4 Developmental and Pediatric Lung Disease
The diagnostic approach to pediatric lung biopsy differs somewhat from that in the adult patient. Many of the usual questions that arise in adult pulmonary pathology are replaced by separate issues involving abnormal development, altered lung growth due to prematurity, genetic disease, and infections secondary to an immature immune system.1 The spectrum of diseases observed in the pediatric lung biopsy differs from its adult counterpart and it is important to approach these biopsies with knowledge of lung development and anatomy. In addition, communication with the clinician, the radiologist, and the surgeon is essential, because a tentative list of diagnostic considerations usually can be built on the basis of clinical, radiologic, and intraoperative findings. This chapter covers a spectrum of common and rare entities that the surgical pathologist may encounter when examining biopsied and resected specimens from pediatric patients.
Processing of Pediatric Lung Biopsy Specimens
General processing of lung biopsy specimens is covered in Chapter 2. Recommendations have been published for processing of pediatric biopsy specimens for diffuse lung disease.2 A point worthy of emphasis is that in both diffuse and localized disease of the pediatric lung, infection should always be considered, and a substantial portion (one third to one half) of the surgical lung biopsy specimen should be sent for cultures if the surgeon has not already sent culture samples directly from the operating room. Touch imprints of the biopsy cut surface can be made and rapidly stained with silver stains for fungi or acid-fast stains for mycobacteria, a technique that is particularly useful for processing of lung biopsy specimens from immunosuppressed or immunocompromised patients. A few small pieces should be retained in glutaraldehyde for electron microscopy, which may have utility in the diagnosis of genetic disorders of surfactant metabolism and viral infections. Additional tissue should be snap-frozen and retained for potential molecular diagnosis of genetic disorders or infectious processes. For patients with suspected autoimmune diseases or chronic hemorrhage syndromes, a piece of lung tissue should be frozen in cryomatrix for possible immunofluorescence study. The remaining lung tissue should be expanded with formalin using a tuberculin syringe or other fine needle by transpleural injection, fixed for approximately 10 minutes, and sectioned for histologic examination. Inflation of pediatric lung biopsy is essential to reproduce the in vivo lung architecture and enables microscopic assessment of alveolar growth and development. In addition to standard hematoxylin and eosin (H&E) stains, many cases of diffuse disease benefit from the addition of connective tissue stains, such as trichrome, Verhoeff-van Gieson, or Movat pentachrome stains, for further evaluation of vascular disease or small-airway scarring, both of which may be under-recognized with use of routine stains. Additional staining for microorganisms, iron, glycogen, and alveolar proteinosis should be performed when indicated.
Cysts and Masses
Many pediatric lung biopsies and resections are performed for localized abnormalities within the lungs, and these may be solid or cystic (Box 4-1). A number of clues can be obtained from the history and findings on diagnostic imaging and intraoperative inspection, and these can be helpful in making the correct diagnosis, even before slides have been reviewed. The location of the mass, the presence of cystic or solid areas, the vascular and bronchial supply, and the onset of symptoms can all be useful in narrowing the scope of the differential diagnosis.3
Bronchogenic Cysts
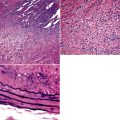
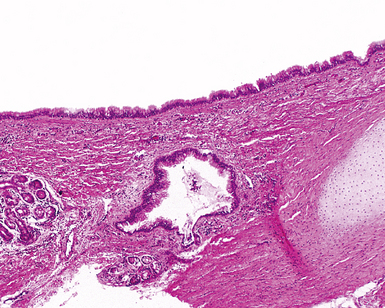
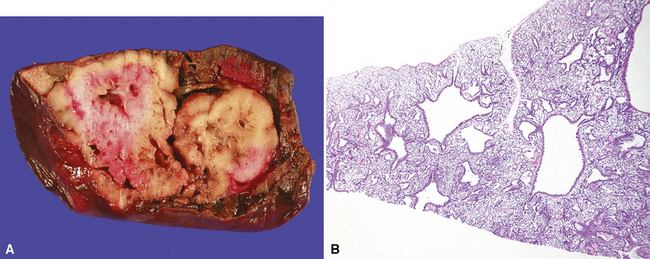
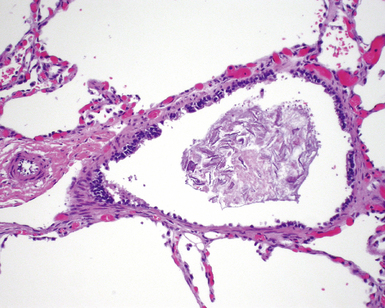
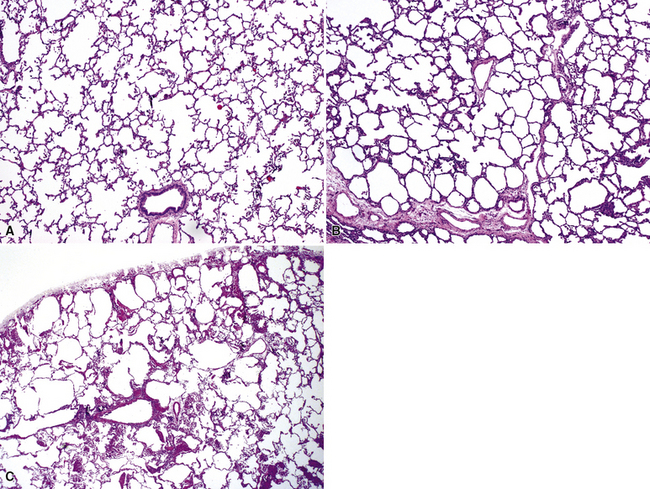
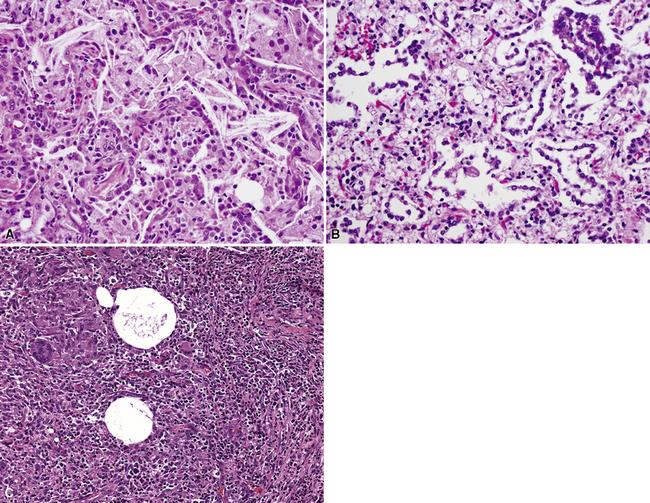
Bronchogenic cysts (or bronchial cysts) are developmental anomalies formed by abnormal budding of the tracheobronchial anlage of the primitive foregut in early development.4,5 They commonly are found in the anterior mediastinum or along the tracheobronchial tree. Less often, they are found within the pulmonary parenchyma, within or below the diaphragm, or even within the pericardium.6,7 Patients with bronchogenic cysts can present with infection or obstruction, although frequently these lesions are an incidental radiologic finding.5 The cyst often is unilocular and lined by ciliated columnar epithelium. Many bronchogenic cysts communicate with the tracheobronchial tree. On occasion, they show squamous metaplasia or mild chronic inflammation.
Considerations in the differential diagnosis for bronchogenic cyst include esophageal duplication cyst for lesions occurring in the mediastinum and congenital pulmonary airway malformations (CPAMs) for lesions occurring within the substance of the lung. The bronchogenic cyst typically has cartilage plates and submucosal glands in the wall, similar to the normal microscopic anatomy of bronchi (Fig. 4-1). These structures may be sparse but can help differentiate this lesion from the esophageal duplication cyst, which lacks these structures and has a double muscular layer in its wall. Both of these entities can be lined by ciliated mucosa. The bronchogenic cyst lacks connection with alveolar tissue, a feature that aids in its distinction from CPAM. On occasion, with inflamed cysts within the lung, the nature of the specific lesion or underlying disorder may be impossible to ascertain. In such situations, a generic diagnosis of “inflamed intraparenchymal cyst,” accompanied by a differential diagnosis summary that includes abscess, CPAM, bronchogenic cyst, esophageal cyst, and intralobar sequestration, is appropriate. Radiologic and clinical features may help in further differentiating among these entities.
Bronchial Atresia
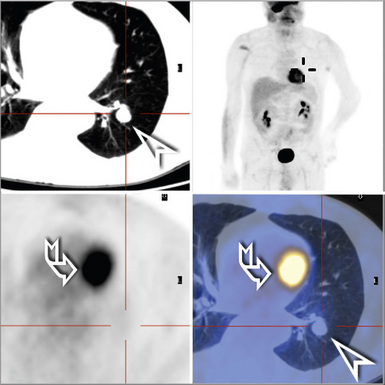
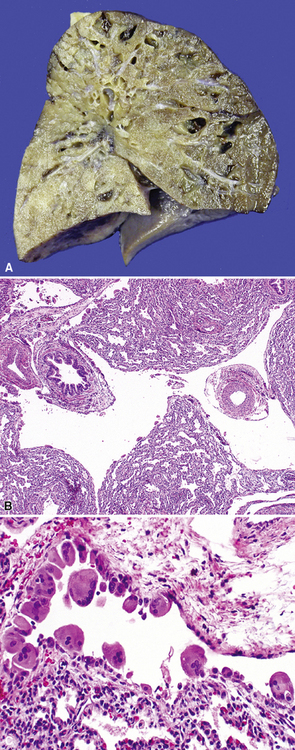
Bronchial atresia is one of the most common forms of congenital pulmonary malformation. Atresia of a segmental or subsegmental bronchus classically results in a central mucus-filled cyst (mucocele) at the point of atresia, dilated distal airways with mucous plugs, and hyperinflated microcystic distal parenchyma (Fig. 4-2). Microscopically, the abnormally developed cystic parenchyma has a pattern identical to that described in type 2 CPAM, consisting of increased numbers of abnormal bronchiolar structures surrounded by variably abundant alveolar spaces which are typically distended and either round or elongated in shape.8,9 Abundant mucus and muciphages typically are noted within proximal airway lumens and adjacent air spaces. Bronchial atresia often is detected prenatally or in infancy as an asymptomatic cystic lesion. Because of interconnection with the normal parenchyma through alveolar pores of Kohn, the abnormal lung distal to a bronchial atresia may potentially become secondarily infected, and presentation in later childhood, adolescence, or adulthood typically is due to symptoms of recurrent pneumonia. The primary considerations in the differential diagnosis are congenital lobar overinflation, which is characterized by normally developed (albeit markedly distended) air spaces, and intralobar sequestration, which is distinguished by the additional finding of aberrant systemic arterial supply.3
Pulmonary Sequestration
Pulmonary sequestration refers to the occurrence of lung tissue that does not communicate with the tracheobronchial tree and that typically has a systemic, rather than pulmonary, arterial supply.10–13 Such lung tissue is therefore “sequestered” from the usual pulmonary airway and vascular connections. These lesions are further subdivided into an extralobar type, which occurs outside the visceral pleura of the adjacent lung, and an intralobar type, which resides within the visceral pleural investment of a lung lobe. Although there is general agreement that extralobar sequestrations represent congenital malformations, the origin of intralobar sequestrations has been more controversial. More frequent diagnosis in adults has led to the theory that they are post-inflammatory lesions with acquired loss of bronchial connection and development of collateral systemic circulation from hypertrophied pulmonary ligament arteries.11 However, intralobar sequestrations diagnosed prenatally or in association with congenital malformations support the concept that the pathogenic mechanisms for extralobar and intralobar sequestrations are similar, and that adult lesions may represent occult malformation detected later in life. Specific features of extralobar and intralobar sequestrations, summarized in Table 4-1, are discussed next.11
Table 4-1 Pulmonary Sequestration: Extralobar versus Intralobar
| Clinical/Historical Feature | Extralobar Sequestration | Intralobar Sequestration |
|---|---|---|
| Location | Outside pleura of lung (“accessory lobe”) Commonly, left lung base |
Within pleura of lung lobe Lower lobe (98%) |
| Gross | Pyramidal structure | Congested, hemorrhagic segment(s) of lobe |
| Age at diagnosis | 60% < 6 months | 50% > 20 years |
| Arterial supply | Systemic | Systemic |
| Origin | Congenital anomaly | Congenital anomaly; possibly acquired in adults |
| Histologic appearance | CPAM type 2 pattern Hyperinflated air spaces |
CPAM type 2 pattern with mucus stasis Hyperinflated, hemorrhagic segment(s) of lobe Inflamed, chronic pneumonia |
CPAM, congenital pulmonary airway malformation.
Modified from Stocker JT. Sequestrations of the lung. Semin Diagn Pathol. 1986;3(2):106–121; and Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12(1):17–37.
