Chapter 61 Developmental Disabilities
Cerebral Palsy
Clinical Features
Cerebral palsy (CP) is a disorder of the development of movement and posture that causes activity limitation; it is attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain (Bax et al., 2007; Rosenbaum et al., 2007). The motor disorders in CP are often accompanied by mental retardation (52%), epilepsy (45%), ophthalmological defects (e.g., vision impairment, motility problems, strabismus) (28%), hearing impairment (especially in those with very low birth weight, kernicterus, neonatal meningitis, or severe hypoxic-ischemic insults) (12%), and speech, oromotor impairments, and language disorders (38%) (Ashwal et al., 2004). Oral-motor problems including feeding difficulties, swallowing dysfunction, and drooling can affect nutrition and growth, oral health, respiration, and self-esteem (Ashwal et al., 2004). The disturbances of movement and posture include spasticity, hypotonia, ataxia, and dyskinesia. The likelihood and severity of the associated deficits generally parallel the severity of the motor impairment.
In birth cohorts from developed countries, the prevalence of CP is 1 to 2 per 100 live births and 100 in 1000 surviving infants less than 28 weeks gestational age (Paneth et al., 2006). This is comparable to figures from the ninth and most recent report from the western Swedish study on the prevalence and origin of CP in a population-based study of 88,371 live births occurring from 1995 to 1998 that reported a prevalence of 1.92 per 1000 live births. (Himmelmann et al., 2005). Gestational age-specific prevalence was 77 per 1000 for children born before 28 weeks. The prevalence of CP was 20% in those born at 24 to 26 weeks of gestation (Ancel et al., 2006), 40 per 1000 for children born at 28 to 31 weeks, 7 per 1000 for children born at 32 to 36 weeks, and 1.1 per 1000 for children born after 36 weeks. Spastic hemiplegic CP occurred in 38%, diplegic CP in 35%, quadriplegic CP in 6%, dyskinetic CP in 15%, and ataxic CP in 6%. For the first time in this series of long-term cohort studies, hemiplegia was the most common CP type, owing to the decline in preterm diplegia. There was also an increase in full-term dyskinetic CP compared with pre-1994 cohorts.
Studies of childhood cognitive and behavioral outcome in children with CP demonstrate an increased frequency of attention deficit hyperactivity disorder (ADHD) (20%) as well as other learning issues, particularly nonverbal learning disabilities (NVLDs), developmental coordination disorders (DCDs), and visuospatial problems. Those born at 23 to 25 weeks gestation have the highest frequency of CP and cognitive difficulties (Johnson et al., 2009; Vohr, 2010). During the 1990s, increased disability in very preterm infants was notable and probably due to increased survival rates (Wilson-Costello et al., 2005). However, since 2000, neurodevelopmental impairment has decreased. Improved outcomes reflect increased antenatal steroid use and cesarean section delivery, as well as decreases in sepsis, severe cerebral ultrasonographic abnormalities, and postnatal steroid use (Wilson-Costello et al., 2007). Preterm infants followed into adulthood show increased numbers of neurological signs in very preterm–born adults compared to term-born controls and are strongly associated with lower intelligence quotient (IQ) (Allin et al., 2006a). Furthermore, young adults who were born very preterm have different personality styles from their term-born peers; they are at an increased risk for psychiatric difficulties (Allin et al., 2006b). Even with these caveats, outcome has significantly improved over the past 2 decades.
Diagnosis and Etiology
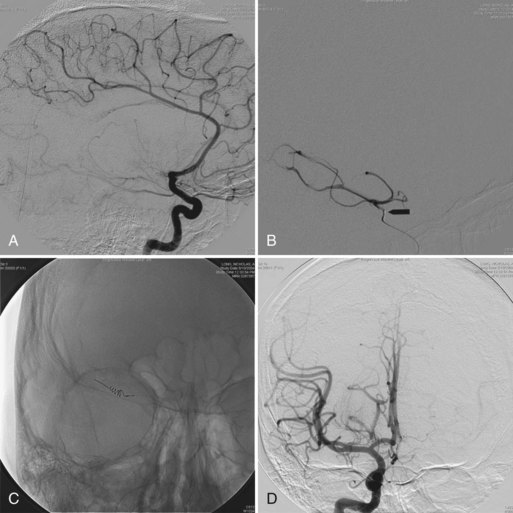
A history of delayed motor milestones and the clinical examination are the basis for the diagnosis of CP. Both the Gross Motor Function Classification System (GMFCS), which defines five levels of motor function (Palisano et al., 1997), and a measure of upper-extremity function such as the ABILHAND-Kids or the Bimanual Fine Motor Function Classification are useful in diagnosing CP. The National Perinatal Collaborative Project, monitoring more than 50,000 pregnancies, documented that among the 200 children with CP, the most important predictors of CP were maternal intellectual impairment, birth weight less than 2000 g, a malformation of any organ system, and breech presentation or delivery (Croen et al., 2001). Commonly used indicators of perinatal hypoxia-ischemia such as abnormal fetal heart rate, presence of meconium, and low Apgar scores did not predict CP and were relatively common in healthy infants. Almost 95% of infants with 5-minute Apgar scores of 3 did not have CP. Even when the Apgar scores are low, the CP risk is minimal unless the infant continues to have neurological problems (e.g., lethargy, hypotonia, poor suck, seizures) in the newborn nursery. In addition to perinatal hypoxic ischemic encephalopathy, other documented causes of CP in the term infant include brain malformations, genetic and metabolic disorders, intrauterine (Nelson, 2009) and perinatal infection (congenital infections, maternal infection during pregnancy and labor, and neonatal meningitis), and stroke, generally resulting in hemiplegic CP (Nelson, 2007). Specific risk factors for stroke in both term and preterm infants includes history of infertility, oligohydramnios, preeclampsia, prolonged rupture of membranes, cord abnormality, chorioamnionitis, and primiparity. Twin gestation is an independent risk factor for CP in both preterm and term infants, as is a vanishing twin in the survivor (Blickstein, 2002; Bonellie et al., 2005). Among preterm infants, risk factors for CP (Hagberg et al., 2009) include long-term ritodrine tocolysis, severe respiratory distress syndrome, low umbilical cord pH (Malin et al., 2010), postnatal steroid therapy (per contra Kent et al., 2005), patent ductus arteriosus requiring surgical ligation, peri-intraventricular hemorrhage, moderate to severe ventricular dilatation, periventricular leukomalacia (PVL), and need for home oxygen (Locatelli et al., 2010; Takahashi et al., 2005; Tran et al., 2005; Vohr, 2010). In the Swedish cohort study (Himmelman et al., 2005), the origin of CP in children born at term was considered to be prenatal in 38%, peri/neonatal in 35%, and unclassifiable in 27%. Among preterm-born children, the etiology was prenatal 17%, peri/neonatal 49%, and unclassifiable 33%.
Metabolic or genetic causes for CP occur infrequently (i.e., 0%-4%). In almost all such cases, there are atypical complaints such as features in the history of a progressive rather than a static encephalopathy, findings on neuroimaging representative of certain genetic or metabolic disorders, or a family history of childhood neurological disorder with associated “CP”. Metabolic and genetic studies are not routine in the evaluation of the child with CP. Consider metabolic and genetic testing when atypical features exist in the history or clinical examination. Evaluating CSF may be appropriate in children with dyskinetic CP of unknown etiology. Detection of a brain malformation in a child with CP warrants consideration of an underlying genetic or metabolic etiology. Of all CP types, the ataxic form most often has a genetic basis. Dopamine-responsive dystonia may mimic CP and is easily diagnosed and treated when suspected (Chaila et al., 2006). Other genetic or metabolic disorders that may mimic CP include: urea cycle disorders, sialidosis type 1, Sjögren-Larsson disease (fatty aldehyde dehydrogenase deficiency), metachromatic leukodystrophy, Krabbe disease (spastic CP), Lesch-Nyhan syndrome, glutaric aciduria 1 (dyskinetic CP), ataxia telangiectasia, Leigh syndrome and other mitochondrial disorders, and subacute sclerosing panencephalitis (ataxic CP) (García-Cazorla et al., 2009).
Brain imaging is useful in determining the cause and prognosis of CP (Krägeloh-Mann, 2007). Neonatal imaging predicts the localization and extent of PVL as well as the neurological outcome and extent of CP in later childhood (Ancel et al., 2006; Sie et al., 2005). Very low-birth-weight premature infants with either echolucency in the periventricular cerebrum or moderate to severe ventricular enlargement have a greater than 50% risk of developing CP. (Hintz and O’Shea, 2008). However, one-third of infants with CP have no ultrasound abnormalities (Ancel et al., 2006). Ultrasound abnormalities are present in preterms that develop disabling CP but not in preterms (usually male) who ultimately develop mild CP (Paneth, 2001). This suggests a different etiology for CP in these two groups. Hypothyroidism of prematurity is associated with twice the risk of echolucencies on ultrasound, and this group is at increased risk for CP (Leviton et al., 1999).
Based on available data, proposed practice parameters for the evaluation of children with CP include neuroimaging if an etiology is not already established. In a study of 273 children over 35 weeks gestation who underwent neuroimaging, one-third had normal scans; focal infarction was observed in 22% of the children with CP and 45% of those with hemiplegia, with the next most common finding being brain malformation (e.g., hydrocephalus, polymicrogyria, cerebellar abnormalities); 12% of the children had PVL and 5% a history of hypoxia-ischemia (Wu et al., 2006). Magnetic resonance imaging (MRI) demonstrates that brain malformations are relatively common in term infants with CP (10%). PVL is the most common finding in preterm infants with CP (Barkovich, 2006). Oxidative stress and excitotoxicity resulting from excessive stimulation of ionotropic glutamate receptors on immature oligodendrocytes are the most prominent molecular mechanisms for PVL (Fatemi et al., 2009). PVL in term-born children is often associated with neonatal resuscitation and respiratory distress (Tang-Wai et al., 2006), and the degree of disability is generally greater than in the preterm (Krageloh-Mann et al., 1999). Multiple cerebral infarctions are associated with perinatal hypoxic-ischemic encephalopathy, whereas most unilateral infarctions are prenatal in origin (Nelson, 2007; Nelson and Chang, 2008; Volpe, 2009) and warrant evaluation for an underlying coagulopathy (Ashwal et al., 2004). Severe injury to the cerebellum occurs as a complication of extreme prematurity. The clinical features include striking motor impairment and variable degrees of ataxia and athetosis or dystonia, which represent a distinct clinical type of CP. Most children are severely impaired with cognitive, language, and motor delays, and microcephaly is the rule (Bodensteiner et al., 2005). Larger corpus callosum size has been associated with better motor performance in preterms (Rademaker et al., 2004). Outcome studies of children and adolescents born preterm suggest a correlation between the presence of MRI abnormalities, CP, and greater cognitive difficulties (Mirmiran et al., 2004; Woodward et al., 2006). Reduced cortical and cerebellar volumes may result from enhanced apoptosis or excitotoxic damage to highly susceptible immature neurons (Bhutta and Anand, 2001). Experimental models of hypoxic-ischemic encephalopathy indicate that neurons in the neonatal brain are more likely to die by delayed apoptosis extending over days to weeks than those in the adult brain. Neurons die by glutamate-mediated excitotoxicity involving downstream caspase-dependent and caspase-independent cell death pathways (Fatemi et al., 2009).
Prevention and Management
Prevention of CP has focused on prevention of prematurity, optimal treatment during the perinatal period, and the development of neuroprotective agents (Nelson and Chang, 2008). Magnesium sulfate may prevent CP in the very low-birth-weight preterm infant by altering oxygen free radicals, excitatory amino acids, and even cytokines (American College of Obstetricians and Gynecologists Committee on Obstetric Practice, 2010). Indomethacin may prevent intraventricular hemorrhage. Treating transitory hypothyroidism in the preterm may improve outcome, although those preterms with transient hypothyroidism do less well than those who are euthyroid (van Wassenaer et al., 2005). The effectiveness of inhaled nitric oxide is debated, with some studies showing efficacy and others not (Mestan et al., 2005; Nelson and Chang, 2008). A recent study (Takahashi et al., 2005) suggested that long-term ritodrine tocolysis, prolonged hypocarbia, and postnatal steroid therapy (per contra Kent et al., 2005) are associated with an increased risk for CP in the preterm infant, raising the possibility of the need for further changes in pre- and perinatal care. Infections during pregnancy, as well as chorioamnionitis at delivery, are risk factors for CP (Nelson, 2009); the resultant elevated cytokines are the presumed brain toxic agent (Gilstrap and Ramin, 2000; Girard et al., 2009). Improved management aimed at diminishing cytokine production may eventually prevent some cases of CP (Dammann and Leviton, 2004; Girard et al., 2009). The mode of delivery of early preterm infants may affect outcome. Some studies suggest that a policy of elective cesarean section in cases of premature rupture of membranes and breech presentations of fetuses weighing less than 1000 to 1500 g may be beneficial (Gabriel et al., 2004), but others do not (Nelson and Chang, 2008). Intraventricular hemorrhage, which usually occurs within 6 hours after birth and possibly during labor and delivery, may be preventable. Hypothermia treatment of hypoxic-ischemic encephalopathy, generally using a “cool cap,” appears to be improving outcome (Fatemi et al., 2009).
Data from several studies involving children with CP (n = 1918) found that about half (range 35% to 62%) of these children develop epilepsy. However, no evidence exists that electroencephalography (EEG) is useful in determining the etiology of CP (Ashwal et al., 2004) and plays no role in workup or management unless seizures occur. By contrast, since the frequency of associated cognitive and sensory deficits is high in the child with CP, appropriate screening should be undertaken.
Rehabilitative therapy influences outcome. The main goals of treatment are to improve the child’s motor function and modify the environment to improve mobility. Orthopedic surgery is sometimes required (Steinbok, 2006). Dorsal rhizotomy has been used to decrease spasticity and improve gait or make children who are wheelchair bound more comfortable. Botulinum toxin and baclofen also have a role in the treatment of spasticity (Gough et al., 2005; Lundy et al., 2009). Because of the high frequency of oromotor dysfunction, nutrition, growth, and other aspects of swallowing dysfunction should be monitored (Ashwal et al., 2004).
Assessing long-term outcome, Mesterman et al. (2010) accessed by phone 163 of all 204 patients diagnosed with CP (at age 0 to 5 years) between 1975 and 1994 in Tel Aviv. Of the 95 patients between 18 and 30 years at the time of the survey, one-third to one-half required help with activities of daily living like eating, toileting, and dressing, one-third did not walk independently, but 80% had 12 years of schooling or more in either regular or special education. Only five were married, most lived at home, but notably, the majority reported being quite satisfied with their lives (Miller, 2007; Shikako-Thomas et al., 2009). The factors that decrease life expectancy in CP are immobility, profound retardation, and the need for special feeding (Strauss et al., 2007). Mobile children with CP may live well into adulthood.
Intellectual Disability
Clinical Features
The definition of a significant intellectual disability (ID) is a performance two standard deviations or more below the mean on age-appropriate standardized norm-referenced tests. The term global developmental delay (GDD) is usually reserved for young children (typically < 5 years of age), while the term intellectual disability (which will replace mental retardation in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders [DSM 5]) is usually applied to older children, in whom IQ testing results are more reliable (Shevell et al., 2003). The American Association of Intellectual and Developmental Disabilities (formerly known as the American Association of Mental Retardation) defines ID not as an absolute condition but as a construct that assists in educational planning and determines needs in terms of activities of daily living. The diagnosis therefore requires both low IQ (<70) and difficulties in adaptive behavior as expressed in conceptual, social, and practical adaptive skills (www.aaidd.org). This definition links the severity of ID to the degree of community support required to achieve optimal independence (Katz and Lazcano-Ponce, 2008). Mild ID indicates the need for intermittent support; moderate ID for limited support; severe ID for extensive support, and profound ID for pervasive support. The IQ definition of ID uses 100 as the mean and 15 as the standard deviation. Children with mild ID have an IQ of 55 to 69, those with moderate ID have an IQ of 40 to 54, those with severe ID have an IQ of 25 to 39, and those with profound ID have an IQ under 25. The level of functioning of children with an IQ around 60 to 70 varies significantly. About half require special education, and half attend regular school. Those in special schools are more likely to have a personal or family history of language delay. Similarly, IQ scores are only one factor in the differentiation between borderline intellectual function (BIF; IQ 70-84) and mild ID. An individual’s overall IQ score can be misleading because he or she may have strengths or weaknesses that markedly affect functioning, or there may have been even minor measurement errors on the test administration. An individual’s adaptive behaviors and functioning are of great importance when making this determination; an IQ of 70 is not necessarily a reliable cutoff between mild ID and BIF. Someone with an IQ of 69 may potentially function better overall than someone with a borderline range IQ of 71. An assessment of adaptive behaviors is crucial to the diagnostic process here too. Clinical interview with the individual and a collateral contact who knows them well can help assess adaptive functioning, as can standardized measures of adaptive behaviors (e.g., Vineland Adaptive Behavior Scales).
