Design of dosage forms
Peter York
Chapter contents
Principles of dosage form design
Biopharmaceutical aspects of dosage form design
Drug factors in dosage form design
Particle size and surface area
Crystal properties: polymorphism
Principles of dosage form design
Drugs are rarely administered as pure chemical substances alone and are almost always given as formulated preparations or medicines. These can vary from relatively simple solutions to complex drug delivery systems through the use of appropriate additives or excipients in the formulations. The excipients provide varied and specialized pharmaceutical functions. It is the formulation additives that, amongst other things, solubilize, suspend, thicken, preserve, emulsify, modify dissolution, improve the compactability and flavour drug substances to form various medicines or dosage forms.
The principal objective of dosage form design is to achieve a predictable therapeutic response to a drug included in a formulation which is capable of large-scale manufacture with reproducible product quality. To ensure product quality, numerous features are required: chemical and physical stability, with suitable preservation against microbial contamination if appropriate, uniformity of dose of drug, acceptability to users, including both prescriber and patient, as well as suitable packaging and labelling. Ideally, dosage forms should also be independent of patient-to-patient variation, although in practice, this feature remains difficult to achieve. However, recent developments are beginning to accommodate this requirement. These include drug delivery systems that rely on the specific metabolic activity of individual patients and implants that respond, for example, to externally applied sound or magnetic fields to trigger a drug delivery function.
Consideration should be given to differences in the bioavailability of drugs and their bio-fate in patients between apparently similar formulations and possible causative reasons. In recent years, increasing attention has therefore been directed towards eliminating variation in bioavailability characteristics, particularly for medicinal products containing an equivalent dose of a drug substance, as it is recognized that formulation factors can influence their therapeutic performance. To optimize the bioavailability of drug substances, it is often necessary to carefully select the most appropriate chemical form of the drug. For example, such selection should address solubility requirements, drug particle size and physical form and consider appropriate additives and manufacturing aids coupled to selecting the most appropriate administration route(s) and dosage form(s). Additionally, suitable manufacturing processes, labelling and packaging are required.
There are numerous dosage forms into which a drug substance can be incorporated for the convenient and efficacious treatment of a disease. Dosage forms can be designed for administration by alternative delivery routes to maximize therapeutic response. Preparations can be taken orally or injected, as well as being applied to the skin or inhaled, and Table 1.1 lists the range of dosage forms which can be used to deliver drugs by the various administration routes. However, it is necessary to relate the drug substance to the clinical indication being treated before the correct combination of drug and dosage form can be made, since each disease or illness often requires a specific type of drug therapy. In addition, factors governing choice of administration route and the specific requirements of that route which affect drug absorption need to be taken into account when designing dosage forms.
Table 1.1
| Administration route | Dosage forms |
| Oral | Solutions, syrups, suspensions, emulsions, gels, powders, granules, capsules, tablets |
| Rectal | Suppositories, ointments, creams, powders, solutions |
| Topical | Ointments, creams, pastes, lotions, gels, solutions, topical aerosols, foams, transdermal patches |
| Parenteral | Injections (solution, suspension, emulsion forms), implants, irrigation and dialysis solutions |
| Respiratory | Aerosols (solution, suspension, emulsion, powder forms), inhalations, sprays, gases |
| Nasal | Solutions, inhalations |
| Eye | Solutions, ointments, creams |
| Ear | Solutions, suspensions, ointments, creams |
Many drugs are formulated into several dosage forms of varying strengths, each having selected pharmaceutical characteristics which are suitable for a specific application. One such drug is the glucocorticoid prednisolone used in the suppression of inflammatory and allergic disorders. Through the use of different chemical forms and formulation additives, a range of effective anti-inflammatory preparations is available, including tablet, enteric-coated tablet, injections, eye drops and enema. The extremely low aqueous solubility of the base prednisolone and acetate salt makes these forms useful in tablet and slowly absorbed intramuscular suspension injection forms, whilst the soluble sodium phosphate salt enables a soluble tablet form and solutions for eye and ear drops, enema and intravenous injection to be prepared. The analgesic paracetamol is also available in a range of dosage forms and strengths to meet the specific needs of the user, including tablets, dispersible tablets, paediatric soluble tablets, paediatric oral solution, sugar-free oral solution, oral suspension, double-strength oral suspension and suppositories.
In addition, whilst many new drugs based on low molecular weight organic compounds continue to be discovered and transformed into medicinal products, the development of drugs from biotechnology is increasing and the importance of these therapeutic agents is growing. Such active compounds are macromolecular and of relatively large molecular weight, and these include materials such as peptides, proteins and viral components. These drug substances present different and complex challenges in their formulation and processing into medicines due to their alternative biological, chemical and structural properties. Nevertheless, the underlying principles of dosage form design remain applicable. At present, these therapeutic agents are principally formulated into parenteral and respiratory dosage forms although other routes of administration are being considered and researched. Delivery of these biotechnologically-based drug substances via these routes of administration imposes additional constraints upon the selection of appropriate formulation excipients.