Extralobar Sequestration
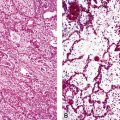
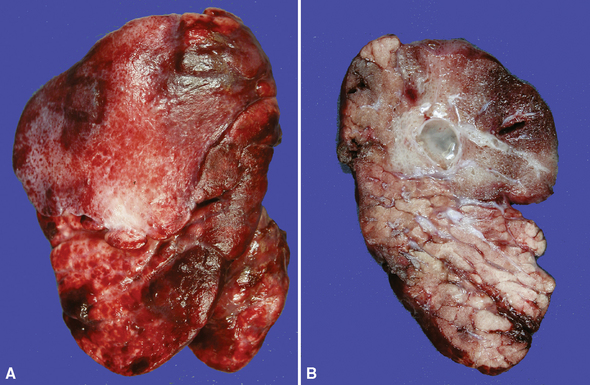
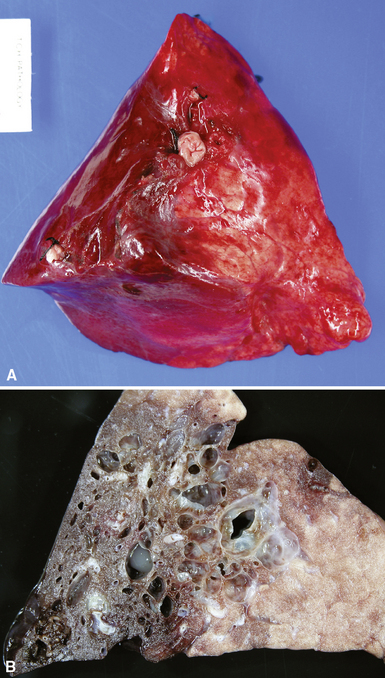
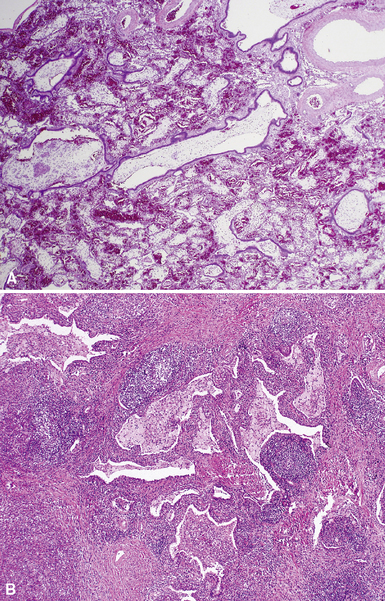
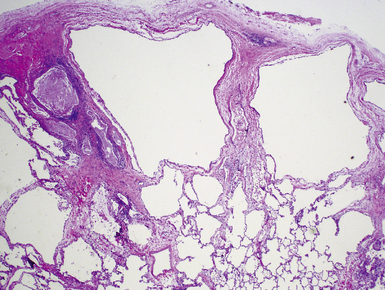
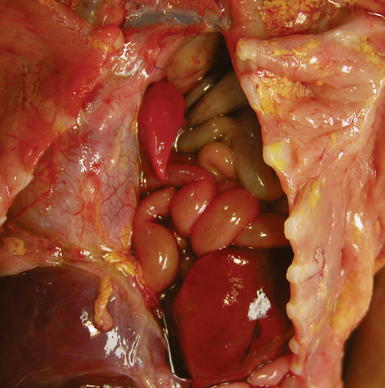
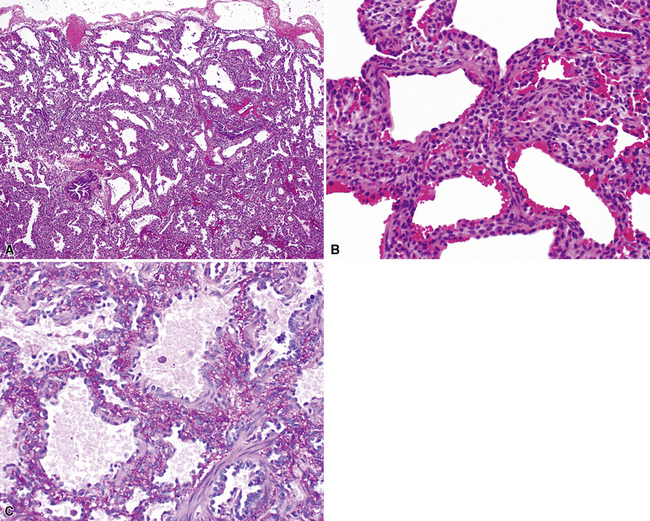
Extralobar sequestrations (ELSs) are thought to arise a a result of abnormal budding from the tracheobronchial anlage. Lying outside the normal lung, these structures appear as “accessory lobes,” completely surrounded by their own visceral pleura.10–15 ELSs usually are found in the lower thoracic cavity but may be found above, within, or below the diaphragm. On gross inspection, they appear as irregularly ovoid or pyramidal portions of lung tissue surrounded by pleura and with a vascular pole at one edge (see Fig. 4-3A). The radiographic or intraoperative finding of a systemic arterial supply confirms the diagnosis of ELS. The systemic artery can arise from a source above or below the diaphragm.16,17 The microscopic appearance may vary but the histologic pattern typically is that of type 2 CPAM and less often resembles that of normal lung11,14,15,18–21 (see Fig. 4-3B and C). Striated muscle occasionally is present within the interstitium of the lesion; this feature is called rhabdomyomatous dysplasia.
Intralobar Sequestration
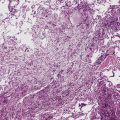
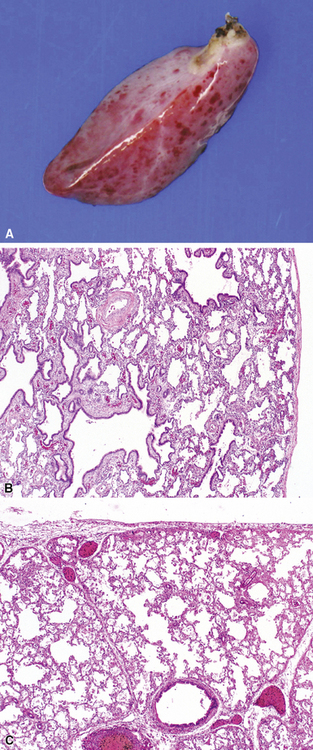
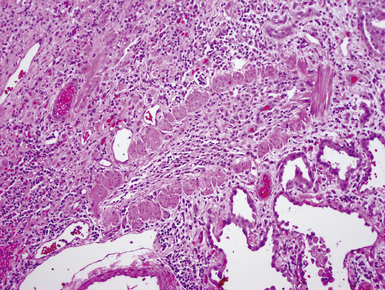
Intralobar sequestrations (ILSs) lie within the parenchyma of the lung. Most ILSs occur in the medial area of a lower lobe, and the abnormal systemic elastic artery can usually be identified in the region of the inferior pulmonary ligament22 (Fig. 4-4A). Radiologic studies can be extremely helpful in supporting the diagnosis.23,24 Various modalities, including computed tomography (CT) and magnetic resonance imaging (MRI), will show a solid or cystic mass that lacks normal bronchovascular patterns. A systemic arterial supply may be confirmed radiologically or at the time of surgery. The gross and microscopic findings are markedly influenced by age at resection and presence or absence of any accrued chronic inflammatory insults (usually secondary infections) within the sequestered lung tissue. In infants with asymptomatic lesions, the lobe contains a segmental region of congestion and hemorrhage due to the high flow of the systemic circulation, accompanied by microcystic parenchyma (see Fig. 4-4B). The maldeveloped parenchyma shows mucus stasis, identical to that in segmental bronchial atresia (Fig. 4-5A). In older children and adults with recurrent pneumonia, the histologic findings are similar in appearance to those in localized bronchiectasis with recurrent infection. Other features include marked acute and chronic inflammation with fibrosis and cyst formation (see Fig. 4-5B).
Congenital Pulmonary Airway Malformations
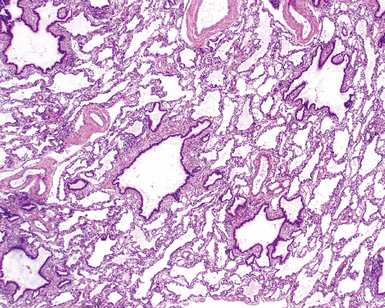
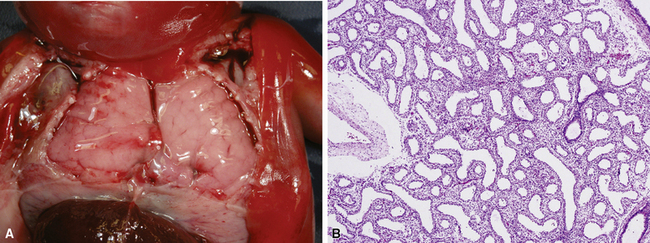
Congenital malformations of the pulmonary airways (i.e., CPAMs), also called congenital cystic adenomatoid malformations (CCAMs), are masses of maldeveloped lung tissue that are classified according to their gross and microscopic appearance.12,25–34 These lesions are identified most commonly in stillborn infants or in newborns with respiratory distress, but they can be discovered in adolescents and rarely in adults.35 Stocker and colleagues initially proposed a classification scheme for CCAM that divided these malformations into three subtypes.36 This scheme was later expanded to five subtypes (types 0 to 4), with a subsequent change in terminology from CCAM to CPAM, acknowledging that not all of these malformations are cystic and not all are adenomatoid.37 The overriding principle of this subclassification is that the dominant morphologic constituent of each type of CPAM reflects the morphology of the normal tracheobronchial tree from proximal to distal—that is, from malformed bronchi, to bronchioles, to distal lung (alveolar) tissue. This construct has provided useful morphologic distinctions and will continue to be revised as the pathogenesis of these lesions is better understood. CPAM classification may be difficult in immature or fetal lungs, and an alternate classification has been proposed for fetal lung resections.33,34
Type 0 CPAM, also called acinar dysplasia, is a rare lesion composed of cartilaginous airways and loose mesenchyme (see later discussion).38–41 Types 1 to 3 have a common general overall appearance of cystic spaces (distorted airways) with intervening structures more or less resembling alveoli (Figs. 4-6 to 4-8). Type 1 CPAM shows larger cysts, having some bronchial differentiation in that they contain ciliated epithelial lining or mucinous-type epithelium or have cartilage in their walls. Type 2 CPAM shows smaller cystic spaces that resemble ectatic irregular bronchioles, evenly separated from each other by alveolar structures, and represents a histologic pattern that implies intrauterine bronchial obstruction, typically from bronchial atresia or pulmonary sequestration. Type 3 CPAM is a rare solid lesion typically encompassing an entire lobe or lung and resembles pulmonary hyperplasia or immature lung in the early canalicular stage of development. Type 4 CPAM has been described as a peripheral large cyst with thin walls and flattened alveolar-type epithelial lining. Existence of this type is controversial, because it may represent unrecognized, undersampled, or completely differentiated examples of cystic pleuropulmonary blastoma (PPB).42–48 Large cysts resembling type 1 or type 4 CCAM should be extensively sampled to exclude the presence of small foci of either malignant primitive spindled cells beneath the alveolar epithelium (“cambium layer”) or immature chondroid foci, diagnostic of cystic (type I) PPB. Immunohistochemical staining for myogenin, desmin, and MyoD1 may be helpful in differentiating the cells of PPB from reactive fibroblastic proliferation or cellular mesenchyme in CPAM. Cystic PPBs have potential for recurrence as high-grade tumors, particularly if incompletely resected.
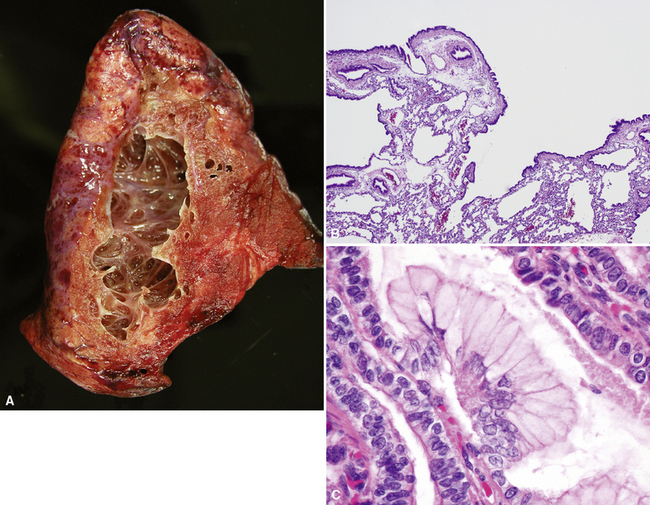
Although the prognosis in most cases is favorable following resection, there are rare reported cases of patients developing carcinomas in association with CPAM.49–55 Many of these lesions are mucinous bronchioloalveolar carcinomas, which has led to the proposal that the mucigenic epithelium in type 1 CPAM is preneoplastic (see Fig. 4-6C). Multifocal bilateral mucinous bronchioloalveolar carcinomas after incomplete resection of CPAM have been described in patients as young as 11 years of age.51 In light of these rare cases, the presence of mucinous epithelium in CPAM and completeness of resection should be documented for follow-up purposes.
