the cerebellar vermis is mildly involved. Seizures are uncommon. Intellectual development usually is normal. Hypertelorism is not a constant finding. Chromosome analysis is invariably normal. The gene, HESX1, is defective in at least some cases (Dattani et al., 1998). No reports of familial cases exist. However, a high incidence of teenage pregnancy and drug abuse in early gestation occurs in mothers of affected infants. Septo-optic–pituitary dysplasia has occurred in an infant of a diabetic mother.
Rhombomeric Deletions and Ectopic Genetic Expression
Rare patients with absence of certain parts of the brain appear in the literature. Only recently, by understanding the families of genes responsible for neural tube segmentation (e.g., HOX, WNT, PAX), have these conditions been understood at the level of molecular embryology. Agenesis of the midbrain and upper pons (metencephalon) with cerebellar hypoplasia are attributable to the EN2 gene, which produces an almost identical malformation in the knockout mouse model (Sarnat et al., 2002). EN1 and WNT1 genes also are essential for development of the mesencephalic and rhombomere 1, but the animal models of these genetic defects produce total agenesis of the cerebellum. Sonic hedgehog also regulates the temporal and spatial precision of the midbrain-hindbrain junction, mediated through Gli activator (Blaess et al., 2006). Absence of the corpus striatum might be due to mutation of the EMX1 gene, which is essential in the programming of the basal telencephalon but not the cerebral cortex (Sarnat and Flores-Sarnat, 2001a). The Chiari malformations, particularly type II, were incompletely explained by mechanical theories of pathogenesis, but a molecular genetic hypothesis of ectopic expression provides a more complete and reasonable explanation (see section on Chiari Malformations) (Sarnat and Flores-Sarnat, 2001b, 2004). Despite documentation of many of these genetic malformations in experimental animal models, definitive confirmation in humans has not occurred.
Agenesis of the Corpus Callosum
The pathogenesis of callosal agenesis relates to the commissural plate; if this plate is not available to guide axons across, the corpus callosum does not develop. Failure of physiological degeneration of a portion of the plate results in a glial barrier to axonal passage and the disappearance or deflection of primordial callosal fibers posteriorly to another destination within their hemisphere of origin (bundle of Probst). Other destinations of callosal axons that are unable to cross the midline at their expected site include passage into the anterior commissure, which can become enlarged as much as four times its normal volume by the addition of these axons; aberrant sites of crossing of individual fibers not forming large bundles; and occasionally, callosal axons descend within the internal capsule with the corticospinal tract as far as the spinal cord, where their termination and function remain unknown (Sarnat 2008).
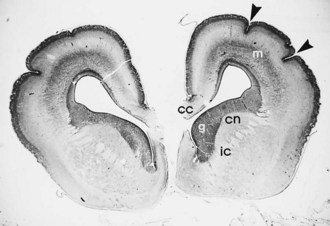
A rare genetic form of callosal agenesis is associated with defective neural crest migration causing aganglionic megacolon (Hirschsprung disease). The cause is a defective human gene, Smad-interacting protein 1 (SMAD1), at the chromosome 2q22-q23 locus (Cacheux et al., 2001). In the absence of a corpus callosum, the lateral ventricles displace laterally, and the third ventricle rises between them (Fig. 60.11). Often the ventricles dilate mildly, but intraventricular pressure is normal. The anomaly may be demonstrable by most imaging techniques. The varying degrees of partial callosal agenesis produce several radiographic variants.
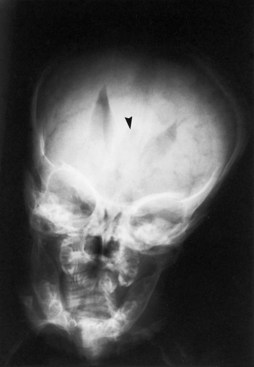
Colpocephaly
1. It appears as a primary malformation, histologically associated with poorly laminated striate cortex, subcortical heterotopia, and defective ependymal lining the occipital horns.
2. It is common in many cases of agenesis of the corpus callosum because of absence of the splenium and hypoplasia of white matter.
3. It may be the acquired result of periventricular leukomalacia, especially in premature infants, because of loss of periventricular white matter in the posterior half of the cerebral hemispheres.
Disorders of Early Neuroblast Migration (8 to 20 Weeks’ Gestation)
Lissencephaly (Agyria, Sometimes with Pachygyria)
Lissencephaly is a failure of development of convolutions in the cerebral cortex because of defective neuroblast migration. The cortex remains smooth, as in the embryonic brain (see Fig. 60.9). The migrations of the cerebellum and the brainstem also usually are involved, but the thalamus and basal ganglia form properly. Structural and metabolic abnormalities of the fetal ependyma may be supplementary factors in disturbing the normal development of radial glial cells.
