Chapter 600 Developmental Disorders of Muscle
Myogenic Regulatory Genes and Genetic Loci of Inherited Diseases of Muscle
A family of four myogenic regulatory genes shares encoding transcription factors of “basic helix-loop-helix” (bHLH) proteins associated with common DNA nucleotide sequences (Table 600-1). These genes direct the differentiation of striated muscle from any undifferentiated mesodermal cell. The earliest bHLH gene to program the differentiation of myoblasts is myogenic factor 5 (Myf5). The second gene, myogenin, promotes fusion of myoblasts to form myotubes. Herculin (also known as MYF6) and MYOD1 are the other two myogenic genes. Myf5 cannot support myogenic differentiation without myogenin, MyoD, and MYF6. Each of these four genes can activate the expression of at least one other and, under certain circumstances, can autoactivate as well. The expression of MYF5 and of herculin is transient in early ontogenesis but returns later in fetal life and persists into adult life. The human locus of the MYOD1 gene is on chromosome 11, very near to the domain associated with embryonal rhabdomyosarcoma. The genes encoding Myf5 and herculin are on chromosome 12 and that for myogenin is on chromosome 1.
Table 600-1 INHERITANCE PATTERNS AND CHROMOSOMAL OR MITOCHONDRIAL LOCI OF NEUROMUSCULAR DISEASES AFFECTING THE PEDIATRIC AGE GROUP
| DISEASE | TRANSMISSION | LOCUS |
|---|---|---|
| Duchenne and Becker muscular dystrophy | XR | Xp21.2 |
| Emery-Dreifuss muscular dystrophy | XR | Xq28 |
| Myotonic muscular dystrophy (Steinert) | AD | 19q13 |
| Facioscapulohumeral muscular dystrophy | AD | 4q35 |
| Limb-girdle muscular dystrophy | AD | 5q |
| Limb-girdle muscular dystrophy | AR | 15q |
| Congenital muscular dystrophy with merosin deficiency | AR | 6q2 |
| Congenital muscular dystrophy (Fukuyama) | AR | 8q31-33 |
| Myotubular myopathy | XR | Xq28 |
| Myotubular myopathy | AR | Unknown |
| Nemaline rod myopathy (NEM1) | AD | 1q21-q23 |
| Nemaline rod myopathy (NEM2) | AR | 2q21.2-q22 |
| Nemaline rod myopathy (NEM3) | AD, AR | 1q42.1 |
| Nemaline rod myopathy (NEM4) | AD | 9q13 |
| Nemaline rod myopathy (NEM5) | AR | 19q13 |
| Congenital muscle fiber-type disproportion | AR, X-linked R | 19p13.2, Xp23.12-p11.4, Xq13.1-q22.1; t(10; 17); sporadic |
| Central core disease | AD | 19q13.1 |
| Myotonia congenita (Thomsen) | AD | 7q35 |
| Myotonia congenita (Becker) | AR | 7q35 |
| Paramyotonia congenita | AD | 17q13.1-13.3 |
| Hyperkalemic periodic paralysis | AD | 17q13.1-13.3 |
| Hyperkalemic periodic paralysis | AD | 1q31-q32 |
| Glycogenosis II (Pompe; acid maltase deficiency) | AR | 17q23 |
| Glycogenosis V (McArdle; myophosphorylase deficiency) | AR | 11q13 |
| Glycogenosis VII (Tarui; phosphofructokinase deficiency) | AR | 1cenq32 |
| Glycogenosis IX (phosphoglycerate kinase deficiency) | XR | Xq13 |
| Glycogenosis X (phosphoglycerate mutase deficiency) | AR | 7p12-p13 |
| Glycogenosis XI (lactate dehydrogenase deficiency) | AR | 11p15.4 |
| Muscle carnitine deficiency | AR | Unknown |
| Muscle carnitine palmityltransferase deficiency 2 | AR | 1p32 |
| Spinal muscular atrophy (Werdnig-Hoffmann; Kugelberg-Welander) | AR | 5q11-q13 |
| Familial dysautonomia (Riley-Day) | AR | 9q31-33 |
| Hereditary motor-sensory neuropathy (Charcot-Marie-Tooth; Dejerine-Sottas) | AD | 17p11.2 |
| Hereditary motor-sensory neuropathy (axonal type) | AD | 1p35-p36 |
| Hereditary motor-sensory neuropathy (Charcot-Marie-Tooth-X) | XR | Xq13.1 |
| Mitochondrial myopathy (Kearns-Sayre) | Maternal; sporadic | Single large mtDNA deletion |
| Mitochondrial myopathy (MERRF) | Maternal | tRNA point mutation at position 8344 |
| Mitochondrial myopathy (MELAS) | Maternal | tRNA point mutation at positions 3243 and 3271 |
AD, autosomal dominant; AR, autosomal recessive; MELAS, mitochondrial encephalopathy lactic acidosis, and stroke; MERRF, myoclonic epilepsy with ragged-red fibers; mtDNA, mitochondrial deoxyribonucleic acid; tRNA, transfer ribonucleic acid; XR, X-linked recessive.