The prevalence of ID in industrialized countries is 0.3% to 3%. Mild ID represents the majority (85%), but roughly 0.4% of the general population is severely intellectually disabled. As a rule, those with severe ID are more likely to have a definable biological cause, whereas those with mild ID tend to come from socially disadvantaged backgrounds and often have a family history of BIF or mild ID (Online Mendelian Inheritance in Man [OMIM] website; Opitz, 2000; Stromme and Magnus, 2000). Environmental factors also play a role. For example, smoking during pregnancy is associated with more than a 50% increase in the prevalence of ID. Other environmental factors may include maternal alcohol use during pregnancy (fetal alcohol syndrome), lead toxicity, and iron deficiency. The risk for recurrence of severe ID is 3% to 10%, and of mild ID, 5% to 40%, suggesting there are many routes to mild ID. The ratio of boys to girls with ID, especially mild ID, is 1.4 : 1. Male excess is present in autism, syndromic X-linked ID (S-XLID) (associated with a specific phenotype), and nonsyndromic X-linked ID (NS-XLID). About 15% of males with ID have X-linked intellectual disability (XLID) (Stevenson and Schwartz, 2009). About 25% of all males with severe ID have XLID, and almost 50% of all cases of mild ID are due to XLID (Partington et al., 2000; Ropers and Hamel, 2005). The recurrence of ID in families with one previous child with severe ID is reported to be between 3% and 9% (Centers for Disease Control and Prevention [CDC], 2009).
Diagnosis and Etiology
Children with ID often have neurological and psychiatric comorbidities. Epidemiological studies suggest that as many as one-fifth of them have epilepsy by the age of 10 years (Airaksinen et al., 2000). The probability of developing epilepsy is fivefold greater for children with severe ID (35%) than for those with mild ID (7%). CP coexists in 6% to 8% of the mildly ID and as many as 30% of the severely ID. Microcephaly occurs in one-fifth of XLID syndromes. Macrocephaly also occurs secondary to increased brain volume or hydrocephalus. Syndromes without hydrocephaly include fragile X, Opitz FG, and Lujan. Syndromes with hydrocephalus include X-linked hydrocephaly-MASA spectrum and hydrocephaly-basal ganglia calcifications syndrome. Children with GDD and ID are at risk for physical disabilities. Impaired vision occurs in 15% to 50% and impaired hearing in about 20%. Thus a formal ophthalmological examination is appropriate. Audiometric assessment can be done by behavioral audiometry or brainstem auditory evoked response (BAER) testing (Shevell et al., 2003). Notably, cutaneous findings are fairly common in XLID, but cardiac anomalies are uncommon (Stevenson and Schwartz, 2009).
An increased prevalence of psychopathology and maladaptive behavior occurs in children with ID. In one epidemiological study, almost 40% of those with ID also had a psychiatric diagnosis, 42% with severe ID and 33% with mild ID (Stromme and Diseth, 2000). The most common comorbid psychiatric diagnoses were ADHD and autistic spectrum disorder (ASD). Overall, autistic features are reported in 9% to 15% of the mildly ID and 12% to 20% of the severely ID. Table 61.1 details specific cognitive and behavioral problems in several common genetically defined ID syndromes, along with the possible neuropathological basis of these disorders.
Table 61.1 Cognitive and Behavioral Problems in Several Genetically Defined Intellectual Disability Syndromes
Etiology is ultimately determined in anywhere from 10% to 81% of children with GDD/ID. Evaluation of ID should be sequential; key elements include the medical, family, and developmental histories, dysmorphology and neurological examinations, and appropriate laboratory and neuroimaging tests (Moeschler and Shevell, 2006; Shevell et al., 2003). The latter can include careful metabolic evaluation together with neuroimaging studies, EEG, cytogenetic studies, and genetic and ophthalmological consultations as appropriate (Mao and Pevsner, 2005; Shevell et al., 2003). Auditory and visual function must be determined, since these are common comorbidities. If a child was born in a locale without universal newborn screening, consider a screening metabolic evaluation that includes a capillary blood gas, serum lactate and ammonia levels, serum amino acids and urine organic acids, and thyroid function studies. An EEG is appropriate when the history suggests possible seizures, paroxysmal behaviors, or an underlying epilepsy syndrome.
Genetic defects are important causes of ID, and recent years have seen important progress in identifying the genes involved in ID. As of June 2010, the entry mental retardation results in 1647 hits on OMIM. If a family history of consanguinity exists or a close family member (sibling, aunt/uncle, or first cousin) is known to have GDD, testing specific to the known disorder should be performed. When there is a family history of unexplained GDD, cytogenetic testing (e.g., high-resolution chromosome studies, testing for subtelomeric deletions, fluorescent in situ hybridization (FISH) and even microarray-based cytogenetics) is appropriate. In the absence of a familial history of GDD, specific historical or physical findings are useful to direct testing (Van Esch et al., 2005). Observed dysmorphic features may prompt specific testing for such entities as Down syndrome, fragile X syndrome (FXS), Rett syndrome, Prader-Willi/Angelman (by FISH), or congenital hypothyroidism (Jones, 2006). Parental consanguinity, documentation of loss or regression of developmental milestones, or unexplained prior parental loss of a child are likely to be caused by a definable disease process, and thus a comprehensive genetic evaluation should be considered (Shevell et al., 2003). Epigenetic factors (e.g., chromatin remodeling) also play a role (Tsankova et al., 2007).
Increasingly sophisticated genetic testing is becoming available. Reviewing the literature on diagnostic studies in ID, Van Karnebeek et al. (2005) found that chromosome abnormalities detectable by classical cytogenetics were diagnostic in about 10% of patients. The median additional benefit of subtelomeric studies was 4.4%, although the rate of subtelomeric abnormalities in ID may be as high as 10% (Lamm et al., 2006; Shao et al., 2008). The yield for FXS screening is higher in those with more severe ID (4%) versus those in the borderline–mild ID group (1.0%) (Mastergeorge et al., 2010). Overall, the etiology of ID can be detected by classical molecular karyotyping and molecular testing for specific disorders in about 20%. Multiplex ligation-dependent probe amplification may be a rapid, accurate, reliable, and cost-effective alternative to FISH for the screening of subtelomeric rearrangements in patients with idiopathic ID (Palomares et al., 2006). Genomic microarrays, also called array comparative genomic hybridization, may also represent one of several novel technologies that allow rapid detection of chromosomal abnormalities such as microdeletions and microduplications from genomic DNA samples (Engels et al., 2007). More than 5000 chromosome copy number variations (CNVs) are now in a Toronto database (Li and Andersson, 2009).
Progress in the clinical and molecular characterization of XLID has outpaced progress in the delineation of ID due to genes on the other 22 chromosomes. Almost half of the estimated 200 XLID genes are identified, and another 20% have been regionally mapped (Stevenson and Schwartz, 2009). The combination of clinical delineation with gene identification and the development of gene panels for screening nonsyndromic XLID minimizes unproductive laboratory testing (Stevenson and Schwartz, 2009). To date, 87 genes have been associated with nonsyndromic or syndromic XLID or both, including FXS, Duchene muscular dystrophy, and Coffin-Lowry syndrome (Stevenson and Schwartz, 2009). Using imaging technologies and more careful scrutiny, some nonsyndromic XLID families were found to have syndromic findings with time. Eighteen genes have been associated with both nonsyndromic XLID and syndromic XLID. This apparent conundrum may be explained by the type of mutation, the location of the alteration in different domains of a gene, or observer inconsistencies (Stevenson and Schwartz, 2009).
Inherited metabolic diseases are responsible for 1% to 5% of unspecified ID, but the mean yield of metabolic investigations is only about 1% (Van Karnebeek et al., 2005). ID is rarely a unique symptom in inborn errors of metabolism. However, some exceptions should be kept in mind, including creatine transporter defect, some patients with adenylosuccinate lyase deficiency, 4-hydroxybutyric aciduria, and Sanfilippo disease type B (García-Cazorla et al., 2009). Table 61.2 lists some of the metabolic disorders that cause isolated ID. Some are potentially treatable.
The identification of ID-associated genes has provided insights into brain function. Two major types of ID genes exist: those leading to dysfunctional neurodevelopmental programs and brain malformations, and those that lead to alterations in molecular synaptic organization and plasticity mechanisms (Vaillend et al., 2008). Some metabolic disorders appear to influence the expression of one or the other group of genes (Stephen et al., 2003). Affected genes have roles in processes such as neuronal outgrowth, synaptic structure and function, and synaptic plasticity and learning and memory. Polymorphisms that predispose to ID but are insufficient to cause symptoms on their own might be present within the protein-coding regions of genes, within their regulatory regions, or in genes that encode small, regulatory ribonucleic acids (RNAs). Allelic variants of genes that are involved in XLID might be candidates for such polymorphisms (Ropers and Hamel, 2005).
Neuroimaging
Computed tomography (CT) contributes to the etiological diagnosis of GDD in approximately 30% of children, with the yield increasing if physical examination findings are present. MRI is more sensitive than CT, with abnormalities found in 48.6% to 65.5% of children with global delay. The chance of detecting an abnormality increases if physical abnormalities, particularly CP, are present (Shevell et al., 2003). With evolving positron emission tomographic (PET) methodology, further inroads into etiology may occur via assessment of glucose metabolism, serotonin synthesis, and receptor status, such as γ-aminobutyric acid (GABA) (Sundaram et al., 2005). Both structural and functional research imaging are likely to contribute (Gothelf et al., 2005).
Management
Available evidence demonstrates the benefits of early intervention through a variety of programs, at least with respect to short-term outcomes, and suggests that early diagnosis of a child with global delay may improve long-term outcome (Shevell et al., 2003). The management of children with ID focuses on finding the appropriate educational setting for children with mild ID, vocational training for those with moderate ID, and determining home or institutional placement for those with severe and profound ID.
Advances in genetic diagnosis have had immediate benefits for families, allowing for carrier testing, genetic counseling, prenatal diagnosis, and preimplantation genetic diagnosis. Some of the gene discoveries have also pointed to potential strategies for treatment. For example, newborn screening suggests that FXS occurs in 1 in 5000 males (Coffee et al., 2009). The mGluR5 hypothesis for FXS has led to the use of mGluR5 antagonists for the treatment of FXS. For example, R-baclofen, a GABA agonist that downregulates mGluR5 activity, has been anecdotally helpful in FXS; controlled trials are in progress. Minocycline lowers matrix metalloproteinase 9 (MMP9) levels at the synapse, which are upregulated in FXS. Minocycline treatment in patients with FXS and a recent survey of 50 patients treated with minocycline revealed improvements in behavior and language based on parent questionnaire in approximately 70% (Schneider et al., 2009).
Autistic Spectrum Disorders
Clinical Features
The diagnosis of an autistic spectrum disorder (ASD) currently requires the early onset of a triad of deficits: impaired sociability, impaired verbal and nonverbal communication, and restricted activities and interests (Rapin and Tuchman, 2006). The range of disabilities seen among children on the spectrum cannot be overemphasized. Indeed, the DSM-5 will emphasize the dimensional nature of the disorder, quantifying each feature as mild, moderate, or severe. ASD will become a dyadic disorder (social/communication deficits and fixated interests and repetitive behaviors) (Box 61.1) and will be diagnosed as with or without a language disorder, since not all ASD children have language disorders. Pragmatic language skills, however, will be incorporated into the social domain. The changes are based on research results which thus far have failed to document either pervasive developmental disorder–not otherwise specified (PDD-NOS) or Asperger disorder as separate biological entities. In addition, removing the age criteria allows for inclusion of children, generally those functioning at a higher intellectual level, who present outside of the current age cutoff.
Box 61.1 Tentative DSM-5 Criteria for an Autistic Spectrum Disorder Diagnosis
1. Clinically significant, persistent deficits in social communication and interactions, as manifest by all of the following:
2. Restricted, repetitive patterns of behavior, interests, and activities, as manifested by at least TWO of the following:
3. Symptoms must be present in early childhood (but may not become fully manifest until social demands exceed limited capacities)
The reported frequency of ASD has increased significantly in the past 2 decades. In the 1990s, the estimated frequency was about 1 per 1000, but the current estimate is about 1 per 100 or even higher. ASDs are 4 times higher in males. The reported increase is attributed to changes in diagnostic practices, with emphasis on the spectrum aspect of the disorder, different referral patterns (more evaluations of language delayed male toddlers), greater availability of services prompting increased utility of diagnosis, loosening of age at diagnosis requirement, and increased public awareness (Frombonne, 2005; Hertz-Picciotto and Delwiche, 2009; Newschaffer et al., 2007). In addition, some children who would have been previously and perhaps correctly diagnosed with a developmental language disorder are receiving ASD diagnoses (Bishop et al., 2008). However, these variables may not completely explain the increase. Environmental factors cannot be discounted (Szpir, 2006), although the theory that the measles, mumps, rubella (MMR) vaccine plays any role in the increase has been completely discredited (Observations, 2010). Both advanced maternal and paternal age may play a role in increasing the frequency of autism (Durkin et al., 2008). The high concordance in monozygotic twins, the increased risk for recurrence in siblings (≈5%-10%), a broader autistic phenotype in families with an autistic proband, which includes anxiety and mood as well as social style and obsessive characteristics (Daniels et al., 2008), and the association with a number of genetic disorders (Box 61.2) support a hereditary basis in many cases (Gillberg, 2010).
Box 61.2
Double Syndromes
Medical Disorders Associated with Autistic Spectrum Disorder Phenotypes
Modified from Gillberg, C., 2010. Double syndromes: autism associated with genetic, medical and metabolic disorders. In: Nass, R.D., Frank, Y. (Eds.), Cognitive and Behavioral Manifestations of Pediatric Diseases. Oxford University Press, New York, pp. 100-110.
The symptoms of ASD, like other developmental disorders, often change with age. Sibship studies provide the basis for investigations of the behavioral manifestation of autism during the first year of life (Elsabbagh and Johnson, 2009). Atypical eye contact, decreased response to name, poor imitation, absent social smile, passive temperament, and delayed language are early markers of the disorder (Zwaigenbaum et al., 2005). Sibling studies suggest there are both early-onset (clear by 12 months) and late-onset (clear by 18 months) forms of autism (Elsabbagh and Johnson, 2009; Zwaigenbaum et al., 2009). Approximately a third of autistic children regress between ages 1 and 3 years, with a peak at 2 years (Lord et al., 2004). Conversely, some toddlers and preschoolers with typical symptoms of autism may no longer appear autistic by school age. They may seem a bit odd and have peculiarities of language prosody and pragmatics. Social skills may be tenuous. NVLDs or ADHD may become the school-age diagnosis (Helt et al., 2008; Lord et al., 2006). Most, however, retain the typical features of autism, particularly those who are intellectually impaired. The natural history of Asperger disorder is less well documented. Often the diagnosis is delayed until late childhood, adolescence, or even adulthood because language is not delayed. NVLDs or ADHD may be the apparent presenting complaint (Martin and Klin, 2004). The characteristic overfocus in a particular or sometimes peculiar interest area may escalate over time, but it may also be the key to a special form of adult success. Outcome studies of children diagnosed with ASD suggest that although 40% show overall improvement during adolescence, as many as a third may deteriorate (Howlin, 2005; Rapin and Tuchman, 2006). The onset of seizures can cause the deterioration. Psychiatric comorbidities commonly develop: depression, bipolar disorder, anxiety, schizophrenia, and even catatonia (Billstedt et al., 2005; Hutton et al., 2008). A minority of patients, usually the higher-functioning group, improve significantly during adolescence. Approximately two-thirds of adults with autism show poor social adjustment (limited independence in social relations), and half require institutionalization. Autistic adults who are not intellectually impaired tend to improve more than those who are intellectually impaired. Prior to the broadening of the diagnostic category, even though higher-functioning individuals with autism and Asperger disorder had the best outcome, only 15% to 30% had fair to good outcomes, and only 5% to 15% became competitively employed, led independent lives, married, and raised families. Psychiatric problems were common in this group. Probably, some “odd” adults go undiagnosed in childhood and adolescence, thus increasing the proportion of those with ASD who ultimately function in the mainstream. Some are highly productive and original in their work (Billstedt et al., 2007; Seltzer et al., 2003). A recent report that assessed outcome in a cohort with normal IQ documented better adult functioning. Half of the participants were rated as “very good” or “good” on a global outcome measure. However, there was considerable variability in measured cognitive ability over time. Over half the sample had large gains or losses in cognitive ability of greater than one standard deviation (Farley et al., 2009).
