Chapter 576 Development and Function of the Gonads
Genetic Control of Embryonic Gonadal Differentiation
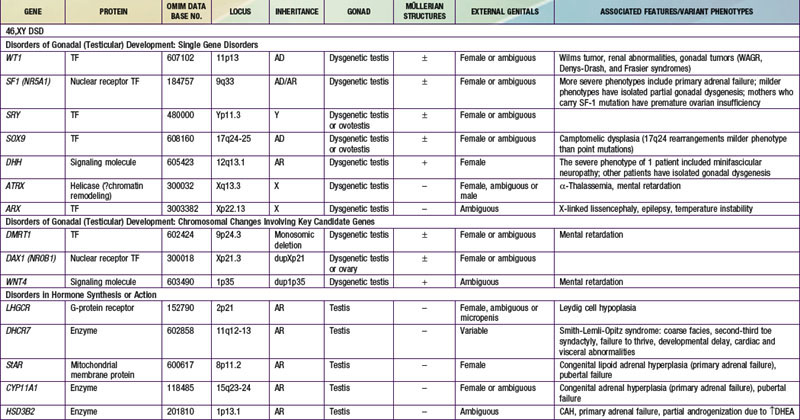
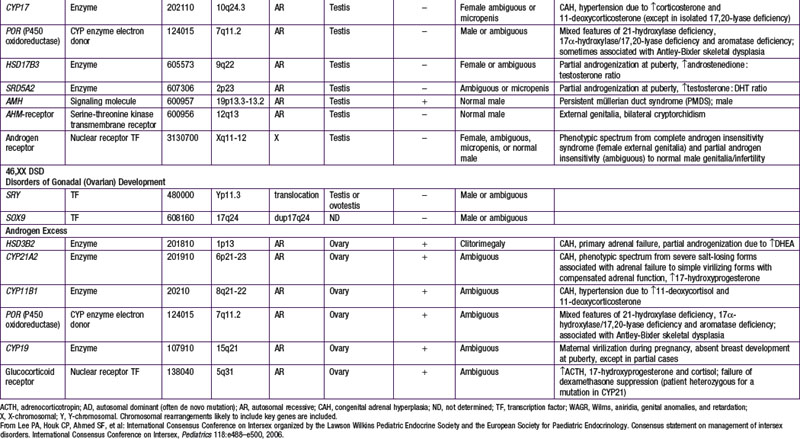
When genetic recombination events on sex chromosomes extend beyond the pseudoautosomal region, X- and Y-specific DNA may be transferred between the chromosomes. Such aberrant recombinations result in X chromosomes carrying SRY, resulting in XX males, or Y chromosomes that have lost SRY, resulting in XY females. SRY acts as a transcriptional regulator to increase cellular proliferation, attract interstitial cells from adjacent mesonephros into the genital ridge, and stimulate testicular Sertoli cell differentiation. Sertoli cells act as an organizer of steroidogenic and germ cell lines and produce antimüllerian hormone (AMH) that causes the female duct system to regress. These cells express low levels of SRY. For additional genes involved in sex development, see Table 576-1.
Function of the Testes
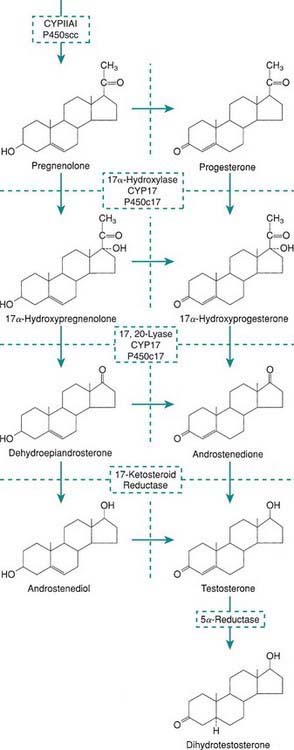
Levels of placental chorionic gonadotropin peak at 8-12 wk of gestation and stimulate the fetal Leydig cells to secrete testosterone, the main hormonal product of the testis. Testosterone is then converted by the enzyme 5α-reductase to its more potent metabolite, dihydrotestoterone. This early period is critical for normal and complete virilization of the XY fetus. Defects in this process lead to different forms of atypical male development (Chapter 582.2). After virilization occurs, fetal levels of testosterone decrease but are maintained at lower levels in the latter half of pregnancy by luteinizing hormone (LH) secreted by the fetal pituitary; this LH-mediated testosterone secretion is required for continued penile growth, and to some degree also for testicular descent.
Within specific target cells, 6-8% of testosterone is converted by 5α-reductase to dihydrotestosterone, a more potent androgen (Fig. 576-1), and about 0.3% is acted on by aromatase to produce estradiol. Approximately half of circulating testosterone is bound to sex hormone–binding globulin (SHBG) and half to albumin; only 2% circulates in the free form. Plasma levels of SHBG are low at birth, rise rapidly during the 1st 10 days of life, and then remain stable until the onset of puberty. Thyroid hormone may play a role in this physiologic increase because neonates with athyreosis (absence of the thyroid gland) have very low levels of SHBG.