600.1 Myotubular Myopathy
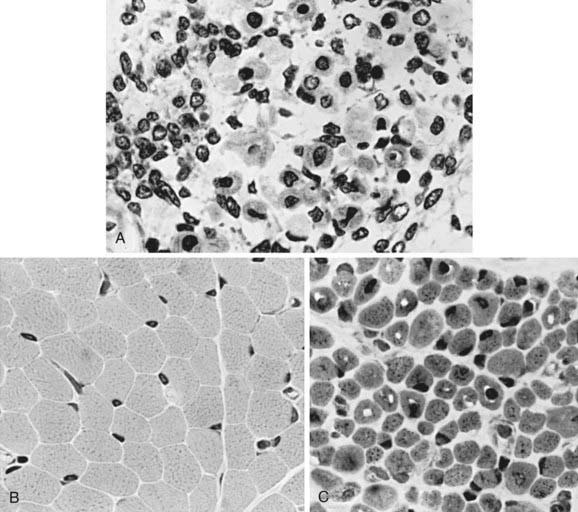
The term myotubular myopathy implies a maturational arrest of fetal muscle during the myotubular stage of development at 8-15 wk of gestation. It is based on the morphologic appearance of myofibers: A row of central nuclei lies within a core of cytoplasm; contractile myofibrils form a cylinder around this core (Fig. 600-1). Many challenge this interpretation and use the more neutral term centronuclear myopathy when referring to this myopathy. This term is nonspecific because internal nuclei occur in many unrelated myopathies.
Pathogenesis
The defective gene of the X-linked form and 3 genes of the autosomal recessive form are known.
Diagnosis
The muscle biopsy findings are diagnostic at birth, even in premature infants. More than 90% of muscle fibers are small and have centrally placed, large vesicular nuclei in a single row. Spaces between nuclei are filled with sarcoplasm containing mitochondria. Histochemical stains for oxidative enzymatic activity and glycogen reveal a central distribution as in fetal myotubes. The cylinder of myofibrils shows mature histochemical differentiation with adenosine triphosphatase stains. The connective tissue of muscle spindles, blood vessels, intramuscular nerves, and motor end plates are mature. Ultrastructural features in neonatal myotubular myopathy, other than those that define the disease, are also mature. Vimentin and desmin show strong immunoreactivity in muscle fibers in myotubular myopathy and no demonstrable activity in normal term neonatal muscle. The molecular genetic marker in blood is available and is useful not only for confirming the diagnosis but also for early prenatal diagnosis. The differentiation from other forms of congenital myopathy is noted in Table 600-2.
Dowling JJ, Vreede AP, Low SE, et al. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5:e1000372.
Lepper C, Conway SJ, Fan C-M. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627-631.
Pierson CR, Tomczak K, Agrawal P, et al. X-linked myotubular and centronuclear myopathies. J Neuropathol Exp Neurol. 2005;64:555-564.
600.2 Congenital Muscle Fiber-Type Disproportion
Diagnosis
CMFTD is diagnosed by muscle biopsy that shows disproportion in size and relative ratios of histochemical fiber types: Type I fibers are uniformly small, and type II fibers are hypertrophic; type I fibers are more numerous than type II fibers. Degeneration of myofibers and other primary myopathic features are absent. The biopsy is diagnostic at birth. Differentiating features from other congenital myopathies are noted in Table 600-2.
Clarke NF, Kolski H, Dye D, et al. Mutations in TPM3 are a common cause of congenital fiber type disproportion. Ann Neurol. 2008;63:327-329.
Clarke NF, North KN. Congenital fiber type disproportion—30 years on. J Neuropathol Exp Neurol. 2003;62:977-989.
Laing NC, Clarke NF, Dye DE, et al. Actin mutations are one cause of congenital fibre type disproportion. Ann Neurol. 2004;56:689-694.
Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682-2688.
Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: Clinical, morphological and genetic studies in 63 patients. Brain. 2004;127:439-451.
600.3 Nemaline Rod Myopathy
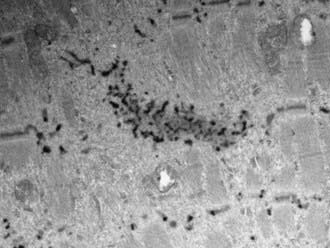
Nemaline rods (derived from the Greek nema, meaning “thread”) are rod-shaped, inclusion-like abnormal structures within muscle fibers. They are difficult to demonstrate histologically with conventional hematoxylin-eosin stain but are easily seen with special stains. They are not foreign inclusion bodies but rather consist of excessive Z-band material with a similar ultrastructure (Fig. 600-2). Chemically, the rods are composed of actin, α-actinin, tropomyosin-3, and the protein nebulin. Nemaline rod formation may be an unusual reaction of muscle fibers to injury because these rod structures have rarely been found in other diseases. They are most abundant in the congenital myopathy known as nemaline rod disease. Most rods are within the myofibrils (cytoplasmic), but intranuclear rods are occasionally demonstrated by electron microscopy.