Specific Clinical Features
Intelligence and Cognition
IQ is not one of the defining criteria for an ASD diagnosis (Matson and Shoemaker, 2009). Although most children with ASDs are intellectually disabled (70%-85%), some have average or even superior intellectual ability. If Asperger disorder is included as an ASD, the rate of ID drops considerably. IQ, however, is a key predictor of long-term outcome in autism, especially for those with an IQ of less than 50, who generally fare poorly. Munson et al. (2008), however, have recently suggested that distinct subtypes of autism exist that differ in severity of intellectual ability, patterns of cognitive strengths and weaknesses, and severity of autism symptoms. Normal IQ is a positive prognostic sign.
Communication
Verbal and nonverbal communication disturbances are a cardinal feature of ASD. Tager-Flusberg et al. (2009) have suggested using expressive language developmental benchmarks assessed with particular parent report and standardized measurement tools as a way of quantifying language capacity in preschool autistic children (e.g., preverbal communication, first words, word combinations, sentences, complex language). Different types of linguistic problems are seen (Rapin and Tuchman, 2006), and the severity of the language deficit generally parallels IQ. In lower-functioning ASD children, language, if present at all, is echolalic; repetitive stereotyped phrases are common. Some script from videos they watch over and over. Some children fail to process language and despite normal hearing, appear deaf (verbal auditory agnosia) (Deonna et al., 2009). Higher-functioning children sometimes talk too much—they talk to talk (semantic and pragmatic deficits) but do not use language to communicate (pragmatics). These children are often extremely literal and concrete. Prosody is frequently impaired, as evidenced by mechanical, excessively rapid, monotonic, high-pitched, and/or poorly modulated speech.
Long-term prognosis correlates with language skills at age 5 to 6 years. Those with conversational language do significantly better than children with little or no language. Nonverbal cognitive ability at age 2 was generally the strongest predictor of age 5 language, while at age 3 communication scores were a stronger predictor of age 5 language for children with autism. Early joint attention as well as vocal and motor imitation skills were more impaired in children who did not develop language by age 5 (but had relatively strong nonverbal cognitive skills) than in children who did develop language by 5 (Thurm et al., 2007).
Abnormalities of play are part of the autistic child’s communication disorder. Play ranges from repetitive stereotypic and indiscriminate sensory use of objects (mouthing, rubbing, etc.), to functional object use and imitation, to mechanical play with puzzles, to interactive play with adults like running, catching, and tickling games, to activities involving their circumscribed areas of interest (e.g., board games, computers, electronic games, high-level word and number tasks). Often, engaging in these activities is pleasurable. Pretend play is often rudimentary, even in higher-functioning young children, involving, for example, simple role taking (Waterhouse et al., 1996). Dress-up for itself alone without additional pretending is common.
Social Skills
Social dysfunction is a cardinal feature of the ASDs. The aloof child most resembles the popular notion of autism. Such children do not follow their parents around, run to greet them, or seek comfort. These children tend to be of low intelligence, have poor verbal and nonverbal communication skills, and exhibit little symbolic play. Passive children are generally somewhat higher functioning overall. These children do not make social approaches, but they accept them when made by others. They engage in some pretend play and join in games, but they take a passive role (e.g., the baby in the game of mothers and fathers). Children who are interactive but odd make spontaneous social approaches to others, but in a peculiar manner. They tend to talk at other people; their persistence may become annoying. Pragmatic language skills are impaired; for example, questions are conversation openers. Many persons on the autistic spectrum are relatively unaware of their social ineptitude except to the extent others tease them, but some are not (Waterhouse et al., 1996). Several books written by high-functioning persons with ASD highlight this (e.g., Grandin, 1995; Willey, 1999).
Restricted Range of Behaviors, Interests, and Activities
A restricted range of behaviors, interests, and activities is the third cardinal feature of autism. In lower-functioning children, these tend to consist of repetitive stereotyped behaviors like twirling, rocking, flapping, licking, and opening and closing doors. Tic disorders and obsessive-compulsive disorders may be the main manifestation of a restricted range of behaviors, interests, and activities in higher-functioning children (Nass and Leventhal, 2004). Many such children have difficulty making transitions. Not only do they often not focus, they also become overfocused. Some children with exceptional mathematical talents, for example, may meet the criteria for a diagnosis of an ASD or Asperger disorder and grow up to be single-minded, peculiar, asocial chess or mathematics geniuses (Baron-Cohen et al., 2003).
Diagnosis and Etiology
Evaluation
The clinical history and observations of the child are the basis for the diagnosis of an ASD. Several questionnaires for parents are available for quantifying the diagnostic criteria. For very young children, the Modified Checklist for Autism in Toddlers (M-CHAT) has been used (Robins et al., 2001). The Social Responsiveness Scale (Constantino, 2005), the Guilliam Autism Rating Scale (GARS) (Guilliam, 2006a), and the Guilliam Asperger Disorder Scale (GADS) (Guilliam, 2006b) are all quick standardized diagnostic measures, as are the Current and Lifetime Social Communication Questionnaires (SCQs) (Rutter et al., 2005), which are excellent screens. The Childhood Autism Rating Scale (CARS) (Schopler et al., 2000) can be used to quantify office observations. The Autism Diagnostic Observation Scale (ADOS) (Lord et al., 1999) and the Autism Diagnostic Interview–Revised (ADI–R) (Rutter et al., 2003) are the gold-standard diagnostic measures but are time consuming and require special training to administer. Neither is probably necessary in the typical clinical setting. Studies comparing ADOS and ADI diagnoses to that of an experienced clinician show excellent concordance.
The standard neurological examination is generally normal, although children with ASDs are often clumsy. Macrocephaly occurs in about a third of children with autism and generally becomes apparent around the age of 1 to 3 years. The fact that macrocephaly is not present at birth suggests there is an increased rate of brain growth in the first years of life that diminishes and may become subnormal in later childhood; macrocephaly in adults with autism is less common than in autistic children (Sacco et al., 2007). Tuberous sclerosis is probably the most common definable cause of autism; skin examination requires careful attention (Gillberg, 2010). Examination for dysmorphisms may be helpful in making genetic diagnoses like FXS, velocardiofacial syndrome, and Smith-Magenis syndrome (see Box 61.2). Hearing impairment may be excluded by a formal audiology assessment. EEG, including a sleep record or overnight video-EEG monitoring, is appropriate to exclude subclinical seizures when language comprehension is impaired or developmental regression has occurred (Kagan-Kushnir et al., 2005). Mild to severe epilepsy and partial and generalized seizures occur in up to one-third of patients with autism by early adulthood. Infancy (infantile spasms) and the peripubertal period are particularly vulnerable periods. The onset of seizures may play a role in adolescent deterioration. Children who are intellectually impaired have a higher risk for epilepsy, but seizures occur in high-functioning children as well. Epileptiform abnormalities on EEG and early epilepsy occur more frequently in children who undergo an early regression (Canitano et al., 2005). The relationship between epilepsy and autism is a subject of considerable disagreement (Tuchman, 2009), and the efficacy of treating epileptiform EEG activity in ASD is hotly debated (Lagae, 2009).
The extent of the evaluation for an underlying metabolic disorder depends on clinical suspicions and the relevance to family counseling. Box 61.2 lists some disorders that can be associated with an ASD phenotype. ASDs occur in approximately 20% of children presenting with intellectual disability and 10% of children with cerebral palsy.
Etiology
Neuropsychological
Three main neurocognitive phenotypes have been advanced to explain autism: underdeveloped theory of mind (TOM), weak central coherence, and executive dysfunction (Frith, 2003). Problems with TOM allude to the inability of those on the autistic spectrum to put themselves in another’s place and to understand that others might have different thoughts than they do. Complementing the TOM explanation of autism is an actively evolving view that mirror neurons are abnormal in the ASDs and underlie the inability to understand others’ emotions (Dapretto et al., 2006). One problem with the TOM hypothesis is that high-functioning autistics can often solve TOM paradigms. Executive dysfunction refers to difficulties with problem solving, planning, and organizing (Hill, 2004; Kenworthy et al., 2008), which are common in, but not specific to, autism. Weak coherence refers to difficulty integrating information into a meaningful whole. Although a detail-oriented, piecemeal-oriented style is common in autism, weak coherence does not clearly explain social difficulties. An inability to see the big picture may interfere with the ability to read emotional facial expressions. Baron-Cohen (2002) has suggested that autism represents the extreme male brain with empathizing weaknesses and systematizing strengths.
Genetics
ASDs are quite heterogeneous with respect to etiology. An identifiable genetic or metabolic disorder is found in 10% to 15% (see Box 61.2). Although over 15 autistic-susceptible gene loci have been extensively studied, the molecular pathogenesis of ASD is still not well understood (Battaglia and Carey, 2006; Schaefer and Mendelsohn, 2008; Sebat et al., 2007). Epigenetic factors affect the relationship between these genes and ASD pathogenesis (Tsankova et al., 2007.). Family members with the same mutation may not manifest autistic symptoms that could be, for example, due to gender effects or environmental factors. In most people with ASDs, extensive genetic testing does not reveal a cause, suggesting that inheritance is multifactorial. Mutations in genes encoding synaptic adhesion molecules like neuroligin, neurexin, CNTNAP (contactin-associated protein), and CADM1 (cell-adhesion molecule 1) suggest that impaired synaptic function underlies ASDs (Miller et al., 2005). However, knockout mouse models of these mutations do not show the full range of autistic symptoms. This could mean that gain of function as well as loss of function arising from these mutations are required for the full ASD picture. Endoplasmic reticulum (ER) stress due to these mutations may cause a trafficking disorder of synaptic receptors like GABAB receptors, resulting in impaired synaptic function and signal transduction. This theory provides for epigenetic factors playing a role as well (Momoi et al., 2010).
Neuropathology
Based on the core symptoms of autism, neuropathological abnormalities would be anticipated and are found in regions important to social function (frontal lobe, superior temporal cortex, parietal cortex and amygdale, language function (language cortex), and repetitive behaviors and stereotypies (orbital frontal cortex and caudate) (Amaral et al., 2008; Bauman and Kemper, 2005; Casanova and Trippe, 2009; Casanova et al., 2006; Herbert et al., 2002, 2005; Pardo and Eberhart, 2007; Schumann et al., 2010; Vargas et al., 2005). Functional imaging studies demonstrate that neural systems related to social functioning, like emotional face recognition and TOM, are abnormal (Corbett et al., 2009). Abnormalities of mirror neurons are also seen when subjects imitate and observe emotions (Rizzolatti and Fabbri-Destro, 2010).
Increased total brain volume, primarily due to increased white matter, is the most frequently replicated imaging finding (Verhoeven et al., 2010). Very young children with autism (18 months to 4 years) have a 5% to10% increase in brain volume, especially in the frontal lobe compared to controls, which parallels the increasing head circumference during this period. In contrast to other white-matter structures, both volume and density of the corpus callosum are reduced (Hardan et al., 2008; Minshew, 2009), perhaps resulting in decreased interhemispheric communication (Williams and Minshew, 2007). Imaging studies also highlight the dissociation between white-matter tract overgrowth and gray-matter dendritic and synaptic underdevelopment. Spectroscopy studies suggest that the gray matter is abnormal and dendritic arborization and synaptosome density reduced. Some investigators speculate that gray-matter abnormalities trigger the white-matter overgrowth (Williams and Minshew, 2007).
The white-matter abnormalities result in abnormality of connectivity, the current leading explanation for the pathogenesis of autism. At the cytoarchitectonic level, minicolumns that determine connectivity are abnormal, especially in the dorsolateral prefrontal cortex (Casanova et al., 2006). As a result and well delineated on diffusion tensor (DT) imaging (Keller et al., 2007; Sundaram et al., 2008), short-range connectivity is increased, and long-range connectivity is decreased (Williams and Casanova, 2010). The hyperconnected local networks may become partially isolated and acquire novel functional properties. By contrast, the decrease in long-range connections could explain the problems with top-down control and integration (Williams and Casanova, 2010).
Management
Preschool children with ASDs should receive special education in a therapeutic nursery or a home-based behavioral modification program (Handelman and Harris, 2002). Applied behavior analysis (ABA), which uses multiple prompting paradigms, reinforcement schedules, and imitation and modeling, and Treatment and Education of Autistic and Related Communication-Handicapped Children (TEACHH), to a lesser degree, have been validated as effective therapies in children with ASD ranging in age from 1 to 7 years (Callahan et al., 2010; Eikeseth, 2009). Early social intervention (ESI) therapies are under development (Dunn, 2005). Social skills groups and video modeling (video vignettes on TV or computer) appear to be effective therapies (Reichow and Volkmar, 2010). Animated mechanicals may enhance the emotional skills of children with ASD (Golan et al., 2010). Table 61.3 lists medications that have been helpful in some children.
| Hyperactivity and inattention | Psychostimulants (methylphenidate; dextroamphetamine); clonidine (Catapres) |
| Obsessive-compulsive behaviors | Tricyclics: Clomipramine (Anafranil) |
| Selective serotonin reuptake inhibitors: Fluoxetine (Prozac), sertraline (Zoloft), paroxetine (Paxil), fluvoxamine (Luvox) | |
| Atypical neuroleptics: Risperidone (Risperdal), olanzapine (Zyprexa), ziprasidone (Geodon) | |
| Aggressive and impulsive behaviors | Mood stabilizers: Carbamazepine (Tegretol), divalproex sodium (Depakote), gabapentin (Neurontin), topiramate (Topamax), lithium (Lithium) |
| Clonidine | |
| Beta-blockers: Propranolol (Inderal) | |
| Anxiolytics: Buspirone (BuSpar) | |
| Tics/stereotypies | Clonidine, clonazepam (Klonopin), pimozide (Orap), haloperidol (Haldol), risperidone, baclofen (Lioresal), deep brain stimulation |
| Self-mutilation | Naloxone (Narcan), propranolol, fluoxetine, clomipramine, lithium |
| Psychosis | Neuroleptics: Haloperidol decanoate, risperidone, olanzapine, ziprasidone |
| Seizures | Depakote, adrenocorticotropic hormone (ACTH) |
Modified from Soorya, L., Kiarashi, J., Hollander, E., 2008. Psychopharmacologic interventions for repetitive behaviors in autism spectrum disorders. Child Adolesc Psychiatr Clin N Am 17, 753-771.
Learning Disabilities
Dyslexia
Clinical Features
The best-studied and probably the most common LD is dyslexia. It occurs in as many as 10% of school-aged children and in 80% of LDs. Males are more often affected. Developmental dyslexia is an unexpected difficulty in learning to read. The definition contains both exclusive and inclusive criteria. As with other LDs, major neurological abnormalities are not present, but minor abnormalities (soft signs) may be detected (Denckla, 1985) (Table 61.4). Major sensory functions must be normal. Normal intelligence is a requirement for a traditional diagnosis. The dyslexic child must have been in a social and educational environment conducive to learning to read. Studies of inner-city elementary school children show that enrichment programs can help some nonreaders become readers.
Table 61.4 Common Soft Signs Associated with Learning Disabilities
| Cranial nerves | Head turns with eyes |
| Mouth opens when eyes open | |
| Difficulty with grimace | |
| Motor | Excess upper-extremity posturing on stressed gait |
| Excess overflow during finger tapping and sequencing | |
| Unsustained one-foot stand | |
| Difficulty with hopping | |
| Excess choreiform movements with arms extended | |
| Cerebellar | Dysrhythmic rapid alternating movements |
| Excess overflow during rapid alternating movements | |
| Ballistic finger-nose-finger test | |
| Difficulty with tandem gait | |
| Sensory | Extinction on double simultaneous stimuli |
| Poor finger localization | |
| DTR | Minor reflex asymmetries |
DTR, Deep tendon reflexes.