The cytoarchitecture of the neocortex in lissencephaly takes one of two forms. In the first, a four-layered sequence develops. The outermost layer is a widened molecular zone; layer 2 contains neurons corresponding to those of normal laminae III, V, and VI; layer 3 is cell-sparse; and layer 4 contains heterotopic neurons that have migrated incompletely. Decreased brain size leads to microcephaly with widened ventricles, representing a fetal stage rather than pressure from hydrocephalus, and an uncovered sylvian fossa representing lack of operculation. The second form of cortical architectural abnormality in lissencephaly is disorganized clusters of neurons with haphazard orientation, forming no definite layers or predictable pattern. Type 2 lissencephaly is associated with several closely related genetic syndromes: Walker-Warburg syndrome, Fukuyama muscular dystrophy, muscle-eye-brain disease of Santavuori, and Meckel-Grüber syndrome, the latter often associated with posterior encephalocele (see Fig. 60.9).
Miller-Dieker Syndrome (Type 1 Lissencephaly)
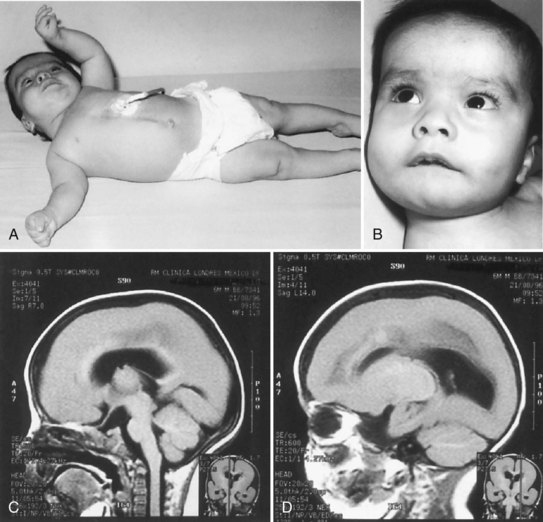
Miller-Dieker syndrome is a familial lissencephaly characterized clinically by microcephaly and a peculiar facies that includes micrognathia, high forehead, thin upper lip, short nose with anteverted nares, and low-set ears (Fig. 60.12). Neurologically, the children are developmentally delayed in infancy and mentally retarded, lack normal responsiveness to stimuli, initially exhibit muscular hypotonia that later evolves into spasticity and opisthotonos, and develop intractable seizures. Death before 1 year of age is common. The EEG often shows focal or multifocal spike-wave discharges that later become bisynchronous bursts of diffuse paroxysmal activity, and extremely high-voltage diffuse rhythmic theta and beta activity. At autopsy, the original cases showed lack of gyral development in the cerebral cortex. Later patients with the typical craniofacial features and clinical course showed gyral development, although the convolutions were abnormal, and pachygyria predominated. The term Miller-Dieker syndrome as originally proposed was to distinguish this syndrome from other cases of lissencephaly without the clinical and dysmorphic facial features. A microdeletion at the chromosome 17p13.3 locus is demonstrable by high-resolution studies in most patients with Miller-Dieker syndrome, and family members of the original patients show the defect (Chong et al., 1997). The responsible gene is LIS1. Histological examination of the brain in Miller-Dieker syndrome confirms the presence of a severe disorder of neuroblast migration, as in other cases of lissencephaly.
X-Linked Recessive Lissencephaly with Abnormal Genitalia
The most recently defined genetic form of lissencephaly is due to a mutation in the ARX gene in both the mouse and human (Kitamura et al., 2002). It generally is associated with microencephaly and global cerebellar hypoplasia but also with abnormal genitalia. The brain malformation is the most severe of the lissencephalies, and affected children are profoundly mentally retarded and have epilepsy.
Subcortical Laminar Heterotopia (Band Heterotopia) and Bilateral Periventricular Nodular Heterotopia
Subcortical laminar heterotopia and bilateral periventricular nodular heterotopia both result from X-linked recessive traits occurring almost exclusively in females. Both disorders present clinically as severe seizure disorders in childhood, although they are often associated also with mental retardation and other neurological deficits. In subcortical laminar heterotopia, a band of gray matter heterotopia within the subcortical white matter lies parallel to the overlying cerebral cortex but separated from it by white matter. Histologically, it lacks lamination, as is the normal cortex, and consists of disoriented neurons and glial cells and fibers with poorly organized architecture. The few male fetuses that have not spontaneously aborted have been born with lissencephaly, in addition, and even more severe neurological deficits. The defective gene and its transcription product in subcortical laminar heterotopia are known; the latter is called doublecortin (Gleeson et al., 1999). In bilateral periventricular nodular heterotopia, islands of neurons and glial cells occur in the subependymal regions around the lateral ventricles and are neuroepithelial cells that have matured in their site of origin without migrating (Eksioglu et al., 1996). The gene responsible is filamin-1. MRI best demonstrates both conditions, but they are also detectable by CT.