Figure 600-2 Electron micrograph of the muscle from a patient shown in Figure 600-4. Nemaline rods (nr) are seen within many myofibrils. They are identical in composition to the normal Z bands (z) (×6,000).
Clinical Manifestations
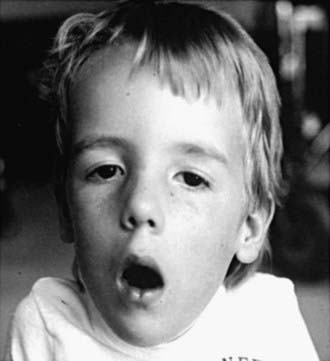
Neonatal, infantile, and juvenile forms of the disease are known. The neonatal form is severe and usually fatal because of respiratory failure since birth. In the infantile form, generalized hypotonia and weakness, which can include bulbar-innervated and respiratory muscles, and a very thin muscle mass are characteristic (Fig. 600-3). The head is dolichocephalic, and the palate high arched or even cleft. Muscles of the jaw may be too weak to hold it closed (Fig. 600-4). Decreased fetal movements are reported by the mother, and neonates suffer from hypoxia and dysphagia; arthrogryposis may be present. Infants with severe neonatal and infantile nemaline myopathy have facies and phenotype that are nearly indistinguishable from those of neonatal myotonic dystrophy, but their mothers have normal facies. The juvenile form is the mildest and is not associated with respiratory failure, but the phenotype, including facial involvement, is similar.

600.4 Central Core, Minicore, and Multicore Myopathies
The disease is characterized pathologically by central cores within muscle fibers in which only amorphous, granular cytoplasm is found with an absence of myofibrils and organelles. Histochemical stains show a lack of enzymatic activities of all types within these cores. The serum CK value is normal in central core disease except during crises of malignant hyperthermia (Chapter 603.2). Central core disease is consistently associated with malignant hyperthermia, which can precede the diagnosis of central core disease. All patients should have special precautions with pretreatment by dantrolene before an anesthetic agent is administered.
600.5 Myofibrillar Myopathies
A unique autosomal recessive myopathy in Cree native infants is characterized by severe generalized muscular hypertonia that is not relieved by neuromuscular blockade, hence is myopathic in origin. Most die in infancy of respiratory insufficiency due to diaphragmatic involvement. The muscle biopsy shows findings similar to many other myofibrillar myopathies (Fig. 600-5).
600.9 Benign Congenital Hypotonia
The diagnosis is one of exclusion after results of laboratory studies, including muscle biopsy and imaging of the brain with special attention to the cerebellum, are normal (see Table 599-2). No known molecular genetic basis for this syndrome has been identified. The differential diagnosis is noted in Table 599-3.
Laugel V, Cossee M, Matis J, et al. Diagnostic approach to neonatal hypotonia: retrospective study on 144 neonates. Eur J Pediatr. 2008;167:517-523.
Mintz-Itkin R, Lerman-Sagie T, Zuk L, et al. Does physical therapy improve outcome in infants with joint hypermobility and benign hypotonia? J Child Neurol. 2009;24:714-719.
600.10 Arthrogryposis
Arthrogryposis multiplex congenita is not a disease but is a descriptive term that signifies numerous congenital contractures (Chapter 674).
Compton AG, Cooper ST, Hill PM, et al. The syntrophin-dysbrevin subcomplex in human neuromuscular disorders. J Neuropathol Exp Neurol. 2005;64:350-361.
Goebel HH. Congenital myopathies in the new millennium. J Child Neurol. 2005;20:94-101.
Merlini L, Angelin A, Tiepolo T, et al. Cyclosporin-A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc Natl Acad Sci U S A. 2008;105:5225-5229.
Nicole S, Vicart S, Davoine CS, et al. Mutations of perlecan, the major proteoglycan of basement membranes, cause Schwartz-Jampel syndrome: a new mechanism for myotonia? Acta Myologica. 2001;20:130.
Relaix F, Rocancourt D, Mansouri A, et al. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435:948-953.
Sarnat HB. Ontogenesis of striated muscle, ed 3. Polin RA, Fox WW, editors. Neonatal and fetal medicine: physiology and pathophysiology, vol 2. WB Saunders, Philadelphia, 2003;1849-1870.
Wallgren-Pattersson C. Gene table: Congenital myopathies. Eur J Paediatr Neurol. 2005;9:27-28.