It is therefore apparent that before a drug substance can be successfully formulated into a dosage form, many factors must be considered. These can be broadly grouped into three categories:
High-quality and efficacious medicines will be formulated and prepared only when all these factors are considered and related to each other. This is the underlying principle of dosage form design.
Biopharmaceutical aspects of dosage form design
Biopharmaceutics can be regarded as the study of the relationship between the physical, chemical and biological sciences applied to drugs, dosage forms and drug action. Clearly, understanding the principles of this subject is important in dosage form design, particularly with regard to drug absorption, as well as drug distribution, metabolism and excretion. In general, a drug substance must be in solution before it can be absorbed via absorbing membranes and epithelia of the skin, gastrointestinal tract and lungs into body fluids. Drugs are absorbed in two general ways: by passive diffusion and by carrier mediated transport mechanisms. In passive diffusion, which is thought to control the absorption of many drugs, the process is driven by the concentration gradient existing across the cellular barrier, with drug molecules passing from regions of high to low concentration. Lipid solubility and degree of ionization of the drug at the absorbing site influence the rate of diffusion. Recent research into carrier mediated transport mechanisms has provided much information and knowledge, providing guidance in some cases for the design of new drug molecules. Several specialized transport mechanisms are postulated, including active and facilitated transport. Once absorbed, the drug can exert a therapeutic effect either locally or at a site of action remote from the site of administration. In the latter case, the drug has to be transported in body fluids (as shown in Fig. 1.1).
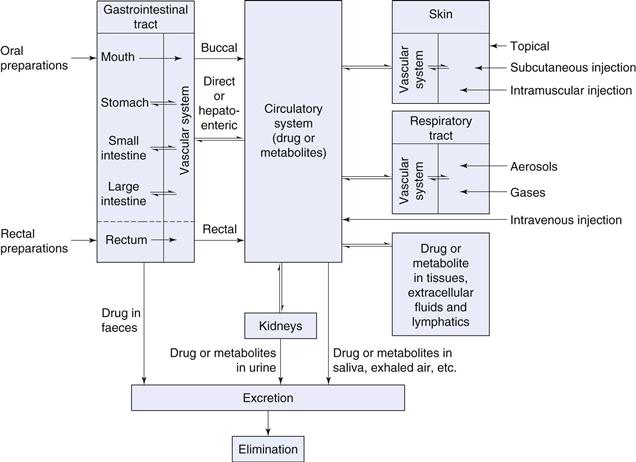
Fig. 1.1 Pathways a drug may take following the administration of a dosage form by different routes.
When the dosage form is designed to deliver drugs via the buccal, respiratory, rectal, intramuscular or subcutaneous routes, the drug passes directly into the circulation blood from absorbing tissues, whilst the intravenous route provides the most direct route of all. When delivered by the oral route, onset of drug action will be delayed because of required transit time in the gastrointestinal tract prior to absorption, the absorption process and factors associated with hepatoenteric blood circulation. The physical form of the oral dosage form will also influence absorption rate and onset of action, with solutions acting faster than suspensions, which in turn generally act faster than capsules and tablets. Dosage forms can thus be listed in order of time of onset of therapeutic effect (see Table 1.2). However, all drugs irrespective of their delivery route remain foreign substances to the human body and distribution, metabolic and elimination processes commence immediately following drug absorption until the drug is eliminated from the body via the urine, faeces, saliva, skin or lungs in unchanged or metabolized form.
Table 1.2
Variation in time of onset of action for different dosage forms
| Time of onset of action | Dosage forms |
| Seconds | Intravenous injections |
| Minutes | Intramuscular and subcutaneous injections, buccal tablets, aerosols, gases |
| Minutes to hours | Short-term depot injections, solutions, suspensions, powders, granules, capsules, tablets, modified-release tablets |
| Several hours | Enteric-coated formulations |
| Days to weeks | Depot injections, implants |
| Varies | Topical preparations |
Routes of drug administration
The absorption pattern of drugs varies considerably between individual drug substances as well as between the different administration routes. Dosage forms are designed to provide the drug in a suitable form for absorption from each selected route of administration. The following discussion considers briefly the routes of drug administration and whilst dosage forms are mentioned, this is intended only as an introduction since they will be dealt with in greater detail later in this book.