Modified from Denckla, M.B., 1985. Revised neurological examination for subtle signs. Pharmacol Bull 21, 773-789.
Diagnosis and Etiology
Children with dyslexia should have a formal neuropsychological evaluation to determine their cognitive strengths and weaknesses and to exclude comorbid problems (e.g., ADHD) that might affect treatment. Deficits in phonological awareness frequently underlie reading difficulties and persist even into adolescence (Shaywitz et al., 1999) and adulthood. Measures that assess phonological functioning (e.g., segmenting words [say cowboy without the boy, say smack without the m], word and nonword blending, sound matching of first and last syllables) best differentiate dyslexic from normal readers. The double-deficit hypothesis of developmental dyslexia proposes that deficits in phonological processing and naming speed represent independent sources of dysfunction in dyslexia (Vukovic and Siegel, 2006). Although phonological processing issues appear to be the primary and/or most common cause of dyslexia, neuropsychological studies have identified other deficit clusters in dyslexics. For example, Crews et al. (2009) identified three dyslexia subtypes: (1) no language or memory deficit, (2) global language and memory deficit, and (3) global memory deficit. Few children fail to read because of visual perceptual difficulties or extraocular motility problems. However, processing by the lateral geniculate magnocellular system (important for monitoring motion, stereopsis, spatial localization, depth, and figure-ground perception) may not appropriately modify the information received from the fast parvocellular system (crucial for color perception, object recognition, and high-resolution form perception) (Amitay et al., 2002; Angélique et al., 2002).
The standard neurological examination is normal. Routine imaging is normal and unnecessary, except perhaps in children with atypical features (Box 61.3).
Dyslexia is often genetic (Kovas et al., 2007). Dyslexia-susceptibility-1-candidate-1 (DYX1C1) was the first gene reported to be associated with dyslexia, possibly with a memory-deficit dyslexia phenotype (Dahdouh et al., 2009). Several genes that affect neuronal migration have also been implicated in dyslexia (e.g., KIAA0319, DCDC2) (Igo et al., 2006; Meng, 2005; Paracchini et al., 2006; Schumacher et al., 2007).
Pathological studies suggest that those with dyslexia have both atypical planum temporale asymmetries and areas of cortical dysplasia, reflecting abnormal neuronal migration particularly in the left hemisphere (Galaburda et al., 2006). Structural imaging demonstrates that in about two-thirds of normal adults, the left planum temporale is larger than the right, but by contrast, only 25% of dyslexics have this same left/right planum asymmetry (Eckert and Leonard, 2000). Dyslexics with atypical asymmetry tend to have more severe language and/or reading deficits. In a group of children with dyslexia with or without ADHD, the presence of an extra sulcus in the left pars triangularis was associated with poor expressive language ability. In those with adequate expressive language functioning, left pars triangularis length related to phonological awareness, phonological short-term memory, and rapid automatic naming (RAN). Right pars triangularis length related to RAN and semantic processing (Kibby et al., 2009). Interhemispheric transfer of information may be abnormal in dyslexia (Beaton et al., 2006). Structural differences of the corpus callosum exist in normal versus dyslexic readers. Theoretically, the splenium is critical because it contains axons linking the planum temporale and angular gyrus.
Functional imaging studies demonstrate that fluent reading requires functional integrity of three left hemisphere regions—an inferior frontal region and two posterior systems (a temporal-parietal system and a ventral occipital-temporal system). Developmentally, the temporal-parietal system predominates initially and is required for learning to integrate the printed word with its phonological and semantic features. The occipital-temporal system constitutes a late-developing rapid sight word identification system that underlies word recognition in skilled readers. Disruption of both posterior systems may occur in developmental dyslexia. In contrast to normal readers, dyslexics may rely on left and right inferior frontal and right posterior regions (Blau et al., 2010; Pugh et al., 2000). Thus they make inefficient use of the posterior system. Functional imaging also implicates the cerebellum, an area that other studies suggest is crucial for language functioning (Fulbright et al., 1999).
Connectivity abnormalities in dyslexics occur in two areas associated with working memory. Within a “phonological” left-lateralized prefrontal network, increased functional connectivity occurs in left prefrontal and inferior parietal regions. Within an “executive” bilateral frontoparietal network, dyslexics showed a decreased connectivity pattern in bilateral dorsolateral prefrontal and posterior parietal regions and increased connectivity in the left angular gyrus, left hippocampal cortex, and right thalamus (Wolf et al., 2010). Abnormalities of very short-range connectivity (e.g., angular gyrus, striate cortex), in association with larger gyri, may explain reading difficulties (Casanova et al., 2010; Silani et al., 2005).
Management
Although dyslexia does not disappear, most children with early reading problems learn to read at average to above-average levels if they are diagnosed by the age of 8 to 9 years (or third to fourth grade) and evidence-based reading instruction is provided. Children diagnosed later, even if remediated, are likely to continue to have reading problems. Three out of four children with reading problems at the end of third grade are still having trouble in seventh grade. In the Connecticut Longitudinal Study, dyslexic children (diagnosed after the third grade) never caught up to average or superior high school readers (Shaywitz et al., 1999). Early identification and provision of evidence-based reading instruction, systematic, phonetic, and multisensory approaches such as the Orton-Gillingham or Wilson methods can reduce the percentage of children reading below grade level in fourth grade from 37% to 6% (Bakker, 2006). However, large population studies suggest that some degree of reading disability persists into adulthood in most, and occupational attainment is lower in some (Undheim, 2009). The magnitude of phonological impairment alone does not appear to fully predict reading outcome. Phonological deficits appear to interact with other cognitive factors like nonverbal IQ and linguistic skills, particularly syntactic processing in determining long-term outcome (Peterson et al., 2009; Wiseheart et al., 2009).
Compensated readers, who are accurate but not fluent, demonstrate a relative underactivation in posterior neural systems for reading located in left parietotemporal and occipitotemporal regions. Persistently poor readers, who are both not fluent and less accurate, activate posterior reading systems but engage them differently from nonimpaired readers; they rely more on memory-based rather than analytic word identification strategies (Shaywitz et al., 2003). The majority of high-risk responder children benefit from systematic reading instruction and develop adequate reading abilities with successful recruitment of temporoparietal and visual association areas for reading (Simos et al., 2005). In another study correlating outcome with anatomy, 8- to 10-year-old poor readers had significantly lower fractional anisotropy (FA) in the left anterior centrum semiovale than good readers; 100 hours of intensive remedial instruction resulted in improved decoding ability and increased FA, consistent with enhanced myelination (Keller and Just, 2009). Although vision problems can interfere with reading, they are not the cause of dyslexia. Eye exercises, behavioral vision therapy, and special tinted filters or lenses are not effective treatments for dyslexia (American Academy of Pediatrics, 2009).
Nonverbal Learning Disabilities
Clinical Features
Mycklebust and Johnson coined the term nonverbal learning disabilities almost 50 years ago. It described a predominantly social emotional disorder that in many ways resembled the current concepts of the ASDs but included some spatial difficulties as well, particularly with personal and extrapersonal space. In that regard, Palombo (2006) recently proposed four subtypes of NVLD that variously involve social cognitive impairments (reciprocal social relations, verbal and nonverbal language, affective processing), social imperception (nonlinguistic perceptual deficits), and various nonverbal neuropsychological deficits (attention, executive function). By contrast, Rourke and colleagues (1995, 2002) and others (Drummond et al., 2005; Pelletier et al., 2001) focus on problems in math, visuoperceptual skills, sensorimotor functioning, and reasoning abilities in their descriptions of NVLD. Math difficulties are part of Rourke’s definition of NVLDs, since the presence of poor performance in arithmetic calculations relative to average or above-average reading (decoding) and spelling defined his original research with NVLD cohorts.
Other studies suggest that math is not necessarily a problem in NVLDs (Forrest, 2004) and that it is largely a diagnosis of visual-spatial disabilities. Social emotional problems had only a secondary place. Despite the fact that NVLD does not appear in the DSM-IV and goes by various names, including right hemisphere syndrome and visuospatial learning disability, the reported frequency ranges from 1% to 10% and even higher (Nass et al., 2006).
Diagnosis and Etiology
Children with NVLDs often present with different symptoms at different ages, probably reflecting both changes in school demands and changes in the central nervous system (Drummond et al., 2005; Rourke et al., 2002). In elementary school, social difficulties, inflexibility, graphomotor and gross motor coordination difficulties, sensorimotor problems, and inattention tend to predominate. In junior and senior high school, academic problems become apparent in mathematics and science, as well as in high-level reading comprehension and written composition, despite good verbal skills. Social issues continue, and problem-solving and organizational skills are relatively impaired. In young adults, psychiatric problems may be prominent. Depending on the strength of verbal skills, academics are not necessarily a problem.
At the neuropsychological assessment, the basis for the identification of children with NVLDs is usually their higher verbal as compared with performance IQ scores. This is an indicator of their visuospatial and visuomotor difficulties, which are an extremely common feature of the disorder. A complete neuropsychological evaluation is required for diagnosis. Some screening measures have been developed (Cornoldi et al., 2003). The simple-to-administer “Draw a Person” exercise (see Box 61.8) offers the clinician easy assessment of visuospatial abilities in the office. Although most highly related to difficulties in math, the visuospatial disabilities inherent in an NVLD may be the underlying cause of academic difficulties.
The overlap between NVLDs and Asperger disorder is widely debated (Klin et al., 2000; Martin and Klin, 2004; Ryburn et al., 2009). Differentiating socioemotional LD/right hemisphere LD/NVLD from Asperger disorder based on the lack of restrictive behaviors and the presence of greater verbal than performance IQ in the former, Barton and colleagues (2004) compared the two groups on several face-processing tasks. About one-third of the subjects had normal famous face recognition, normal perception of facial configuration, normal imagery for famous faces, and good performance on standard neuropsychological tests of face perception and memory, while two-thirds did not (although their deficits were not of the same magnitude as those seen in prosopagnosic subjects). NVLD subjects were in both groups. Box 61.4 indicates other domains that are commonly affected.
Neuroanatomically, the NVLDs are a reflection of right hemisphere dysfunction (Gross-Tsur et al., 1995; Manoach et al., 1995; Rourke et al., 2002). Thus, like the varied deficit patterns after acquired right hemisphere lesions in adults, NVLDs would not be expected to be a single entity but rather many different disorders with some overlapping features (Heilman and Valenstein, 2009). Frontal lobe dysfunction may underlie some of the deficits in reasoning skills, attention, and executive function, as well as social judgment. Parietal dysfunction may underlie the spatial deficits, paralinguistic problems, and sensorimotor problems. Rourke suggests that right hemisphere dysfunction is sufficient to produce NVLDs and that white-matter abnormalities necessarily produce NVLDs (Rourke, 1995; Rourke et al., 2002). According to Rourke, the NVLD phenotype is common in a number of pediatric and neurological disorders, many of which specifically affect the white matter (Box 61.5).
Box 61.5 Disorders Associated with a Nonverbal Learning Disorder Profile
Visuospatial difficulties, central to NVLD, are hallmark deficits in such genetically divergent syndromes as Turner syndrome, Williams syndrome (WS) (Gray et al., 2006), VCF syndrome, and neurofibromatosis (see Table 61.1 and Box 61.5). WS is perhaps the best studied in terms of correlating the visuospatial issues with neuropathology using neuroimaging. WS is associated with decreased overall brain volume, particularly in the cerebrum and brainstem, with relative volume preservation of the cerebellum and the superior temporal gyrus. The ratio of frontal to posterior (parietal and occipital) tissue is greater than in controls. WS has relative preservation of cerebral gray-matter volume and disproportionate reduction in cerebral white-matter volume. However, within the cerebral gray-matter tissue compartment, the right occipital lobe shows excess volume loss (Reiss et al., 2000). These latter features could underlie the visuospatial difficulties. Notably, face processing is preserved in WS patients.
Management
Suggested management for NVLDs often trades on developing verbal strengths to compensate for the nonverbal deficits. Because these children have difficulties dealing with novel or complex situations, the emphasis is on systematic learning. Programs to develop and strengthen visual spatial skills are available. Education must provide appropriate strategies for frequently occurring troublesome situations. Generalization of learned strategies should be encouraged. Appropriate nonverbal behaviors with peers, instinctual for most children, may need to be explained and practiced. Social groups are often helpful (Burger, 2004). Individual specific learning issues require individualized management (e.g., dyscalculia).
Developmental Coordination Disorders
Diagnosis
A wide range of motoric difficulties are often considered synonymous with DCDs, including clumsiness, gross motor difficulties, fine motor difficulties, visuomotor problems and decreased dexterity, dysgraphia, dyspraxia, and even adventitious movements (Box 61.6). DCDs often co-occur with ADHD, with ADHD and perceptual problems (dubbed DAMP), with ASDs, with developmental language disorders, and with dyslexia (Smits-Engelsman et al., 2003; Zwicker et al., 2009). Longitudinal studies suggest that the frequency of DCD changes with age and that the presence or absence of early neurological problems increases the frequency (Hadders-Algra, 2002; Hadders-Algra et al., 2004). Moreover, longitudinal studies have reported that DCD does not necessarily resolve itself and continues into adolescence and adulthood (Losse et al., 1991).
Anomalous dominance/handedness may also co-occur with DCDs. With respect to handedness (manual dominance), most ultimately right-handed children declare after 1 year of age and before 5 years of age. Strong left dominance, especially when established before 1 year of age, should raise concerns that handedness is pathological (reflects left hemisphere dysfunction) (Hiscock and Chapieski, 2004). Many infants appear to be transiently left-handed at 3 to 6 months. The percentage of right-handers (and probably the strength of handedness) increases through age 5 years, when the adult percentage (about 90%) appear to be right-handed. Most right-handers are strongly right-handed, whereas most left-handers are ambidextrous. The dexterity of normal, presumably genetically determined, left-handers is the equal of right-handers.
Evaluation and Etiology
Geuze (2005) suggests that DCDs are characterized by poor postural control (hypotonia or hypertonia, poor distal control, static and dynamic balance), difficulty in motor learning (learning new skills, movement planning, adaptation to change, automatization), and poor sensorimotor coordination (coordination within/between limbs; sequencing of movement; use of feedback, timing, anticipation, strategic planning). In one study, about 75% of children who were judged to have a DCD by a team of specialists (physiatrist, occupational therapist, physical therapist) performed below the 15th percentile on a comprehensive motor battery like the Movement Assessment Battery for Children (MABC) or the Bruininks Oseretsky Test of Motor Proficiency. The remaining 25% had handwriting problems or low muscle tone, issues not measured by even the comprehensive motor battery that was used (Geuze et al., 2001). An evaluation for dyspraxia (the inability to perform learned skilled movement despite the motor abilities to do so) is probably required for a comprehensive assessment, as is a neurological examination for soft signs (Denckla, 1985) (Table 61.5). Dysgraphia, for example, is a common problem for children with DCDs, which can occur for many reasons. It can result from a primary/isolated disorder or exist as a symptom of visuomotor difficulties, dexterity deficits, or dyspraxia (part of a generalized deficit or a material-specific deficit). It can also occur along with dyslexia as a higher-order cognitive disorder (Table 61.6).
| Neurological System Affected | Soft Sign | Age of Appearance or Disappearance |
|---|---|---|
| Cranial nerves | Head does not move with eyes | 6-7 years |
| Sticks tongue out for 10 sec | 6-7 years | |
| Motor | Toe-heel walk | 3 years |
| Heel walk without associated movements | 5 years | |
| Hop 10 times | 5 years | |
| Hops indefinitely | 7 years | |
| One-foot stand for 30 sec | 7 years | |
| No longer drifts up and down with pronated and supinated arms | 3-4 years | |
| Rigid tripod | 5 years | |
| Dynamic tripod | 7-8 years | |
| Choreiform movements | 7-10 years | |
| Athetoid movements | 2-4 years | |
| Cerebellar | Tandem | 6 years |
| No overflow during rapid alternating movements | 7-8 years | |
| Sensory | Stereoagnosis, graphesthesia | 6 years |
| No longer extinguishes on double simultaneous stimulation | 8 years |

Procedural learning for simple sequences of movements is intact in DCDs (Ahonen et al., 2004; Wilson et al., 2003). However, children with DCDs have difficulty generating an accurate visuospatial representation of an intended action, as indicated by deficits in motor imagery and an atypical strategy for mental rotation (Deconinck et al., 2008; Wilson et al., 2004) which relies on an alternative less-efficient object-based strategy. Visual imagery is mainly object based. It requires normal primary and secondary visual cortices. Motor imagery is a copy of real movements, allowed to come to consciousness only because the actual movement intended is inhibited (Decety and Jeannerod, 1995; Wilson et al., 2004).
