[level-membership-for-neurosurgery-category]
CHAPTER 218 Developmental Abnormalities of the Craniocervical Junction
The first anatomic description of “manifestations of occipital vertebrae” was attributed to Meckel in 1815 by Gladstone and Erickson-Powell.1 Its clinical significance became appreciated after the classic radiologic studies on basilar invagination by Chamberlain in 1939.2
The term craniovertebral junction refers to the occipital bone that surrounds the foramen magnum and the atlas and axis vertebrae. Into the 1970s, surgical treatment of conditions affecting the craniovertebral junction consisted of posterior decompression by enlargement of the foramen magnum and removal of the posterior arch of the atlas and axis vertebrae. However, mortality and morbidity rates associated with this treatment were high in patients with irreducible lesions and cervicomedullary compression.3 A physiologic approach based on an understanding of craniovertebral dynamics, the site of encroachment, and stability of the craniovertebral junction was adopted at the University of Iowa Hospitals and Clinics in 1977. Since then, 6000 patients with neurological symptoms and signs secondary to an abnormality in the craniocervical region have been studied. Understanding of the pathology of these abnormalities and their treatment is simplified if one has knowledge of the bony anatomy, biomechanics, and embryology of the region.
Anatomy of the Craniovertebral Junction
Bone-Ligament Complex
By itself, the geometry of the craniovertebral complex is meant to provide mobility at the cost of stability.4 However, the complex is maintained in a clamp-like vise by the craniovertebral musculature, which assists in stabilization of the spine, initiates movements within the joints, and maintains fluidity of the motion complex.
Blood Supply
The blood supply to the odontoid process is via anterior and posterior ascending vessels from the vertebral arteries, with a contribution from the carotid arteries, which form an apical arcade around the alar ligament.5 Thus, the blood supply of the odontoid process is vulnerable with a type II odontoid fracture.6
Parke and coworkers demonstrated pharyngovertebral veins with frequent lymphovenous anastomoses.7 This connection may provide an additional route for septic involvement of the craniovertebral complex, which can result in osteomyelitis of the bone, as well as joint effusions.
Lymphatic Drainage
Lymphatic drainage of the occipitoatlantoaxial joint complex is primarily into the retropharyngeal lymph nodes and then into the upper deep jugular cervical chain.3 These nodes also receive drainage from the nasopharynx, paranasal sinuses, and retropharyngeal area. A retrograde infection may affect the synovial lining of the craniovertebral joint complex and cause an inflammatory effusion, instability, and possible neurological deficit, thereby contributing to the so-called Grisel syndrome.8
Embryology and Development of Craniovertebral Junction Disorders
Congenital anomalies of the base of the skull and the atlanto-occipital region involve both the osseous structures and the nervous system. The frequent occurrence of patterns with various combinations suggests an interrelationship between if not a common cause of the origin and development of these structures.3
The bony cranial base is formed by the process of endochondral ossification in which a cartilaginous framework is first developed and subsequently resorbed, with further deposition of bone based on distorting forces such as brain and eye development.9,10 The clivus is elongated by sutural growth of the spheno-occipital synchondrosis and by further sutural growth along the lateral portion of the base.
The majority of the skull and facial bones develop by intramembranous ossification. Such development bypasses the intermediate cartilaginous stage characteristic of development of the bony cranial base.10,11
The occipital sclerotomes correspond to the segmental nerves that group together to form the hypoglossal nerve, which passes through the bone via the individual foramina.6,10 The first two occipital sclerotomes ultimately form the basiocciput. The third sclerotome is responsible for the exoccipital center as it forms the jugular tubercles.11,12 The key to understanding the craniovertebral junction is the embryology of the fourth occipital sclerotome, which in lower animals is called the proatlas. The hypocentrum of the fourth occipital sclerotome forms the anterior tubercle of the clivus. The centrum of the proatlas itself forms the apical cap of the dens, as well as the apical ligament. The neural arch component of the proatlas divides into a ventral-rostral component and a caudal-dorsal portion. The ventral portion forms the U-shaped anterior margin of the foramen magnum, as well as the occipital condyles and the midline occipital condyle. The cruciate ligament and the alar ligaments are condensations of the lateral portion of the proatlas. The caudal division of the neural arch of the proatlas forms the lateral atlantal masses of C1, as well as the superior portion of the posterior arch of the atlas.
During embryogenesis, the hypocentrum of the second spinal sclerotome disappears. The centrum forms the body of the axis vertebra, and the neural arches develop into facets and the posterior arch of the axis. Thus, the body of the dens arises from the first sclerotome, whereas a terminal portion of the odontoid process arises from the proatlas. The most inferior portion of the body of the axis is formed by the second spinal sclerotome. At birth, the odontoid process is separated from the body of the axis vertebra by a cartilaginous band that represents a vestigial disk and is referred to as the neural central synchondrosis.6,13 It lies below the level of the superior articular facets of the axis and does not represent the anatomic base of the dens. This synchondrosis is present in most children younger than 3 to 4 years and disappears by 8 years of age. In most instances, the odontoid process is seen at birth but does not fuse to the base of the axis.14 The tip of the odontoid is not ossified at birth and thus is not seen on lateral radiographs. It is represented by a separate ossification center, which is usually seen at 3 years of age and fuses with the remainder of the dens by the age of 12 years.
Expansion of the posterior fossa occurs as a result of a combination of endochondral resorption, sutural growth, and bony accretion.15 Growth of the basion elongates the basiocciput and lowers the frontal margins of the foramen magnum. There is a comparably matched resorptive drift downward and backward at the opisthion as a result of downward displacement of the cerebellum, together with rotation of the occipital and temporal lobes of the brain.16
Because of the forward inclination of the top-heavy cranial end of the fetus, the stability of the craniovertebral articulation must be maintained by the geometry of the articular surfaces of the craniovertebral junction, as well as by the ligamentous attachments and, more importantly, by the heavy development of the dorsal and lateral cervical musculature, which provides a clamping action on the craniovertebral region.3,4
Significant advances in recent years have occurred with the discovery of developmental control genes.9–11,15–18 Two families of regulatory genes have been implicated in the subsequent development of the sclerotomal portions of the somites during their resegmentation to form the vertebral primordia and in specification of the identity of each vertebra. Common to all these genes are phylogenetically highly conserved DNA sequences termed homeovox (Hox genes) and paired vox (Pax genes) sequences. They promote the production of proteins that modulate morphogenesis by influencing the transcription of specific downstream genes.
Teratogen-induced disturbances in Hox gene expression and mutations in Hox genes can cause alterations in both the number and identity of the cervical vertebrae forming at or near the limit of their expression domain. For example, inactivation of the Hox-D3 gene results in mutant mice with assimilation of the atlas to the basiocciput.19 The sensitivity of the occipitocervical junction to disturbances in Hox gene expression might prove to be the underlying cause of malformations in this region. Pax genes are expressed in diverse cell types and contribute to development of the early nervous system. Control of resegmentation of the sclerotomes to establish the intervertebral boundaries seems to be independently regulated by two genes in the Pax family.17,20
Implications of Craniovertebral Abnormalities
Congenital diseases involving connective tissue, such as Morquio’s syndrome and other mucopolysaccharidoses, as well as Down syndrome, can lead to severe atlantoaxial subluxation.3,14,21 There is a high incidence of anterior and posterior spina bifida of C1 and also os odontoideum in these instances. It is possible that because of abnormal, excessive head movements in the embryo between days 50 and 53, the process of chondrification is impaired, thereby resulting in anterior and later posterior spina bifida of C1.16
Os odontoideum was previously thought to be congenital and was described as a failure of fusion between the centrum component of C1 and that of C2. However, this radiographic abnormality always has a hypoplastic dens, and the neural central synchondrosis is a definite visible entity.13 To be congenital, the defect would have to be below the superior slopes of the C2 facets.6 Os odontoideum most likely originates as odontoid trauma that may predate birth but certainly occurs before the age of 4 years, with subsequent distraction and separation of the odontoid fragments.3,22 The superior segment is distracted upward by the apical and alar ligaments and receives its blood supply from the descending branch of the occipital artery, which courses along the apical ligament. This subsequently leads to incompetence of the cruciate ligament and further abnormalities.
Spine trauma in children younger than 8 years is mainly centered at the craniovertebral border because of the high fulcrum of neck motion.3 It results in ligamentous injuries more often than fractures. However, odontoid fractures in this age group are usually seen as avulsion injuries with separation of the neural central synchondrosis.
Anomalies and malformations of the caudal occipital sclerotomes are collectively called “manifestations of occipital vertebrae” and result in abnormal bony ridges and outgrowths of the ventral aspect of the foramen magnum.1 At times, segmentation abnormalities of the clivus are recognized and represent failure of the last occipital sclerotome to separate from the basiocciput and clivus. Consequently, bone growth in the anterior aspect of the foramen magnum indents the ventral cervicomedullary junction with age (as the clivus expands inferiorly and dorsally).