Schizencephaly
Schizencephaly is a unilateral or bilateral deep cleft (usually in the general position of the sylvian fissure) but is not a sylvian fissure. This cleft is the full thickness of the hemispheric wall, and no cerebral tissue remains between the meninges and the lateral ventricle (the pial-ependymal seam). If the cerebral cortical walls on either side of the deep cleft are in contact, the condition is closed lip, and if a wide subarachnoid space separates the two walls, it is open lips, but these two variants do not provide a clue to pathogenesis. Schizencephaly is often classified as a neuroblast migratory disorder, but this mechanism is only partially true; it is primarily a disorder of development of the telencephalic flexure (see Fig. 60.6). It may occur either as a Mendelian or sporadic genetic trait or as a fetal deformation of the telencephalic hemisphere at the time of development of the telencephalic flexure (Sarnat and Flores-Sarnat, 2010a); in some cases, it results from porencephaly due to fetal cerebral infarction.
Schizencephaly was thought to be associated with defective expression of the gene, EMX2 (Granata et al., 1997), but this is not the case (Tietjen et al., 2007), and the genetic basis remains unknown. It may be associated with a variable degree of lissencephaly/pachygyria, may be bilaterally symmetrical, or may be asymmetrical and more severe on one side. Schizencephaly is unilateral in half of cases.
Disorders of Cerebellar Development (32 Days’ Gestation to 1 Year Postnatally)
Chiari Malformation
The pathogenesis has been a matter of controversy for many years. Mechanical theories have dominated since the time of Chiari: (1) the traction theory, a result of a tethered spinal cord with traction as the vertebral column grows; (2) the pulsion theory of fetal hydrocephalus pushing the cerebellum and brainstem from above; and (3) the crowding theory in which a small posterior fossa provides insufficient room for the growth of neural structures and causes a “toothpaste tube effect.” The torcula is indeed too low and the volume of the posterior fossa too small, so that this latter explanation is probably a true contributory factor, but only in late gestation as a superimposed secondary influence. A molecular genetic hypothesis of ectopic expression of a segmentation gene in the rhombomeres explains not only the Chiari malformation but also the brainstem anomalies, the myelodysplasia, and the defective basioccipital and supraoccipital bone formation that results in a too-small posterior fossa (Sarnat, 2004a).
Global Cerebellar Hypoplasia
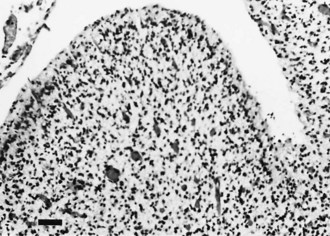
Global cerebellar hypoplasia has diverse causes that include chromosomal and genetically determined diseases, Tay-Sachs disease, Menkes kinky hair disease, some cases of spinal muscular atrophy, and sporadic cases of unknown cause. Histologically, there may be a selective depletion of granule cells or a loss of Purkinje cells and other neuronal elements in addition to granule cells (Fig. 60.13). In selective granule cell depletion, the axons and dendrites of Purkinje cells are deformed.
Focal Cerebellar Dysplasia
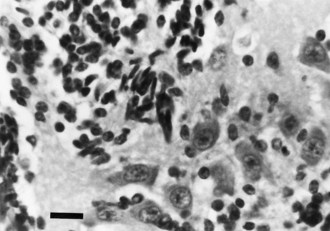
Focal dysplasias and hamartomas of the cerebellar cortex (Fig. 60.14) are often incidental findings at autopsy and are often clinically asymptomatic. More extensive lesions present abnormal cerebellar findings clinically. These small focal malformations are a disorder of neuronal migration programmed as genetic defects or, more commonly, acquired from brief insults during the long period of cerebellar development. Focal ischemic insults and exposure to cytotoxic drugs or viruses are among the more common causes. The granule cells of the cerebellar cortex retain a regenerative capacity lost early in gestation by most other neurons, but the regenerative pattern of lamination in the cerebellar cortex may be imperfect.