Theoretically, the cerebellum, parietal lobe, and basal ganglia could each/all be involved in DCD (Zwicker et al., 2009). The soft neurological signs seen in children with DCD (e.g., dysmetria, dysdiadochokinesia, impaired timing) all suggest cerebellar involvement (Ivry, 2003; Lundy-Ekman et al., 1991). Poor postural control is a hallmark of DCD (O’Hare and Khalid, 2002). Parietal lobe involvement could underlie the visuospatial problems as well as the imagery problems that characterize DCD (Wilson, 2004) (Boxes 61.7 and 61.8). Theoretically, basal ganglia involvement is possible, but the spared simple procedural learning argues against this.
Box 61.7 Signs of Visuospatial Learning Disabilities
1. Can the child easily memorize material such as names, information, and poems?
2. Is the child able to make use of the available space when drawing?
3. Can the child use tools such as scissors, set square, or ruler that require independent and coordinated use of both hands?
4. Does the child understand spoken commands or texts that involve space relationships?
5. Is the child able to execute complex everyday movements such as tying shoelaces?
6. Does the child show good understanding of spatial relationships in calculation; can he or she write numbers in column correctly?
7. Does the child have good spatial orientation abilities?
8. Is the child good at drawing?
9. Can the child easily interact with friends?
10. Has the child reached a good linguistic learning level for his or her age?
11. Has the child reached a good mathematical learning level for his or her age?
12. Is the child competent in learning contexts that rely on visuospatial skills?
13. Is the child distracted easily?
14. Is the child often restless or hyperactive?
15. Is the child a good observer of the environment in which he or she lives?
16. Does the child demonstrate an interest in new objects, and can he or she deal with them?
Treatment
Children with significant DCDs may benefit from process-oriented occupational therapy, motor imagery intervention, and perceptual motor training (Wilson et al., 2002). Recent studies suggest that task-oriented therapy, which is focused on specific task performance, is more effective than deficit-oriented therapy, which is directed toward functioning of the nervous and musculoskeletal system (Polatajko and Cantin, 2006; Wilson, 2005). Computers can facilitate output for those with poor graphomotor skills. Sometimes difficulties are sufficient to require taping of lectures and access to a scribe. Extended time is often required in the classroom setting and for test taking. Lack of activity as well as social/emotional issues and poor self-esteem may place children with DCD at risk for health problems. Physical activity and therapy are priorities (Zwicker et al., 2009).
Dyscalculia
Clinical Features
Developmental dyscalculia (DD) can involve any or all aspects of mathematics, from difficulties representing and manipulating numeric information nonverbally, to learning and remembering arithmetic facts, to executing arithmetic procedures. The prevalence of dyscalculia is approximately 6% (similar to that of dyslexia and ADHD), affecting both genders equally. Indeed, the only clear gender difference in mathematical skills is in the extremely superior range (scoring over 700 on the math SAT in seventh grade), where males outnumber females by more than 10 to 1. A developmental Gerstmann syndrome (right/left disorientation, finger agnosia, dysgraphia, dyscalculia, and sometimes, constructional apraxia) occurs in as many as 2% of school-aged children. The mean IQ of children with dyscalculia is generally normal; one-fourth show symptoms of ADHD and approximately one-fifth are dyslexic. Dyscalculia is common in children with NVLD (Shalev and Gross-Tsur, 2001).
Evaluation and Etiology
Children with neuropsychological signs of both left and right hemisphere dysfunction can have dyscalculia. Both groups have similar problems on arithmetic batteries, but those with left hemisphere dysfunction seem to perform significantly worse in addition, subtraction, complex multiplication, and division and also make more visuospatial errors (Shalev and Gross-Tsur, 2001). Imaging studies show that parietal and frontal abnormalities predominate. Children with DD have been shown to have weaker brain activation in the IPS and inferior frontal gyrus of both hemispheres for approximate calculation than typically achieving children (Kucian et al., 2006). Evidence of parietal dysfunction (Grafman and Romero, 2001; Price et al., 2007) and reduced gray-matter volumes in frontal and parietal areas (Rotzer et al., 2008) are also reported in DD. Deficits in parietal and frontal lobe function in children with DD relate to poor spatial working memory (Rotzer et al., 2009). Several studies implicate deficit in working memory a factor associated with DD (Camos, 2008).
Attention Deficit Hyperactivity Disorder
Clinical Features
The overall defining features of ADHD are inappropriate inattention, impulsivity, distractibility, and hyperactivity for chronological and mental age. Box 61.9 delineates the specific DSM-IV-TR criteria for diagnosis. Empirical data collected since DSM-IV has not supported the presence of subtypes, so the DSM-5 will probably not include subtyping and may or may not differentiate ADD (no hyperactive/impulsive symptoms ever) from ADHD (appreciable hyperactivity symptoms in childhood even if they dissipate). Furthermore, it has become clear that the DSM-IV-TR criteria are inadequate for preschoolers as well as older adolescents and adults. DSM-5 will likely reference “age-related manifestations” (Castellanos, 2009).
Box 61.9
Criteria for Diagnosis of Attention Deficit Hyperactivity Disorder
ADHD Inattention Symptoms
Modified with permission from American Psychiatric Association, 2000. Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text revision. American Psychiatric Association, Washington, DC.
The reported prevalence of ADHD in school-aged children ranges from 1% to 20%. This wide range reflects the lack of a biological marker for the disorder. The variation in prevalence is secondary to differing techniques of ascertainment: parent, child, or teacher perspective; which diagnostic questionnaire is used; age at ascertainment (standards are most clear-cut for the elementary school-aged child); and even country of study (e.g., there is more ADHD in the United States than in the United Kingdom, probably because hyperactivity is a requirement in U.K. criteria) (Nass, 2006). ADHD is more common in males, but when affected, females tend to have less hyperactivity and more severe general cognitive deficits. Although complete recovery from ADHD was thought to be the rule, it now appears that ADHD persists into adulthood in 60% to 70% people diagnosed with ADHD in childhood (Kessler et al., 2005). Social-emotional difficulties in adulthood occur in 40% to 50% (Murphy et al., 2002). Serious psychiatric or antisocial disorders occur in 10%. Substance abuse, particularly alcoholism, is frequent (Riggs et al., 2008). ADHD occurs and can be diagnosed in the toddler (although the false-positive rate is relatively high) and may be four to eight times more common in males than females. Box 61.10 lists criteria appropriate for diagnosing ADHD in preschoolers.
Evaluation and Etiology
History is the primary basis of ADHD diagnosis. Since the disorder must affect functioning in at least two settings, multiple informants are required for diagnosis. In addition to an interview, questionnaires for parents, teachers, and the child or adolescent him/herself are useful for making the diagnosis. Given the high rate of ADHD comorbidities (e.g., anxiety and mood disorders, oppositional defiant disorder, conduct disorders), both narrow-band questionnaires that specifically assess for ADHD and broad-band questionnaires that assess not only for ADHD but for other psychiatric comorbidities as well should be used (Nass, 2006) (Fig. 61.1). The traditional neurological examination is generally normal, although soft signs are common (e.g., synkinesis during repetitive finger tapping and sequencing tasks and choreiform movements with the arms extended). Neuropsychological testing is not necessary unless comorbid learning issues are suspected. The neuropsychological profile reveals a normal IQ, but low scores are common on the Wechsler IQ subtests that demand attention or rapid processing (e.g., digit span, coding, arithmetic, symbol search). Executive frontal lobe functioning—the ability to initiate, inhibit, sustain, and shift attention, working memory, and organizational skills—is often impaired (Crosbie et al., 2007). Measures of spatial working memory, stop task response suppression,* continuous performance tasks (CPT), Stroop naming speed, Reitan trails B score, and mazes score have been found to correlate most highly with a clinical ADHD diagnosis. CPTs like the TOVA (Testing of Variables of Attention Deficit) or the Connors Continuous Performance Test are not useful as isolated diagnostic measures because of the high rate of both false-positive and false-negative results.
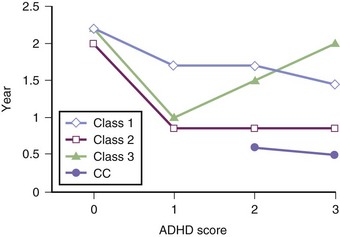
Overall, neuropsychological deficits are consistent with dysfunction of the basal ganglia–frontal circuits. However, these neuropsychological test abnormalities do not really explain the cognitive neurobiology of ADHD, hence the search for endophenotypes (Castellanos and Tannock, 2002; Konrad et al., 2010; Sonuga-Barke, 2010). Some candidates for core deficits of ADHD include failure of inhibitory control, dysregulation of brain systems mediating reward and response cost, and deficits in arousal, activation, and effortful control. Better delineation of endophenotypes would not only enhance understanding of the disorder but would increase the power of genetic research to identify susceptibility genes (Crosbie et al., 2007).
Several medical and environmental causes of ADHD exist (Box 61.11). The pediatric neurologist should take particular note of its high frequency in infants who were born preterm (about 20%) and its relatively high frequency in patients with epilepsy (10%-20%) even before they receive antiepileptic medications.
Box 61.11
Possible Environmental Causes/Contributors to Attention-Deficit Hyperactivity Disorder
Chronic illness: Allergy, asthma, celiac disease, sleep disorders
Endocrine: Thyrotoxicosis, hypothyroidism, generalized resistance to thyroid hormone
Genetic disorders: Turner syndrome, fragile X syndrome
Infectious disease: Otitis media, human immunodeficiency virus
Metabolic disorders: Phenylketonuria
Toxins, pre- and postnatal: Maternal smoking, drugs, alcohol; lead, mercury
From Mill and Petronis, 2008; Nass, 2006; Nass and Frank, 2010.
Dopamine appears to be the primary neurotransmitter involved in ADHD (Cools, 2008; Solanto, 2002). Hence, genetic studies have focused on candidate genes involved in dopaminergic transmission (dopamine transporter and dopamine receptors genes). The dopamine D4 receptor (DRD4) has been the most well studied and is prevalent in the basal ganglia–frontal networks implicated in the pathophysiology of ADHD (Albayrak et al., 2008). Recent data suggest that the genotype may even influence the physiological response to medication in ADHD (Gilbert et al., 2006).
Most structural imaging studies reveal abnormalities in frontal and striatal regions, but some have implicated the cerebellum and corpus callosum (Durston et al., 2004; Seidman, 2005). One recent longitudinal study shows that plasticity may play a role in outcome. Children with ADHD show relative cortical thinning in regions important for attentional control. Children with a worse outcome have “fixed” thinning of the left medial prefrontal cortex, which may compromise the anterior attentional network and encumber clinical improvement. Right parietal cortex thickness normalization in patients with a better outcome may represent compensatory cortical change (Shaw et al., 2006). Cortical volume, surface area, and folding throughout the cerebral cortex appear to be decreased, suggesting the disorder begins prenatally and affects neurogenesis (Wolosin et al., 2009).
Functional neuroimaging studies have assessed the degree of brain activation associated with neuropsychological tasks tapping attention and disinhibition. In many studies, the ADHD and control subjects do equally well on the experimental task. Thus, activation differences indicate group differences in the neural systems used to accomplish the task. Results of most of these studies are consistent with the structural studies locating abnormalities of brain activation in patients with ADHD in fronto–basal ganglia–cerebellar circuits (Biederman and Faraone, 2005; Biederman et al., 2006; MTA Cooperative Group, 2004).
Management
Medication, in particular psychostimulants, is the mainstay of treatment (Greenhill et al., 2002) (Table 61.7). The National Institutes of Health (NIH)-sponsored Multimodal Treatment Study of ADHD (MTA) has shown both behaviorally and by neuropsychological testing that stimulants work, and that they work better than behavioral modification (Biederman et al., 2006; Epstein et al., 2006). Approximately 75% of children respond to stimulants initially. During the initial 14 months of the MTA study, children receiving intensive behavioral management did not do as well with respect to the core symptoms of ADHD (inattention, hyperactivity, impulsivity) as those receiving stimulants, demonstrating that medication is critical to managing ADHD. Those children who had comorbid issues like anxiety or oppositional behavior needed both medication and behavioral management to do well. Some longitudinal data from MTA study suggest that the effectiveness of medication may decrease over time. At the year 3 outcome assessment about 2 years after the study treatment protocol ended, the different treatment groups no longer differed in terms of ADHD symptoms. Those who had received intensive medication management or combined medication and behavioral management did not fare better than those who had received intensive behavioral therapy or community management (whatever their private doctor prescribed). However, three subgroups that did not correspond to the initial treatment groups were identified at year 3. Group 1 had gradual improvement over time, with increasing and significant benefit from medication use at 3 years out (34% of the sample). Group 2 had a larger initial improvement that was maintained over time (52% of the sample). Group 3 returned to baseline symptoms after initial benefit (14% of the sample) (Fig. 61.2). So although the same treatment benefits were not present at year 3, most of the children were doing well nonetheless. Intensive medication management may only make a persistent long-term difference if continued with the same intensity as during the MTA’s initial 14-month study period. In contrast, starting or adding medication at a less-than-optimal intensity and/or too late in a child’s ADHD clinical course, particularly if a child’s behavior is deteriorating, may not only be ineffective but also (if not carefully examined in data analysis) even make medication appear to be associated with worse outcomes. Some children may eventually be able to stop medication, perhaps just because of early intensive treatment. Medication alone and combined therapy (medication and behavioral management) did decrease the number of ADHD children who developed oppositional defiant disorder at the 3-year follow-up (Molina et al., 2009).
Table 61.7 Treatment of Attention-Deficit Hyperactivity Disorder
| Stimulants | Methylphenidate (Ritalin, Focalin, Concerta, Daytrana), dextroamphetamine (Dexedrine, Adderall, Vyvanse) |
| α-Agonists | Clonidine (Catapres), guanfacine (Tenex), Intuniv |
| Antidepressants | SSRIs; tricyclic antidepressants; bupropion (Wellbutrin); trazodone; SSNRIs (venlafaxine [Effexor], duloxetine [Cymbalta]); MAOIs (selegiline [Deprenyl]) |
| Antimanic | Lithium carbonate (Lithium) |
| Mood stabilizers | Carbamazepine (Tegretol), divalproex sodium (Depakote), gabapentin (Neurontin), topiramate (Topamax) |
| Beta-blockers | Propranolol (Inderal), atenolol (Tenormin) |
| Anxiolytics | Buspirone (BuSpar); clonazepam (Klonopin), SSRIs |
| Atypical neuroleptics | Risperidone (Risperdal, Zyprexa) and others |
| Nonstimulants | Atomoxetine (Strattera), modafinil (Provigil) |
MAOI, Monoamine oxidase inhibitor; SSNRI, selective serotonin and norepinephrine reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor.
Data from Biederman and Faraone, 2005; Dopheide and Pliszka, 2009; Swanson et al., 2006.
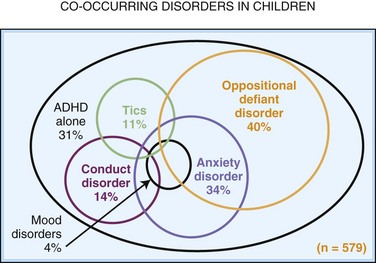
Fig. 61.2 MTA Study outcome of co-occurring disorders in children.
(Data from MTA Cooperative Group, 1999. Arch Gen Pysch 54, 1088.)