Biomechanics
The unique anatomic configuration of the craniovertebral junction creates a distinct biomechanical behavior that differs from that of the other spinal joints. The motion characteristics of the different levels of the craniovertebral junction are due to the geometry of the opposing bones of the vertebrae and the skull base, the shape of the joints, and the arrangements of the ligaments.4,23–25 An intervertebral disk is absent between the occiput, C1, and C2. The ball-and-socket–shaped occiput-C1 joint allows slightly more flexion-extension than the other levels of the cervical spine do, although it is quite rigid in axial rotation and lateral bending.
The biconvex articular surfaces of the C1 and C2 joints allow gliding and wide rotation of C1 around the dens. The atlantoaxial motion segment is the most flexible motion segment in the entire spine with respect to axial rotation in that it allows a bilateral range of motion of 90 degrees. More than half of all cervical axial rotation occurs at the C1-2 motion segment, a point that should be kept in mind when considering atlantoaxial arthrodesis. In a child, the amount of anterior-posterior translation that occurs between the dens and the anterior ring of the atlas is up to 5 mm. When the transverse component of the cruciate ligament has been disrupted, the alar ligaments are still intact.25 Hence, the amount of displacement remains between 5 and 6 mm until the alar ligaments become incompetent. It is only when the alar ligaments and the transverse portion of the cruciate ligament are incompetent that a separation of more than 5 to 6 mm occurs. Beyond the age of 8 years, excursion of the predental space must be limited to 3 mm.26
Failure of one alar ligament results in moderate rotary atlantoaxial instability.27 Bilateral transection of the alar ligaments causes considerably more alterations in craniovertebral motion, and the coupling action is disturbed. The transverse atlantal ligament is the strongest and thickest ligament in the entire spine. It is the predominant stabilizer of the atlas and constrains C1 around the dens. When torn, the transverse ligament is incapable of repair. Because injury to this ligament renders C1 grossly unstable, C1-2 fusion is mandated. The largest extent of rotation occurs at the atlantoaxial joint. When rotation exceeds 40 to 50 degrees, an interlock of the lateral inferior facet of the atlas over the superior articular facet of the axis vertebra occurs. If the transverse ligament becomes deficient, the anterior arch will sublux forward and result in unilateral dislocation and an interlock at less than 40 degrees.13,26 Rotation of more than 30 to 35 degrees produces an angulation of the contralateral vertebral artery.25,28 With greater rotation, the vertebral artery is stretched, and the ipsilateral vessel may demonstrate occlusion.28,29 This phenomenon has an implication in wrestling and football injuries and in certain head rotation maneuvers performed under general anesthesia or forced chiropractic manipulations.30,31 There is a muscle-clamping action that takes place in vivo and produces compressive force across the cervical spine. This muscular action results in the “interlocking stiffening” that maintains alignment during rotation. When the protective muscles are relaxed or inadequately developed, as in the case of a young child under general anesthesia, the craniovertebral junction becomes inherently less stable than that in an adult.3 As muscular development occurs, there is less tendency for instability at the craniovertebral junction.
Biomechanical Comparison of Cervical Orthoses
A halo brace is most effective at constraining cervical motion at all levels, and control of spinal motion is related to the distance of the spinal segment from the region of the rigid skull fixation.3,27 A halo brace provides the best control of motion at the craniovertebral junction, intermediate control of midcervical motion, and the least control of motion at the cervicothoracic junction. This pattern reflects snaking movements of the spine.
Classification of Craniovertebral Junction Abnormalities
A wide variety of congenital, developmental, and acquired abnormalities can occur at the craniovertebral junction, and there may be single or multiple abnormalities in the same individual. The pathology of these abnormalities is extensive.3 A working classification has been provided, but it must be appreciated that there are overlapping causes within this classification (Table 218-1).
TABLE 218-1 Classification of Craniocervical Junction Abnormalities
|
I Congenital anomalies and malformations of the craniocervical junction
|
Epidemiology
Abnormalities of the craniovertebral junction must be suspected in infants with Goldenhar’s syndrome, skeletal dysplasias, and Conradi’s syndrome.3 It should also be suspected in infants with “torticollis.” Diseases such as Down syndrome are associated with a 14% to 20% incidence of atlantoaxial dislocation. In Morquio’s syndrome, a combination of atlantoaxial instability, os odontoideum, and cervicothoracic abnormalities occurs in 30% to 50% of individuals.3,14,32 The incidence of atlas assimilation is 0.25% in the general population. Many such patients have associated segmentation failure of the upper cervical spine.33
We have reviewed syndromes cited as altering the craniovertebral junction in more than 6000 patients seen at our institution. A search of the scientific literature identified genetic syndromes that affect the craniovertebral junction. In addition, the Online Mendelian Inheritance in Man (OMIM) database was used to identify disorders of the craniovertebral junction encountered in various clinical populations. The information recorded includes systemic manifestations of the syndromes, the effect on the craniovertebral junction and cervical vertebrae, inheritance pattern, and genes or genetic loci associated with the disorder. A total of 84 syndromes have been identified that affect the craniovertebral junction.34
Clinical Findings
The most interesting feature of the clinical manifestation of craniovertebral abnormalities is their diversity as a result of compromise of the lower brainstem, cervical spinal cord, cranial nerves, cervical roots, and vascular supply.6 The symptoms of craniovertebral dysfunction may be insidious and be manifested as false localizing signs. Infrequently, rapid neurological progression is followed by sudden death. More often than not, an antecedent history of minor trauma induces a pattern of symptoms and signs that may progress at a galloping pace.
Congenital abnormalities of the craniovertebral junction are frequently associated with an abnormal general physical appearance. The head may be cocked to one side, as in patients with rotary luxation of the atlas on the axis, or the classic triad of Klippel-Feil syndrome (abnormally low hairline posteriorly, limitation of neck motion, and short neck) may be noted. Facial asymmetry and webbing of the neck may occur in conjunction with this syndrome. At times, scoliosis is present. It is not uncommon to see children with a small dysmorphic stature. There is an increased incidence of craniovertebral abnormalities with disease states such as achondroplasia, spondyloepiphyseal dysplasia, and related diseases of dwarfism. The most common neurological symptoms and signs are enumerated in Table 218-2.
TABLE 218-2 Signs and Symptoms of Craniovertebral Anomalies (Insidious or Rapid Onset of Symptoms and Signs)
Neurodiagnostic Imaging
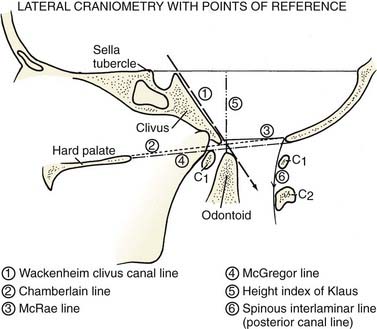
The diagnostic procedure of choice is magnetic resonance imaging (MRI), but plain cervical spine radiographs with lateral flexion and extension views are essential to gain a better understanding of the biomechanics. Numerous craniometric reference lines were described before the advent of computed tomography (CT) and MRI.33,35–38 They were used to locate the position of neural structures with the associated osseous abnormality (Fig. 218-1). Chamberlain’s palato-occipital line joins the hard palate to the posterior edge of the foramen magnum.2 In normal individuals, the tip of the dens should lie below this line or at most 3 to 5 mm above it. Basilar invagination occurs when the dens projects above this line. Wackenheim’s clivus canal line is of greatest significance.37,38 This line is drawn along the clivus and extrapolated into the cervical spinal canal. The odontoid process must be ventral to this line or tangential to it. The odontoid process transects this line in patients with basilar invagination, atlantoaxial dislocation, and anterior occipitoatlantal dislocation. McRae’s foramen magnum line joins the anterior and posterior edges of the foramen magnum.33 The tip of the dens must be below this line. If sagittal space for the cervicomedullary junction is less than 20 mm in a child older than 8 years, a neurological deficit is usually present.
Three-dimensional CT is useful for defining the abnormality, as well as for facilitating coronal and sagittal sectioning.38,39 We have found CT to be invaluable in recognizing the presence or absence of epiphyseal growth plates in toddlers and young children and for assessing the extent of fusion and segmentation defects, as well as “missing” osseous components. Spondylolisthesis and spondylolysis are best appreciated with three-dimensional studies.
Treatment
Several factors have an impact on the specific treatment of abnormalities of the craniovertebral junction6: (1) reducibility of the bony lesion (i.e., the ability to restore anatomic alignment and thereby relieve the compression on neural structures); (2) the mechanics of compression and the direction of encroachment; (3) the cause of the pathologic process, as well as the presence of hindbrain herniation, syrinx, and vascular abnormalities; and (4) the presence of abnormal ossification centers and epiphyseal growth plates.