Craniosynostosis
The development of the cranium and sutures closely relates to neuromeric formation in the neural tube. Mesencephalic neural crest induces most of craniofacial development and is involved in the formation of the rostral two-thirds of the cranial vault and underlying meninges. More than 100 syndromes are identifiable that include a component of craniosynostosis. Several have a genetic basis, including the syndromes of Apert, Crouzon, Pfeiffer, and Saethre-Chotzen; these are related to fibroblast growth factor receptor defects, and in some, specific causative genes such as TWIST are recognized (Flores-Sarnat, 2002b). The genetic bases of the more common isolated craniostenoses of the coronal and sagittal sutures are unknown. Although once thought to be only a cosmetic defect, some believe that untreated isolated synostosis results in neurological impairment. Advances in imaging have enhanced both prenatal and postnatal diagnosis, and parallel advances in craniofacial surgery makes treatment more effective from both a neurological and cosmetic perspective. Distinguishing true craniosynostosis from positional deformities of the head resulting from abnormal prenatal compression by the maternal pelvis and postnatal effects of head position, particularly in premature infants, is important for management.
Bachmann R., Reilmann R., Schwindt W., et al. FLAIR imaging for multiple sclerosis: a comparative MR study at 1.5 and 3.0 Tesla. Eur Radiol. 2006;16:915-921.
Bakshi R., Benedict R.H.B., Bermel R.A., et al. T2 hypointensity in the deep gray matter of patients with multiple sclerosis. A quantitative magnetic resonance imaging study. Arch Neurol. 2002;59:62-68.
Bakshi R., Thompson A.J., Rocca M.A., et al. MRI in multiple sclerosis: current status and future prospects. Lancet Neurology. 2008;7:615-625.
Barth P.G., Blennow G., Lenard H.G., et al. The syndrome of autosomal recessive pontocerebellar hypoplasia, microcephaly and extrapyramidal dyskinesias (pontocerebellar hypoplasia type 2): compiled data from 10 pedigrees. Neurology. 1995;45:311-317.
Beck R.W., Trobe J.D., Moke P.S., et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol. 2003;121:944-949.
Bermel R.A, Bakshi R. The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurology. 2006;5:158-170.
Bitsch A., Kuhlmann T., Stadelmann C., et al. A longitudinal MRI study of histopathologically defined hypointense multiple sclerosis lesions. Ann Neurol. 2001;49:793-796.
Blaess S., Corrales J.D., Joyner A.L. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development. 2006;133:1799-1809.
Bonnevillea F., Moriartya D.M., Belinda S.Y., et al. Whole-Brain N-Acetylaspartate Concentration: Correlation with T2-Weighted Lesion Volume and Expanded Disability Status Scale Score in Cases of Relapsing-Remitting Multiple Sclerosis. AJNR. 2002;23:371-375.
Bourdette D., Simon J. The radiologically isolated syndrome. Is it very early multiple sclerosis? Neurology. 2009;72:780-781.
Brass S.D., Benedict R.H.R, Weinstock-Guttman R., et al. Cognitive impairment is associated with subcortical magnetic resonance imaging grey matter T2 hypointensity in multiple sclerosis. Mult Scler. 2006;12:437-444.
Brex P.A., Ciccarelli O., O’Riordan J., et al. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med. 2002;346:158-164.
Brinkmann V., Pinschewer D.D., Feng L., et al. Fty720: Altered Lymphocyte Traffic Results in Allograft Protection. Transplantation. 2001;72:764-769.
Brown S.A., Warburton D., Brown L.Y., et al. Holoprosencephaly due to mutations in Zic2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180-183.
Cacheux V., Dastot-Le Moal F., Kaariainen H., et al. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum Mol Genet. 2001;10:1503-1510.
Cadavid D., Wolansky L.J., Skumick J., et al. Efficacy of treatment of MS with IFNβ-1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology. 2009;72:1976-1983.
Chen J.T., Kuhlmann T, Jansenc G.H., et al. Voxel-based analysis of the evolution of magnetization transfer ratio to quantify remyelination and demyelination with histopathological validation in a multiple sclerosis. NeuroImage. 2007;36:1152-1158.
Chevassus-au-Louis N., Baraban S.C., Gaïarsa J.-L., et al. Cortical malformations and epilepsy: new insights from animal models. Epilepsia. 1999;40:811-821.
Chong S.S., Pack S.D., Roschke A.V., et al. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet. 1997;6:147-155.
Clark D.C., Mizuguchi M., Antalffy B., et al. Predominant localization of the LIS family of gene products to Cajal-Retzius cells and ventricular neuroepithelium in the developing human cortex. J Neuropathol Exp Neurol. 1997;56:1044-1052.
Coles A.J., Compston D.A.S., Selmaj K.W., et al. Alemtuzumab vs. Interferon beta-1a in early multiple sclerosis. N Eng J Med. 2008;359:1786-1801.