Oral route
The oral route is the most frequently used route for drug administration. Oral dosage forms are intended usually for systemic effects resulting from drug absorption through the various epithelia and mucosa of the gastrointestinal tract. A few drugs, however, are intended to dissolve in the mouth for rapid absorption or for local effect in the tract due to poor absorption by this route or low aqueous solubility. Compared with other routes, the oral route is the simplest, most convenient and safest means of drug administration. However, disadvantages include relatively slow onset of action, possibilities of irregular absorption and destruction of certain drugs by the enzymes and secretions of the gastrointestinal tract. For example, insulin-containing preparations are inactivated by the action of stomach fluids.
Whilst drug absorption from the gastrointestinal tract follows the general principles described later in this book, several specific features should be emphasized. Changes in drug solubility can result from reactions with other materials present in the gastrointestinal tract, as for example the interference of absorption of tetracyclines through the formation of insoluble complexes with calcium, which can be available from foodstuffs or formulation additives. Gastric emptying time is an important factor for effective drug absorption from the intestine. Slow gastric emptying can be detrimental to drugs inactivated by the gastric juices and can delay absorption of drugs more effectively absorbed from the intestine. In addition, since environmental pH can influence the ionization and lipid solubility of drugs, the pH change occurring along the gastrointestinal tract, from a pH as low as 1 in the stomach to approximately 7 or 8 in the large intestine, is important to both the degree and site of drug absorption. Since membranes are more permeable to unionized rather than ionized forms and since most drugs are weak acids or bases, it can be shown that weak acids, being largely unionized, are well absorbed from the stomach. In the small intestine (pH from around 4 to 6.5), with its extremely large absorbing surface, both weak acids and weak bases are well absorbed.
The most popular oral dosage forms are tablets, capsules, suspensions, solutions and emulsions. Tablets are prepared by compaction and contain drugs and formulation additives which are included for specific functions, such as disintegrants which promote tablet break-up into granules and powder particles in the gastrointestinal tract, facilitating drug dissolution and absorption. Tablets are often coated, either to provide a protective barrier to environmental factors for drug stability purposes or to mask unpleasant drug taste, as well as to protect drugs from the acid conditions of the stomach (enteric coating). Increasing use is being made of modified-release tablet products such as fast-dissolving systems and controlled, delayed or sustained-release formulations. Benefits of controlled-release tablet formulations, achieved for example by the use of polymeric-based tablet cores or coating membranes, include reduced frequency of drug-related side-effects and maintaining steady drug-plasma levels for extended periods; important when medications are delivered for chronic conditions or where constant levels are required to achieve optimal efficacy, as in treating angina and hypertension.
Capsules are solid dosage forms containing drug and, usually, appropriate filler(s), enclosed in a hard or soft shell composed primarily of gelatin or other suitable polymeric material. As with tablets, uniformity of dose can be readily achieved and various sizes, shapes and colours of shell are commercially available. The capsule shell readily ruptures and dissolves following oral administration and in most cases drugs are released from capsules faster than from tablets. Recently, increased interest has been shown in filling semi-solid and microemulsion formulations into hard gelatin capsules to provide rapidly dispersing dosage forms for poorly soluble drugs.
Suspensions, which contain finely divided drugs suspended in a suitable vehicle, are a useful means of administering large amounts of drugs that would be inconvenient if taken in tablet or capsule form. They are also useful for patients who experience difficulty in swallowing tablets and capsules and for paediatric use. Whilst dissolution of drugs is required prior to absorption, the fine solid particles in a suspension have a large surface area to present to the gastrointestinal fluids and this facilitates drug dissolution thus aiding absorption and thereby the onset of drug action. Not all oral suspensions, however, are formulated for systemic effects and several are designed for local effects in the gastrointestinal tract. On the other hand, solutions, including formulations such as syrups and linctuses, are absorbed more rapidly than solid dosage forms or suspensions since drug dissolution is not required.
Rectal route
Drugs given rectally in solution, suppository or emulsion form are generally administered for local rather than systemic effects. Suppositories are solid forms intended for introduction into body cavities (usually rectal but also vaginal and urethral) where they melt, releasing the drug. The choice of suppository base or drug carrier can greatly influence the degree and rate of drug release. This route of drug administration is also indicated for drugs inactivated by the gastrointestinal fluids when given orally or when the oral route is precluded, as for example when a patient is vomiting or unconscious. Drugs administered rectally enter the systemic circulation without passing through the liver, an advantage for drugs significantly inactivated by the liver following oral route absorption. Disadvantageously, the rectal route is inconvenient and drug absorption is often irregular and difficult to predict.