Developmental Abnormalities Affecting the Craniovertebral Region
Grisel’s Syndrome
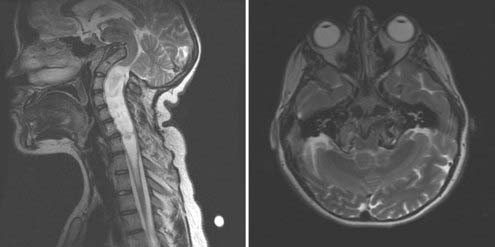
Grisel’s syndrome is defined as an inflammatory, spontaneous subluxation that affects the atlantoaxial joints after parapharyngeal infection.8 It may occur with tonsillitis, mastoiditis, retropharyngeal abscess, and otitis media. Most affected children are younger than 12 years. These children have torticollis, neck pain, or a neurological deficit.40 The severe subluxation may produce symptoms and signs of cervical cord compression (Fig. 218-2).
In 1987, Wilson and coworkers reviewed 62 cases of Grisel’s syndrome,41 including 14 children in whom subluxation developed after tonsillectomy, adenoidectomy, mastoiditis, and resection of a pharyngeal rhabdomyosarcoma. Twelve children had symptoms of pharyngitis or cervical adenitis, 7 had tonsillitis, and 7 had cervical abscesses. There were 5 children with acute rheumatic fever and 4 with acute mastoiditis. Several patients were assigned a diagnosis of nonspecific parapharyngeal infection.
Down Syndrome
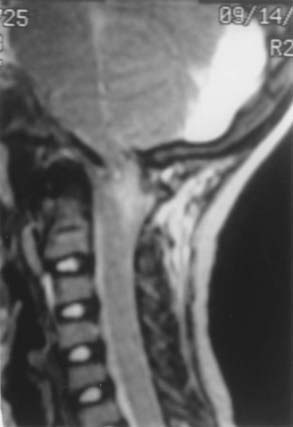
Down syndrome is easily recognized by the characteristic facial features, hypotonia, mental retardation, ligamentous laxity, and transverse palmar creases.3 Almost every organ is involved. This syndrome is the most commonly recognized chromosomal abnormality in humans, with an incidence of 1 in 700 live births. Craniovertebral instability in Down syndrome has received increasing attention since the report by Spitzer and associates of occipitoatlantal dislocation in 9 of 26 patients investigated with Down syndrome. However, atlantoaxial instability in Down syndrome has been described most since the initial publication by Tisher and Martel in 1965.42 The incidence of atlantoaxial instability is 14% to 24% in patients with Down syndrome, although the incidence of symptomatic atlantoaxial instability is believed to be less than 1% (Fig. 218-3). The prevalence of bony abnormalities such as os odontoideum, hypoplastic odontoid process, and rotary atlantoaxial luxation in patients with Down syndrome has caused concern about their participation in the Special Olympics, and it has been recommended that cervical spine radiographs be required for such participants.43 An atlantodental interval of greater than 4.5 mm was thought to require medical attention.44
The lack of attendant neurological symptoms and signs has resulted in the unfortunate belief that these children do not require surgical attention. Minor trauma, as well as upper respiratory infection, has resulted in severe neurological deficits in children with previously recognized pathologic instability without any intervention.4,35,45
There are pessimistic reports on the outcome of arthrodesis of the cervical spine in children with Down syndrome.46–48 Fortunately, excellent surgical results have been reported by Menezes and Ryken,49,50 as well as by Brockmeyer.51
Burke and coworkers studied 32 individuals with Down syndrome and found that only 1 had atlantoaxial instability at the onset of examination.35 However, at the 13-year follow-up, a predental space of greater than 5 mm had developed in 7 children. Two patients underwent atlantoaxial arthrodesis, and 1 succumbed to the disease before treatment. In 3 of the 32 individuals in this study, an odontoid abnormality consistent with os odontoideum developed. Review of the initial radiographs showed that these abnormalities were not present then.
One hundred ten patients were evaluated by the senior author up until 2004.50 Of these 110, 54 underwent surgical treatment. There were 34 males and 20 females ranging in age from 2 to 56 years. A follow-up of 54 patients showed that 36 underwent operative stabilization with a follow-up period of 26 to 118 months. Eighteen patients were treated conservatively.
Worley and colleagues from Duke Medical Center reviewed patients with Down syndrome and new onset of focal weakness.52 Ten cases of a new onset of focal weakness in patients with Down syndrome were encountered in a combined population of 850 from a clinic with a prevalence of 1.2. The median age at diagnosis was 4 years (range, 1 month to 44 years). The cause of the focal weakness was stroke from moyamoya in 2 patients, vascular occlusive disease in 1, venous sinus thrombosis in 1, brain abscess in 1, traumatic subdural hematoma in 1, spinal cord injury from cervical stenosis in 1, atlantoaxial instability in 1, and brachial plexus injury in 1. Thus, the differential diagnosis of a new onset of focal weakness in patients with Down syndrome is broad, and this must be kept in mind because of the multiorgan involvement.
The results of combined neurosurgery and orthopedic investigation were reported at the International Society of Pediatric Neurosurgery in Vancouver in 2005. It was recommended that surgical treatment should be offered to all children with Down’s syndrome and neurological dysfunction.50,53,54 Craniovertebral junction instability in association with a dystopic os odontoideum, as well as the combination of bifid anterior and posterior atlantal arches, required surgical attention. Reducible basilar invagination mandated primary stabilization. Atlantoaxial instability with sagittal plane excursion of more than 8 mm required surgical attention.50,51
Proatlas Segmentation Failures or Manifestations of Occipital Vertebrae
Proatlas segmentation failures result in malformation and anomalies of the most caudal occipital sclerotomes. These abnormalities surround the foramen magnum and usually involve the posterior arch of C1.3,15,55–57 Hindbrain herniation is associated with these abnormalities in 32% to 38% of individuals. At times, the proatlas component of the dens may fail to separate from the portion that forms the basiocciput of the clivus. Consequently, the anterior arch of the atlas comes to rest above the body of the axis. Sometimes the proatlas abnormality is united with the clivus and grossly distorts the cervicomedullary junction ventrally. Variations of this anomaly may be observed in the midline, laterally, and at times dorsally (Fig. 218-4).55
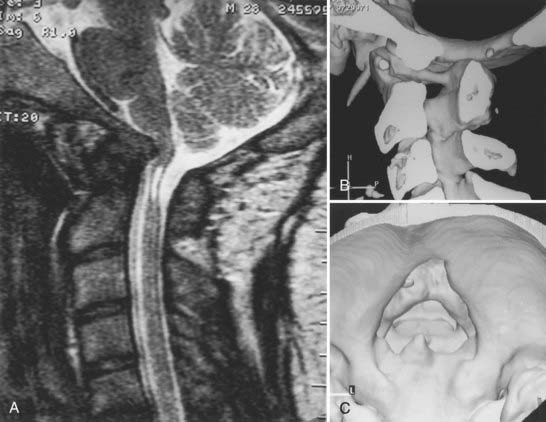
The best definition of the abnormality was achieved with three-dimensional CT combined with MRI. Hindbrain herniation was present when the volume of the posterior fossa was reduced, especially by distortion of the vertical height of the posterior fossa.56 Treatment of this condition consists of precise definition of the abnormality, relief of neurovascular compression, and prevention of recurrence by stabilization. These abnormalities are fairly complex and require careful study of preoperative images.
Assimilation of the Atlas and Klippel-Feil Syndrome
Assimilation of the atlas results from failure of segmentation between the fourth occipital sclerotome and the first spinal sclerotome. It may be unilateral, focal, segmental, or bilateral3 and occurs in conjunction with abnormalities such as Klippel-Feil syndrome (Fig. 218-5A and B). In this case, basilar invagination is a secondary phenomenon.58 Atlas assimilation was encountered in 550 of 6000 patients in the craniovertebral junction database.15 Hindbrain herniation occurred in 38% and was caused by a reduction in the volume of the posterior fossa. The situation is compounded by failure of segmentation of the second and third cervical vertebrae.55,59 When associated with atlas assimilation, atlantoaxial instability occurs as a result of abnormal load placed on the motion segment.3,55,58 Initially, the atlantoaxial instability will be reducible. The sequence of events is that pannus forms around the odontoid process, and at this time the lesion is reducible. However, as the child grows, grooving occurs behind the occipital condyles secondary to upward migration of the axis vertebra. This was described by Menezes in 1987. It is then followed by reducible basilar invagination up to approximately the age of 14 to 15.60 As these children age, the lesion becomes an irreducible basilar invagination. During the phase of reducibility and partial reducibility, a prolific granulation tissue mass sits above the odontoid process surrounding it. It is nature’s attempt to reduce the excursion. This, in turn, compounds the compression on the cervicomedullary junction.3,58,61
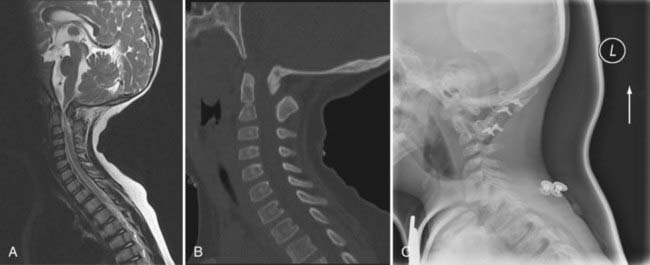
In patients with irreducible basilar invagination under these circumstances, the clivus tends to be horizontally oriented; there is an abnormal groove behind the occipital condyles that pushes up the skull base and results in the horizontal clivus.6 This leads to complete irreducibility. Thus, a child who is being evaluated for atlantoaxial dislocation is more likely than a full-grown adult to have a reducible atlantoaxial dislocation or a reducible basilar invagination.60 Hence, an operative procedure that relies on posterior decompression for “hindbrain herniation” without addressing the potential for instability can lead to unfortunate results.