Comi G., Abramsky O., Arbizu T., et al. Long-term open extension of oral laquinimod in patients with relapsing multiple sclerosis shows favourable safety and sustained low relapse Multiple Sclerosis. Immunomodulation. 2009. September 10, 2009
Confavreux C., Hutchinson M., Hours M.M., et al. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med. 1998;339:285-291.
Curran T., D’Arcangelo G. Role of reelin in the control of brain development. Brain Res Rev. 1998;26:286-294.
Dahl J., Myhr K.M., Daltveit AX., et al. Pregnancy, delivery, and birth outcome in women with multiple sclerosis. Neurology. 2005;65:1961-1963.
Dalton C.M., Brex P.A., Miszkiel K.A., et al. New T2 lesions enable an earlier diagnosis of multiple sclerosis in clinically isolated syndromes. Annals of Neurology. 2003;53:673-676.
Dattani M.T., Martínez-Barbera J.P., Thomas P.Q., et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nature Genet. 1998;19:125-133.
De Stefano N., Battaglini M., Stromillo M.L., et al. Brain damage as detected by magnetization transfer imaging is less pronounced in benign than in early relapsing multiple sclerosis. Brain. 2006;129:2008-2016.
De Stefano N., Matthews P.M., Fu L., et al. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;121:1469-1477.
D’hooghe M.B., Nagels G., Uitdehaag B.M.J. Long-term effects of childbirth in MS. J Neurol Neurosurg Psychiatry. 2010;81:38-41.
Dupuy S., Houser C.R. Developmental changes in GABA neurons of the rat dentate gyrus: an in situ hybridization and birthdating study. J Comp Neurol. 1997;389:402-418.
Dutt S., Matasci M., Sommer L., Zimmermann D.R. Guidance of neural crest cell migration: the inhibitory function of the chondroitin sulfate proteoglycan, versican. Sci World J. 2006;6:1114-1117.
Eksioglu Y.Z., Scheffere I.E., Cardenas P., et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16:77-87.
Filippi M., Agosta F. Magnetization Transfer MRI in Multiple Sclerosis. J Neuroimaging. 2007;17(Suppl. s1):S22-S26.
Filippi M., Cercignani M., Inglese M., et al. Diffusion tensor magnetic resonance imaging in multiple sclerosis. Neurology. 2001;56:304-311.
Flores-Sarnat L. Hemimegalencephaly: Part 1. Genetic, clinical and imaging aspects. J Child Neurol. 2002;17:373-384.
Flores-Sarnat L. New insights into craniosynostosis. Semin Pediatr Neurol. 2002;9:274-291.
Flores-Sarnat L. Neurocutaneous syndromes and hemimegalencephaly. In: Curatolo P., Riva D. Neurocutaneous Syndromes in Children. John Libbey Eurotext and Fondazione Pierfranco e Luisa Mariani; 2006:55-72.
Flores-Sarnat L. Hemimegalencephaly syndrome. In: Sarnat H.B., Curatolo P. Handbook of Clinical Neurology, vol. 87. Malformations of the Nervous System. Edinburgh: Elsevier; 2008:153-176.
Flores-Sarnat L., Sarnat H.B., Dávila-Gutiérrez G., Álvarez A. Hemimegalencephaly: Part 2. Neuropathological aspects suggesting a disorder of cellular lineage. J Child Neurol. 2003;18:776-785.
Gauthier S.A., Berger A.M., Liptak Z., et al. Rate of Brain Atrophy in Benign vs Early Multiple Sclerosis. Arch Neurol. 2009;66:234-237.
Gleeson J.G., Minnerath S.H., Fox J.W., et al. Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol. 1999;45:146-153.
Golden J.A. Holoprosencephaly. A defect in brain patterning. J Neuropathol Exp Neurol. 1998;57:991-999.
Goodkin D.E., Rudick R.A., Medendorp S.V., et al. Low-dose (7.5 mg) oral methotrexate reduces the rate of progression in chronic progressive multiple sclerosis. Annals of Neurology. 1995;37:30-40.
Granata T., Farina L., Faiella A., et al. Familial schizencephaly associated with EMX2 mutation. Neurology. 1997;48:1403-1406.
Gressens P. Pathogenesis of migration disorders. Curr Opin Neurol. 2006;19:135-140.
Gronseth G.S., Ashman E.J. Practice parameter: the usefulness of evoked potentials in identifying clinically silent lesions in patients with suspected multiple sclerosis (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;54:1720-1725.
Hahn J.S., Barnes P.D., Clegg N.J., et al. Septopreoptic holoprosencephaly: a mild subtype associated with midline craniofacial anomalies. Am J Neuroradiol. 2010;31:1596-1601.