Parenteral route
A drug administered parenterally is one injected via a hollow needle into the body at various sites and to varying depths. The three main parenteral routes are subcutaneous, intramuscular and intravenous. Other routes, such as intracardiac and intrathecal, are used less frequently. The parenteral route is preferred when rapid absorption is essential, as in emergency situations or when patients are unconscious or unable to accept oral medication, and in cases when drugs are destroyed, inactivated or poorly absorbed following oral administration. In general, blood levels attained are more predictable than those achieved by oral dosage forms.
Injectable preparations are usually sterile solutions or suspensions of drugs in water or other suitable physiologically acceptable vehicles. As referred to previously, drugs in solution are rapidly absorbed and thus suspension injections are slower acting than solution injections. In addition, since body fluids are aqueous, by using drugs suspended in oily vehicles, a preparation exhibiting slower absorption characteristics can be formulated to give a depot preparation, providing a reservoir of drug which is slowly released into the systemic circulation. Such preparations are administered by intramuscular injection deep into skeletal muscles (e.g. several penicillin-containing injections). Alternatively, depot preparations can be achieved by subcutaneous implants or pellets, which are compacted or moulded discs of drug placed in loose subcutaneous tissue under the outer layers of the skin. Such systems include solid microspheres, biodegradable polymeric microspheres (e.g. polylactide co-glycollic acid homo- and copolymers) containing proteins or peptides (e.g. human growth hormone and leuprolide). More generally, subcutaneous injections are aqueous solutions or suspensions which allow the drug to be placed in the immediate vicinity of blood capillaries. The drug then diffuses into the capillaries. Inclusion of vasoconstrictors or vasodilators in subcutaneous injections will clearly influence blood flow through the capillaries, thereby modifying the capacity for absorption. This principle is often used in the administration of local anaesthetics with the vasoconstrictor adrenaline, which delays drug absorption. Conversely, improved drug absorption can result when vasodilators are included. Intravenous administration involves injection of sterile aqueous solutions directly into a vein at an appropriate rate. Volumes delivered can range from a few millilitres, as in emergency treatment or for hypnotics, up to litre quantities, as in replacement fluid treatment or nutrient feeding.
Given the generally negative patient acceptance of this important route of drug delivery, primarily associated with pain and inconvenience, recent developments to help with self-injection by patients have focused on ‘needle-free’ injection systems and devices which propel drug in aqueous solution or powder form at high velocity directly through the external layers of the skin.
Topical route
Drugs are applied topically, that is to the skin, mainly for local action. Whilst this route can also be used for systemic drug delivery, percutaneous absorption is often poor and erratic, although several transdermal patches delivering drug for systemic distribution (e.g. fentanyl patches for severe pain management and nicotine patches for cessation of smoking) are available. Drugs applied to the skin for local effect include antiseptics, antifungals and anti-inflammatory agents, as well as skin emollients for protective effects.
Pharmaceutical topical formulations – ointments, creams and pastes – are composed of drug in a suitable semi-solid base which is either hydrophobic or hydrophilic in character. The bases play an important role in determining the drug release character from the formulation. Ointments are hydrophobic, oleaginous-based dosage forms, whereas creams are semi-solid emulsions. Pastes contain more solids than ointments and thus are stiffer in consistency. For topical application in liquid form other than solution, lotions, suspensions of solids in aqueous solution or emulsions are used.
Application of drugs to other topical surfaces such as the eye, ear and nose is common and ointments, creams, suspensions and solutions are utilized. Ophthalmic preparations are required, amongst other features, to be sterile. Nasal dosage forms include solutions or suspensions delivered by drops or fine aerosol from a spray. Ear formulations in general are viscous to prolong contact with affected areas.
Respiratory route
The lungs provide an excellent surface for absorption when the drug is delivered in gaseous, aerosol mist or ultrafine solid particle form. For drug particles presented to the lungs as an aerosol, particle size largely determines the extent to which they penetrate the alveolar region, the zone of rapid absorption. Drug particles that are in the region 0.5–1 µm diameter reach the alveolar sacs. Particles smaller than this range are either exhaled or, if larger, deposited upon larger bronchial airways. This delivery route is particularly useful for the direct treatment of asthmatic problems, using both powder aerosols (e.g. salmeterol xinafoate) and pressurized metered dose aerosols containing the drug in liquefied inert propellant (e.g. salbutamol sulphate inhaler). Importantly, this delivery route is being increasingly recognized as a useful means of administering the therapeutic agents emerging from biotechnology requiring systemic distribution and targeted delivery, such as peptides and proteins.
Drug factors in dosage form design
Each type of dosage form requires careful study of the physical and chemical properties of drug substances to achieve a stable, efficacious product. These properties, such as dissolution, crystal size and polymorphic form, solid-state stability and drug-additive interaction, can have profound effects on the physiological availability and physical and chemical stability of the drug. By combining such information and knowledge with those from pharmacological and biochemical studies, the most suitable drug form and additives can be selected for the formulation of chosen dosage forms.