The Klippel-Feil triad consists of a short neck, webbed neck, and low hairline with limitation of neck motion.61–63 There may be associated deafness, a high arched palate, facial palsies, and cardiovascular abnormalities. Abnormal rib fusions and scoliosis are common, and 30% of individuals have genitourinary tract abnormalities.64,65
Basilar Invagination
Basilar invagination is a primary developmental defect that implies prolapse of the vertebral column into the base of the skull. It is frequently associated with developmental anomalies of the region, such as occipitalization, assimilation, and block vertebra.3,13,14,33 The common manifestations of neurodysgenesis, such as a Chiari I malformation and syringohydromyelia, occur in 25% to 30% of patients with basilar invagination (Fig. 218-6).
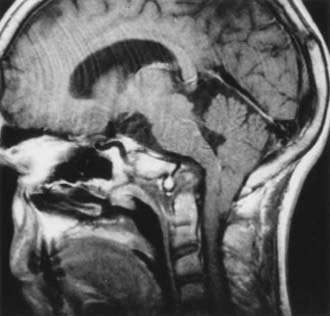
The term basilar invagination was used by Chamberlain in 1939 as a synonym for platybasia.2 In addition, basilar impression has been used interchangeably with the latter. Basilar impression is an acquired form of basilar invagination caused by softening of the skull, and it occurs in diseases such as osteogenesis imperfecta, rickets, hyperparathyroidism, Paget’s disease, Hurler’s syndrome, and acro-osteolysis (Hajdu-Cheney syndrome).32,66–72 Platybasia refers to an abnormal obtuse basal angle formed by the clivus and the anterior skull base. There are no symptoms or signs attributable to platybasia alone. It is not a measure of basilar invagination, although it may be associated with invagination.13
There are two types of basilar invagination. In the ventral variety there is shortening of the basiocciput, so the clivus is short and horizontally oriented and thus displaces the plane of the foramen magnum in an upward direction in relation to the spinal column.6 In the paramesial type, condylar hypoplasia may be present, and the clivus becomes dorsally displaced into the posterior fossa and may be of normal length.3 The occipital hypoplasia may be unilateral and leads to torticollis.38 The distinction between these two types is not clinically rigid because a mixture often occurs.
Basilar invagination is commonly associated with an abnormal odontoid process invaginating into the posterior fossa. Of significance is the fact that the body of the axis becomes elongated and the true odontoid process is small.3,14,15 Of greater significance is the abnormal clivus-odontoid articulation. The resultant abnormal clivus-canal angle produces an indentation on the pons, medulla, or cervicomedullary junction in a ventral manner.
The Chiari malformation is associated with basilar invagination in about 25% to 30% of individuals.3,60 In this situation, ventral compression of the cervicomedullary junction by the bony abnormality should be relieved by ventral decompression, which must be performed before any posterior surgical procedure. If a posterior procedure is done before relieving the ventral compression, 30% of individuals have an unfavorable result. The reason for this poor outcome is that the ventral abnormality acts as a peg that indents into the pontomedullary or cervicomedullary junction when the patient is positioned for the posterior procedure. In addition, fusion in a flexed position compromises the surgeon’s ability to perform a satisfactory ventral decompression. The ability to reduce the invagination is related to age, as previously described for atlas assimilation. The presence of syringohydromyelia with hindbrain herniation and basilar invagination should not sway the treating neurosurgeon to perform a posterior operative procedure. In most cases, the syringohydromyelia disappears once the ventral abnormality has been corrected.39
Anomalies of the Odontoid Process
Aplasia-Hypoplasia of the Dens
Dens aplasia-hypoplasia has several degrees of expression, and a rudimentary dens may be present or completely absent.13,15 Theoretically, the cruciate ligament is incompetent, thereby leading to atlantoaxial instability. This is quite common in patients with atlas assimilation and failure of segmentation of C2 and C3. In these individuals, the body of the axis is abnormal, but the dens itself may be hypoplastic. Significant vascular compromise from stretching and distortion of the vertebral arteries has been seen with such lesions.3,29 Chronic atlantoaxial dislocation in this situation may result in the formation of granulation tissue at the site of luxation and compression of the cervicomedullary junction.
Os Odontoideum
Os odontoideum refers to an independent bone cranial to the axis in the place of the dens. It is not an isolated dens but exists apart from a small hypoplastic dens. Radiographically, an os odontoideum has rounded, smooth cortical borders separated by a variable gap from a small odontoid process.3,13 It is usually located in the position of the normal odontoid tip or near the basiocciput in the area of the foramen magnum, where it may fuse with the clivus. The gap between the free ossicle and the axis usually extends above the level of the superior facets of the axis. This leads to incompetence of the cruciate ligament and, subsequently, atlantoaxial instability.
Fielding and Griffin described two types of os odontoideum22: the orthotopic variety, in which the ossicle lies in the position of the normal dens and moves in unison with the atlas and axis vertebrae, and the dystopic variety, in which the ossicle lies near the inferior end of the clivus, fuses with the occipital bone, and moves in unison with the clivus (Fig. 218-7A and B).
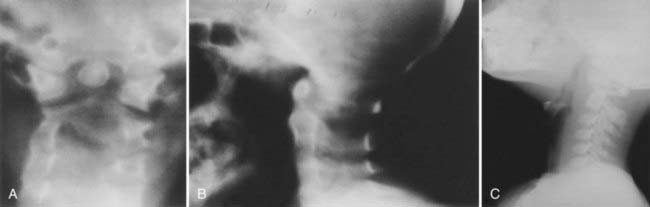
Evidence favors an unrecognized fracture in the region of the base of the odontoid as the most common cause of an os odontoideum.4,15,22 It has been seen in patients who have been shown to have a complete odontoid process in early childhood with the subsequent development of os odontoideum, in one of identical twins who experienced cervical trauma, and in patients with gross ligamentous laxity such as those with Down or Morquio’s syndrome.14,35,72
Basilar Impression and Bone-Softening Syndrome
Basilar impression arises as a consequence of constitutive skeletal disease. These so-called bone-softening disorders may be congenital, as in osteogenesis imperfecta, spondyloepiphyseal dysplasia, acro-osteolysis, Hurler’s syndrome, and achondroplasia, or they may be associated with Paget’s disease, osteomalacia, hyperparathyroidism, and renal rickets.32,66–70,73–75
Anatomically and radiographically, there is infolding of the squama occipitalis; as a result, the posterior fossa floor becomes elevated, and the margin of the foramen magnum curves upward.76,77 The basiocciput becomes foreshortened and elevated, and the clivus becomes thinned, truncated, and horizontally oriented, conditions creating an acute craniovertebral angle. Anteroposteriorly, the petrous portion of the temporal bone is also deformed. These changes ultimately permit the clivus-atlas-odontoid complex to assume an abnormally rostral location within the foramen magnum and further restrict the space within the posterior fossa (Fig. 218-8).
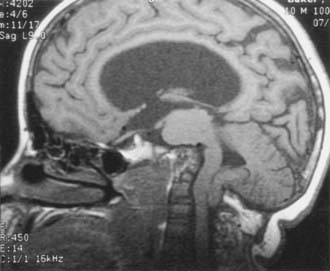
The rostral extent of invagination dictates the attendant neurological manifestations.73–75 The brainstem is elevated and splayed over the ventrally situated clivus-atlas-odontoid complex. In addition to causing mechanical compression, it acts as a fulcrum by which traction is applied to the caudal brainstem and the rostral cervical spinal cord and produces bulbar dysfunction and myelopathy.6 The lower cranial nerves are stretched and distorted as the brainstem is forced upward, thereby resulting in characteristic lower cranial nerve palsies.71,74 Cerebellar involvement may be primary and be caused by compression from infolding of the foramen magnum or secondary and be caused by hindbrain herniation (Fig. 218-9). This acquired form of hindbrain herniation may lead to the development of syringohydromyelia. Neurological dysfunction may thus be coupled with hydrocephalus, syringohydromyelia, and hindbrain herniation.