Side effects limit treatment (e.g., weight loss, rebound depression or irritability, flat affect) in 5% to 10%. Guidelines for stimulant use exist (Erenberg, 2006; Pliszka et al., 2006) (Fig. 61.3). Many different formulations of the stimulants exist, with differing duration of action (Fig. 61.4). Neither tics nor Tourette syndrome are contraindications to stimulant use in ADHD, although they may affect medication choices (Pliszka et al., 2006) (Fig. 61.5). Comorbid mood or anxiety disorders also affect their use (Brown, 2000; Pliszka et al., 2006). In addition to stimulants, other medications can be used to treat ADHD. Strattera is an accepted nonstimulant treatment, as are the α-agonists (clonidine, guanfacine, Intuniv). Tricyclics, selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, bupropion, and modafinil have all been used, generally in those who failed to respond to stimulants (see Table 61.7).
Fig. 61.3 Algorithm for the psychopharmacological treatment of ADHD.
(From Pliszka, S.R., Crismon, M.L., Hughes, C.W., et al., 2006. The Texas Children’s Medication Algorithm Project: revision of the algorithm for pharmacotherapy of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 45, 642-657.)
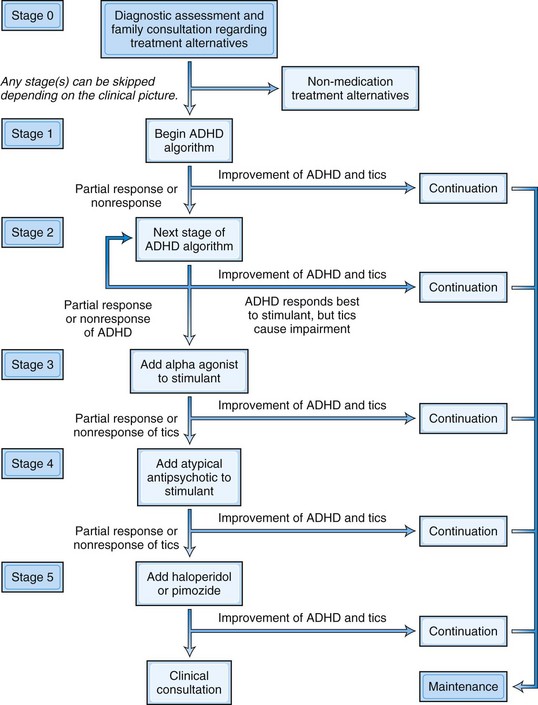
Fig. 61.5 Algorithm for the psychopharmacological treatment of ADHD and comorbid tic disorder.
(From Pliszka, S.R., Crismon, M.L., Hughes, C.W., et al., 2006. The Texas Children’s Medication Algorithm Project: revision of the algorithm for pharmacotherapy of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 45, 642-657.)
Ahonen T., Kooistra L., Viholainen H., et al. Developmental motor learning disability. In: Dewey D., Tupper D. Developmental Motor Disorders. New York: Guilford Press; 2004:265-290.
Airaksinen E.M., Matilainen R., Mononen T., et al. A population-based study on epilepsy in mentally retarded children. Epilepsia. 2000;41:1214-1220.
Albayrak O.S., Friedel B.G., Schimmelmann A., et al. Genetic aspects in attention-deficit hyperactivity disorder. J Neural Transm. 2008;115:305-315.
Allin M., Rooney M., Cuddy M., et al. Personality in young adults who are born preterm. Pediatrics. 2006;117:309-316.
Allin M., Rooney M., Griffiths T., et al. Neurological abnormalities in young adults born preterm. J Neurol Neurosurg Psychiatry. 2006;77:495-499.
Amaral D., Schumann C.M., Nordahl C.W. Neuroanatomy of autism. Trends Neurosci.. 2008;31:137-143.
Amaral D.G., Shcumann C.M., Nordahl C.W. Neuroanatomy of autism. Trends Neurosci. 2008;31:137-145.
American Academy of Pediatrics, Section on Ophthalmology, Council on Children Pediatric Ophthalmology, Strabismus and American Association of Certified with Disabilities, American Academy of Ophthalmology, American Association for Orthoptists. Learning disabilities, dyslexia, and vision. Pediatrics. 2009;124:837-844.
American College of Obstetricians, Gynecologists Committee on Obstetric Practice, Society for Maternal-Fetal Medicine. Magnesium sulfate before anticipated preterm birth for neuroprotection, Committee Opinion No. 455. Obstet Gynecol, 115. 2010;3:669-671.
American Psychiatric Association. Diagnostic and Statistical Manual, text revised, fourth ed. Washington, DC: American Psychiatric Association. 2000.
Amitay S., Ben-Yehudah G., Banai K., et al. Disabled readers suffer from visual and auditory impairments but not from a specific magnocellular deficit. Brain. 2002;125:2272-2285.
Ancel P., Livinec F., Larroque M.B., et al. EPIPAGE Study Group, Cerebral palsy among very preterm children in relation to gestational age and neonatal ultrasound abnormalities: the EPIPAGE cohort study. Pediatrics. 2006;117:828-835.
Angélique D., Hendriks W., Kolka H. Evidence for magnocellular involvement in the identification of flanked letters. Neuropsychologia. 2002;40:1881-1890.
Ashwal S., Russman B.S., Blasco P.A., et al. Practice Parameter: Diagnostic assessment of the child with cerebral palsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2004;62:851-863.
Bakker D.J. Treatment of developmental dyslexia: a review. Pediatr Rehabil. 2006;9:3-13.
Barkovich A.J. MR imaging of the neonatal brain. Neuroimaging Clin N Am. 2006;16:117-135. viii-ix
Baron-Cohen S. The extreme male brain theory of autism. Trends Cogn Sci. 2002;6:248-254.
Baron-Cohen S., Wheelwirght S., Lawson J., et al. The exact mind. In: Goswami U., editor. Handbook of Cognitive Development. London: Blackwell; 2003:491-508.
Barton J., Cherkasova M.V., Hefter R., et al. Are patients with social developmental disorders prosopagnosic? Perceptual heterogeneity in the Asperger and socio-emotional processing disorders. Brain. 2004;127:1706-1716.
Battaglia A., Carey J.C. Etiologic yield of autistic spectrum disorders: a prospective study. Am J Med Genet C Semin Med Genet. 2006;142C(1):3-7.
Bauman M.L., Kemper T.L. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005;23:183-187.
Bax M., Flodmark O., Tydeman C. From syndrome toward disease. The definition and classification of cerebral palsy. Dev Med Child Neurol. 2007;49:39-42.
Beaton A., Edwards R., Peggie A. Dyslexia and across-hands finger localization deficits. Neuropsych, e-document. 2006.
Bhutta A.T., Anand K.J. Abnormal cognition and behavior in preterm neonates linked to smaller brain volumes. Trends Neurosci. 2001;24:129-132.
Biederman J., Faraone V. ADHD. Lancet. 2005;366:237-245.
Biederman J., Arnsten A.F., Faraone S.V., et al. New developments in the treatment of ADHD. J Clin Psychiatry. 2006;67:148-159.
Billstedt E., Gillberg C., Gillberg C. Autism after adolescence: population-based 13- to 22-year old follow-up study of 120 individuals with autism diagnosed in childhood. J Autism Dev Disord. 2005;35:351-360.
Billstedt E., Gillberg I.C., Gillberg C. Autism in adults: symptom patterns and early childhood predictors. Use of the DISCO in a community sample followed from childhood. J Child Psychol Psychiatr. 2007;48:717-729.
Bishop D., Andrew J.O., Whitehouse A.J., et al. Autism and diagnostic substitution: evidence from a study of adults with a history of developmental language disorder. Dev. Med. Child Neurol.. 2008;50:341-345.
Blau V., Reithler J., van Atteveldt N., et al. Deviant processing of letters and speech sounds as proximate cause of reading failure: a functional magnetic resonance imaging study of dyslexic children. Brain. 2010;133(Pt 3):868-879.
Blickstein I. Cerebral palsy in multifoetal pregnancies. Dev Med Child Neurol. 2002;44:352-355.
Bodensteiner J.B., Johnsen S.D. Cerebellar injury in the extremely premature infant: newly recognized but relatively common outcome. J Child Neurol. 2005;20:139-142.
Bonellie S.R., Currie D., Chalmers J. Comparison of risk factors for cerebral palsy in twins and singletons. Dev Med Child Neurol. 2005;47:587-591.
Brown T. Attention Deficit Disorders and Comorbidities in Children, Adolescents and Adults. Washington, DC: American Psychiatric Press; 2000.
Burger N. A Special Kind of Brain. London: Jessica Kingsley Publishers; 2004.
Callahan K., Shukla-Mehta S., Magee S., et al. ABA versus TEACCH: the case for defining and validating comprehensive treatment models in autism. J Autism Dev Disord. 2010;40:74-88.
Camos V. Low working memory capacity impedes both efficiency and learning of number transcoding in children. J Exp Child Psychol. 2008;99:37-57.
Campbell L., Daly E., Toal F., et al. Brain and behaviour in children with 22q11.2 deletion syndrome: a volumetric and voxel-based morphometry MRI study. Brain. 2006;129:1218-1228.
Canitano R., Luchetti A., Zappella M. Epilepsy, electroencephalographic abnormalities, and regression in children with autism. J Child Neurol. 2005;20:27-31.
Casanova M.F., Buxhoeveden D.P., Switala A.E., et al. Minicolumnar pathology in autism. Neurology. 2002;58:428-432.
Casanova M.F., Imke A.J., van Kooten A.E., et al. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006;112:287-303.
Casanova M., Trippe J. Radial cytoarchitecture and patterns of cortical connectivity in autism. Phil Trans R Soc B. 2009;364(1522):1433-1436.
Casanova M.F., El-Baz A.S., Giedd J., et al. Increased white matter gyral depth in dyslexia: implications for corticocortical connectivity. J Autism Dev Disord. 2010;40:21-29.
Castellanos F.X., Tannock R. Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci. 2002;3:617-628.
Castellanos F.X., http://www.dsm5.org/Pages/Default.aspx, 2009.
Centers for Disease Control and Prevention. Intellectual disability. Available at http://www.cdc.gov/ncbddd/dd/mr3.htm, 2009.
Chaila E., Dominick J., McCabe H., et al. Broadening the phenotype of childhood-onset dopa-responsive dystonia. Arch Neurol. 2006;63:1185-1188.
Coffee B., Keith K., Albizua I., et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. American Journal of Human Genetics. 2009;85(4):503-514.
Constantino J. Social Responsiveness Scale (SRS). Los Angeles: Western Psychological Services; 2005.
Cools R. Role of dopamine in the motivational and cognitive control of behavior. Neuroscientist. 2008;14:381-389.
Corbett B.A., Carmean V., Ravizza S., et al. A functional and structural study of emotion and face processing in children with autism. Psychiatry Research. 2009;173(3):196-205.
Cornoldi C., Venneri A., Marconato F., et al. A rapid screening measure for the identification of visuospatial learning disability in schools. J Learning Disabilities. 2003;36:299-306.
Crews K., D’Amato J., Rik C. Subtyping children’s reading disabilities using a comprehensive neuropsychological measure. Int J Neurosci. 2009;119(10):1615-1639.
Croen L.A., Grether J.K., Curry C.J., et al. Congenital abnormalities among children with cerebral palsy: more evidence for prenatal antecedents. J Pediatr. 2001;138:804-810.
Crosbie J., Perusse D., Barr C.L., et al. Validating psychiatric endophenotypes: childhood attention deficit hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46:894-921.
Dahdouh F., Anthoni H., Tapia-Paez I., et al. Further evidence for DYX1C1 as a susceptibility factor for dyslexia. Psychiatr Genet. 2009;19(2):59-63.
Dammann O., Leviton A. Inflammatory brain damage in preterm newborns—Dry numbers, wet lab, and causal inferences. Early Hum Dev. 2004;79:1-15.
Daniels J.L., Forssen U., Hultman C.M., et al. Parental psychiatric disorders associated with autism spectrum disorders in the offspring. Pediatrics. 2008;121:1357-1362.
Dapretto M., Davies M.S., Pfeifer J.H., et al. Understanding emotions in others: mirror neuron dysfunction in children with autism spectrum disorders. Nature Neurosci. 2006;9:28-30.
Decety J., Jeannerod M. Mentally simulated movements in virtual reality: Does Fitts’s law hold in motor imagery? Behav Brain Res. 1995;72:127-134.
Deconinck F.J.A., Spitaels L., Fias W., et al. ‘Is developmental coordination disorder a motor imagery deficit?’. J Clin Exp Neuropsychol. 2008;31(6):720-730.
Denckla M.B. Revised neurological examination for subtle signs. Pharmacol Bull. 1985;21:773-789.
Deonna T., Prelaz-Girod A.C., Mayor-Dubois C., et al. Sign language in Landau-Kleffner syndrome. Epilepsia. 2009;50(Suppl. 7):77-82.
Dopheide J.A., Pliszka S.R. Attention-deficit-hyperactivity disorder: an update. Pharmacotherapy. 2009;29(6):66-79.
Drummond C.R., Ahmad S.A., Rourke B.P. Rules for classification of younger children with nonverbal learning disabilities and basic phonological processing disabilities. Arch Clin Neuropsychol. 2005;20:171-182.
Dunn M. S.O.S Social Skills in Our Schools: A Social Skills Program for Children with Pervasive Developmentally Disorders, Including High-Functioning Autism and Asperger Syndrome, and Their Typical Peers. Port Chester, NY: National Professional Resources, Inc.; 2005.
Durkin M.S., Maenner M.J., Newschaffer C.J., et al. Advanced parental age and the risk of autism spectrum disorder. Am J Epidemiol. 2008;168:1268-1276.
Durston S., Hulshoff Pol H.E., Schnack H.G., et al. Magnetic resonance imaging of boys with attention-deficit/hyperactivity disorder and their unaffected siblings. J Am Acad Child Adolesc Psychiatry. 2004;43:332-340.
Eckert M., Leonard C. The planum temporale in reading. Ment Retard Dev Disabil Res Rev. 2000;6:198-206.
Eikeseth S. Outcome of comprehensive psycho-educational interventions for young children with autism. Res Dev Disabil. 2009;30:158-178.
Elsabbagh M., Johnson M. Getting answers from babies about autism. Trends Cogn Sci. 2009;14(2):77-87.
Engels H.A., Brockschmidt A., Hoischen C., et al. DNA microarray analysis identifies candidate regions and genes in unexplained mental retardation. Neurology. 2007;68:743-750.
Epstein J., Conners K., Hervey A., et al. The Multimodal Treatment Study of ADHD (MTA) Cooperative Study Group. Assessing medication effects in the MTA study using neuropsychological outcomes. J Child Psychol Psychiatry. 2006;47:446-454.
Erenberg G. Tics and stimulants. Semin Pediatr Neurol. 2006;12:217-230.
Farley M., McMahon W.M., Fombonne E., et al. Twenty-year outcome for individuals with autism and average or near-average cognitive abilities. Autism Res. 2009;2:109-118.
Fatemi A., Wilson M.A., Johnston M.V. Hypoxic-ischemic encephalopathy in the term infant. Clin Perinatol. 2009;36(4):835-858.
Forrest B. The utility of math difficulties, internalizing psychopathology and visuo spatial deficits in identifying children with NVLD: evidence for a visual spatial disability. Child Neuropsychol. 2004;10:129-146.
Frith U. Autism: Explaining the Enigma, second ed. Oxford, UK: Blackwell; 2003.
Frombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry. 2005;66(Suppl. 10):3-8.
Fulbright R.K., Jenner A.R., Mencl W.E., et al. The cerebellum’s role in reading: a functional MR imaging study. AJNR Am J Neuroradiol. 1999;20:1925-1930.
Greenwood Genetic Center. XLID update. Available at http://www.ggc.org/xlmr.htm, 2011.
Gabriel R., Grolier F., Graesslin O. Can obstetric care provide further improvement in the outcome of preterm infants? Eur J Obstet Gynecol Reprod Biol. 2004;117(Suppl. 1):25-28.
Galaburda A., LoTurco J., Ramos F., et al. From genes to behavior in developmental dyslexia. Nat Neurosci. 2006;9:1213-1217.
García-Cazorla A., Wolf N.I., Serrano M., et al. Inborn errors of metabolism and motor disturbances in children. J Inherit Metab Dis. 2009;32:618-629.
García-Cazorla A., Wolf N.I., Serrano M., et al. Mental retardation and inborn errors of metabolism. J Inherit Metab Dis. 2009;32:597-608.
Geuze R.H. Postural control in children with developmental coordination disorder. Neural Plast. 2005;12:183-196. discussion 263-272
Geuze R.H., Jongmans M., Schoemaker M., et al. Developmental coordination disorder. Hum Movement Sci. 2001;20:1-5.