Hahn J.S., Pinter J.D. Holoprosencephaly: genetic, neuroradiological and clinical advances. Semin Pediatr Neurol. 2002;9:309-319.
Hauser S.L., Waubant E., Arnold D.L. B-Cell Depletion with Rituximab in Relapsing–Remitting Multiple Sclerosis. N Engl J Med. 2008;358:676-688.
Hevner R.F. Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J Neuropathol Exp Neurol. 2007;66:101-109.
Houtchens, M.K., Glanz, B., Weiner, H.L. Cognitive Phenotype in Multiple Sclerosis. J Multiple Sclerosis. In Press.
Israely I., Costa R.M., Xie C.W., et al. Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr Biol. 2004;14:1657-1663.
Jaquier M., Klein A., Boltshauser E. Spontaneous pregnancy outcome after prenatal diagnosis of anencephaly. BJOG. 2006;133:951-953.
Johansson C.B., Momma S., Clarke D.L., et al. Identification of a neural stem cell in the adult mammalian nervous system. Cell. 1999;96:25-34.
Jouet M., Kenwrick S. Gene analysis of L1 neural cell adhesion molecule in prenatal diagnosis of hydrocephalus. Lancet. 1995;345:161-162.
Kappos L., Antel J., Comi G., et al. Oral Fingolimod (FTY720) for Relapsing Multiple Sclerosis. N Engl J Med. 2006;355:1124-1140.
Kappos L., Radue E.-W., O’Connor P., et al. A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N Engl J Med. 2010;362:387-401.
Kelley R.L., Roessler E., Hennekam R.C., et al. Holoprosencephaly in RSH/Smith-Lemli-Opitz syndrome: does abnormal cholesterol metabolism affect the function of Sonic hedgehog? Am J Med Genet. 1996;66:78-84.
Kelly V.M., Nelson L.M., Chakravarty E.F. Obstetric outcomes in women with multiple sclerosis and epilepsy. Neurology. 2009;73:1831-1836.
Kendler A., Golden J.A. Progenitor cell proliferation outside the ventricular and subventricular zones during human brain development. J Neuropathol Exp Neurol. 1996;55:1253-1258.
Kitamura K., Yanazawa M., Sugiyama N., et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359-369.
Lebrun C., Bensa C., Debouverie M., et al. Association between clinical conversion to multiple sclerosis in radiologically isolated syndrome and magnetic resonance imaging, cerebrospinal fluid, and visual evoked potential. Arch Neurol. 2009;66:841-846.
Losseff N.A., Webb S.L., O’Riordan J.I., et al. Spinal cord atrophy and disability in multiple sclerosis. A new reproducible and sensitive MRI method with potential to monitor disease progression. Brain. 1996;119:701-708.
Lynch S.A., Bond P.M., Copp A.J., et al. A gene for autosomal dominant sacral agenesis maps to the holoprosencephaly region at 7q36. Nat Genet. 1995;11:93-95.
Marcorelles P., Laquerrière A., Adde-Michel C., et al. Evidence for tangential migration disturbances in human lissencephaly resulting from a deficit in LIS1, DCX and ARX genes. Acta Neuropathol. 2010;120:503-515.
Marín-Padilla M. Cajal-Retzius cells and the development of the neocortex. Trends Neurosci. 1998;21:64-71.
Marquardt T., Pfaff S.L. Cracking the transcriptional code for cell specification in the neural tube. Cell. 2001;106:1-20.
Mathiesen H.K., Jonsson A., Tscherning T., et al. Correlation of Global N-Acetyl Aspartate With Cognitive Impairment in Multiple Sclerosis. Arch Neurol. 2006;63:533-536.
Mikol D.D., Barkhof F., Chang P., et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label. Lancet Neurology. 2008;7:903-914.
Miller D.H., Weinshenker B.G., Filippi M., et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. 2008;14:1157-1174.
Miller D.M., Weinstock-Guttman B., Lee J.-C., et al. A meta-analysis of methylprednisolone in recovery from multiple sclerosis exacerbations. Mult Scler. 2000;6:267-273.
Minneboo A., Uitdehaag B.M.J., Ader H.J., et al. Patterns of enhancing lesion evolution in multiple sclerosis are uniform within patients. Neurology. 2005;65:56-61.
Mione M.C., Cavanagh J.F.R., Harris B., Parnavelas J.G. Cell fate specification and symmetrical/asymmetrical divisions in the developing cerebral cortex. J Neurosci. 1997;17:2018-2029.
Mueller B.A., Zhang J., Critchlow C.W. Birth outcomes and need for hospitalization after delivery among women with multiple sclerosis. Am J Obstet Gynecol. 2002;186:446-452.