The pathogenesis of secondary invagination and osteochondrodysplasia remains obscure.74 Certainly, there is inherent bone fragility, with the load-bearing chondrocranium unable to maintain the weight of the cranium and its contents. Thus, both the skull base and the cranial vault are deformed. It is possible that recurrent microfractures in the region of the foramen magnum underlie the progressive infolding of the posterior skull base.75 At surgery, it is not uncommon to find proliferation of bone composing the skull base, a finding reminiscent of exuberant callus.76 The high metabolic activity of the reactive bone is confirmed by radioactive bone scanning and is indicative of chronic active bone remodeling.
Osteogenesis imperfecta is noted for its wide clinical and genetic variability; given the low tensile strength and load-bearing properties of bone in these syndromes, it is surprising that basilar impression is not more common. The various types of osteogenesis imperfecta have different clinical manifestations. Infants born with type II osteogenesis imperfecta have the most severe bone fragility and a high propensity for fractures because of a reduction in type I collagen synthesis. Hence, neonates with this variant rarely survive past infancy. In the other subtypes, abnormalities of the atlas and axis region and upper cervical buckling lead to quadriparesis.77–79 In young individuals with basilar impression or upper cervical abnormalities, we recommend a custom-fitted modified Minerva brace to “shore up” the skull and prevent progression of the secondary invagination.76,80 This must be documented with MRI.76
Surgical intervention, regardless of the approach, was well tolerated in our series, with a 100% fusion rate. Neural decompression was accomplished in 32 individuals. All these symptomatic patients improved, with good fusion. However, although the functional improvement persisted, basilar invagination progressed in 80% of patients according to imaging criteria despite successful fusion. In all these patients the entire fusion mass migrated rostrally as a result of further squamous occipital and petrous bone infolding.3,6,76 Additionally, the posterior fusion mass tends to act as a fulcrum on which the lever arm of the anterior skull base turns, thereby exacerbating the ventral compression. In all patients who were managed with a Minerva orthosis, the brace provided ventral cranial base stability, thus affording relief of symptoms and preventing further skeletal deformity.
Skeletal Dysplasias
Skeletal dysplasias are divided into five large categories: osteochondral dysplasia, dysostosis, idiopathic osteolysis, chromosomal aberrations, and primary metabolic abnormalities.68,75 Osteochondral dysplasias and dysostosis account for the largest and most complex entities.
The primary metabolic and chromosomal abnormalities are numerous. Metabolic abnormalities include problems with calcium or phosphorus metabolism such as rickets and pseudohypoparathyroidism. Abnormalities of calcium and phosphorus metabolism lead to bone softening and a secondary form of invagination. This may be paramesial, which is common with achondroplasia, in which case the sagittal diameter of the foramen magnum is preserved and the transverse diameter is markedly reduced (Fig. 218-10). In addition, an inward bending of the exoccipital bone results in further invagination and the creation of a dural shelf that compresses the dorsal cervicomedullary junction. Thus, in patients with these syndromes, posterior decompression with duraplasty is necessary.75,81
Atlantoaxial instability occurs with increasing incidence in the skeletal dysplasias. Sleep apnea is a major symptom, as is progressive spastic quadriparesis.82,83 There is inward bending of the posterior arch of the atlas that adds to the paramesial foramen magnum stenosis.75 Before embarking on therapy, one must ascertain that proper attention has been directed to hydrocephalus, if present.
Mucopolysaccharidosis
The mucopolysaccharidoses are primary metabolic abnormalities of complex carbohydrate metabolism. These inheritable storage diseases are often manifested as dwarfism, mental retardation, macrocephaly, corneal clouding, and skeletal dysplasia. Generalized ligamentous laxity is thought to contribute to the atlantoaxial luxation described in a variety of mucopolysaccharidoses. Death commonly occurs as a result of Morquio’s syndrome (mucopolysaccharidosis type IV) by the age of 7 years secondary to cervical myelopathy, as well as effects on the respiratory system and resultant hypoxia. Holzgreve and coworkers described atlantoaxial instability in a review of 13 patients with type IV mucopolysaccharidosis.72
Aryanpur J, Hurko O, Francomano C, et al. Craniocervical decompression for cervicomedullary compression in pediatric patients with achondroplasia. J Neurosurg. 1990;73:375-382.
Burke SW, French HA, Roberts E. Chronic atlantoaxial instability in Down’s syndrome. J Bone Joint Surg Am. 1985;67:1356-1360.
McAllion SJ, Paterson CR. Causes of death in osteogenesis imperfecta. J Clin Pathol. 1996;49:627-630.
McRae DL, Barnum AS. Occipitalization of the atlas. AJR Am J Roentgenol. 1953;70:23-46.
Menezes AH. Specific entities affecting the craniocervical region: Down’s syndrome. Childs Nerv Syst. 2008;24:1165-1168.
Menezes AH. Craniocervical developmental anatomy and its implications. Childs Nerv Syst. 2008;24:1109-1122.
Menezes AH, Fenoy KA. Remnants of occipital vertebrae: proatlas segmentation abnormalities. Neurosurgery. 2009;64:945-953.
Menezes AH, Foltz GD. Transoral approach to the ventral craniocervical border. Oper Tech Neurosurg. 2005;8:150-157.
Panjabi MM, Dvorák J, Duranceau J, et al. Three-dimensional movements of the upper cervical spine. Spine. 1988;13:726-730.
Parke WW, Rothman RH, Brown MD. The pharyngovertebral veins. An anatomical rationale for Grisel’s syndrome. J Bone Joint Surg Am. 1984;66:568.
Pauli RM, Horton VK, Glinski LP, et al. Prospective assessment of risks for cervicomedullary junction compression in infants with achondroplasia. Am J Hum Genet. 1995;56:32-744.
Sawin PD, Menezes AH. Basilar invagination in osteogenesis imperfecta and related osteochondrodysplasias: medical and surgical management. J Neurosurg. 1997;86:950-960.
Schiff DCM, Parke WW. The arterial blood supply of the odontoid process (dens). Anat Rec. 1972;172:399-400.
Selecki BR. The effects of rotation of the atlas on the axis: experimental work. Med J Aust. 1969;1:1012-1015.
Shen FH, Samartzis D, Herman J, et al. Radiographic assessment of segmental motion at the atlantoaxial junction in the Klippel-Feil patient. Spine. 2006;31:171-177.
Smoker WRK, Khanna G. Imaging the craniocervical junction. Childs Nerv Syst. 2008;24:1123-1145.
VonTorklus D, Gehle W. The upper cervical spine. Regional anatomy, pathology and traumatology. In: Allen M, editor. A Systemic Radiological Atlas and Textbook. New York: Grune & Stratton; 1972:1-99.
Werne S. Studies in spontaneous atlas dislocation. The craniovertebral joints. Acta Orthop Scand Suppl. 1957;23:11-83.
1 Gladstone J, Erickson-Powell W. Manifestation of occipital vertebra and fusion of atlas with occipital bone. J Anat Physiol. 1914-1915;49:190-199.
2 Chamberlain WE. Basilar impression (platybasia). Yale J Biol Med. 1938/1939;11:487.
3 Menezes AH. Congenital and acquired abnormalities of the craniovertebral junction. In: Youmans J, editor. Neurological Surgery. 4th ed. Philadelphia: WB Saunders; 1995:1035-1089.
4 Goel VK, Clark CR, Gallaes K, et al. Moment-rotation relationships of the ligamentous occipito-atlanto-axial complex. J Biomechanics. 1988;21:673-680.
5 Schiff DCM, Parke WW. The arterial blood supply of the odontoid process (dens). Anat Rec. 1972;172:399-400.
6 Menezes AH. Developmental and acquired abnormalities of the craniovertebral junction. In: Crockard HA, Hayward RD, Hoff JT, editors. Neurosurgery—The Scientific Basis of Clinical Practice. 3rd ed. London: Blackwell Scientific; 2000:1120-1145.
7 Parke WW, Rothman RH, Brown MD. The pharyngovertebral veins. An anatomical rationale for Grisel’s syndrome. J Bone Joint Surg Am. 1984;66:568.
8 Grisel P. Enucleation de l’atlas et torticollis nasopharyngien. Presse Med. 1930;38:50-56.
9 Christ B, Wilting J. From somites to vertebral column. Ann Anat. 1992;174:23-32.
10 Dietrich S, Kessel M. The vertebral column. In: Thorogood P, editor. Embryos, Genes and Birth Defects. Chichester, UK: Wiley; 1997:281-302.
11 Kessel M, Gruss P. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell. 1991;67:89-104.
12 Keynes RJ, Stern C. Mechanisms of vertebrate segmentation. Development. 1988;103:413-429.
13 VonTorklus D, Gehle W. The upper cervical spine. Regional anatomy, pathology and traumatology. In: Allen M, editor. A Systemic Radiological Atlas and Textbook. New York: Grune & Stratton; 1972:1-99.
14 Stevens JM, Chong WK, Barber C, et al. A new appraisal of abnormalities of the odontoid process associated with atlantoaxial subluxation and neurological disability. Brain. 1994;117:133-148.