Pulmonary Interstitial Emphysema
Pulmonary interstitial emphysema (PIE) results from dissection of air into the interstitial connective tissue of the lung. Rupture of alveoli or disruption of airway walls often is responsible for this phenomenon.12,56–59 Air accumulates in the interstitium along bronchovascular bundles and interlobular septa, creating cystic spaces that may at first resemble tissue architecturally torn during sectioning (Fig. 4-9A and B). The usual clinical scenario is that of a premature infant with neonatal respiratory distress syndrome receiving mechanical ventilation. Acute PIE usually resorbs over time, but the chronic form persists as cystic lesions lined by fibrous tissue or multinucleate giant cells (see Fig. 4-9C). Grossly, the process can involve both lungs diffusely or may be localized to one or two lobes. Multiple small cysts can be seen to extend along interlobular septa.
Peripheral Cysts Secondary to Lung Maldevelopment
Hypoplastic lungs, and those damaged in the neonatal period, are susceptible to persistent alterations in alveolar growth. This maldevelopment frequently appears as alveolar enlargement or cysts, particularly in the subpleural areas and peripheral lobules. Microscopic examination reveals irregular air space enlargement with fibrovascular walls lined by alveolar cells (Fig. 4-10). Peripheral cysts have been described in the lungs of several patients with Down syndrome.60–65
Pulmonary Hyperlucency
Several conditions may lead to the radiologic appearance of hyperlucency (Box 4-2). Clinical history and knowledge of the indication for resection are helpful, because the pathologic findings can be extremely subtle histologically. The clinical presentation may include shortness of breath, tachypnea, wheezing, or cough, typically in infants. The chest radiograph shows marked lobar enlargement with displacement of the mediastinum. The two most commonly occurring histologic patterns are congenital lobar overinflation (so-called congenital lobar emphysema) (in 70% of the cases) and polyalveolar lobe (30%).
Congenital Lobar Overinflation
Congenital lobar overinflation (CLO), or congenital lobar emphysema, occurs when there is overdistention of the normal alveolar parenchyma12,66,67 (Fig. 4-11). The etiology is variable, but the underlying cause frequently is a partial or intermittent high-grade obstruction of the bronchus supplying the affected lobe.68 Bronchomalacia may result in collapse of the lobar bronchus with expiration, resulting in progressive air-trapping in the affected lobe. The obstruction can occur as a result of other intrinsic factors such as bronchial stenosis, abnormal “kinked” bronchial anatomy, mucosal webs, or mucous plugging. Alternatively, congenital lobar emphysema can be extrinsic as a result of various vascular or neoplastic etiologies. Approximately one half of the cases are idiopathic. The upper lobe is involved in nearly all cases. Lower lobe involvement is highly unusual except in acquired cases in patients with previous hyaline membrane disease or bronchopulmonary dysplasia (BPD). Some cases may arise secondary to trauma from tracheal suctioning during respiratory support.69
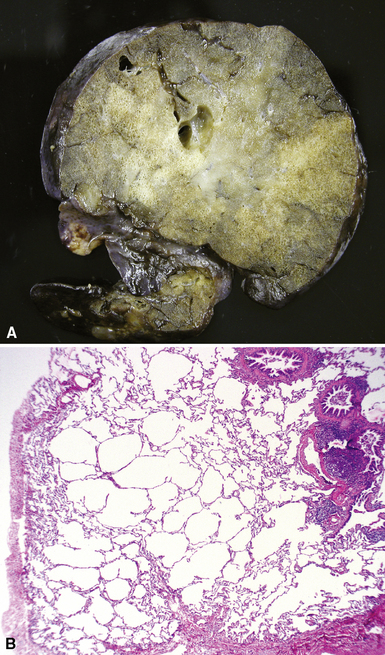
On gross examination, CLO is characterized by a markedly enlarged lobe, which generally retains its basic shape. Alveoli, alveolar ducts, and respiratory bronchioles typically are dilated on histologic examination. Unlike bronchial atresia and CPAM, CLO shows otherwise normal alveolar development, with appropriate numbers of bronchiolar structures and appropriate alveolar septation. The source of obstruction is identified only occasionally by gross and microscopic examination70–72 (Fig. 4-12).
Polyalveolar Lobe
Polyalveolar lobe occurs when there is an increase in the regional number of alveoli relative to the corresponding conducting airways and arteries.73–75 Whereas the arteries and airways in these lungs are normal, the alveolar regions are enlarged by an increased number of nearly normal alveoli. The diagnosis can be made by radial alveolar counts, which are performed by counting the number of alveoli transected by a line drawn from the respiratory bronchiole to the nearest acinar edge (pleura or septum).76 The normal count varies with age but should be between 5 and 10 for infants, and 10 and 12 for young children. Radial alveolar counts in polyalveolar lobe will be approximately two to three times that number (Fig. 4-13).
Disorders of Lung Development
Acinar Dysplasia
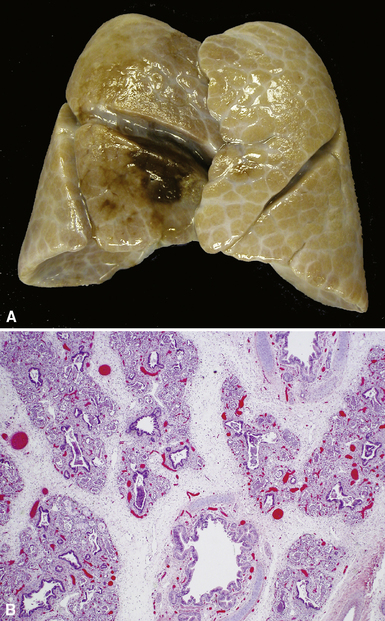
Described in 1986, acinar dysplasia is a rare severe diffuse developmental lung disorder resulting in marked deficiency of acinar development, identical to the entity described as type 0 CPAM.38–41 The lungs are small, with accentuation of small lobules by white interlobular septa (Fig. 4-14A). Microscopically, the lobular bronchi are surrounded by only few primitive air spaces, with virtually no alveolar development (see Fig. 4-14B). This disorder is uniformly fatal within the first few hours of life and typically is diagnosed at autopsy. Although acinar dysplasia is presumed to be of genetic origin, the etiology is unknown.
Congenital Alveolar Dysplasia
Congenital alveolar dysplasia also is a rare diffuse developmental lung disorder with incomplete air space development.77 Relative to those in acinar dysplasia, the air spaces are more numerous and exhibit greater complexity, but completely mature alveoli are lacking. The air spaces show primary septation but insufficient secondary septation, generally resembling the saccular stage of development (Fig. 4-15). This disorder can be difficult to distinguish from prematurity of lungs with superimposed injury and remodeling due to prolonged ventilation, and on a practical level, the diagnosis is reserved for term infants, in whom the disorder is more likely to be a developmental abnormality, rather than an acquired impairment of alveolar growth. The etiology is unknown.
Pulmonary Hypoplasia
Pulmonary hypoplasia refers to abnormally small size of the lungs due to limitation of intrauterine development. Although primary forms of hypoplasia exist, this term most commonly refers to the secondary forms of lung hypoplasia in which physical compression of the lungs, by extrathoracic or intrathoracic processes, limits intrauterine growth. Common causes include congenital diaphragmatic hernia (Fig. 4-16), intrauterine chylothorax or pleural effusion, osteochondrodysplasia with small thorax, neuromuscular disorders with poor respiratory effort, and intrathoracic or intra-abdominal lesions with mass effect on thoracic contents.78 Grossly, lung hypoplasia can be documented by low lung-to-body weight ratio or by low lung volumes.79,80 Microscopically, the lobules are small, with a reduced radial alveolar count. Hypoplastic lungs are prone to development of hyaline membrane disease, even at term gestation. Beyond the postnatal period, the simplification of lobules manifests on biopsy as alveolar enlargement and simplification, identical in appearance to chronic neonatal lung disease due to prematurity (see the discussion in section “Alveolar Growth Abnormalities”).
Pulmonary Hyperplasia
Pulmonary hyperplasia refers to enlargement of the lungs due to increased mass and volume, typically due to high-grade obstruction of the larynx (laryngeal atresia) or trachea (tracheal compression).81,82 Morphologically, the air spaces are abnormally elongated and increased in number (Fig. 4-17).
Vascular Disorders
Alveolar Capillary Dysplasia with Misalignment of Pulmonary Veins
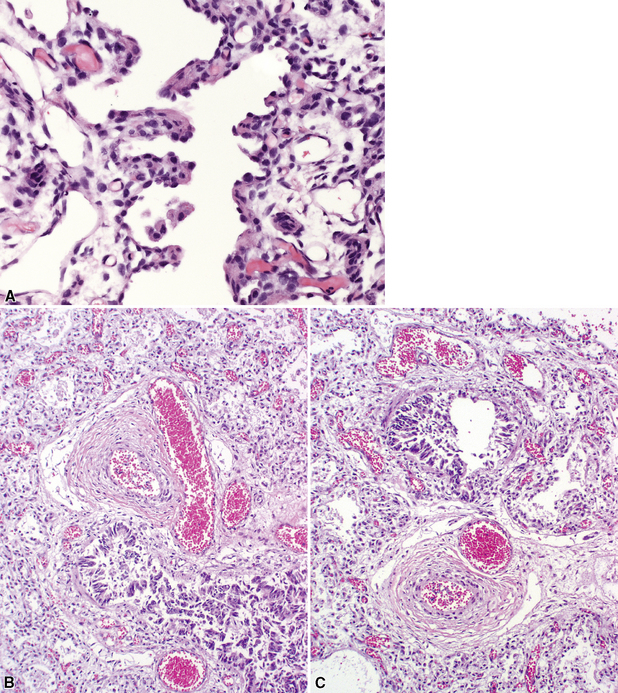
Alveolar capillary dysplasia is a disease characterized by a distinctive pattern of diffuse pulmonary vascular maldevelopment (Fig. 4-18). The disease typically manifests clinically soon after birth with profound respiratory distress, often after a brief asymptomatic period.83–85 The clinical picture is one of severe persistent pulmonary hypertension, and survival beyond the neonatal period is rare.86–88 Some cases are associated with other visceral malformations, and familial cases have been reported as well, supporting a genetic etiology.89 Histopathologic examination reveals abnormal lobular architecture with a diminished number of capillaries within alveolar walls. These alveolar capillaries are abnormally located within the central portion of the septa, rather than adjacent to the alveolar epithelial cells. The pulmonary arteries show marked medial hypertrophy, and the smaller branches show increased muscle, including the arterioles within the alveolar walls. Additional features include misalignment of the pulmonary veins, with congested pulmonary veins abnormally located adjacent to pulmonary arteries within bronchovascular sheaths and congested venules accompanying the hypertrophied arterioles within the lobular parenchyma. Of note, some pulmonary veins may lie in their normal location within interlobular septa, and misalignment of small pulmonary veins and venules within the lobules is more reliably identified. Lymphangiectasia is a variable feature.
Congenital Pulmonary Lymphangiectasis
Congenital pulmonary lymphangiectasis is a disease of newborns that manifests with dyspnea and cyanosis and often is lethal.90–95 Panlobar diffuse ectasia of lymphatic channels along normal lymphatic routes (bronchovascular bundles, interlobular septa, and subpleural regions) is characteristic (Fig. 4-19). Localized lymphangiectasis occasionally is observed in adults and children and usually is found as an incidental radiographic abnormality.96,97 Of note, chronic heart failure or pulmonary venous obstruction can lead to secondary lymphangiectasis, which has a similar histologic appearance, and clinical correlation is important in distinguishing between primary and secondary forms of lymphangiectasia.98
Diffuse Pulmonary Lymphangiomatosis
Diffuse pulmonary lymphangiomatosis is a rare disease, occurring in children or young adults, in which the normal lymphatic regions show an increased number of complex lymphatic channels.93,95,99,100 The patients generally present with dyspnea and occasionally with hemoptysis. Microscopic examination reveals increased numbers of anastomosing lymphatic channels with interspersed fibroblasts, collagen, and small vessels distributed along the usual lymphatic routes of the lung. The lymphatics can be more easily observed with use of connective tissue stains and immunohistochemical stains for keratin (to demonstrate these structures in negative relief), or CD31 (which stains the lymphatic endothelium) (Fig. 4-20). Some cases have an associated hemorrhagic “kaposiform” spindle cell component.