Whilst comprehensive property evaluation will not be required for all types of formulations, those properties which are recognized as important in dosage form design and processing are listed in Table 1.3. Also listed in Table 1.3 are the stresses to which the formulation might be exposed during processing and manipulation into dosage forms, as well as the procedures involved. Variations in physicochemical properties, occurring for example between batches of the same material or resulting from alternative treatment procedures, can modify formulation requirements as well as processing and dosage form performance. For instance, the fine milling of poorly aqueous soluble drug substances can modify their wetting and dissolution characteristics, important properties during granulation and product performance respectively. Careful evaluation of these properties and understanding of the effects of these stresses upon these parameters are therefore important in dosage form design and processing as well as product performance.
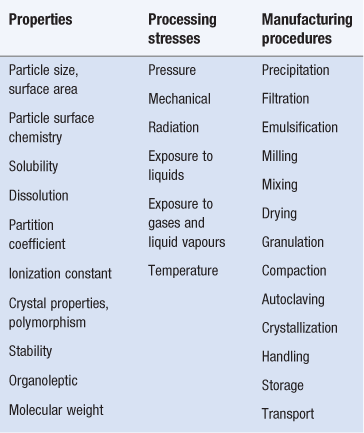
Particle size and surface area
Particle size reduction results in an increase in the specific surface (i.e. surface area per unit weight) of powders. Drug dissolution rate, absorption rate, dosage form content uniformity and stability are all dependent to varying degrees on particle size, size distribution and interactions of solid surfaces. In many cases, for both drugs and additives, particle size reduction is required to achieve the desired physicochemical characteristics.
It is now generally recognized that poorly aqueous soluble drugs showing a dissolution rate-limiting step in the absorption process will be more readily bioavailable when administered in a finely subdivided form with a larger surface than as a coarse material. Examples include griseofulvin, tolbutamide, indomethacin and nifedipine. The fine material, often of micrometre or submicrometre (nanometre) size, with large specific surface, dissolves at faster rates which can lead to improved drug absorption by passive diffusion. With many of the new drugs being introduced exhibiting extremely low aqueous solubility, alternative formulation strategies to enhance drug dissolution are being used, such as co-precipitates of drug and adjuvant particles, complexation with hydrophilic polymers or oligosaccharides, or the formation of co-crystals with hydrophilic templating compounds.
Rates of drug dissolution can be adversely affected, however, by unsuitable choice of formulation additives, even though solids of appropriate particle size are used. Tableting lubricant powders, for example, can impart hydrophobicity to a formulation and inhibit drug dissolution. Fine powders can also increase air adsorption or static charge, leading to wetting or agglomeration problems. Micronizing drug powders can lead to changes in crystallinity and particle surface energy which cause reduced chemical stability. Drug particle size also influences content uniformity in solid dosage forms, particularly for low-dose formulations. It is important in such cases to have as many particles as possible per dose to minimize potency variation between dosage units. Other dosage forms are also affected by particle size, including suspensions (for controlling flow properties and particle interactions), inhalation aerosols (for optimal penetration of drug particles to absorbing mucosa) and topical formulations (for freedom from grittiness).
Solubility
All drugs, regardless of their administration route, must exhibit at least limited aqueous solubility for therapeutic efficacy. Thus relatively insoluble compounds can exhibit erratic or incomplete absorption, and it might be appropriate to use a more soluble salt or other chemical derivatives. Alternatively, micronizing, complexation or solid dispersion techniques might be employed. Solubility, and especially degree of saturation in the vehicle, can also be important in the absorption of drugs already in solution in liquid dosage forms, since precipitation in the gastrointestinal tract can occur, modifying bioavailability.
Solubilities of acidic or basic compounds are pH-dependent and can be altered by forming salts, with different salts exhibiting different equilibrium solubilities. However, the solubility of a salt of a strong acid is less affected by changes in pH than the solubility of a salt of a weak acid. In the latter case, when pH is lower, the salt hydrolyses to an extent dependent on pH and pKa, resulting in decreased solubility. Reduced solubility can also occur for slightly soluble salts of drugs through the common ion effect. If one of the ions involved is added as a different, more soluble salt, the solubility product can be exceeded and a portion of the drug precipitates.
Dissolution
As mentioned above, for a drug to be absorbed it must first be dissolved in the fluid at the site of absorption. For example, an orally administered drug in tablet form is not absorbed until drug particles are dissolved or solubilized by the fluids at some point along the gastrointestinal tract, depending on the pH-solubility profile of the drug substance. Dissolution describes the process by which the drug particles dissolve.