15 Menezes AH. Craniocervical developmental anatomy and its implications. Childs Nerv Syst. 2008;24:1109-1122.
16 David KM, McLachlan JC, Aiton JF, et al. Cartilaginous development of the human craniovertebral junction as visualized by a new three-dimensional computer reconstruction technique. J Anat. 1998;192:269-277.
17 Koseki H, Wallin J, Wilting J, et al. A role of Pax-1 as a mediator of notochordal signals during the dorsoventral specification of vertebrae. Development. 1993;119:649-660.
18 Lufkin T, Mark M, Hart CP, et al. Homeotic transformation of the occipital bones of the skull by ectopic expression of a homeobox gene. Nature. 1992;359:835-841.
19 Condie BG, Capecchi MR. Mice homozygous for a targeted disruption of Hoxd-3 (Hox-4.1) exhibit anterior transformations of the first and second cervical vertebrae, the atlas and the axis. Development. 1993;119:579-595.
20 Kimura Y, Matsunami H, Inoue T, et al. Cadherin-11 expressed in association with mesenchymal morphogenesis in the head, somite and limb bud of early mouse embryos. Dev Biol. 1995;169:347-358.
21 Menezes AH. Specific entities affecting the craniocervical region: syndromes affecting the craniocervical junction. Childs Nerv Syst. 2008;24:1155-1163.
22 Fielding JW, Griffin PP. Os odontoideum. An acquired lesion. J Bone Joint Surg Am. 1974;56:187-190.
23 Panjabi MM, Dvorák J, Duranceau J, et al. Three-dimensional movements of the upper cervical spine. Spine. 1988;13:726-730.
24 Panjabi MM, Dvorák J, Crisco JJIII, et al. Flexion, extension and lateral bending of the upper cervical spine in response to alar ligament transections. J Spinal Disord. 1991:4157-4167.
25 Werne S. Studies in spontaneous atlas dislocation. The craniovertebral joints. Acta Orthop Scand Suppl. 1957;23:11-83.
26 Fielding JW, Cochron GB, Lawsing JFIII, et al. Tears of the transverse ligament of the atlas. A clinical and biomechanical study. J Bone Joint Surg Am. 1974;56:1683-1691.
27 Dickman CA, Crawford NR, Paramore CG. Biomechanical characteristics of C1-2 cable fixations. J Neurosurg. 1996;85:316-322.
28 Selecki BR. The effects of rotation of the atlas on the axis: experimental work. Med J Aust. 1969;1:1012-1015.
29 Bhatnagar M, Sponseller PD, Carroll CIV, et al. Pediatric atlantoaxial instability presenting as cerebral and cerebellar infarcts. J Pediatr Orthop. 1991;11:103-107.
30 Miyachi S, Okamura K, Watanabe M, et al. Cerebellar stroke due to vertebral artery occlusion after cervical spine trauma. Two case reports. Spine. 1994;19:83-88.
31 Okawara S, Nibbelink D. Vertebral artery occlusion following hyperextension and rotation of the head. Stroke. 1974;5:640-642.
32 Blaw ME, Langer LO. Spinal cord compression in Morquio-Brailsford’s disease. J Pediatr. 1969;74:593-600.
33 McRae DL, Barnum AS. Occipitalization of the atlas. AJR Am J Roentgen. 1953;70:23-46.
34 Menezes AH. Craniovertebral junction database analysis: incidence, classification, presentation and treatment algorithms. Childs Nerv Syst. 2008;24:1101-1108.
35 Burke SW, French HA, Roberts E. Chronic atlantoaxial instability in Down’s syndrome. J Bone Joint Surg Am. 1985;67:1356-1360.
36 McGregor M. The significance of certain measurements of the skull in the diagnosis of basilar impression. Br J Radiol. 1948;21:171-181.
37 Wackenheim A. Radiologic diagnosis of congenital forms, intermittent forms and progressive forms of stenosis of the spinal canal of the level of the atlas. Acta Radiol Diagn (Stockh). 1969;9:481-486.
38 Smoker WRK, Khanna G. Imaging the craniocervical junction. Childs Nerv Syst. 2008;24:1123-1145.
39 Menezes AH, Foltz GD. Transoral approach to the ventral craniocervical border. Oper Tech Neurosurg. 2005;8:150-157.
40 Wetzel FT, LaRocca H. Grisel’s syndrome. A review. Clin Orthop Rel Res. 1989;240:141-152.
41 Wilson BC, Jarvis BL, Handon RCJr. Nontraumatic subluxation of the atlantoaxial joint: Grisel’s syndrome. Ann Otol Rhinol Laryngol. 1987;96:705-708.
42 Tisher J, Martel W. Dislocation of the atlas in mongolism. Radiology. 1965;84:904-906.
43 American Academy of Pediatrics. Committee on Sports Medicine. Atlantoaxial instability in Down syndrome. Pediatrics. 1984;74:152-154.
44 Winnell J, Burke SW. Sports participation of children with Down syndrome. Orthop Clin North Am. 2003;34:439-443.
45 Msall ME, Reese ME, DiGaudio K, et al. Symptomatic atlantoaxial instability associated with medical and rehabilitative procedures in children with Down syndrome. Pediatrics. 1990;85:447-449.
46 Arlet V, Rigault P, Padovani JP, et al. Atlantoaxial instability in children with trisomy 21: atlantoaxial (C1-C2) or occipitoaxial (O-C2) arthrodesis? Rev Chir Orthop Reparatrice Appar Mot. 1992;78:240-247.
47 Rizzolo S, Lemos MJ, Mason DE. Posterior spinal arthrodesis for atlantoaxial instability in Down syndrome. J Pediatr Orthop. 1995;15:543-548.
48 Segal LS, Drummond DS, Zanotti RM, et al. Complications of posterior arthrodesis of the cervical spine in patients who have Down’s syndrome. J Bone Joint Surg Am. 1991;73:1547-1554.
49 Menezes AH, Ryken TC. Craniovertebral abnormalities in Down’s syndrome. Pediatr Neurosurg. 1992;18:24-33.
50 Menezes AH. Specific entities affecting the craniocervical region: Down’s syndrome. Childs Nerv Syst. 2008;24:1165-1168.
51 Brockmeyer D. Down syndrome and craniovertebral instability. Topic review and treatment recommendations. Pediatr Neurosurg. 1999;31:71-77.
52 Worley G, Shbarou R, Heffner AN, et al. New onset focal weakness in children with Down syndrome. Am J Med Genet A. 2004;128:15-18.
53 Taggard DA, Menezes AH, Ryken TC. Treatment of Down syndrome–associated craniovertebral junction abnormalities. J Neurosurg Spine. 2000;93:205-213.
54 Tredwell SJ, Newman DE, Lockitch G. Instability of the upper cervical spine in Down’s syndrome. J Pediatr Orthop. 1990;10:602-606.
55 Menezes AH, Fenoy KA. Remnants of occipital vertebrae: proatlas segmentation abnormalities. Neurosurgery. 2009;64:945-953.
56 Nishikawa M, Sakamoto H, Hakuba A, et al. Pathogenesis of Chiari malformation: a morphometric study of the posterior cranial fossa. J Neurosurg. 1997;86:40-47.
57 Komatsu Y, Shibata T, Yasuda S, et al. Atlas hypoplasia as a cause of high cervical myelopathy. Case report. J Neurosurg. 1993;79:917-919.
58 Gholve PA, Hosalka HS, Ricchetti ET, et al. Occipitalization of the atlas in children. J Bone Joint Surg Am. 2007;89:571-578.
59 Shen FH, Samartzis D, Herman J, et al. Radiographic assessment of segmental motion at the atlantoaxial junction in the Klippel-Feil patient. Spine. 2006;31:171-177.
60 Menezes AH. Primary craniovertebral anomalies and the hindbrain herniation syndrome (Chiari I): data base analysis. Pediatr Neurosurg. 1995;23:260-269.
61 Pizzutillo PD, Woods M, Nicholson L, et al. Risk factors in Klippel-Feil syndrome. Spine. 1994;19:2110-2116.
62 Marin-Padilla M, Marin-Padilla T. Morphogenesis of experimentally induced Arnold-Chiari malformation. J Neurol Sci. 1981;50:29-55.
63 Palant DI, Carter BL. Klippel-Feil syndrome and deafness. Am J Disabled Child. 1972;123:218-221.
64 Pueschel SM, Scola F. Atlantoaxial instability in individuals with Down’s syndrome: epidemiologic, radiographic and clinical studies. Pediatrics. 1987;80:555-560.
65 Müller F, O’Rahilly R. Occipitocervical segmentation in staged human embryos. J Anat. 1994;185:251-258.
66 Aryanpur J, Hurko O, Francomano C, et al. Craniocervical decompression for cervicomedullary compression in pediatric patients with achondroplasia. J Neurosurg. 1990;73:375-382.