Pulmonary Arteriovenous Malformations
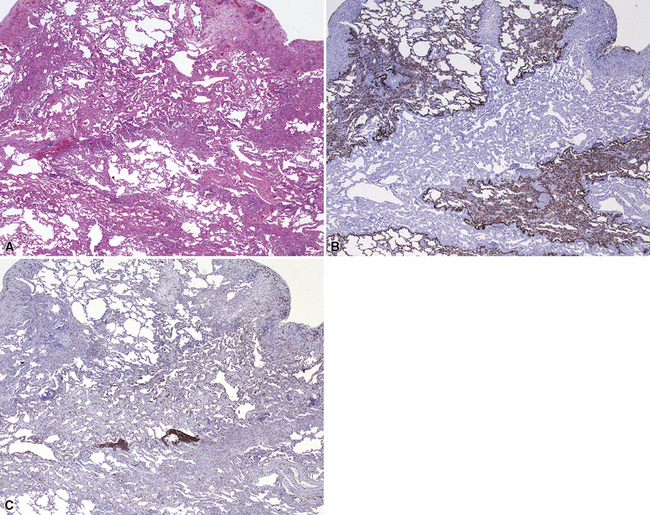
Pulmonary arteriovenous malformations (PAVMs) are defined as direct connections between branches of the pulmonary artery and the pulmonary vein.101 Common signs and symptoms are dyspnea, hemoptysis, palpitations, and chest pain. Initial clinical presentation of pulmonary arteriovenous malformations is fairly rare in young children and infants and tends to occur in older children and adults. The diagnosis can be made on clinical and radiologic grounds, followed by pathologic confirmation. Grossly, the malformations can be single or multiple and show ectatic vessels scattered amidst lung parenchyma (Fig. 4-21). Microscopic examination reveals dilated vessels and vascular tangles. The vessels are irregular and are not always in their usual position adjacent to bronchioles, in the case of pulmonary arteries, or in the interlobular septa, in the case of pulmonary veins. Otolaryngologic examination is suggested in patients with PAVM to rule out Osler-Weber-Rendu disease, because approximately one third of patients with single PAVM and one half of patients with multiple PAVMs will have this disease.101,102 Rare cases of multiple small PAVMs and polysplenia have been described in young children.103,104
Complications of Prematurity
Hyaline Membrane Disease
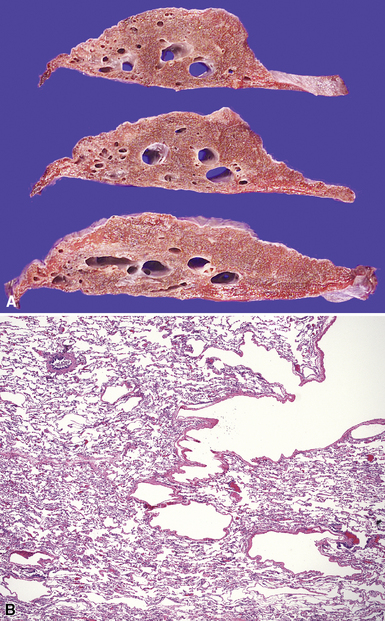
Hyaline membrane disease is a form of acute lung injury seen in neonates and is the pathologic correlate of neonatal respiratory distress syndrome (RDS). Hyaline membrane disease arises as a result of surfactant deficiency due to prematurity.105 Although surfactant granules can be observed in lung cells at a gestational age of 20 weeks, surfactant is not produced in sufficient amounts until 34 weeks. Lack of surfactant can result either from prematurity or, less commonly, from inadequate resorption of lung liquid at birth leading to a dilutional deficiency. Surfactant deficiency results in increased alveolar surface tension, with subsequent resistance to inflation and alveolar collapse at end-expiration. In this process, the alveoli become injured,106 presumably as a result of shear stresses on the alveolar walls. Increases in either respiratory effort or mechanical ventilation pressures can increase the severity of the injury. This injury in turn leads to diffuse alveolar damage, which is similar in appearance to that observed in adult cases of acute respiratory distress syndrome (ARDS).107
Grossly, the lungs are firm, red, and consolidated, without significant aeration. Microscopic examination reveals the presence of homogeneous lightly eosinophilic linear material closely adherent to the alveolar surface (Fig. 4-22A). These hyaline membranes may look relatively uniform, but they are actually composed of a myriad of materials, including cytoplasm and nucleoplasm of dead cells, plasma transudate, and amniotic fluid. Hyaline membranes form within 3 to 4 hours of birth and are well developed by 12 to 24 hours. An interesting finding in jaundiced infants with acute lung injury is the presence of yellow hyaline membranes secondary to bilirubin staining108 (see Fig. 4-22B). Complications of BPD have become relatively rare as a result of improvements in therapy, including surfactant replacement and advances in mechanical ventilation and oxygen therapy.109 Of note, if numerous neutrophils accompany hyaline membranes, the possibility of acute infection should be considered, because these are not usual components of hyaline membrane disease.
Bronchopulmonary Dysplasia
BPD is a chronic lung disease that occurs in a proportion of children who require respiratory support in the neonatal period.107,110–112 As the clinical treatment of prematurity has evolved, the pathologic appearance of this disease has changed.109 The designation was first used for the chronic lung disease that developed subsequently in patients with previous hyaline membrane disease.113 Examination of the lung in “classic” BPD showed a variegated pattern, with some lobules showing alveolar fibrosis and collapse, whereas adjacent lobules showed overdistention (Fig. 4-23A).114–116 This appearance of the chronic disease was explained by observations in the pre-existing hyaline membrane disease. Early cases of hyaline membrane disease showed areas of necrotizing bronchiolitis with poor aeration of distal alveolar parenchyma. These areas were subsequently spared from the continuous alveolar insult related to oxygen therapy and mechanical ventilation. As the bronchioles and hyaline membranes healed, airway and interstitial fibroblast proliferation occurred (see Fig. 4-23B). After fibrosis of the injured areas ensued, and healing of the bronchiole was complete, patchy fibrosis was evident in the affected regions, and nearly normal, overdistended alveoli were seen in the spared regions (see Fig. 4-23C and D).
In current practice, neonates at risk for hyaline membrane disease are treated with respiratory support and surfactant replacement therapy. This strategy obviates the need for intense oxygen therapy and the mechanical stress that occurred historically. Nevertheless, it is proposed that the lower levels of oxygen treatment in the current regimen result in a generalized uniform alveolar injury.109 The injured alveoli continue to show maturation by thinning of their septa; however, there is a lack of additional subdivision and branching of the alveolar units of the lobule, thereby leading to an alveolar simplification, so-called “new” BPD.109,112,117 This lack of normal maturation results in a decrease in the absolute numbers of alveoli. The alveolar walls may be of normal thickness or may be mildly fibrotic. As in polyalveolar lobe, radial alveolar counts can be used in BPD to assess the number of alveoli within lobules.76,109
Pediatric Interstitial Lung Disease
Attempts to classify pediatric interstitial lung disease with the same categories used in adults can result in difficulties in classification and even misclassification.1 Several of the patterns of disease are similar in adults and in children, but the underlying etiology and prognosis may be different in pediatric cases.118–123 So-called usual interstitial pneumonia (UIP) is virtually nonexistent in children.124 Despite these differences, recognition of patterns of interstitial disease remains as important in the diagnosis of diffuse lung disease in children as it is in adults125 (Box 4-3 and Table 4-2). The subtypes of interstitial lung disease are described separately in this section. Even if a precise diagnosis or underlying etiologic disorder cannot be established histologically, the pattern of injury should guide further diagnostic evaluation and in many cases will help to determine prognosis. Assessment of inflammation, and exclusion of infection, may be helpful in evaluating the potential role of steroid therapy also. Practically speaking, the most important diseases to recognize in the neonatal period are the abnormalities of alveolar development and growth, congenital disorders of surfactant metabolism, and alveolar capillary dysplasia. The most important diseases to consider in older children with chronic diffuse lung disease include obliterative bronchiolitis, collagen vascular disease, and hypersensitivity pneumonitis.
Box 4-3 Differential Diagnosis of Pediatric Diffuse Lung Disease
Data from Swensen SJ, Hartman TE, Mayo JR, et al. Diffuse pulmonary lymphangiomatosis: CT findings. J Comput Assist Tomogr. 1995;19(3):348–352; Coren ME, Nicholson AG, Goldstraw P, et al. Open lung biopsy for diffuse interstitial lung disease in children. Eur Respir J. 1999;14(4):817–821; Fan LL, Mullen AL, Brugman SM, et al. Clinical spectrum of chronic interstitial lung disease in children. J Pediatr. 1992;121(6):867–872; Deutsch GH, Young LR, Deterding RR, et al: Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1120–1128; and Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157(4 Pt 1):1301–1315.
| Histologic Finding | Consider |
|---|---|
| Diffuse type II pneumocyte hyperplasia | Acute lung injury (proliferative diffuse alveolar damage, viral pneumonitis) Genetic disorders of surfactant metabolism |
| Alveolar macrophages Siderophages |
Pulmonary capillaritis Collagen vascular disease Coagulopathy with recurrent hemorrhage Chronic congestive vasculopathy (PVS, VOD, left ventricular obstruction) Idiopathic pulmonary hemosiderosis |
| Foamy (vacuolated) macrophages | Aspiration pneumonia Metabolic storage disorders Genetic disorders of surfactant metabolism Airway obstruction, postobstructive pneumonia Endogenous lipoid pneumonia in peripheral lung cysts |
| With eosinophils and fibrin | Eosinophilic pneumonia |
| Lightly pigmented or nonpigmented | Desquamative interstitial pneumonia Drug reaction |
| Granular proteinaceous material | Genetic disorders of surfactant metabolism Pulmonary alveolar proteinosis Pneumocystis infection Infection (especially viral pneumonitis with epithelial necrosis) |
| Organizing pneumonia | Infection Collagen vascular disease Hypersensitivity pneumonitis Idiopathic |
| Alveolar septal thickening by lymphocytes | |
| Bronchiolocentric | Hypersensitivity pneumonitis Infection (e.g., viral bronchiolitis) Aspiration pneumonia Cystic fibrosis Collagen vascular disease Primary ciliary dyskinesia |
| Nonspecific interstitial pneumonia | Collagen vascular disease Hypersensitivity pneumonitis Genetic disorders of surfactant metabolism (older children) |
| Lymphocytic interstitial pneumonia | Primary immunodeficiency HIV infection Sjögren syndrome |
| Follicular bronchiolitis | Primary immunodeficiency (e.g., CVID) HIV infection Epstein-Barr virus infection Collagen vascular disease |
| Alveolar septal thickening By fibrosis |
Bronchopulmonary dysplasia |
| By increased cellularity | Pulmonary interstitial glycogenosis (infantile cellular interstitial pneumonia) |
| By edema or muscular arterioles | Vascular/cardiogenic disease |
| With few central capillaries | Alveolar capillary dysplasia with misalignment of pulmonary veins |
CVID, common variable immunodeficiency; PVS, pulmonary vein stenosis; VOD, veno-occlusive disease.
Alveolar Growth Abnormalities
Alveolar growth abnormalities are a group of disorders characterized morphologically by the presence of enlarged and simplified air spaces, indicating incomplete or altered alveolarization, either due to prenatal or postnatal factors. A common example of an alveolar growth abnormality, previously discussed, is chronic neonatal lung disease due to prematurity (see earlier section on BPD) (Fig. 4-24). However, sometimes a similar pattern is seen on lung biopsy from infants and young children, despite history of term gestation or lack of respiratory distress syndrome in the neonatal period. Considerations in the differential diagnosis in term infants include pulmonary hypoplasia; disordered lung growth due to underlying chromosomal syndromes (e.g., Down syndrome, other trisomies), malformation syndromes, or congenital heart disease; and disordered lung growth due to poor postnatal alveolarization in the setting of severe neonatal illness. In some patients, impaired alveolarization is considered to be multifactorial—for example, a premature infant with Down syndrome and atrioventricular canal. This morphologic category, described as alveolar growth abnormalities, accounts for the largest subset of diffuse lung disease in infants,123 and attention to alveolar architecture in a well-inflated lung biopsy specimen is essential for diagnosis. Generally speaking, this type of abnormality is likely to be underappreciated by pathologists who interpret predominantly adult lung biopsy specimens, because of the similarity of the enlarged infant air spaces to those in normal adult lung tissue or in emphysematous lung. Lack of interstitial fibrosis or inflammation in most cases also prevents recognition of impaired alveolar architecture as the primary abnormality. Other pathologic findings commonly associated with alveolar growth problems include pulmonary interstitial glycogenosis (discussed next) and secondary pulmonary arteriopathy. In infants with chronic lung disease due to prematurity, these findings may help to explain exacerbation of disease or clinical severity that is disproportionate to that expected for gestational age.