Neema M., Stankiewicz J., Arora A., et al. T1- and T2-based MRI measures of diffuse gray matter and white matter damage in patients with multiple sclerosis. J Neuroimaging. 2007;17:16S-21S.
O’Connor P., Filippi M., Amason B. 250 µg or 500 µg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurology. 2009;8:889-897.
O’Connor P.W., Li D., Freedman M.S., et al. A Phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology. 2006;66:894-900.
Okuda D.T., Mowry E.M., Beheshtian M.D., et al. Incidental MRI anomalies suggestive of multiple sclerosis. Neurology. 2009;72:800-805.
O’Riordan J.I., Thompson A.J., Kingsley D.P., et al. The prognostic value of brain MRI in clinically isolated syndromes of the CNS. A 10-year follow-up. Brain. 1998;121:495-503.
Plawner L.L., Delgado M.R., Miller V.S., et al. Neuroanatomy of holoprosencephaly as predictor of function: beyond the face predicting the brain. Neurology. 2002;59:1058-1066.
Rakic P. Radial versus tangential migration of neuronal clones in the developing cerebral cortex. Proc Natl Acad Sci U S A. 1995;92:11323-11327.
Rammohan K.W., Shoemaker J. Emerging multiple sclerosis oral therapies. Neurology. 2010;74:S47-S53.
Rashid W., Miller D.H. Recent Advances in Neuroimaging of Multiple Sclerosis. Semin Neurol. 2008;28:046-055.
Rho J.M., Storey T.W. Molecular ontogeny of major neurotransmitter receptor systems in the mammalian central nervous system: norepinephrine, dopamine, serotonin, acetylcholine and glycine. J Child Neurol. 2001;16:271-281.
Roessler E., Belloni E., Gaudenz K., et al. Mutations in the human sonic hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357-360.
Rovaris M., Gass A., Bammer R., et al. Diffusion MRI in multiple sclerosis. Neurology. 2005;65:1526-1532.
Roy N., Mahadevan N., McLean M., et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell. 1995;80:167-178.
Rudick R.A, Fisher E., Lee J.-C., et al. Use of the brain parenchymal fraction to measure whole brain atrophy in relapsing-remitting MS. Neurology. 1999;53:1698.
Sarnat H.B. Molecular genetic classification of central nervous system malformations. J Child Neurol. 2000;15:675-687.
Sarnat H.B. Regional ependymal upregulation of vimentin in Chiari II malformation, aqueductal stenosis and hydromyelia. Pediatr Devel Pathol. 2004;7:48-60.
Sarnat H.B. Watershed infarcts in the fetal and neonatal brainstem. An aetiology of central hypoventilation, dysphagia, Möbius syndrome and micrognathia. Eur J Paediatr Neurol. 2004;8:71-87.
Sarnat H.B. Embryology and malformations of the forebrain commissures. In: Sarnat H.B., Curatolo P. Handbook of Clinical Neurology, vol. 87. Malformations of the Nervous System. Edinburgh: Elsevier; 2008:67-87.
Sarnat H.B., Benjamin D.R., Siebert J.R., et al. Agenesis of the mesencephalon and metencephalon with cerebellar hypoplasia: putative mutation in the EN2 gene. Report of 2 cases in early infancy. Pediatr Dev Pathol. 2002;5:54-68.
Sarnat H.B., Born D.E. Synaptophysin immunocytochemistry with thermal intensification: a marker of terminal axonal maturation in the human fetal nervous system. Brain Dev. 1999;21:41-50.
Sarnat H.B., Flores-Sarnat L. Neuropathological research strategies in holoprosencephaly. J Child Neurol. 2001;16:918-931.
Sarnat H.B., Flores-Sarnat L. A new classification of malformations of the nervous system: an integration of morphological and molecular genetic criteria as patterns of genetic expression. Eur J Paediatr Neurol. 2001;5:57-64.
Sarnat H.B., Flores-Sarnat L. Cajal-Retzius and subplate neurons: their role in cortical development. Eur J Paediatr Neurol. 2002;6:91-97.
Sarnat H.B., Flores-Sarnat L. Integrative classification of morphology and molecular genetics in central nervous system malformations. Am J Med Genet. 2004;126A:386-392.
Sarnat H.B., Flores-Sarnat L. Embryology of the neural crest: its inductive role in the neurocutaneous syndromes. J Child Neurol. 2005;20:637-643.
Sarnat, H.B., Flores-Sarnat, L., 2010a. Telencephalic flexure and developmental disorders of the sylvian fissure. [Submitted.]
Sarnat H.B., Flores-Sarnat L., Trevenen C. Synaptophysin immunoreactivity in the human hippocampus and neocortex from 6 to 41 weeks of gestation. J Neuropathol Exp Neurol. 2010;69:234-245.