67 Cheney WD. Acro-osteolysis. Am J Roentgenol Ther Nucl Med. 1965;94:595-607.
68 Dubousset J. Cervical abnormalities in osteochondroplasia. Basic Life Sci. 1988;48:215-217.
69 Gertner JM, Root L. Osteogenesis imperfecta. Orthop Clinic North Am. 1990;21:151-162.
70 Hajdu N, Kauntze R. Cranioskeletal dysplasia. Br J Radiol. 1948;21:42-48.
71 Harkey HL, Crockard HA, Stevens JM, et al. The operative management of basilar impression in osteogenesis imperfecta. Neurosurgery. 1990;27:782-786.
72 Holzgreve W, Grope H, VonFigwrak E. Morquio syndrome. Clinical findings in 11 patients with mucopolysaccharidosis type IVA and two with mucopolysaccharidosis type IVB. Hum Genet. 1981;57:360-365.
73 Fremion AS, Garg BP, Kalsbeck J. Apnea as the sole manifestation of cord compression in achondroplasia. J Pediatr. 1984;104:398-401.
74 Hecht JT, Butler IJ. Neurologic morbidity associated with achondroplasia. J Child Neurol. 1990;5:84-97.
75 Ryken TC, Menezes AH. Cervicomedullary compression in achondroplasia. J Neurosurg. 1994;81:43-48.
76 Sawin PD, Menezes AH. Basilar invagination in osteogenesis imperfecta and related osteochondrodysplasias: medical and surgical management. J Neurosurg. 1997;86:950-960.
77 Menezes AH. Specific entities affecting the craniocervical region. Osteogenesis imperfecta and related osteochondrodysplasias: medical and surgical management of basilar impression. Childs Nerv Syst. 2008;24:1169-1172.
78 McAllion SJ, Paterson CR. Causes of death in osteogenesis imperfecta. J Clin Pathol. 1996;49:627-630.
79 Menezes AH. Osteogenesis imperfecta [editorial]. J Neurosurg. 2006;105:359-360.
80 Hunt TE, Dekaban AS. Modified head-neck support for basilar invagination with brain-stem compression. J Can Med Assoc. 1982;126:947-948.
81 Bagley CA, Pindrik JA, Brookland MJ, et al. Cervicomedullary decompression for foramen magnum stenosis in achondroplasia. J Neurosurg. 2006;104(suppl 3):166-172.
82 Pauli RM, Horton VK, Glinski LP, et al. Prospective assessment of risks for cervicomedullary junction compression in infants with achondroplasia. Am J Hum Genet. 1995;56:32-744.
83 Wassman ERJr, Rimoin DL. Cervicomedullary compression with achondroplasia. J Pediatr. 1988;113:411.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 218 Developmental Abnormalities of the Craniocervical Junction
The first anatomic description of “manifestations of occipital vertebrae” was attributed to Meckel in 1815 by Gladstone and Erickson-Powell.1 Its clinical significance became appreciated after the classic radiologic studies on basilar invagination by Chamberlain in 1939.2
The term craniovertebral junction refers to the occipital bone that surrounds the foramen magnum and the atlas and axis vertebrae. Into the 1970s, surgical treatment of conditions affecting the craniovertebral junction consisted of posterior decompression by enlargement of the foramen magnum and removal of the posterior arch of the atlas and axis vertebrae. However, mortality and morbidity rates associated with this treatment were high in patients with irreducible lesions and cervicomedullary compression.3 A physiologic approach based on an understanding of craniovertebral dynamics, the site of encroachment, and stability of the craniovertebral junction was adopted at the University of Iowa Hospitals and Clinics in 1977. Since then, 6000 patients with neurological symptoms and signs secondary to an abnormality in the craniocervical region have been studied. Understanding of the pathology of these abnormalities and their treatment is simplified if one has knowledge of the bony anatomy, biomechanics, and embryology of the region.
Anatomy of the Craniovertebral Junction
Bone-Ligament Complex
By itself, the geometry of the craniovertebral complex is meant to provide mobility at the cost of stability.4 However, the complex is maintained in a clamp-like vise by the craniovertebral musculature, which assists in stabilization of the spine, initiates movements within the joints, and maintains fluidity of the motion complex.
Blood Supply
The blood supply to the odontoid process is via anterior and posterior ascending vessels from the vertebral arteries, with a contribution from the carotid arteries, which form an apical arcade around the alar ligament.5 Thus, the blood supply of the odontoid process is vulnerable with a type II odontoid fracture.6
Parke and coworkers demonstrated pharyngovertebral veins with frequent lymphovenous anastomoses.7 This connection may provide an additional route for septic involvement of the craniovertebral complex, which can result in osteomyelitis of the bone, as well as joint effusions.
Lymphatic Drainage
Lymphatic drainage of the occipitoatlantoaxial joint complex is primarily into the retropharyngeal lymph nodes and then into the upper deep jugular cervical chain.3 These nodes also receive drainage from the nasopharynx, paranasal sinuses, and retropharyngeal area. A retrograde infection may affect the synovial lining of the craniovertebral joint complex and cause an inflammatory effusion, instability, and possible neurological deficit, thereby contributing to the so-called Grisel syndrome.8
Embryology and Development of Craniovertebral Junction Disorders
Congenital anomalies of the base of the skull and the atlanto-occipital region involve both the osseous structures and the nervous system. The frequent occurrence of patterns with various combinations suggests an interrelationship between if not a common cause of the origin and development of these structures.3
The bony cranial base is formed by the process of endochondral ossification in which a cartilaginous framework is first developed and subsequently resorbed, with further deposition of bone based on distorting forces such as brain and eye development.9,10 The clivus is elongated by sutural growth of the spheno-occipital synchondrosis and by further sutural growth along the lateral portion of the base.
The majority of the skull and facial bones develop by intramembranous ossification. Such development bypasses the intermediate cartilaginous stage characteristic of development of the bony cranial base.10,11
The occipital sclerotomes correspond to the segmental nerves that group together to form the hypoglossal nerve, which passes through the bone via the individual foramina.6,10 The first two occipital sclerotomes ultimately form the basiocciput. The third sclerotome is responsible for the exoccipital center as it forms the jugular tubercles.11,12 The key to understanding the craniovertebral junction is the embryology of the fourth occipital sclerotome, which in lower animals is called the proatlas. The hypocentrum of the fourth occipital sclerotome forms the anterior tubercle of the clivus. The centrum of the proatlas itself forms the apical cap of the dens, as well as the apical ligament. The neural arch component of the proatlas divides into a ventral-rostral component and a caudal-dorsal portion. The ventral portion forms the U-shaped anterior margin of the foramen magnum, as well as the occipital condyles and the midline occipital condyle. The cruciate ligament and the alar ligaments are condensations of the lateral portion of the proatlas. The caudal division of the neural arch of the proatlas forms the lateral atlantal masses of C1, as well as the superior portion of the posterior arch of the atlas.
During embryogenesis, the hypocentrum of the second spinal sclerotome disappears. The centrum forms the body of the axis vertebra, and the neural arches develop into facets and the posterior arch of the axis. Thus, the body of the dens arises from the first sclerotome, whereas a terminal portion of the odontoid process arises from the proatlas. The most inferior portion of the body of the axis is formed by the second spinal sclerotome. At birth, the odontoid process is separated from the body of the axis vertebra by a cartilaginous band that represents a vestigial disk and is referred to as the neural central synchondrosis.6,13 It lies below the level of the superior articular facets of the axis and does not represent the anatomic base of the dens. This synchondrosis is present in most children younger than 3 to 4 years and disappears by 8 years of age. In most instances, the odontoid process is seen at birth but does not fuse to the base of the axis.14 The tip of the odontoid is not ossified at birth and thus is not seen on lateral radiographs. It is represented by a separate ossification center, which is usually seen at 3 years of age and fuses with the remainder of the dens by the age of 12 years.
Expansion of the posterior fossa occurs as a result of a combination of endochondral resorption, sutural growth, and bony accretion.15 Growth of the basion elongates the basiocciput and lowers the frontal margins of the foramen magnum. There is a comparably matched resorptive drift downward and backward at the opisthion as a result of downward displacement of the cerebellum, together with rotation of the occipital and temporal lobes of the brain.16
Because of the forward inclination of the top-heavy cranial end of the fetus, the stability of the craniovertebral articulation must be maintained by the geometry of the articular surfaces of the craniovertebral junction, as well as by the ligamentous attachments and, more importantly, by the heavy development of the dorsal and lateral cervical musculature, which provides a clamping action on the craniovertebral region.3,4
Significant advances in recent years have occurred with the discovery of developmental control genes.9–11,15–18 Two families of regulatory genes have been implicated in the subsequent development of the sclerotomal portions of the somites during their resegmentation to form the vertebral primordia and in specification of the identity of each vertebra. Common to all these genes are phylogenetically highly conserved DNA sequences termed homeovox (Hox genes) and paired vox (Pax genes) sequences. They promote the production of proteins that modulate morphogenesis by influencing the transcription of specific downstream genes.