Pulmonary Interstitial Glycogenosis (Infantile Cellular Interstitial Pneumonitis)
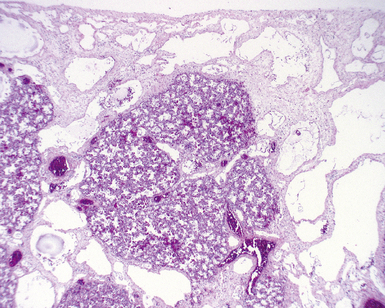
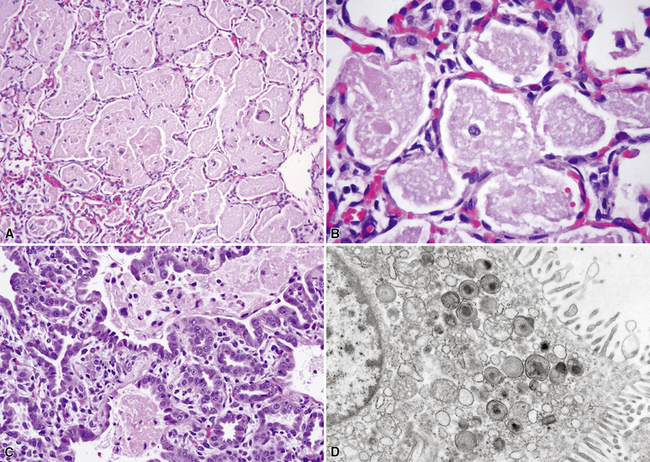
Pulmonary interstitial glycogenosis (PIG), also called infantile cellular interstitial pneumonitis, is a disorder that occurs in infants younger than than 6 months of age, with the highest frequency in neonates. These infants develop tachypnea and respiratory distress, and chest radiographs may show bilateral interstitial infiltrates. Microscopic evaluation shows variable thickening of the alveolar septa due to a proliferation of round to oval bland mesenchymal cells (Fig. 4-25), with a paucity of inflammatory cells.126 The spindle cells contain cytoplasmic glycogen granules demonstrable by their periodic acid/Schiff (PAS)–positive, diastase-digestible staining characteristics.127,128 The process may be diffuse or patchy, with great variability in severity. It is vastly underreported in the medical literature and can be identified in the setting of a wide variety of associated conditions, including chronic neonatal lung disease due to prematurity, hypoplasia, congenital heart disease, and even congenital cystic malformations. Although initially proposed to be a genetic or developmental condition,129 recognition of the many associated conditions has led to the concept that PIG is a reactive response to injury unique to the infant lung, perhaps reflecting differences in the proliferative capacity of the growing neonatal lung. Affected patients sometimes require ventilatory support, but they tend to show good recovery from their disease over the course of weeks, and steroid therapy has been used with symptomatic improvement in some cases. Prognosis generally is thought to relate to the severity of any underlying associated pathology, such as chronic neonatal lung disease.
Genetic Disorders of Surfactant Metabolism
The genetic disorders of surfactant metabolism have been associated with several different histologic patterns, including pulmonary alveolar proteinosis (PAP), chronic pneumonitis of infancy (CIP), desquamative interstitial pneumonia (DIP), nonspecific interstitial pneumonia (NSIP), and idiopathic pulmonary fibrosis.130,131 The dominant histologic features probably depend on the genotype and the age at presentation. Generally speaking, neonates and infants have more abundant alveolar proteinosis material and more prominent alveolar epithelial hyperplasia, whereas older children and adolescents have less conspicuous globular proteinosis material, less epithelial hyperplasia, more abundant cholesterol clefts, and more abundant fibrosis. Presentation in the neonatal period is typical of surfactant protein B gene mutations and ABCA3 mutations. Presentation in later infancy, childhood, or adolescence is more typical of ABCA3 mutations or surfactant protein C gene mutations.
Pulmonary Alveolar Proteinosis
PAP is characterized by the accumulation of granular-appearing proteinaceous material within the alveolar spaces132 and is subdivided into congenital, acquired, and secondary forms.
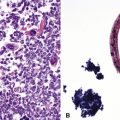
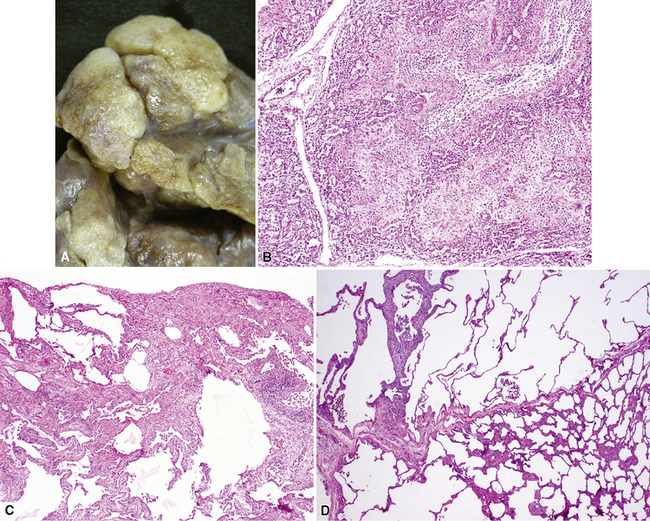
Congenital forms of pulmonary alveolar proteinosis are progressive fatal diseases caused by defects in surfactant production and metabolism, and have been reported from mutations of the surfactant protein B gene and the ABCA3 gene, the latter probably related to packaging and secretion of surfactant proteins133–137 (Fig. 4-26A to C). This form of PAP typically is accompanied by prominent diffuse type 2 alveolar epithelial hyperplasia, increased alveolar macrophages, and occasional cholesterol clefts. Over a period of weeks, diffuse interstitial widening and chronic remodeling of air spaces can be recognized. Electron microscopy may be helpful in assessing lamellar body ultrastructure.138,139 Presence of multivesicular bodies associated with SFTPB gene mutations or dense bodies associated with ABCA3 mutations (see Fig. 4-26D) help to confirm the diagnostic impression based on histologic pattern, although normal lamellar bodies do not exclude a genetic disorder of surfactant metabolism. Definitive characterization of disease also requires correlation with mutation testing. Considerations in the differential diagnosis for congenital PAP include acquired PAP and secondary PAP.
Acquired PAP is unusual in infants and children but may be observed in adolescents. Acquired PAP is thought to arise as a result of autoantibodies to granulocyte-macrophage colony-stimulating factor (GM-CSF) and is more common in adults. Of interest, an identical pattern of disease has been described in children with genetic mutations in the GM-CSF receptor.140,141 The same pattern is occasionally seen in children and adolescents with underlying systemic disorders such as leukemia, bone marrow transplantation, and collagen vascular diseases, despite absence of autoantibodies to GM-CSF. PAP in this setting generally is considered to be a problem of macrophage dysfunction and impaired surfactant recycling, although the mechanism of disease is not clear.
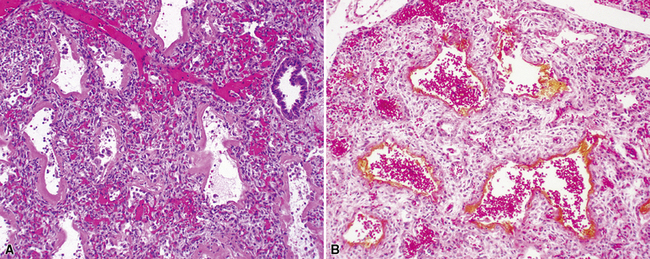
Finally, secondary PAP is observed in infants and children with infections causing extensive alveolar epithelial necrosis. The most common infections implicated are those caused by respiratory syncytial virus, cytomegalovirus, and parainfluenza virus. Occasionally, it is possible to recognize increased inflammation or viral cytopathic changes such as multinucleate giant cells in secondary PAP (Fig. 4-27). Affected patients frequently are immunosuppressed with severe combined immunodeficiency syndrome, leukemia, or lysinuria.142–145 Pneumocystis infection also should be excluded in this clinical setting.
Chronic Pneumonitis of Infancy
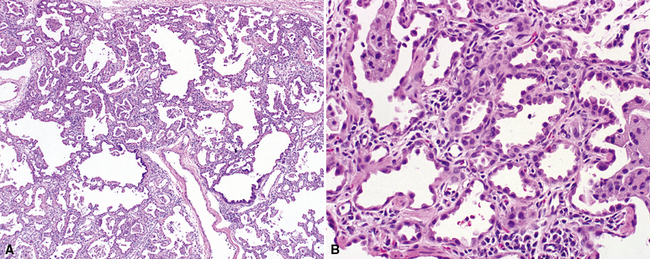
Initially recognized in 1992 and later named by Katzenstein and colleagues in 1995, CPI is a pattern of diffuse interstitial lung disease in infants and young children, which has now been recognized to be associated with the genetic disorders of surfactant metabolism, most commonly mutations in the surfactant protein C gene (SFTPC)146–149 (Fig. 4-28). Microscopically, the lungs show alveolar septal thickening by fibroblastic spindle cells, and marked type II pneumocyte hyperplasia. Inflammation and fibrosis are sparse, although conspicuous remodeling of air spaces and interstitial extension of airway smooth muscle are commonly seen. Consolidation by air space macrophages, proteinaceous material, and cholesterol clefts is a frequent finding. Prognosis generally is poor, with development of chronic lung disease or death in a majority of affected infants. Surfactant protein C gene mutations are inherited in an autosomal dominant fashion, and pulmonary fibrosis has been recognized in some families of infants with CPI.
Desquamative Interstitial Pneumonia
In adults, DIP generally is a smoking-related illness resulting in filling of the alveoli with lightly pigmented macrophages. In children, a DIP pattern evokes an array of possible diagnoses including the genetic disorders of surfactant metabolism, particularly surfactant protein B and ABCA3 defects, various viral infections, aspiration, drug reactions, and inhalational injury. In some cases of DIP occurring in early childhood, stabilization has been achieved with systemic corticosteroid treatment.118,125
Nonspecific Interstitial Pneumonia
The term nonspecific interstitial pneumonia (NSIP) is used to describe idiopathic pulmonary diseases that show a uniform expansion of the alveolar septa by inflammation, fibrosis, or both.150 NSIP is a relatively commonly observed interstitial disease pattern in children,118 but further investigation often allows identification of a specific etiologic disorder. Presence of mild to moderate diffuse lymphocyte infiltrates (NSIP pattern) in the pediatric population raises various diagnostic possibilities including chronic disease due to the genetic disorders of surfactant metabolism (ABCA3, SFTPC), viral pneumonitis, hypersensitivity pneumonitis, and collagen vascular diseases.
Lymphocytic Interstitial Pneumonia and Follicular Bronchiolitis
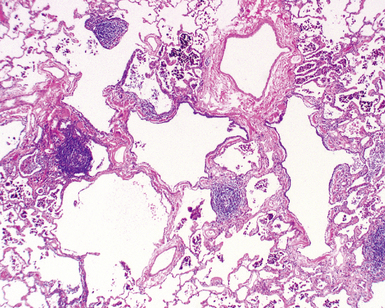
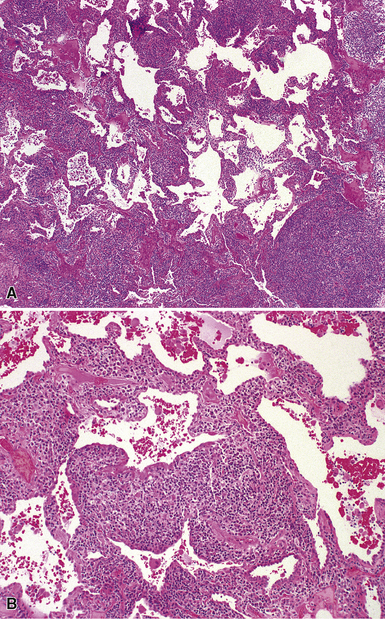
Follicular bronchiolitis and lymphocytic interstitial pneumonia in children is histologically identical to that observed in adults. The presence of lymphocyte aggregates with germinal centers surrounding bronchioles is characteristic of follicular bronchiolitis (Fig. 4-29), whereas a robust and diffuse lymphocytic interstitial infiltrate of the alveolar walls is the key finding in lymphocytic interstitial pneumonia (Fig. 4-30). Both patterns represent forms of pulmonary lymphoid hyperplasia, and may be observed in the same biopsy. Follicular bronchiolitis can be observed in patients with immune deficiencies such as common variable immune deficiency or hypogammaglobulinemia,125,151 collagen vascular diseases such as juvenile rheumatoid arthritis or Sjögren syndrome,125,152–154 or acquired immunodeficiency due to maternal-fetal transmission of human immunodeficiency virus (HIV).155–160 Follicular bronchiolitis also should prompt consideration of Epstein-Barr virus infection.
Hypersensitivity Pneumonitis (Extrinsic Allergic Alveolitis)
Hypersensitivity pneumonitis in children is clinically and histologically similar to that seen in adults. The patient generally presents with exercise intolerance and cough. Although the list of antigens in the adult form of the disease is relatively broad as a result of a vast array of occupational exposures, a majority of the cases in children involve either bird antigens (70%) or molds (15%).161,162 Histologically, hypersensitivity pneumonitis is characterized by a diffuse interstitial lymphocytic infiltrate with bronchiolocentric accentuation and scattered poorly formed granulomas. The air spaces may show consolidation, with macrophages or organizing pneumonia.