Sarnat, H.B. Flores-Sarnat, L., 2012. Congenital aqueductal stenosis associated with aplasia or hypoplasia of the dorsal median septum of the midbrain. A disorder of genetic gradients along the axes of the neural tube.
Sarnat H.B., Menkes J.H. How to construct a neural tube. J Child Neurol. 2000;15:110-124.
Schuldiner M., Eiges R., Eden A., et al. Induced neuronal differentiation of human embryonic stem cells. Brain Res. 2001;913:201-205.
Sicotte N.L., Liva S.M., Klutch R., et al. Treatment of multiple sclerosis with the pregnancy hormone estriol. Annals of Neurology. 2002;52:421-428.
Sicotte N.L., Voskuhl RR, Bouvier S., et al. Comparison of Multiple Sclerosis Lesions at 1.5 and 3.0 Tesla. Investigative Radiology. 2003;38:423-427.
Simeone A. Towards the comprehension of genetic mechanisms controlling brain morphogenesis. Trends Neurosci. 2002;25:119-121.
Simeone T.A., Donevan S.D., Rho J.M. Molecular biology and ontogeny of GABAA and GABAB receptors in the mammalian central nervous system. J Child Neurol. 2003;18:39-48.
Simon E.M., Hevner R.F., Pinter J.D., et al. The middle interhemispheric variant of holoprosencephaly. Am J Neuroradiol. 2002;23:151-156.
Stenager E., Stenager E.N., Jensen K. Effect of pregnancy on the prognosis for multiple sclerosis. A 5-year follow up investigation. Acta Neurologica Scandinavica. 1994;90:305-308.
Takano T., Sawai C.h., Takeushi Y. Radial and tangential neuronal migration disorder in ibotenate-induced cortical lesions in hamsters: immunohistochemical study of reelin, vimentin, and calretinin. J Child Neurol. 2004;19:107-115.
Tavazzia E., Dwyera M.G., Weinstock-Guttmanc B., et al. Quantitative diffusion weighted imaging measures in patients with multiple sclerosis. NeuroImage. 2007;36:746-754.
Taylor H.S., Block K., Bick D.P., et al. Mutation analysis of the EMX2 gene in Kallmann’s syndrome. Fertil Steril. 1999;72:910-914.
Thomas L.B., Gates M.A., Steindler D.A. Young neurons from the adult subependymal zone proliferate and migrate along an astrocyte, extracellular matrix-rich pathway. Glia. 1996;17:1-14.
Tietjen I., Bodell A., Apse K., et al. Comprehensive EMX2 genotyping of a large schizencephaly case series. Am J Med Genet. 2007;143:1313-1316.
Traboulsee A., Dehmeshki J., Peters K.R., et al. Disability in multiple sclerosis is related to normal appearing brain tissue MTR histogram abnormalities. Mult Scler. 2003;9:566-573.
Truyen L., van Waesberghe J.H.T.M., van Walderveen M.A.A., et al. Accumulation of hypointense lesions (“black holes”) on T1 spin-echo MRI correlates with disease progression in multiple sclerosis. Neurology. 1996;47:1469-1476.
Ulfig N. Calcium-binding proteins in the human developing brain. Adv Anat Embryol Cell Biol. 2002;165(III-IX):1-92.
Voskuhl R.R., Palaszynski K. Sex hormones in experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neuroscientist. 2001;7:258-270.
Vukusic S., Confavreux C. Pregnancy and multiple sclerosis: the children of PRIMS. Clinical Neurology and Neurosurgery. 2006;108:266-270.
Vukusic S., Hutchinson M., Hours M., et al. Pregnancy and multiple sclerosis (the PRIMS study): clinical predictors of post-partum relapse. Brain. 2004;127(Pt 6):1353-1360.
Werring D.J., Brassat D., Droogan A.G., et al. The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosis. A serial diffusion MRI study. Brain. 2000;123:1667-1676.
Yang J.-S., Xua L.-Y., Xiaoa R.G., et al. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-β in Lewis rats. J Neuroimmunology. 2004;156:3-9.
Yew D.T., Luo C.B., Heizmann C.W., Chan W.Y. Differential expression of calretinin, calbindin D28K and parvalbumin in the developing human cerebellum. Brain Res Dev Brain Res. 1997;103:37-45.
Zheng C., Heintz N., Hatten M.E. CNS gene encoding astrotactin, which supports neuronal migration along glial fibers. Science. 1996;272:417-419.
Zivadinov R. Can imaging techniques measure neuroprotection and remyelination in multiple sclerosis? Neurology. 2007;68(Suppl. 3):S72-S82.