Teratogen-induced disturbances in Hox gene expression and mutations in Hox genes can cause alterations in both the number and identity of the cervical vertebrae forming at or near the limit of their expression domain. For example, inactivation of the Hox-D3 gene results in mutant mice with assimilation of the atlas to the basiocciput.19 The sensitivity of the occipitocervical junction to disturbances in Hox gene expression might prove to be the underlying cause of malformations in this region. Pax genes are expressed in diverse cell types and contribute to development of the early nervous system. Control of resegmentation of the sclerotomes to establish the intervertebral boundaries seems to be independently regulated by two genes in the Pax family.17,20
Implications of Craniovertebral Abnormalities
Congenital diseases involving connective tissue, such as Morquio’s syndrome and other mucopolysaccharidoses, as well as Down syndrome, can lead to severe atlantoaxial subluxation.3,14,21 There is a high incidence of anterior and posterior spina bifida of C1 and also os odontoideum in these instances. It is possible that because of abnormal, excessive head movements in the embryo between days 50 and 53, the process of chondrification is impaired, thereby resulting in anterior and later posterior spina bifida of C1.16
Os odontoideum was previously thought to be congenital and was described as a failure of fusion between the centrum component of C1 and that of C2. However, this radiographic abnormality always has a hypoplastic dens, and the neural central synchondrosis is a definite visible entity.13 To be congenital, the defect would have to be below the superior slopes of the C2 facets.6 Os odontoideum most likely originates as odontoid trauma that may predate birth but certainly occurs before the age of 4 years, with subsequent distraction and separation of the odontoid fragments.3,22 The superior segment is distracted upward by the apical and alar ligaments and receives its blood supply from the descending branch of the occipital artery, which courses along the apical ligament. This subsequently leads to incompetence of the cruciate ligament and further abnormalities.
Spine trauma in children younger than 8 years is mainly centered at the craniovertebral border because of the high fulcrum of neck motion.3 It results in ligamentous injuries more often than fractures. However, odontoid fractures in this age group are usually seen as avulsion injuries with separation of the neural central synchondrosis.
Anomalies and malformations of the caudal occipital sclerotomes are collectively called “manifestations of occipital vertebrae” and result in abnormal bony ridges and outgrowths of the ventral aspect of the foramen magnum.1 At times, segmentation abnormalities of the clivus are recognized and represent failure of the last occipital sclerotome to separate from the basiocciput and clivus. Consequently, bone growth in the anterior aspect of the foramen magnum indents the ventral cervicomedullary junction with age (as the clivus expands inferiorly and dorsally).
Biomechanics
The unique anatomic configuration of the craniovertebral junction creates a distinct biomechanical behavior that differs from that of the other spinal joints. The motion characteristics of the different levels of the craniovertebral junction are due to the geometry of the opposing bones of the vertebrae and the skull base, the shape of the joints, and the arrangements of the ligaments.4,23–25 An intervertebral disk is absent between the occiput, C1, and C2. The ball-and-socket–shaped occiput-C1 joint allows slightly more flexion-extension than the other levels of the cervical spine do, although it is quite rigid in axial rotation and lateral bending.
The biconvex articular surfaces of the C1 and C2 joints allow gliding and wide rotation of C1 around the dens. The atlantoaxial motion segment is the most flexible motion segment in the entire spine with respect to axial rotation in that it allows a bilateral range of motion of 90 degrees. More than half of all cervical axial rotation occurs at the C1-2 motion segment, a point that should be kept in mind when considering atlantoaxial arthrodesis. In a child, the amount of anterior-posterior translation that occurs between the dens and the anterior ring of the atlas is up to 5 mm. When the transverse component of the cruciate ligament has been disrupted, the alar ligaments are still intact.25 Hence, the amount of displacement remains between 5 and 6 mm until the alar ligaments become incompetent. It is only when the alar ligaments and the transverse portion of the cruciate ligament are incompetent that a separation of more than 5 to 6 mm occurs. Beyond the age of 8 years, excursion of the predental space must be limited to 3 mm.26
Failure of one alar ligament results in moderate rotary atlantoaxial instability.27 Bilateral transection of the alar ligaments causes considerably more alterations in craniovertebral motion, and the coupling action is disturbed. The transverse atlantal ligament is the strongest and thickest ligament in the entire spine. It is the predominant stabilizer of the atlas and constrains C1 around the dens. When torn, the transverse ligament is incapable of repair. Because injury to this ligament renders C1 grossly unstable, C1-2 fusion is mandated. The largest extent of rotation occurs at the atlantoaxial joint. When rotation exceeds 40 to 50 degrees, an interlock of the lateral inferior facet of the atlas over the superior articular facet of the axis vertebra occurs. If the transverse ligament becomes deficient, the anterior arch will sublux forward and result in unilateral dislocation and an interlock at less than 40 degrees.13,26 Rotation of more than 30 to 35 degrees produces an angulation of the contralateral vertebral artery.25,28 With greater rotation, the vertebral artery is stretched, and the ipsilateral vessel may demonstrate occlusion.28,29 This phenomenon has an implication in wrestling and football injuries and in certain head rotation maneuvers performed under general anesthesia or forced chiropractic manipulations.30,31 There is a muscle-clamping action that takes place in vivo and produces compressive force across the cervical spine. This muscular action results in the “interlocking stiffening” that maintains alignment during rotation. When the protective muscles are relaxed or inadequately developed, as in the case of a young child under general anesthesia, the craniovertebral junction becomes inherently less stable than that in an adult.3 As muscular development occurs, there is less tendency for instability at the craniovertebral junction.
Biomechanical Comparison of Cervical Orthoses
A halo brace is most effective at constraining cervical motion at all levels, and control of spinal motion is related to the distance of the spinal segment from the region of the rigid skull fixation.3,27 A halo brace provides the best control of motion at the craniovertebral junction, intermediate control of midcervical motion, and the least control of motion at the cervicothoracic junction. This pattern reflects snaking movements of the spine.
Classification of Craniovertebral Junction Abnormalities
A wide variety of congenital, developmental, and acquired abnormalities can occur at the craniovertebral junction, and there may be single or multiple abnormalities in the same individual. The pathology of these abnormalities is extensive.3 A working classification has been provided, but it must be appreciated that there are overlapping causes within this classification (Table 218-1).
TABLE 218-1 Classification of Craniocervical Junction Abnormalities
|
I Congenital anomalies and malformations of the craniocervical junction
|
Epidemiology
Abnormalities of the craniovertebral junction must be suspected in infants with Goldenhar’s syndrome, skeletal dysplasias, and Conradi’s syndrome.3 It should also be suspected in infants with “torticollis.” Diseases such as Down syndrome are associated with a 14% to 20% incidence of atlantoaxial dislocation. In Morquio’s syndrome, a combination of atlantoaxial instability, os odontoideum, and cervicothoracic abnormalities occurs in 30% to 50% of individuals.3,14,32 The incidence of atlas assimilation is 0.25% in the general population. Many such patients have associated segmentation failure of the upper cervical spine.33
We have reviewed syndromes cited as altering the craniovertebral junction in more than 6000 patients seen at our institution. A search of the scientific literature identified genetic syndromes that affect the craniovertebral junction. In addition, the Online Mendelian Inheritance in Man (OMIM) database was used to identify disorders of the craniovertebral junction encountered in various clinical populations. The information recorded includes systemic manifestations of the syndromes, the effect on the craniovertebral junction and cervical vertebrae, inheritance pattern, and genes or genetic loci associated with the disorder. A total of 84 syndromes have been identified that affect the craniovertebral junction.34
Clinical Findings
The most interesting feature of the clinical manifestation of craniovertebral abnormalities is their diversity as a result of compromise of the lower brainstem, cervical spinal cord, cranial nerves, cervical roots, and vascular supply.6 The symptoms of craniovertebral dysfunction may be insidious and be manifested as false localizing signs. Infrequently, rapid neurological progression is followed by sudden death. More often than not, an antecedent history of minor trauma induces a pattern of symptoms and signs that may progress at a galloping pace.
Congenital abnormalities of the craniovertebral junction are frequently associated with an abnormal general physical appearance. The head may be cocked to one side, as in patients with rotary luxation of the atlas on the axis, or the classic triad of Klippel-Feil syndrome (abnormally low hairline posteriorly, limitation of neck motion, and short neck) may be noted. Facial asymmetry and webbing of the neck may occur in conjunction with this syndrome. At times, scoliosis is present. It is not uncommon to see children with a small dysmorphic stature. There is an increased incidence of craniovertebral abnormalities with disease states such as achondroplasia, spondyloepiphyseal dysplasia, and related diseases of dwarfism. The most common neurological symptoms and signs are enumerated in Table 218-2.
TABLE 218-2 Signs and Symptoms of Craniovertebral Anomalies (Insidious or Rapid Onset of Symptoms and Signs)