During dissolution, the drug molecules in the surface layer dissolve, leading to a saturated solution around the particles to form the diffusion layer. Dissolved drug molecules then pass throughout the dissolving fluid to contact absorbing mucosa and are absorbed. Replenishment of diffusing drug molecules in the diffusion layer is achieved by further drug dissolution and the absorption process continues. If dissolution is fast or the drug remains in solution form, the rate of absorption is primarily dependent upon its ability to traverse the absorbing membrane. If, however, drug dissolution is slow due to its physicochemical properties or formulation factors, then dissolution may be the rate-limiting step in absorption and influences drug bioavailability. The dissolution of a drug is described in a simplified manner by the Noyes–Whitney equation:
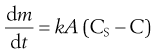 (1.1)
(1.1)
where 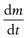 is the dissolution rate, k is the dissolution rate constant, A is the surface area of dissolving solid, CS is the drug’s solubility and C is the concentration of drug in the dissolution medium at time t. The equation reveals that dissolution rate can be raised by increasing the surface area (reducing particle size) of the drug, by increasing the solubility of the drug in the diffusion layer and by increasing k which in this equation incorporates the drug diffusion coefficient and the diffusion layer thickness. During the early phases of dissolution, CS > C and if the surface area, A, and experimental conditions are kept constant then k can be determined for compacts containing drug alone. The constant k is termed the intrinsic dissolution rate constant and is a characteristic of each solid drug compound in a given solvent under fixed hydrodynamic conditions.
is the dissolution rate, k is the dissolution rate constant, A is the surface area of dissolving solid, CS is the drug’s solubility and C is the concentration of drug in the dissolution medium at time t. The equation reveals that dissolution rate can be raised by increasing the surface area (reducing particle size) of the drug, by increasing the solubility of the drug in the diffusion layer and by increasing k which in this equation incorporates the drug diffusion coefficient and the diffusion layer thickness. During the early phases of dissolution, CS > C and if the surface area, A, and experimental conditions are kept constant then k can be determined for compacts containing drug alone. The constant k is termed the intrinsic dissolution rate constant and is a characteristic of each solid drug compound in a given solvent under fixed hydrodynamic conditions.
Drugs with k values below 0.1 mg−1 cm−2 usually exhibit dissolution rate-limiting absorption. Particulate dissolution can also be examined where an effort is made to control A, and formulation effects can be studied.
Dissolution rate data, when combined with solubility, partition coefficient and pKa results, provide an insight into the potential in vivo absorption characteristics of a drug. However, in vitro tests only have significance when they are related to in vivo results. Once such a relationship has been established, in vitro dissolution tests can be used as a predictor of in vivo behaviour. The importance of dissolution testing has been widely recognized by official compendia, as well as drug regulatory authorities, with the inclusion of dissolution specifications using standardized testing procedures for a range of preparations.
A guide for predicting the intestinal absorption of drugs for orally administered medicines based on the solubility, dissolution and permeability of drugs, the Biopharmaceutics Classification System (BCS) was established in 1995. This system has proved extremely useful in aiding the design of oral medicines and has recently been extended to incorporate drug absorption and transport and the effects of metabolism.
Partition coefficient and pKa
As pointed out earlier, for relatively insoluble compounds the dissolution rate is often the rate-determining step in the overall absorption process. Alternatively, for soluble compounds the rate of permeation across biological membranes is the rate-determining step. Whilst dissolution rate can be changed by modifying the physicochemical properties of the drug and/or altering the formulation composition, the permeation rate is dependent upon the size, relative aqueous and lipid solubility and ionic charge of drug molecules, factors which can be altered through molecular modifications. The absorbing membrane acts as a lipophilic barrier to the passage of drugs which is related to the lipophilic nature of the drug molecule. The partition coefficient, for example between oil and water, is a measure of lipophilic character.
The majority of small molecular weight drugs are weak acids or bases and, depending on the pH, exist in an ionized or unionized form. Membranes of absorbing mucosa are more permeable to unionized forms of drugs than to ionized species because of the greater lipid solubility of the unionized forms and the highly charged nature of the cell membrane which results in the binding or repelling of the ionized drug, thereby decreasing penetration.
The dominating factors that therefore influence the absorption of weak acids and bases are the pH at the site of absorption and the lipid solubility of the unionized species. These factors, together with the Henderson–Hasselbalch equations for calculating the proportions of ionized and unionized species at a particular pH, constitute the pH-partition theory for drug absorption. However, these factors do not describe completely the process of absorption since certain compounds with low partition coefficients and/or which are highly ionized over the entire physiological pH range show good bioavailability and therefore other factors are clearly involved.