Eosinophilic Pneumonia
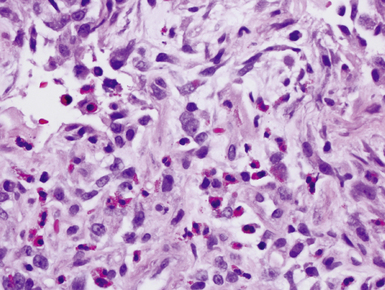
Eosinophilic pneumonia in children is similar in appearance to that in adults. The causes also are similar, with many cases being secondary to drug reactions, parasite infections, or systemic diseases.163 Many cases are idiopathic. The histologic triad of eosinophils, macrophages, and fibrin filling the alveolar spaces usually is easily appreciated in acute eosinophilic pneumonia (Fig. 4-31).
Aspiration Injury
Children with abnormal swallowing function, or gastroesophageal reflux, may aspirate food or gastric fluids into the lung, resulting in aspiration injury. Diagnosis using oil red O stains for lipophages on bronchoalveolar lavage specimens has been suggested. Although this method is relatively sensitive, it is not very specific, since many other diseases including storage diseases, resolving hemorrhage, and resolving pneumonia can result in increased lipophages.164 The histologic features associated with aspiration are variable and often nonspecific, making aspiration a difficult diagnosis to make with certainty. Accumulation of intra-alveolar foamy macrophages and cholesterol clefts occasionally is observed (Fig. 4-32A). Presence of aspirated food particles or interstitial lipid vacuoles, as in exogenous lipoid pneumonia due to mineral oil aspiration (see Fig. 4-32B), will point to a specific diagnosis. Chronic airway irritation can result in follicular bronchiolitis or organizing pneumonia with granulation tissue plugs occurring within airway lumina. In severe chronic cases, bronchiectasis can occur.165 Aspirated food particles may elicit a surrounding granulomatous reaction.166 An interesting but unusual interaction has been described in infants in whom aspiration of fat or oils occurs coincident with the development of infection by rapid-growing mycobacteria, resulting in a lipoid pneumonia with granulomas.167 Acid-fast bacteria can be demonstrated within the lipid droplets in such cases (see Fig. 4-32C).
Obliterative Bronchiolitis
In children, airway obliteration may follow any form of chronic small- and large-airway injury, particularly that resulting in airway mucosal necrosis followed by fibrosis. Common etiologic conditions include preceding severe viral bronchiolitis (e.g., adenovirus or influenza infection), chronic aspiration injury, Stevens-Johnson syndrome, chronic graft-versus-host disease in bone marrow transplant recipients, and chronic airway rejection in lung transplant recipients168 (Fig. 4-33). Asthma and cystic fibrosis also may result in focal obliterative bronchiolitis.
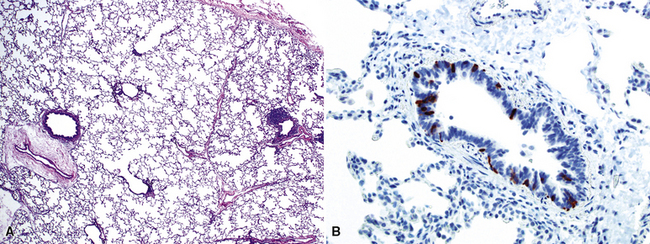
Neuroendocrine Cell Hyperplasia of Infancy (Persistent Tachypnea of Infancy)
Neuroendocrine cell hyperplasia of infancy (NEHI) is a more recently described pathologic correlate to the clinical syndrome of persistent tachypnea of infancy.169–171 Patients with NEHI are infants and young children with clinical signs and symptoms of chronic tachypnea and hypoxia, often with chronic oxygen requirement, and evidence of hyperinflated lungs on the chest radiograph and patchy perihilar ground-glass opacity on the chest CT scan.172 Morphologically, the lung biopsy tissue appears almost normal, with only mild lymphoid hyperplasia and subtle alveolar duct expansion (Fig. 4-34). The virtually “normal” biopsy specimen and presence of appropriate alveolarization should prompt further evaluation for NEHI. The diagnosis is made by identifying increased numbers of airway neuroendocrine cells and large neuroepithelial bodies on special stains (e.g., bombesin immunohistochemistry). It remains unknown whether this is a disorder with genetic predisposition or a reactive condition secondary to other forms of small- or large-airway injury. Some children have a history of preceding viral bronchiolitis, and familial cases have been recognized.
Vascular Disease as a Cause of Interstitial Lung Disease
Vascular diseases, especially those related to congenital heart defects, can mimic interstitial lung disease (ILD) clinically and radiologically.119,120,173–175 Although many of these cases are identified before biopsy, it is important to consider a vascular cause in analysis of a wedge biopsy specimen from a patient being evaluated for ILD.
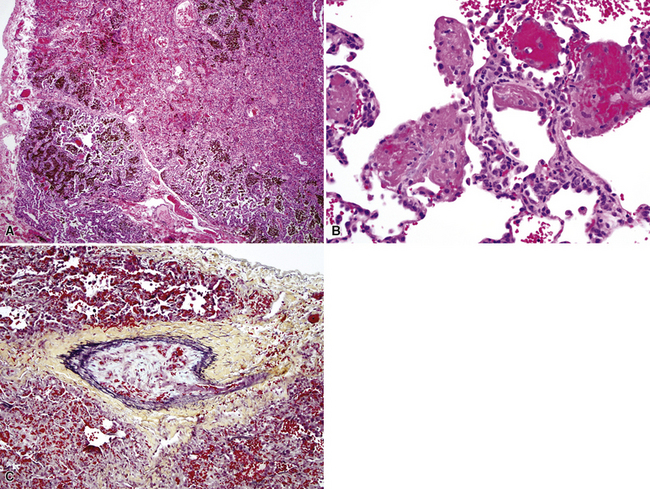
Hemorrhage Syndromes in Children
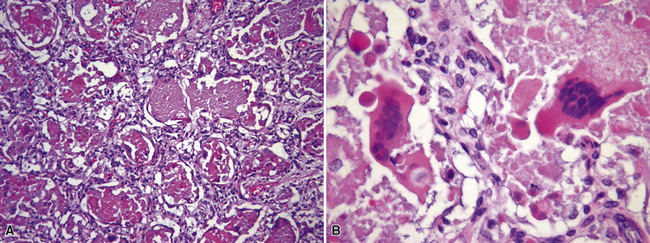
Chronic recurrent hemorrhage in children may result from recurrent episodes of pulmonary vasculitis, often small-vessel vasculitides such as capillaritis (Fig. 4-35A and B).176,177 Other considerations in the differential diagnosis include chronic recurrent hemorrhage due to coagulopathy, chronic hemorrhage due to pulmonary arteriopathy, chronic congestive vasculopathy and pulmonary veno-occlusive disease (see Fig. 4-35C), and idiopathic pulmonary hemosiderosis. Chronic pulmonary hemorrhage has been described in milk aspiration (Heiner syndrome). Of note, idiopathic pulmonary hemosiderosis is a clinicopathologic diagnosis applied when abundant hemosiderin-laden macrophages are present on biopsy, without other associated pathologic changes, and the clinical etiology remains unknown.
1 Fan L.L., Langston C. Pediatric interstitial lung disease: children are not small adults. Am J Respir Crit Care Med.. 2002;165(11):1466-1467.
2 Langston C., Patterson K., Dishop M.K., et al. A protocol for the handling of tissue obtained by operative lung biopsy: recommendations of the Child Pathology Co-operative Group. Pediatr Dev Pathol.. 2006;9:173-180.
3 Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg.. 2003;12(1):17-37.
4 Aktogu S., Yuncu G., Halilcolar H., et al. Bronchogenic cysts: clinicopathological presentation and treatment. Eur Respir J.. 1996;9(10):2017-2021.
5 Cioffi U., Bonavina L., De Simone M., et al. Presentation and surgical management of bronchogenic and esophageal duplication cysts in adults. Chest.. 1998;113(6):1492-1496.
6 Coselli M.P., de Ipolyi P., Bloss R.S., et al. Bronchogenic cysts above and below the diaphragm: report of eight cases. Ann Thorac Surg.. 1987;44(5):491-494.
7 Kanemitsu Y., Nakayama H., Asamura H., et al. Clinical features and management of bronchogenic cysts: report of 17 cases. Surg Today.. 1999;29(11):1201-1205.
8 Riedlinger W.F., Vargas S.O., Jenning R.W., et al. Bronchial atresia is common to extralobar sequestration, intralobar sequestration, congenital cystic adenomatoid malformation, and lobar emphysema. Pediatr Dev Pathol.. 2006;9:361-373.
9 Kunisaki S.M., Fauza D.O., Nemes L.P., et al. Bronchial atresia: the hidden pathology within a spectrum of prenatally diagnosed lung masses. J Pediatr Surg.. 2006;41(1):61-65.
10 Louie H.W., Martin S.M., Mulder D.G. Pulmonary sequestration: 17-year experience at UCLA. Am Surg.. 1993;59(12):801-805.
11 Stocker J.T. Sequestrations of the lung. Semin Diagn Pathol.. 1986;3(2):106-121.
12 Stocker J.T., Drake R.M., Madewell J.E. Cystic and congenital lung disease in the newborn. Perspect Pediatr Pathol.. 1978;4:93-154.
13 Stocker J.T., Kagan-Hallet K. Extralobar pulmonary sequestration: analysis of 15 cases. Am J Clin Pathol.. 1979;72(6):917-925.
14 Aulicino M.R., Reis E.D., Dolgin S.E., et al. Intra-abdominal pulmonary sequestration exhibiting congenital cystic adenomatoid malformation. Report of a case and review of the literature. Arch Pathol Lab Med.. 1994;118(10):1034-1037.
15 Morad N.A., al-Malki T., e-Tahir M. Intra-abdominal pulmonary sequestration: diagnostic difficulties. Pathology.. 1997;29(2):218-220.
16 Chan Y.F., Oldfield R., Vogel S., et al. Pulmonary sequestration presenting as a prenatally detected suprarenal lesion in a neonate. J Pediatr Surg.. 2000;35(9):1367-1369.
17 Rosado-de-Christenson M.L., Frazier A.A., Stocker J.T., et al. From the archives of the AFIP. Extralobar sequestration: radiologic-pathologic correlation. Radiographics.. 1993;13(2):425-441.
18 Zangwill B.C., Stocker J.T. Congenital cystic adenomatoid malformation within an extralobar pulmonary sequestration. Pediatr Pathol.. 1993;13(3):309-315.
19 Conran R.M., Stocker J.T. Extralobar sequestration with frequently associated congenital cystic adenomatoid malformation, type 2: report of 50 cases. Pediatr Dev Pathol.. 1999;2(5):454-463.
20 Cass D.L., Crombleholme T.M., Howell L.J., et al. Cystic lung lesions with systemic arterial blood supply: a hybrid of congenital cystic adenomatoid malformation and bronchopulmonary sequestration. J Pediatr Surg.. 1997;32(7):986-990.
21 Fraggetta F., Cacciaguerra S., Nash R., et al. Intra-abdominal pulmonary sequestration associated with congenital cystic adenomatoid malformation of the lung: just an unusual combination of rare pathologies? Pathol Res Pract.. 1998;194(3):209-211.
22 Frazier A.A., Rosado-De-Christenson M.L., Stocker J.T., et al. Intralobar sequestration: radiologic-pathologic correlation. Radiographics.. 1997;17(3):725-745.
23 Zylak C.J., Eyler W.R., Spizarny D.L., et al. Developmental lung anomalies in the adult: radiologic-pathologic correlation. Radiographics.. 2002;22(Spec No):S25-S43.
24 Au V.W., Chan J.K., Chan F.L. Pulmonary sequestration diagnosed by contrast enhanced three-dimensional MR angiography. Br J Radiol.. 1999;72(859):709-711.
25 Bale P.M. Congenital cystic malformation of the lung. A form of congenital bronchiolar (“adenomatoid”) malformation. Am J Clin Pathol.. 1979;71(4):411-420.
26 Benning T.L., Godwin J.D., Roggli V.L., et al. Cartilaginous variant of congenital adenomatoid malformation of the lung. Chest.. 1987;92(3):514-516.
27 Cloutier M.M., Schaeffer D.A., Hight D. Congenital cystic adenomatoid malformation. Chest.. 1993;103(3):761-764.
28 Luck S.R., Reynolds M., Raffensperger J.G. Congenital bronchopulmonary malformations. Curr Probl Surg.. 1986;23(4):245-314.
29 Miller R.K., Sieber W.K., Yunis E.J. Congenital adenomatoid malformation of the lung. A report of 17 cases and review of the literature. Pathol Annu.. 1980;15(Pt 1):387-402.
30 Moerman P., Fryns J.P., Vandenberghe K., et al. Pathogenesis of congenital cystic adenomatoid malformation of the lung. Histopathology.. 1992;21(4):315-321.
31 Nakamura Y. Pulmonary disorders in infants. Acta Pathol Jpn.. 1993;43(7–8):347-359.
32 Rosado-de-Christenson M.L., Stocker J.T. Congenital cystic adenomatoid malformation. Radiographics.. 1991;11(5):865-886.