Gilbert D., Wang Z., Sallee F., et al. Dopamine transporter genotype influences the physiological response to medication in ADHD. Brain. 2006;129:2038-2046.
Gillberg C. Double syndromes: Autism associated with genetic, medical and metabolic disorders. In: Nass R.D., Frank Y. Cognitive and Behavioral Manifestations of Pediatric Diseases. New York: Oxford University Press; 2010:100-110.
Gilstrap L.C., Ramin S.M. Infection and cerebral palsy. Semin Perinatol. 2000;24:200-203.
Girard S., Kadhim H., Roy M., et al. Role of perinatal inflammation in cerebral palsy. Pediatr Neurol. 2009;40:168-174.
Golan O., Ashwin E., Granader Y., et al. Simon. Enhancing emotion recognition in children with autism spectrum conditions: an intervention using animated vehicles with real emotional faces. J Autism Dev Disord. 2010;40(3):269-279.
Gothelf D., Furfaro J.A., Penniman L., et al. The contribution of novel brain imaging techniques to understanding the neurobiology of mental retardation and developmental disabilities. Ment Retard Dev Disabil Res Rev. 2005;11:331-339.
Gough M., Fairhurst C., Shortland A.P. Botulinum toxin and CP: time for reflection? Dev Med Child Neurol. 2005;47:709-712.
Grafman J., Romero S. Appearances may not be deceiving: calculation deficits due to a brain structure abnormality in neurologically normal children. Brain. 2001;124:1681-1682.
Grandin T. Thinking in Pictures. New York: Vintage Press; 1995.
Gray A., Karmiloff-Smith V., Funnell A., et al. In-depth analysis of spatial cognition in Williams syndrome: a critical assessment of the role of the LIMK1 gene. Neuropsychologia. 2006;44:679-685.
Greenhill L., Beyer D.H., Finkleson J., et al. Guidelines and algorithms for the use of methylphenidate in children with attention-deficit/hyperactivity disorder. J Attention Disord. 2002;6(Suppl. 1):89-100.
Gross-Tsur V., Shalev R., Manor O., et al. Developmental right hemisphere syndrome: clinical spectrum of NLD. J Learn Disabil. 1995;28:80-86.
Guilliam J. Guilliam Asperger Disorder Scale. Upper Saddle River, NJ: GADS, Pearson Education, Inc.; 2006.
Guilliam J. Guilliam Autism Rating Scale. Upper Saddle River, NJ: GARS, Pearson Education, Inc.; 2006.
Hadders-Algra M. Two distinct forms of minor neurological dysfunction: perspectives emerging from a review of data of the Groningen Perinatal Project. Dev Med Child Neurol. 2002;44:561-571.
Hadders-Algra M., Mavinkurve-Groothuis A.M., Groen S.E., et al. Quality of general movements and the development of minor neurological dysfunction at toddler and school age. Clin Rehabil. 2004;18:287-299.
Hagberg G., Henrik J.E., Maria B.L. Injury to the preterm brain and cerebral palsy: clinical aspects, molecular mechanisms, unanswered questions, and future research directions. J Child Neurol. 2009;24(9):1064-1084.
Handelman J., Harris S. Special Needs Project. Santa Barbara, CA: Preschool Education Programs for Children with Autism; 2002.
Hardan A., Pabalan M., Gupta N., et al. Can children with autism recover? If so, how? Neuropsychol Rev. 2008;18(4):339-366.
Heilman K., Valenstein E. Clinical Neuropsychology. New York: Oxford University Press; 2009.
Helt M., Kelley E., Kinsbourne M., et al. Can children with autism recover? If so, how? Neuropsychol Rev. 2008;18:339-366.
Herbert M.R., Ziegler D.A., Deutsch C.K., et al. Brain asymmetries in autism and developmental language disorder: a nested whole-brain analysis. Brain. 2005;128:213-226. 2005
Herbert M., Harris R., Adrien G.J., et al. Abnormal asymmetry in language association cortex in autism. Ann Neurol. 2002;52:588-596.
Hertz-Picciotto I., Delwiche L. The rise in autism and the role of age at diagnosis. Epidemiology. 2009;20(1):84-90.
Hill E. Executive dysfunction in autism. Trends Cogn Sci. 2004;8:260-269.
Himmelmann K., Hagberg G., Beckung E., et al. The changing panorama of cerebral palsy in Sweden, IX, Prevalence and origin in the birth-year period 1995-1998. Acta Paediatr. 2005;94(3):287-294.
Hintz S.R., O’Shea M. Neuroimaging and neurodevelopmental outcomes in pre-term infants. Semin Perinatol. 2008;32:11-19.
Hiscock M., Chapieski L. Hand preference, manual asymmetry and manual skill. In: Dewey D., Tupper D. Developmental Motor Disorders. NY: Guilford Press; 2004:353-388.
Howlin P. Outcome of autistic spectrum disorder. In: Volkmar F.R., Paul R., Klin A., et al. Handbook of Autism and Pervasive Developmental Disorders. Hoboken, NJ: Wiley; 2005:215-227.
Hutton J., Goode S., Murphy M., et al. New-onset psychiatric disorders in individuals with autism. Autism. 2008;12(4):373-390.
Igo R., Chapman N., Berninger V.W., et al. Genomewide scan for real-word reading subphenotypes of dyslexia: novel chromosome 13 locus and genetic complexity. Am J Med Genet. 2006;141:15-27.
Ivry R.B. Cerebellar involvement in clumsiness and other developmental disorders. Neural Plast. 2003;10:141-153.
Johnson S., Hennessy E., Smith R., et al. Academic attainment and special educational needs in extremely preterm children at 11 years of age: the EPICure study. Arch Dis Child Fetal Neonatal Ed. 2009;94(4):F283-F289.
Jones K. Smith’s Recognizable Patterns of Human Malformations. Philadelphia: WB Saunders; 2006.
Kagan-Kushnir T., Roberts S.W., Snead O.C.3rd. Screening electroencephalograms in autism spectrum disorders: evidence-based guideline. J Child Neurol. 2005;20:197-206.
Katz G., Lazcano-Ponce E. Intellectual disability: definition, etiological factors, classification, diagnosis, treatment and prognosis. Salud Publica Mex. 2008;50(Suppl 2):s132-s141.
Keller T.A., Kana R.K., Just M.A. A developmental study of the structural integrity of white matter in autism. Neuroreport. 2007;18(1):23-27.
Keller T.A., Just M.A. Altering cortical connectivity: remediation-induced changes in the white matter of poor readers. Neuron. 2009;64(5):624-631.
Kent A., Lomas F., Hurrion E., et al. Antenatal steroids may reduce adverse neurological outcome following chorioamnionitis: neurodevelopmental outcome and chorioamnionitis in premature infants. J Paediatr Child Health. 2005;41:186-190.
Kenworthy L., Verys B., Anthony L., et al. Understanding executive control in ASD in the lab and in the real world. Neuropsychology. 2008;18:320-338.
Kessler R.C., Adler L.A., Barkley R., et al. Patterns and predictors of attention-deficit/hyperactivity disorder persistence into adulthood: results from the national comorbidity survey replication. Biol Psychiatry. 2005;57:1442-1451.
Kibby M., Kroese Y., Judith M., et al. The pars triangularis in dyslexia and ADHD: a comprehensive approach. Brain Lang. 2009;111(1):46-54.
Klin A., Volkmar F., Sparrow S. Asperger Syndrome. New York: Guilford Press; 2000.
Konrad K., Dempfle A., Friedel S., et al. Familiality and molecular genetics of attention networks in ADHD. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(1):148-158.
Kovas Y., Haworth C.M., Dale P.S., et al. The genetic and environmental origins of learning abilities and disabilities in the early school years. Monogr Soc Res Child Dev. 2007;72(vii):1-144.
Krägeloh-Mann I. The role of neuroimaging in cerebral palsy. Dev Med Child Neurol. 2007;49:144-151.
Krägeloh-Mann I., Toft P., Lunding J., et al. Brain lesions in preterms: origin, consequences and compensation. Acta Paediatr. 1999;88:897-908.
Kucian K., Loenneker T., Dietrich T., et al. Impaired neural networks for approximate calculation in dyscalculic children: A functional MRI study. Behav Brain Funct. 2006;2:31.
Lagae L. Rational treatment options with AEDs and ketogenic diet in Landau-Kleffner syndrome: Still waiting after all these years. Epilepsia. 2009;50(Suppl 7):59-62.
Lamm A.C., Lamm S.T., Lai K.K., et al. High rate of detection of subtelomeric aberration by using combined MLPA and subtelomeric FISH approach in patients with moderate to severe mental retardation. Clin Biochem. 2006;39:196-202.
Leviton A., Paneth N., Reuss M.L., et al. Hypothyroxinemia of prematurity and the risk of cerebral white matter damage. J Pediatr. 1999;134:706-711.
Li M., Andersson H. Clinical application of micro-array based molecular cytogenetics: an emerging new era of genomic medicine. J Pediatr. 2009;155:311-317.
Locatelli A., Andreani M., Pizzardi A., et al. Antenatal variables associated with severe adverse neurodevelopmental outcome among neonates born at less than 32 weeks. Eur J Obstet Gynecol Reprod Biol. 2010;152:143-147.
Locatelli A., Ghidini A., Assi F., et al. Which factors affect the occurrence of severe cerebral lesions in preterm neonates who are born with intrauterine infection? Am J Obstet Gynecol. 2008;199(4):404. e1-5
Lord C., Risi S., DiLavore P.S., et al. Autism from 2 to 9 years of age. Arch Gen Psychiatry. 2006;63:694-701.
Lord C., Rutter M., DiLavore P., et al. Autism Diagnostic Observation Schedule—WPS Edition. Los Angeles: Western Psychological Services; 1999.
Lord C., Shulman C., DiLavore P. Regression and word loss in autistic spectrum disorders. J Child Psychol Psychiatry. 2004;45:936-955.
Losse A., Henderson S.E., Elliman D., et al. Clumsiness in children—do they grow out of it? A 10-year follow-up study. Dev Med Child Neurol. 1991;33:55-68.
Lundy C.T., Doherty G.M., Fairhurst C.B. Botulinum toxin type A injections can be an effective treatment for pain in children with hip spasms and cerebral palsy. Dev Med Child Neurol. 2009;51(9):705-710.
Lundy-Ekman L., Ivry R.B., Keele S., et al. Timing and force control deficits in clumsy children. J Cogn Neurosci. 1991;3:367-376.
Malin G.L., Morris R.K., Khan K.S. Strength of association between umbilical cord pH and perinatal and long term outcomes: systematic review and meta-analysis. BMJ. 2010;13:340-356.
Manoach D., Sandson T., Weintraub S. The developmental social-emotional processing disorder is associated with right hemispheric abnormalities. Neuropsychiatry Neuropsychol Behav Neurol. 1995;8:99-105.
Mao R., Pevsner J. The use of genomic microarrays to study chromosomal abnormalities in mental retardation. Ment Retard Dev Disabil Res Rev. 2005;11:279-285.
Martin T., Klin A. When Asperger’s syndrome and a NVLD look alike. J Dev Behav Pediatr. 2004;25:59.
Mastergeorge A., Au J., Hagerman R. Fragile X: a family of disorders. In: Nass R., Frank Y. Cognitive and Behavioral Abnormalities of Pediatric Diseases. New York: Oxford University Press; 2010:170-187.
Matson J., Shoemaker M. Intellectual disability and its relationship to autism spectrum disorders. Res Dev Disabil. 2009;30(6):1107-1114.
Meng H., Shelley D., Smith S.D., et al. DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc Natl Acad Sci U S A. 2005;102:17053-17058.
Meng H., Smith S.D., Hager K., et al. DCDC2is associated with reading disability and modulates neuronal development in the brain. Proc Natl Acad Sci USA. 2005;102:18763.
Mestan K., Marks K.L., Hecox K., et al. Neurodevelopmental outcomes of premature infants treated with inhaled nitric oxide. N Engl J Med. 2005;353:23-32.
Mesterman R., Leitner Y., Yifat R., et al. Cerebral palsy long-term medical, functional, educational, and psychosocial outcomes. J Child Neurol. 2010;25:36.
Mill J., Petronis A. Pre- and peri-natal environmental risks for attention-deficit hyperactivity disorder (ADHD): the potential role of epigenetic processes in mediating susceptibility. J Child Psychol Psychiatry. 2008;49:1020-1030.
Miller G. Extreme Prematurity: Practices. New York: Bioethics and the Law Cambridge University Press; 2007.
Miller M.T., Strömland K., Ventura L., et al. Autism associated with conditions characterized by developmental errors in early embryogenesis: a mini review. Int J Dev Neurosci. 2005;23:201-219.
Minshew N. Corpus callosum volume in children with autism. Psychiatry Research. 2009;174(1):57-61.
Mirmiran M., Barnes P.D., Keller K., et al. Neonatal brain magnetic resonance imaging before discharge is better than serial cranial ultrasound in predicting cerebral palsy in very low birth weight preterm infants. Pediatrics. 2004;114(4):992-998.
Moeschler J.B., Shevell M. American Academy of Pediatrics Committee on Genetics, Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics. 2006;117(6):2304-2316.
Molina B., Hinshaw S.P., Swanson J.M., et al. MTA at 8 years: prospective follow-up of children treated for combined-type ADHD in a multisite study. J Am Acad Child Adolesc Psychiatry. 2009;48(5):484-500.
Momoi T., Fujita E., Senoo H., et al. Genetic factors and epigenetic factors for autism: endoplasmic reticulum stress and impaired synaptic function. Cell Biol Int. 2010;34(1):13-19.
MTA Cooperative Group. National Institute of Mental Health Multimodal Treatment Study of ADHD follow-up: 24-month outcomes of treatment strategies for attention-deficit hyperactivity disorder. Pediatrics. 2004;113:754-761.
Munson J., Dawson G., Sterling L., et al. Evidence for latent classes of IQ in young children with autism spectrum disorder. Am J Ment Retard. 2008;113(6):439-452.
Murphy K.R., Barkeley R.A., Bush T. Young adults with ADHD: Subtype differences in comorbidities, educational and clinical history. J Nerv Ment Disord. 2002;190:147-157.
Nass R., Leventhal F. The Tourette-autistic spectrum. In: Rizzo M., Eslinger P. Principles and Practice of Behavioral Neurology and Neuropsychology. New York: WB Saunders; 2004:917-978.
Nass R. Evaluation and assessment issues in the diagnosis of attention deficit hyperactivity disorder. Semin Pediatr Neurol. 2006;12:200-216.
Nass R., Leventhal F., Solodow W., et al. Non-verbal learning disabilities: frequency in an LD school. Thalamus. 2006;24:17-36.
Nelson K.B., Chang T. Is cerebral palsy preventable? Curr Opin Neurol. 2008;21:129-135.
Nelson K.B. Perinatal ischemic stroke. Stroke. 2007;38:742-745.
Nelson K.B. Infection in pregnancy and cerebral palsy. Dev Med Child Neurol. 2009;52:253-254.
Newschaffer C.J., Croen L.A., Daniels J., et al. The epidemiology of autism spectrum disorders. Annual Rev Public Health. 2007;28:235-258.
O’Hare A., Khalid S. The association of abnormal cerebellar function in children with developmental coordination and reading difficulties. Dyslexia. 2002;8:234-248.
2010 Observations, Lancet retracts Wakefield’s MMR paper. BMJ. 2010:340.
Opitz J.M. Vision and insight in the search for gene mutations causing nonsyndromal mental deficiency. Neurology. 2000;55:328-330.
Palisano R., Rosenbaum P., Walter S., et al. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol. 1997;39:214-223.
Palomares M., Delicado A., Lapunzina P., et al. MLPA vs multiprobe FISH: comparison of two methods for the screening of subtelomeric rearrangements in 50 patients with idiopathic mental retardation. Clin Genet. 2006;69:228-233.
Palombo J. NVLD. New York: WW Norton; 2006.
Paneth N. Cerebral palsy in term infants-birth or before birth? J Pediatr. 2001;138:791-792.
Paneth N., Hong T., Korzeniewski S. The descriptive epidemiology of cerebral palsy. Clin Perinatol. 2006;33:251-267.