Crystal properties: polymorphism
Practically all drug substances are handled in powder form at some stage during manufacture into dosage forms. However, for those substances composed of, or containing, powders or compacted powders in the finished product, the crystal properties and solid-state form of the drug must be carefully considered. It is well recognized that drug substances can be amorphous (i.e. without regular molecular lattice arrangements), crystalline, anhydrous, at various degrees of hydration or solvated with other entrapped solvent molecules, as well as varying in crystal hardness, shape and size. In addition, many drug substances can exist in more than one form with different molecular packing arrangements in the crystal lattice. This property is termed polymorphism and different polymorphs may be prepared by manipulation of conditions of particle formation during crystallization such as solvent, temperature and rate of cooling. It is known that only one form of a pure drug substance is stable at a given temperature and pressure, with the other forms, termed metastable, converting at different rates to the stable crystalline form. The different polymorphs vary in physical properties such as dissolution and solid-state stability, as well as processing behaviour in terms of powder flow and compaction during tableting in some cases.
These different crystalline forms can be of considerable importance in relation to ease or difficulty of formulation and as regards stability and biological activity. As might be expected, higher dissolution rates are obtained for metastable polymorphic forms; for example, the alternative polymorphic forms of rifaximin exhibit different in vitro dissolution rates and bioavailability. In some cases, amorphous forms are more active than crystalline forms.
The polypeptide hormone insulin, widely used in the regulation of carbohydrate, fat and protein metabolism, also demonstrates how differing degrees of activity can result from the use of different crystalline forms of the same agent. In the presence of acetate buffer, zinc combines with insulin to form an extremely insoluble complex of the proteinaceous hormone. This complex is an amorphous precipitate or crystalline product depending on environmental pH. The amorphous form, containing particles of no uniform shape and smaller than 2 µm, is absorbed following intramuscular or subcutaneous injection and has a short duration of action, whilst the crystalline product, consisting of 10–40 µm sized rhombohedral crystals, is more slowly absorbed and has a longer duration of action. Insulin preparations which are intermediate in duration of action are prepared by taking physical mixtures of these two products.
Polymorphic transitions can also occur during milling, granulating, drying and compacting operations (e.g. transitions during milling for digoxin and spironolactone). Granulation can result in solvate formation or, during drying a solvent or water molecule(s) may be lost to form an anhydrous material. Consequently, the formulator must be aware of these potential transformations which can result in undesirable modified product performance, even though routine chemical analyses may not reveal any changes. Reversion from metastable forms, if used, to the stable form may also occur during the lifetime of the product. In suspensions, this may be accompanied by changes in the consistency of the preparation which affects its shelf-life and stability. Such changes can often be prevented by additives, such as hydrocolloids and surface-active agents.
Stability
The chemical aspects of formulation generally centre on the chemical stability of the drug and its compatibility with the other formulation ingredients. In addition, it should be emphasized that the packaging of the dosage form is an important factor contributing to product stability and must be an integral part of stability testing programmes. It has been mentioned previously that one of the principles of dosage form design is to ensure that the chemical integrity of drug substances is maintained during the usable life of the product. At the same time, chemical changes involving additives and any physical modifications to the product must be carefully monitored to optimize formulation stability.
In general, drug substances decompose as a result of the effects of heat, oxygen, light and moisture. For example, esters such as aspirin and procaine are susceptible to solvolytic breakdown, whilst oxidative decomposition occurs for substances such as ascorbic acid. Drugs can be classified according to their sensitivity to breakdown:
1. stable in all conditions (e.g. kaolin)
2. stable if handled correctly (e.g. aspirin)
3. only moderately stable even with special handling (e.g. vitamins)
4. very unstable (e.g. certain antibiotics in solution form).
Whilst the mechanisms of solid-state degradation are complex and often difficult to analyse, a full understanding is not a prerequisite in the design of a suitable formulation containing solids. For example, in cases where drug substances are sensitive to hydrolysis, steps such as minimum exposure to moisture during preparation, low moisture content specifications in the final product and moisture-resistant packaging can be used. For oxygen-sensitive drugs, antioxidants can be included in the formulation and, as with light-sensitive materials, suitable packaging can reduce or eliminate the problem. For drugs administered in liquid form, the stability in solution as well as the effects of pH over the physiological range of 1–8 should be understood. Buffers may be required to control the pH of the preparation to improve stability; where liquid dosage forms are sensitive to microbial attack, preservatives are required.
In these formulations, and indeed in all dosage forms incorporating additives, it is also important to ensure that the components, which may include additional drug substances as in multivitamin preparations, do not produce chemical interactions themselves. Interactions between drug(s) and added excipients such as antioxidants, preservatives, suspending agents, colourants, tablet lubricants and packaging materials do occur and must be checked for during the design of formulations. Over recent years, data from thermal analysis techniques, particularly microcalorimetry and differential scanning calorimetry (DSC), when critically examined, have been found useful in rapid screening for possible drug-additive and drug-drug interactions. For example, DSC has revealed that the widely used tableting lubricant magnesium stearate interacts with aspirin and should be avoided in formulations containing this drug.