33 Cha I., Adzick N.S., Harrison M.R., et al. Fetal congenital cystic adenomatoid malformations of the lung: a clinicopathologic study of eleven cases. Am J Surg Pathol.. 1997;21(5):537-544.
34 Kreiger P.A., Ruchelli E.D., Mahboubi S., et al. Fetal pulmonary malformations: defining histopathology. Am J Surg Pathol.. 2006;30(5):643-649.
35 Avitabile A.M., Greco M.A., Hulnick D.H., et al. Congenital cystic adenomatoid malformation of the lung in adults. Am J Surg Pathol.. 1984;8(3):193-202.
36 Stocker J.T., Madewell J.E., Drake R.M. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol.. 1977;8(2):155-171.
37 Stocker J.T. Congenital and developmental diseases. In: Hammar S.P., editor. Pulmonary Pathology. New York: Springer-Verlag; 1994:155-190.
38 Rutledge J.C., Jensen P. Acinar dysplasia: a new form of pulmonary maldevelopment. Hum Pathol.. 1986;17(12):1290-1293.
39 Chambers H.M. Congenital acinar aplasia: an extreme form of pulmonary maldevelopment. Pathology.. 1991;23:69-71.
40 Davidson L.A., Batman P., Fagan D.G. Congenital acinar dysplasia: a rare cause of poulmonary hypoplasia. Histopathology.. 1998;32(1):57-59.
41 Moerman P., Vanhole C., Devlieger H., Fryns J.P. Severe primary pulmonary hypoplasia (“acinar dysplasia”) in sibs: a genetically determined mesodermal defect? J Med Genet.. 1998;35(11):964-965.
42 Federici S., Domenichelli V., Tani G., et al. Pleuropulmonary blastoma in congenital cystic adenomatoid malformation: report of a case. Eur J Pediatr Surg.. 2001;11(3):196-199.
43 MacSweeney F., Papagiannopoulos K., Goldstraw P., et al. An assessment of the expanded classification of congenital cystic adenomatoid malformations and their relationship to malignant transformation. Am J Surg Pathol.. 2003;27(8):1139-1146.
44 Dehner L.P. Pleuropulmonary blastoma is THE pulmonary blastoma of childhood. Semin Diagn Pathol.. 1994;11(2):144-151.
45 Manivel J.C., Priest J.R., Watterson J., et al. Pleuropulmonary blastoma. The so-called pulmonary blastoma of childhood. Cancer.. 1988;62(8):1516-1526.
46 Merriman T.E., Beasley S.W., Chow C.W., et al. A rare tumor masquerading as an empyema: pleuropulmonary blastoma. Pediatr Pulmonol.. 1996;22(6):408-411.
47 Indolfi P., Casale F., Carli M., et al. Pleuropulmonary blastoma: management and prognosis of 11 cases. Cancer.. 2000;89(6):1396-1401.
48 Priest J.R., Williams G.M., Hill D.A., et al. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol.. 2009;44:14-30.
49 Sheffield E.A., Addis B.J., Corrin B., et al. Epithelial hyperplasia and malignant change in congenital lung cysts. J Clin Pathol.. 1987;40(6):612-614.
50 Benjamin D.R., Cahill J.L. Bronchioloalveolar carcinoma of the lung and congenital cystic adenomatoid malformation. Am J Clin Pathol.. 1991;95(6):889-892.
51 Kaslovsky R.A., Purdy S., Dangman B.C., et al. Bronchioloalveolar carcinoma in a child with congenital cystic adenomatoid malformation. Chest.. 1997;112(2):548-551.
52 Usui Y., Takabe K., Takayama S., et al. Minute squamous cell carcinoma arising in the wall of a congenital lung cyst. Chest.. 1991;99(1):235-236.
53 Ribet M.E., Copin M.C., Soots J.G., et al. Bronchioloalveolar carcinoma and congenital cystic adenomatoid malformation. Ann Thorac Surg.. 1995;60(4):1126-1128.
54 Ota H., Langston C., Honda T., et al. Histochemical analysis of mucous cells of congenital adenomatoid malformation of the lung: insights into the carcinogenesis of pulmonary adenocarcinoma expressing gastric mucins. Am J Clin Pathol.. 1998;110(4):450-455.
55 Granata C., Gambini C., Balducci T., et al. Bronchioloalveolar carcinoma arising in congenital cystic adenomatoid malformation in a child: a case report and review on malignancies originating in congenital cystic adenomatoid malformation. Pediatr Pulmonol.. 1998;25(1):62-66.
56 Yao J.L., Fasano M., Morotti R., et al. Demonstration of communication between alveolus and interstitium in persistent interstitial pulmonary emphysema: case report. Pediatr Dev Pathol.. 1999;2(5):484-487.
57 Zimmermann H. Progressive interstitial pulmonary lobar emphysema. Eur J Pediatr.. 1982;138(3):258-262.
58 Stocker J.T., Madewell J.E. Persistent interstitial pulmonary emphysema: another complication of the respiratory distress syndrome. Pediatrics.. 1977;59(6):847-857.
59 Brewer L.L., Moskowitz P.S., Carrington C.B., et al. Pneumatosis pulmonalis: a complication of the idiopathic respiratory distress syndrome. Am J Pathol.. 1979;95(1):171-190.
60 Cooney T.P., Thurlbeck W.M. Pulmonary hypoplasia in Down’s syndrome. N Engl J Med.. 1982;307(19):1170-1173.
61 Cooney T.P., Wentworth P.J., Thurlbeck W.M. Diminished radial count is found only postnatally in Down’s syndrome. Pediatr Pulmonol.. 1988;5(4):204-209.
62 Gonzalez O.R., Gomez I.G., Recalde A.L., et al. Postnatal development of the cystic lung lesion of Down syndrome: suggestion that the cause is reduced formation of peripheral air spaces. Pediatr Pathol.. 1991;11(4):623-633.
63 Gyves-Ray K., Kirchner S., Stein S., et al. Cystic lung disease in Down syndrome. Pediatr Radiol.. 1994;24(2):137-138.
64 Schloo B.L., Vawter G.F., Reid L.M. Down syndrome: patterns of disturbed lung growth. Hum Pathol.. 1991;22(9):919-923.
65 Tyrrell V.J., Asher M.I., Chan Y. Subpleural lung cysts in Down’s syndrome. Pediatr Pulmonol.. 1999;28(2):145-148.
66 Hislop A., Reid L. New pathological findings in emphysema of childhood. 2. Overinflation of a normal lobe. Thorax.. 1971;26(2):190-194.
67 Mani H., Suarez E., Stocker J.T. The morphologic spectrum of infantile lobar emphysema: a study of 33 cases. Paediatr Respir Rev.. 2004;5(suppl A):S313-S320.
68 Stanger P., Lucas R.V.Jr, Edwards J.E. Anatomic factors causing respiratory distress in acyanotic congenital cardiac disease. Special reference to bronchial obstruction. Pediatrics.. 1969;43(5):760-769.
69 Miller K.E., Edwards D.K., Hilton S., et al. Acquired lobar emphysema in premature infants with bronchopulmonary dysplasia: an iatrogenic disease? Radiology.. 1981;138(3):589-592.
70 Newman B., Yunis E. Lobar emphysema associated with respiratory syncytial virus pneumonia. Pediatr Radiol.. 1995;25(8):646-648.
71 Nathan L., Leveno K.J., Carmody T.J.3rd, et al. Meconium: a 1990s perspective on an old obstetric hazard. Obstet Gynecol.. 1994;83(3):329-332.
72 Swaminathan S., Quinn J., Stabile M.W., et al. Long-term pulmonary sequelae of meconium aspiration syndrome. J Pediatr.. 1989;114(3):356-361.
73 Hislop A., Reid L. New pathological findings in emphysema of childhood. 1. Polyalveolar lobe with emphysema. Thorax.. 1970;25(6):682-690.
74 Tapper D., Schuster S., McBride J., et al. Polyalveolar lobe: anatomic and physiologic parameters and their relationship to congenital lobar emphysema. J Pediatr Surg.. 1980;15(6):931-937.
75 Munnell E.R., Lambird P.A., Austin R.L. Polyalveolar lobe causing lobar emphysema of infancy. Ann Thorac Surg.. 1973;16(6):624-628.
76 Emery J.L., Mithal A. The number of alveoli in the terminal respiratory unit of man during late intrauterine life and childhood. Arch Dis Child.. 1960;35:544-557.
77 MacMahon H.E. Congenital alveolar dysplasia: a developmental anomaly involving pulmonary alveoli. Pediatrics.. 1948;2:43-57.
78 Greenough A. Factors adversely affecting lung growth. Paediatr Respir Rev.. 2000;1(4):314-320.
79 Langston C., Kida K., Reed M., Thurlbeck W.M. Human lung growth in late gestation and in the neonate. Am Rev Respir Dis.. 1984;129:607-613.
80 DePaepe M.E., Friedman R.M., Gundogan F., Pinar H. Postmortem lung weight/body weight standards for term and preterm infants. Pediatr Pulmonol.. 2005;40:445-448.
81 Wigglesworth J.S., Desai R., Hislop A.A. Fetal lung growth in congenital laryngeal atresia. Pediatr Pathol.. 1987;7:515-525.
82 Silver M.M., Thurston W.A., Patrick J.E. Perinatal pulmonary hyperplasia due to laryngeal atresia. Hum Pathol.. 1988;19:110-113.
83 Wagenvoort C.A. Misalignment of lung vessels: a syndrome causing persistent neonatal pulmonary hypertension. Hum Pathol.. 1986;17(7):727-730.
84 Janney C.G., Askin F.B., Kuhn C.3rd. Congenital alveolar capillary dysplasia—an unusual cause of respiratory distress in the newborn. Am J Clin Pathol.. 1981;76(5):722-727.
85 Langston C. Misalignment of pulmonary veins and alveolar capillary dysplasia. Pediatr Pathol.. 1991;11(1):163-170.
86 Tibballs J., Chow C.W. Incidence of alveolar capillary dysplasia in severe idiopathic persistent pulmonary hypertension of the newborn. J Paediatr Child Health.. 2002;38(4):397-400.
87 Alameh J., Bachiri A., Devisme L., et al. Alveolar capillary dysplasia: a cause of persistent pulmonary hypertension of the newborn. Eur J Pediatr.. 2002;161(5):262-266.
88 Haraida S., Lochbuhler H., Heger A., et al. Congenital alveolar capillary dysplasia: rare cause of persistent pulmonary hypertension. Pediatr Pathol Lab Med.. 1997;17(6):959-975.
89 Boggs S., Harris M.C., Hoffman D.J., et al. Misalignment of pulmonary veins with alveolar capillary dysplasia: affected siblings and variable phenotypic expression. J Pediatr.. 1994;124:125-128.
90 Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 13-1992. A full-term newborn boy with chronic respiratory distress. N Engl J Med.. 1992;326(13):875-884.
91 Noonan J.A., Walters L.R., Reeves J.T. Congenital pulmonary lymphangiectasis. Am J Dis Child.. 1970;120(4):314-319.
92 Hunter W.S., Becroft D.M. Congenital pulmonary lymphangiectasis associated with pleural effusions. Arch Dis Child.. 1984;59(3):278-279.
93 Hilliard R.I., McKendry J.B., Phillips M.J. Congenital abnormalities of the lymphatic system: a new clinical classification. Pediatrics.. 1990;86(6):988-994.
94 Brown M., Pysher T., Coffin C.M. Lymphangioma and congenital pulmonary lymphangiectasis: a histologic, immunohistochemical, and clinicopathologic comparison. Mod Pathol.. 1999;12(6):569-575.
95 Faul J.L., Berry G.J., Colby T.V., et al. Thoracic lymphangiomas, lymphangiectasis, lymphangiomatosis, and lymphatic dysplasia syndrome. Am J Respir Crit Care Med.. 2000;161(3 Pt 1):1037-1046.
96 Wagenaar S.S., Swierenga J., Wagenvoort C.A. Late presentation of primary pulmonary lymphangiectasis. Thorax.. 1978;33(6):791-795.
97 Verlaat C.W., Peters H.M., Semmekrot B.A., et al. Congenital pulmonary lymphangiectasis presenting as a unilateral hyperlucent lung. Eur J Pediatr.. 1994;153(3):202-205.
98 France N.E., Brown R.J. Congenital pulmonary lymphangiectasis. Report of 11 examples with special reference to cardiovascular findings. Arch Dis Child.. 1971;46(248):528-532.
99 Tazelaar H.D., Kerr D., Yousem S.A., et al. Diffuse pulmonary lymphangiomatosis. Hum Pathol.. 1993;24(12):1313-1322.
100 Swensen S.J., Hartman T.E., Mayo J.R., et al. Diffuse pulmonary lymphangiomatosis: CT findings. J Comput Assist Tomogr.. 1995;19(3):348-352.
101 Burke C.M., Safai C., Nelson D.P., et al. Pulmonary arteriovenous malformations: a critical update. Am Rev Respir Dis.. 1986;134(2):334-339.