Clinical patterns of pubertal changes vary widely (Chapters 12 and 555 covering pubertal maturation). In 95% of boys, enlargement of the genitals begins between 9.5 and 13.5 yr, reaching maturity at 13-17 yr. In a minority of normal boys, puberty begins after 15 yr of age. In some boys, pubertal development is completed in less than 2 yr, but in others it may take longer than 4.5 yr. The adolescent growth spurt occurs later in boys than in girls.
Function of the Ovaries
The most important estrogens produced by the ovary are estradiol-17 (E2) and estrone (E1); estriol is a metabolic product of these 2, and all 3 estrogens may be found in the urine of mature females. Estrogens also arise from androgens produced by the adrenal gland and the gonads (see Fig. 568-1). This conversion explains why in certain types of disorders of sex differentiation in males, feminization occurs at puberty. In 17-ketosteroid reductase deficiency, for example, the enzymatic block results in markedly increased secretion of androstenedione, which is converted in the peripheral tissues to estradiol and estrone. These estrogens, in addition to those directly secreted by the testis, result in gynecomastia. Estradiol produced from testosterone in the complete androgen insensitivity syndrome causes complete feminization in XY individuals.
The average age at menarche in American girls is 12.5-13 yr, but the range of “normal” is wide, and 1-2% of normal girls have not menstruated by 16 yr of age. The age at onset of pubertal signs varies, with recent studies suggesting earlier ages than previously thought, especially in the U.S. African-American population (Chapter 555). Menarche generally correlates closely with skeletal age. Maturation and closure of the epiphyses is at least partially estrogen dependent, as demonstrated by a very tall 28 yr old, normally masculinized male with continued growth due to incomplete closure of the epiphyses, who proved to have complete estrogen insensitivity owing to an estrogen-receptor defect.
Bergada I, Bergada C, Campo S. Role of inhibins in childhood and puberty. J Pediatr Endocrinol Metab. 2001;14:343-353.
Biason-Lauber A, Konrad D, Navratil F, et al. A WNT4 mutation associated with Müllerian-duct regression and virilization in a 46,XX woman. N Engl J Med. 2005;351:792-798.
Bouvattier C, Carel JC, Lecointre C, et al. Postnatal changes of T, LH, and FSH in 46,XY infants with mutations in the AR gene. J Pediatr Endocrinol Metab. 2002;87:29-32.
Habert R, Lejeune H, Saez J. Origin, differentiation and regulation of fetal and adult Leydig cells. Mol Cell Endocrinol. 2001;179:47-74.
Hughes IA. Female development—all by default? N Engl J Med. 2004;351:748-750.
Josso N, diClemente N, Gouedard L. Anti-müllerian hormone and its receptors. Mol Cell Endocrinol. 2001;179:25-32.
Koopman P. The genetics and biology of vertebrate sex determination. Cell. 2001;105:843-847.
Ostrer H. Sex determination: lessons from families and embryos. Clin Genet. 2001;59:207-215.
Pierik FH, Vreeburg JTM, Stijnen T, et al. Serum inhibin B as a marker of spermatogenesis. J Clin Endocrinol Metab. 1998;83:3110-3114.
Quigley CA. The postnatal gonadotropin and sex steroid surge: insights from the androgen insensitivity syndrome (editorial). J Clin Endocrinol Metab. 2002;87:24-28.
Rajpert-De Meyts E. Expression of anti-müllerian hormone during normal and pathological gonadal development: association with differentiation of Sertoli and granulosa cells. J Clin Endocrinol Metab. 1999;84:3836-3844.
Rey RA, Belville C, Nihoul-Fékété C, et al. Evaluation of gonadal function in 107 intersex patients by means of serum anti-müllerian hormone measurements. J Clin Endocrinol Metab. 1999;84:627-631.
Sequera AM, Fideleff HL, Boquete HR, et al. Basal ultra sensitive LH assay: a useful tool in the early diagnosis of male pubertal delay? J Pediatr Endocrinol Metab. 2002;15:589-596.
Stenvers KL, Findlay JK. Inhibins: from reproductive hormones to tumor suppressors. Trends Endocrinol Metab. 2010;21:174-180.
Swerdloff RS, Wang C, Cunningham G, et al. Long-term pharmacokinetics of transdermal testosterone gel in hypogonadal men. J Clin Endocrinol Metab. 2000;85:4500-4510.
Teixeira J, Maheswaran S, Donahue PK. Müllerian inhibiting substance: an instructive development hormone with diagnostic and possible therapeutic applications. Endocr Rev. 2001;22:657-674.