Paracchini S., Thomas A., Castro S., et al. The chromosome 6p22 haplotype associated with dyslexia reduces the expression of KIAA0319, a novel gene involved in neuronal migration. Hum Mol Genet. 2006;15:1659-1666.
Pardo C.A., Eberhart C.G. The neurobiology of autism. Brain Pathol. 2007;17:434-447.
Partington M., Mowat D., Einfeld S., et al. Genes on the X chromosome are important in undiagnosed mental retardation. Am J Med Genet. 2000;92:57-61.
Pelletier P.M., Ahmad S., Rourke B.P. Classification rules for basic phonological processing disabilities and nonverbal learning disabilities: formulation and external validity. Child Neuropsychol. 2001;7:84-98.
Peterson R.L., Pennington B.F., Shriberg L.D., et al. What influences literacy outcome in children with speech sound disorder? J Speech Lang Hearing Res. 2009;52(5):1175-1188.
Pliszka S.R., Crismon M.L., Hughes C.W., et al. Texas Consensus Conference Panel on Pharmacotherapy of Childhood Attention Deficit Hyperactivity Disorder. The Texas Children’s Medication Algorithm Project: revision of the algorithm for pharmacotherapy of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2006;45(6):642-657.
Polatajko H., Cantin N. Developmental coordination disorder. Semin Pediatr Neurol. 2006;12:250-255.
Price G.R., Holloway I., Rasanen P., et al. Impaired parietal magnitude processing in developmental dyscalculia. Curr Biol. 2007;17:24-29.
Pugh K., Mencl W.E., Jenner A.R., et al. Functional neuroimaging studies of reading and reading disability (developmental dyslexia). Ment Retard Dev Disabil Res Rev. 2000;6:207-213.
Rademaker K.J., Lam J.N., Van Haastert I.C., et al. Larger corpus callosum size with better motor performance in prematurely born children. Semin Perinatol. 2004;28:279-287.
Rapin I., Tuchman R. Autism. New York: Cambridge University Press; 2006.
Reichow B., Volkmar F.R. Social skills interventions for individuals with autism: evaluation for evidence-based practices within a best evidence synthesis framework. J Autism Dev Disord. 2010;40(2):149-166. 2010 Feb
Reiss A.L., Eliez S., Schmitt J.E., et al. IV, Neuroanatomy of Williams syndrome: a high-resolution MRI study. J Cogn Neurosci. 2000;12(Suppl. 1):65-73.
Renieri A., Mari F., Mencarelli M.A., et al. Diagnostic criteria for the Zappella variant of Rett syndrome (the preserved speech variant). Brain Dev. 2009;31:208-216.
Riggs P., Levin F., Green A.I., et al. Comorbid psychiatric and substance abuse disorders: recent treatment research. Substance Abuse. 2008;29(3):51-63.
Rizzolatti G., Fabbri-Destro M. Mirror neurons: from discovery to autism. Exp Brain Res. 2010;200:223-237.
Robertson L., Hall S.E., Jacoby P., et al. The association between behavior and genotype in Rett syndrome using the Australian Rett Syndrome Database. Am J Med Genet Neuropsychiatr Genet. 2006;141B:177-183.
Robins D., Fein D., Barton M., et al. The modified checklist for autism in toddlers. J Autism Dev Disord. 2001;31:131-144.
Ropers H.H., Hamel B.C.J. X-linked mental retardation. Nat Rev Genet. 2005;6:46-57.
Rosenbaum P., Paneth N., Leviton A., et al. A report: the definition and classification of cerebral palsy. Dev Med Child Neurol. 2007;Suppl. 49:8-14.
Rotzer S., Kucian K., Martin E., et al. Optimized voxel-based morphometry in children with developmental dyscalculia. Neuroimage. 2008;39:417-422.
Rotzer S., Loenneker T., Kucia K., et al. Dysfunctional neural network of spatial working memory contributes to developmental dyscalculia. Neuropsychologia. 2009;47:2859-2865.
Rourke B.P. Syndrome of Nonverbal Learning Disabilities: Neurodevelopmental Manifestations. NY: Guilford Press; 1995.
Rourke B.P., Ahmad S., Collins D., et al. Child clinical/pediatric neuropsychology: some recent advances. Ann Rev Psychol. 2002;53:309-339.
Rutter M., Bailey A., Lord C. Social Communication Questionnaire (SCQ)—WPS Edition. Los Angeles: Western Psychological Services; 2005.
Rutter M., LeCouteur A., Lord C. Autism Diagnostic Interview, Revised (ADI-R)—WPS Edition. Los Angeles: Western Psychological Services; 2003.
Ryburn B., Anderson V., Wales R. Asperger syndrome: how does it relate to non-verbal learning disability? J Neuropsychol. 2009;3(Pt 1):107-123.
Sacco R., Militerni R., Frolli A., et al. Clinical, morphological, and biochemical correlates of head circumference in autism. Biol Psychiatry. 2007;62:1038-1047.
Schaefer G.B., Mendelsohn N.J., Professional Practice and Guidelines Committee. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders. Genet Med. 2008;10:301-305.
Schneider A., Hagerman R.J., Hessl D. Fragile X syndrome—from genes to cognition. Dev Disabil Res Rev. 2009;15:333-342.
Schopler E., Reichler R., Rochen B., et al. Childhood Autism Rating Scale (CARS)—WPS Edition. Los Angeles: Western Psychological Services; 2000.
Schumacher J., Hoffmann P., Schmal C., et al. Genetics of dyslexia: the evolving landscape. J Med Genet. 2007;44:289-297.
Schumann C.M., Bloss C.S., Barnes C.C., et al. Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. J Neurosci. 2010;30(12):4419-4427.
Sebat J., Lakshmi B., Malhotra D., et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445-449.
Seidman L. Structural imaging in ADHD. Biol Psychiatry. 2005;57:1263-1272.
Seltzer M., Krauss M.W., Shattuck P., et al. The symptoms of autism spectrum disorders in adolescence and adulthood. J Autism Dev Disord. 2003;33:565-581.
Shalev R., Gross-Tsur V. Developmental dyscalculia. Pediatr Neurol. 2001;16:337-342.
Shao L., Shaw C.A., Lu X.Y., et al. Identification of chromosome abnormalities in subtelomeric regions by microarray analysis: a study of 5,380 cases. Am J Med Genet A. 2008;146A(17):2242-2251.
Shaw P., Lerch J., Greenstein D., et al. Longitudinal mapping of cortical thickness and clinical outcome in children and adolescents with attention-deficit hyperactivity disorder. Arch Gen Psychiatry. 2006;63:540-549.
Shaywitz S.E., Fletcher J.M., Holahan J.M., et al. Persistence of dyslexia: the Connecticut longitudinal study at adolescence. Pediatrics. 1999;104:1351-1359.
Shaywitz S., Shaywitz B., Fulbright R., et al. Neural systems for compensation and persistence: young adult outcome of childhood reading disability. Biol Psychiatry. 2003;54:25-33.
Shevell M., Ashwal S., Donley D., et al. Practice parameter: evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2003;60:367-380.
Shikako-Thomas K., Lach L., Majnemer A., et al. Quality of life from the perspective of adolescents with cerebral palsy: “I just think I’m a normal kid, I just happen to have a disability”. Qual Life Res. 2009;18:825-832.
Sie L.T., Hart A.A., van Hof J., et al. Predictive value of neonatal MRI with respect to late MRI findings and clinical outcome, A study in infants with periventricular densities on neonatal ultrasound. Neuropediatrics. 2005;36:78-89.
Silani G., Frith U., Demonet J.F., et al. Brain abnormalities underlying altered activation in dyslexia: a voxel based morphometry study. Brain. 2005;128:2453-2461.
Simos M., Panagiotis G., Fletcher J., et al. Early development of neurophysiological processes involved in normal reading and reading disability: a magnetic source imaging study. Neuropsychology. 2005;19:787-798.
Smits-Engelsman B.C., Wilson P.H., Westenberg Y., et al. Fine motor deficiencies in children with developmental coordination disorder and learning disabilities: an underlying open-loop control deficit. Hum Move Sci. 2003;22:495-513.
Solanto M. Dopamine dysfunction in AD/HD: integrating clinical and basic neuroscience research. Behav Brain Res. 2002;130:65-71.
Sonuga-Barke E. Disambiguating inhibitory dysfunction in attention-deficit/ hyperactivity disorder: toward the decomposition of developmental brain phenotypes. Biol Psychiatry. 2010;67:599-601.
Steinbok P. Selection of treatment modalities in children with spastic cerebral palsy. Neurosurg Focus. 2006;21:e4.
Stephen G., Kalhler C., Fahey M. Metabolic disorders and mental retardation. Am J Med Genet C Semin Med Genet. 2003;117C:31-41.
Stevenson R., Schwartz C. X-linked intellectual disability: unique vulnerability of the male genome. Dev Disabil Res Rev. 2009;15:361-368.
Strauss D., Shavelle R., Reynolds R., et al. Survival in cerebral palsy in the last 20 years: signs of improvement? Dev Med Child Neurol. 2007;49(2):86-92.
Stromme P., Diseth T.H. Prevalence of psychiatric diagnoses in children with mental retardation: data from a population-based study. Dev Med Child Neurol. 2000;42:266-270.
Stromme P., Magnus P. Correlations between socioeconomic status, IQ and aetiology in mental retardation: a population-based study of Norwegian children. Soc Psychiatry Psychiatr Epidemiol. 2000;35:12-18.
Sundaram S.K., Chugani H., Chugani T., et al. Positron emission tomography methods with potential for increased understanding of mental retardation and developmental disabilities. Ment Retard Dev Disabil Res Rev. 2005;11:325-330.
Sundaram S.K., Kumar A., Makki M.I., et al. Diffusion tensor imaging of frontal lobe in autism spectrum disorder. Cereb Cortex. 2008;18:2659-2665.
Swanson J.M., Greenhill L.L., Lopez F.A., et al. Modafinil film-coated tablets in children and adolescents with attention-deficit/hyperactivity disorder: results of a randomized, double-blind, placebo-controlled, fixed-dose study followed by abrupt discontinuation. J Clin Psychiatry. 2006;67:137-147.
Szpir M. Tracing the origins of autism: a spectrum of new studies. Environ Health Perspect. 2006;114(7):A412-A418.
Tager-Flusberg H., Rogers S., Cooper J., et al. Defining spoken language benchmarks and selecting measures of expressive language development for young children with autism spectrum disorders. J Speech Lang Hearing Res. 2009;52(3):643-652.
Takahashi R., Yamada M., Takahashi T., et al. Risk factors for cerebral palsy in preterm infants. Early Hum Dev. 2005;81:545-553.
Tang-Wai R., Webster R., Shevell M. A clinical and etiologic profile of spastic diplegia. Pediatr Neurol. 2006;34:212-218.
Thurm A., Lord C., Lee L., et al. Predictors of language acquisition in preschool children with autism spectrum disorders. J Autism Dev Disord. 2007;37(9):1721-1734.
Tran U., Gray P.H., O’Callaghan M.J. Neonatal antecedents for cerebral palsy in extremely preterm babies and interaction with maternal factors. Early Hum Dev. 2005;81:555-561.
Tsankova N., Renthal W., Kumar A., et al. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:357-363.
Tuchman R. CSWS-related autistic regression versus autistic regression without CSWS. Epilepsia. 2009;50(Suppl 7):18-20.
Undheim A.M. A thirteen-year follow-up study of young Norwegian adults with dyslexia in childhood: reading development and educational levels. Dyslexia. 2009;15(4):291-303.
Vaillend C., Poirier R., Larroche S. Genes, plasticity and mental retardation. Behav Brain Res. 2008;192:88-105.
Van Esch H., Bauters M., Ignatius J., et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442-453.
Van Karnebeek C.D., Jansweijer M.C., Leenders A.G., et al. Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness. Eur J Hum Genet. 2005;13:6-25.
van Wassenaer A.G., Westera J., Houtzager B.A., et al. Ten-year follow-up of children born at <30 weeks’ gestational age supplemented with thyroxine in the neonatal period in a randomized, controlled trial. Pediatrics. 2005;116:e613-e618.
Vargas D.L., Nascimbene C., Krishnan C., et al. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67-81.
Verhoeven J., De Cock P., Lagae L., et al. Neuroimaging of autism. Neuroradiology. 2010;52(1):3-14.
Vohr B. Outcome of prematurity. In: Nass R., Frank Y. Cognitive and Behavioral Abnormalities of Pediatric Disorders. New York: Oxford University Press, 2010.
Volpe J. Neonatal Neurology. Philadelphia: WB Saunders; 2009.
Vukovic R.K., Siegel L.S. The double-deficit hypothesis: a comprehensive analysis of the evidence. J Learn Disabil. 2006;39:25-47.
Waterhouse L., Morris R., Allen D., et al. Diagnosis and classification in autism. J Autism Dev Disord. 1996;26:59-86.
Willey L. Pretending to Be Normal. London: Jessica Kingsley Publishers; 1999.
Williams D.L., Minshew N.J. Understanding autism and related disorders: what has imaging taught us? Neuroimaging Clin N Am. 2007;17(4):495-509.
Williams E.L., Casanova M.F. Autism and dyslexia: a spectrum of cognitive styles as defined by minicolumnar morphometry. Medical Hypotheses. 2010;74(1):59-62.
Wilson P.H. Practitioner review: approaches to assessment and treatment of children with DCD: an evaluative review. J Child Psychol Psychiatry. 2005;46:806-823.
Wilson P.H., Maruff P., Lum J. Procedural learning in children with developmental coordination disorder. Hum Move Sci. 2003;22:515-526.
Wilson P.H., Maruff P., Butson M., et al. Internal representation of movement in children with developmental coordination disorder: a mental rotation task. Dev Med Child Neurol. 2004;46:754-759.
Wilson P. Visuospatial, kinesthetic, visuomotor integration visuoconstructive disorders. In: Dewey D., Tupper D. Developmental Motor Disorders. NY: Guilford Press; 2004:291-312.
Wilson P., Thomas P., Maruff P. Motor imagery training ameliorates motor clumsiness in children. J Child Neurol. 2002;27:491-499.
Wilson-Costello D., Friedman H., Minich N., et al. Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics. 2005;115:997-1003.
Wilson-Costello D., Friedman H., Minich N., et al. Improved neurodevelopmental outcomes for extremely low birth weight infants in 2000-2002. Pediatrics. 2007;119:37-45.
Wiseheart R., Altmann L.J.P., Park H., et al. Sentence comprehension in young adults with developmental dyslexia. Ann Dyslexia. 2009;59(2):151-167.
Wolf R.C., Sambataro F., Lohr C., et al. Functional brain network abnormalities during verbal working memory performance in adolescents and young adults with dyslexia. Neuropsychologia. 2010;48(1):309-318.
Wolosin S.1., Marin E., Richardson J.G., et al. Abnormal cerebral cortex structure in children with ADHD. Hum Brain Mapping. 2009;30:175-184.
Woodward L.J., Anderson P.J., Austin N.C., et al. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N Engl J Med. 2006;355(7):685-694.
Wu Y.W., Escobar G.J., Grether J.K., et al. Chorioamnionitis and cerebral palsy in term and near-term infants. JAMA. 2006;290:2677-2684.
Wu Y.W., Shah S.J., Newman T.B., et al. Cerebral palsy in a term population: risk factors and neuroimaging findings. Ann Neurol. 2006;60:S115.
Zwaigenbaum L., Bryson S., Lord C., et al. Clinical assessment and management of toddlers with suspected autism spectrum disorder: insights from studies of high-risk infants. Pediatrics. 2009;123(5):1383-1391.
Zwaigenbaum L., Bryson S., Rogers T., et al. Behavioral manifestations of autism in the first year of life. Int J Dev Neurosci. 2005;23:143-152.
Zwicker J.G., Missiuna C., Boyd L.A. Neural correlates of developmental coordination disorder: a review of hypotheses. J Child Neurol. 2009;24(10):1273-1281.
* Stop tasks present equally probable stimuli (e.g., an X and an O) with the instruction to press a corresponding key as quickly as possible depending on which letter appears, creating a tendency to respond on most trials. On a minority of trials (25% typically), a signal (e.g., a tone) indicates that the child is not to respond. Timing of the tone is varied to estimate the speed of the inhibition process (how much warning does the child need to interrupt the response, which is independent of the speed of the response output process).