102 Sawyer S.M., Menahem S., Chow C.W., Robertson C.F. Progressive cyanosis in a child with hereditary hemorrhagic telangiectasia (Osler–Weber–Rendu disease). Pediatr Pulmonol.. 1992;13(2):124-127.
103 Kapur S., Rome J., Chandra R.S. Diffuse pulmonary arteriovenous malformation in a child with polysplenia syndrome. Pediatr Pathol Lab Med.. 1995;15(3):463-468.
104 Papagiannis J., Kanter R.J., Effman E.L., et al. Polysplenia with pulmonary arteriovenous malformations. Pediatr Cardiol.. 1993;14(2):127-129.
105 Farrell P.M., Avery M.E. Hyaline membrane disease. Am Rev Respir Dis.. 1975;111(5):657-688.
106 Ikegami M., Jacobs H., Jobe A. Surfactant function in respiratory distress syndrome. J Pediatr.. 1983;102(3):443-447.
107 O’Brodovich H.M., Mellins R.B. Bronchopulmonary dysplasia. Unresolved neonatal acute lung injury. Am Rev Respir Dis. 1985;132(3):694-709.
108 Morgenstern B., Klionsky B., Doshi N. Yellow hyaline membrane disease. Identification of the pigment and bilirubin binding. Lab Invest. 1981;44(6):514-518.
109 Husain A.N., Siddiqui N.H., Stocker J.T. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol.. 1998;29(7):710-717.
110 Bonikos D.S., Bensch K.G., Northway W.H., et al. Bronchopulmonary dysplasia: the pulmonary pathologic sequel of necrotizing bronchiolitis and pulmonary fibrosis. Hum Pathol.. 1976;7:643-666.
111 Nickerson B.G. Bronchopulmonary dysplasia. Chronic pulmonary disease following neonatal respiratory failure. Chest.. 1985;87(4):528-535.
112 Jobe A.H., Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med.. 2001;163(7):1723-1729.
113 Northway W.H.Jr, Rosan R.C., Porter D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med.. 1967;276(7):357-368.
114 Northway W.H.Jr. Bronchopulmonary dysplasia: then and now. Arch Dis Child.. 1990;65(10 Spec No):1076-1081.
115 Stocker J.T. Pathologic features of long-standing “healed” bronchopulmonary dysplasia: a study of 28 3- to 40-month-old infants. Hum Pathol.. 1986;17(9):943-961.
116 Northway W.H.Jr, Moss R.B., Carlisle K.B., et al. Late pulmonary sequelae of bronchopulmonary dysplasia. N Engl J Med.. 1990;323(26):1793-1799.
117 Coalson J.J. Pathology of new bronchopulmonary dysplasia. Semin Neonatol.. 2003;8(1):73-81.
118 Coren M.E., Nicholson A.G., Goldstraw P., et al. Open lung biopsy for diffuse interstitial lung disease in children. Eur Respir J.. 1999;14(4):817-821.
119 Fan L.L., Mullen A.L., Brugman S.M., et al. Clinical spectrum of chronic interstitial lung disease in children. J Pediatr.. 1992;121(6):867-872.
120 Fan L.L., Langston C. Chronic interstitial lung disease in children. Pediatr Pulmonol.. 1993;16(3):184-196.
121 Fan L.L., Deterding R.R., Langston C. Pediatric interstitial lung disease revisited. Pediatr Pulmonol.. 2004;38:369-378.
122 Clement A., ERS Task Force. Task force on chronic interstitial lung disease in immunocompetent children. Eur Respir J.. 2004;24:686-697.
123 Deutsch G.H., Young L.R., Deterding R.R., et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med.. 2007;176:1120-1128.
124 Katzenstein A.L., Myers J.L. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med.. 1998;157(4 Pt 1):1301-1315.
125 Nicholson A.G., Kim H., Corrin B., et al. The value of classifying interstitial pneumonitis in childhood according to defined histological patterns. Histopathology.. 1998;33(3):203-211.
126 Schroeder S.A., Shannon D.C., Mark E.J. Cellular interstitial pneumonitis in infants. A clinicopathologic study. Chest.. 1992;101(4):1065-1069.
127 Canakis A.M., Cutz E., Manson D., et al. Pulmonary interstitial glycogenosis: a new variant of neonatal interstitial lung disease. Am J Respir Crit Care Med.. 2002;65(11):1557-1565.
128 Smets K., Dhaene K., Schelstraete P., et al. Neonatal pulmonary interstitial glycogen accumulation disorder. Eur J Pediatr.. 2004;163:408-409.
129 Onland W., Molenaar J.J., Leguit R.J., et al. Pulmonary interstitial glycogenosis in identical twins. Pediatr Pulmonol.. 2005;40:362-366.
130 Cole F.S., Hamvas A., Nogee L.M. Genetic disorders of neonatal respiratory function. Pediatr Res.. 2001;50:157-162.
131 Whitsett J.A., Wert S.E., Xu Y. Genetic disorders of surfactant homeostasis. Biol Neonate.. 2005;87:283-287.
132 Rosen S.H., Castleman B., Liebow A.A. Pulmonary alveolar proteinosis. N Engl J Med.. 1958;258(23):1123-1142.
133 Nogee L.M., de Mello D.E., Dehner L.P., et al. Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med.. 1993;328(6):406-410.
134 deMello D.E., Heyman S., Phyelps D.S., et al. Ultrastructure of lung in surfactant protein B deficiency. Am J Respir Cell Mol Biol.. 1994;11:230-239.
135 Nogee L.M., Garnier G., Dietz H.C., et al. A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J Clin Invest.. 1994;93(4):1860-1863.
136 Shulenin S., Nogee L.M., Annilo T., et al. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med.. 2004;350(13):1296-1303.
137 Bullard J.E., Wert S.E., Whitsett J.A., et al. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med.. 2005;172:1026-1031.
138 Tryka A.F., Wert S.E., Mazursky J.E., et al. Absence of lamellar bodies with accumulation of dense bodies characterizes a novel form of congenital surfactant defect. Pediatr Dev Pathol.. 2000;3(4):335-345.
139 Edwards V., Cutz E., Viero S., et al. Ultrastructure of lamellar bodies in congenital surfactant deficiency. Ultrastruct Pathol.. 2005;29:503-509.
140 Martinez-Moczygemba M., Doan M.L., Elidemir O., et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRα gene in the X chromosome pseudoautosomal region 1. J Exp Med.. 2008;205(12):2711-2716. Epub 2008 Oct 27
141 Suzuki T., Sakagami T., Rubin B.K., et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med.. 2008;205(12):2703-2710. Epub 2008 Oct 27
142 Bedrossian C.W., Luna M.A., Conklin R.H., et al. Alveolar proteinosis as a consequence of immunosuppression. A hypothesis based on clinical and pathologic observations. Hum Pathol.. 1980;11(5 suppl):527-535.
143 Nachajon R.V., Rutstein R.M., Rudy B.J., et al. Pulmonary alveolar proteinosis in an HIV-infected child. Pediatr Pulmonol.. 1997;24(4):292-295.
144 Samuels M.P., Warner J.O. Pulmonary alveolar lipoproteinosis complicating juvenile dermatomyositis. Thorax.. 1988;43(11):939-940.
145 Parto K., Svedstrom E., Majurin M.L., et al. Pulmonary manifestations in lysinuric protein intolerance. Chest.. 1993;104(4):1176-1182.
146 Fisher M., Roggli V., Merten D., et al. Coexisting endogenous lipoid pneumonia, cholesterol granulomas, and pulmonary alveolar proteinosis in a pediatric population. Pediatr Pathol.. 1992;12:365-383.
147 Katzenstein A.L., Gordon L.P., Oliphant M., et al. Chronic pneumonitis of infancy. A unique form of interstitial lung disease occurring in early childhood. Am J Surg Pathol.. 1995;19(4):439-447.
148 Nogee L.M., Dunbar A.E.3rd, Wert S.E., et al. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med.. 2001;344(8):573-579.
149 Tredano M., Griese M., Brasch F., et al. Mutation in SFTPC in infantile pulmonary alveolar proteinsosis with or without fibrosing lung disease. Am J Med Genet A.. 2004;126A:18-26.
150 Katzenstein A.L., Fiorelli R.F. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol.. 1994;18(2):136-147.
151 Yousem S.A., Colby T.V., Carrington C.B. Follicular bronchitis/bronchiolitis. Hum Pathol.. 1985;16(7):700-706.
152 Athreya B.H., Doughty R.A., Bookspan M., et al. Pulmonary manifestations of juvenile rheumatoid arthritis. A report of eight cases and review. Clin Chest Med.. 1980;1(3):361-374.
153 Uziel Y., Hen B., Cordoba M., et al. Lymphocytic interstitial pneumonitis preceding polyarticular juvenile rheumatoid arthritis. Clin Exp Rheumatol.. 1998;16(5):617-619.
154 Lovell D., Lindsley C., Langston C. Lymphoid interstitial pneumonia in juvenile rheumatoid arthritis. J Pediatr.. 1984;105(6):947-950.
155 Grieco M.H., Chinoy-Acharya P. Lymphocytic interstitial pneumonia associated with the acquired immune deficiency syndrome. Am Rev Respir Dis.. 1985;131(6):952-955.
156 Kornstein M.J., Pietra G.G., Hoxie J.A., et al. The pathology and treatment of interstitial pneumonitis in two infants with AIDS. Am Rev Respir Dis.. 1986;133(6):1196-1198.
157 Teirstein A.S., Rosen M.J. Lymphocytic interstitial pneumonia. Clin Chest Med.. 1988;9(3):467-471.
158 Pitt J. Lymphocytic interstitial pneumonia. Pediatr Clin North Am.. 1991;38(1):89-95.
159 Marchevsky A., Rosen M.J., Chrystal G., et al. Pulmonary complications of the acquired immunodeficiency syndrome: a clinicopathologic study of 70 cases. Hum Pathol.. 1985;16(7):659-670.
160 Rubinstein A., Morecki R., Silverman B., et al. Pulmonary disease in children with acquired immune deficiency syndrome and AIDS-related complex. J Pediatr.. 1986;108(4):498-503.
161 Fan L.L. Hypersensitivity pneumonitis in children. Curr Opin Pediatr.. 2002;14(3):323-326.
162 Yee W.F., Castile R.G., Cooper A., et al. Diagnosing bird fancier’s disease in children. Pediatrics.. 1990;85(5):848-852.
163 Oermann C.M., Panesar K.S., Langston C., et al. Pulmonary infiltrates with eosinophilia syndromes in children. J Pediatr.. 2000;136(3):351-358.
164 Colombo J.L., Hallberg T.K. Recurrent aspiration in children: lipid-laden alveolar macrophage quantitation. Pediatr Pulmonol.. 1987;3(2):86-89.
165 Annobil S.H., Morad N.A., Kameswaran M., et al. Bronchiectasis due to lipid aspiration in childhood: clinical and pathological correlates. Ann Trop Paediatr.. 1996;16(1):19-25.
166 Gadol C.L., Joshi V.V., Lee E.Y. Bronchiolar obstruction associated with repeated aspiration of vegetable material in two children with cerebral palsy. Pediatr Pulmonol.. 1987;3(6):437-439.
167 Annobil S.H., Benjamin B., Kameswaran M., et al. Lipoid pneumonia in children following aspiration of animal fat (ghee). Ann Trop Paediatr.. 1991;11(1):87-94.
168 Moonnumakal S.P., Fan L.L. Bronchiolitis obliterans in children. Curr Opin Pediatr.. 2008;20(3):272-278.
169 Deterding R.R., Fan L.L., Morton R., et al. Persistent tachypnea of infancy (PTI)—a new entity. Pediatr Pulmonol.. 2001;32(suppl 23):72-73.
170 Deterding R.R., Pye C., Fan L.L., Langston C. Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol.. 2005;40:157-165.
171 Cutz E., Yeger H., Pan J. Pulmonary neuroendocrine cell system in pediatric lung disease—recent advances. Pediatr Dev Pathol.. 2007;10(6):419-435.
172 Brody A.S., Crotty E.J. Neuroendocrine cell hyperplasia of infancy (NEHI). Pediatr Radiol.. 2006;36:1328.
173 Sondheimer H.M., Lung M.C., Brugman S.M., et al. Pulmonary vascular disorders masquerading as interstitial lung disease. Pediatr Pulmonol.. 1995;20(5):284-288.
174 Rabinovitch M., Haworth S.G., Castaneda A.R., et al. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation.. 1978;58:1107-1122.
175 Haworth S.G. Pulmonary vascular disease in different types of congenital heart disease: implications for interpretation of lung biopsy findings in early childhood. Br Heart J.. 1984;52:557-571.
176 Susarla S.C., Fan L.L. Diffuse alveolar hemorrhage syndromes in children. Curr Opin Pediatr.. 2007;19:314-320.
177 Fullmer J., Langston C., Dishop M.K., Fan L.L. Pulmonary capillaritis in children: a review of eight cases with comparison to other alveolar hemorrhage syndromes. J Pediatr.. 2005;146:376-381.