Organoleptic properties
Modern medicines require that pharmaceutical dosage forms are acceptable to the patient. Unfortunately, many drug substances in use today are unpalatable and unattractive in their natural state and dosage forms containing such drugs, particularly oral preparations, may require the addition of approved flavours and/or colours.
The use of flavours applies primarily to liquid dosage forms intended for oral administration. Available as concentrated extracts, solutions, adsorbed onto powders or microencapsulated, flavours are usually composed of mixtures of natural and synthetic materials. The taste buds of the tongue respond quickly to bitter, sweet, salt or acid elements of a flavour. Unpleasant taste can be overcome by using water-insoluble derivatives of drugs which have little or no taste. An example is the use of amitriptyline pamoate, although other factors, such as bioavailability, must remain unchanged. If an insoluble derivative is unavailable or cannot be used, a flavour or perfume can be used. However, unpleasant drugs in capsules or prepared as coated particles or tablets may be easily swallowed, avoiding the taste buds.
Selection of flavour depends upon several factors but particularly on the taste of the drug substance. Certain flavours are more effective at masking various taste elements; for example, citrus flavours are frequently used to combat sour or acid-tasting drugs. Solubility and stability of the flavour in the vehicle are also important. In addition, the age of the intended patient should also be considered, since children for example prefer sweet tastes, as well as the psychological links between colours and flavours (e.g. yellow colour is associated with lemon flavour). Sweetening agents may also be required to mask bitter tastes. Sucrose continues to be used but alternatives, such as sodium saccharin which is 200–700 times sweeter depending on concentration, are available. Sorbitol is recommended for diabetic preparations.
Colours are employed to standardize or improve an existing drug colour, to mask a colour change or complement a flavour. Whilst colours are obtained both from natural sources (e.g. carotenoids) or synthesized (e.g. amaranth), the majority used are synthetically produced. Dyes may be aqueous (e.g. amaranth) or oil soluble (e.g. Sudan IV) or insoluble in both (e.g. aluminium lakes). Lakes, which are generally calcium or aluminium complexes of water-soluble dyes, are particularly useful in tablets and tablet coatings because of greater stability to light than corresponding dyes, which also vary in their stability to pH and reducing agents. However, in recent years, the inclusion of colours in formulations has become extremely complex because of the banning of many traditionally used colours in many countries.
Other drug properties
At the same time as ensuring that dosage forms are chemically and physically stable and are therapeutically efficacious, it is also relevant to establish that the selected formulation is capable of efficient and, in most cases, large-scale manufacture. In addition to those properties previously discussed such as particle size and crystal form, other characteristics such as hygroscopicity, flowability and compactability are particularly valuable when preparing solid dosage forms where the drugs constitute a large percentage of the formulation. Hygroscopic drugs can require low moisture manufacturing environments and need to avoid water during preparation. Poorly flowing formulations may require the addition of flow agents (e.g. fumed silica). Studies of the compactability of drug substances are frequently undertaken using instrumented tablet machines in formulation laboratories to examine the tableting potential of the material in order to foresee any potential problems during compaction, such as lamination or sticking, which may require modification of the formulation or processing conditions.
Therapeutic considerations in dosage form design
The nature of the clinical indication, disease or illness for which the drug is intended is an important factor when selecting the range of dosage forms to be prepared. Factors such as the need for systemic or local therapy, duration of action required, and whether the drug will be used in emergency situations, need to be considered. In the vast majority of cases a single drug substance is prepared into a number of dosage forms to satisfy both the particular preferences of the patient or physician and the specific needs of a certain clinical situation. For example, many asthmatic patients use inhalation aerosols, from which the drug is rapidly absorbed into the systematic circulation following deep inhalation for rapid emergency relief, and oral products for chronic therapy.
Patients requiring urgent relief from angina pectoris, a coronary circulatory problem, place tablets of nitroglycerin sublingually. This gives rapid drug absorption directly into the blood capillaries under the tongue. Thus, whilst systemic effects are generally obtained following oral and parenteral drug administration, other routes can be employed as the drug and situation demand. Local effects are generally restricted to dosage forms applied directly, such as those applied to the skin, ear, eye, throat and lungs. Some drugs may be well absorbed by one route and not another and must therefore be considered individually.
The age of the patient also plays a role in defining the types of dosage forms made available. Infants generally prefer liquid dosage forms, usually solutions and mixtures, given orally. Also, by having liquid preparations, the amount of drug administered can be readily adjusted by dilution to give the required dose for the particular patient, taking weight, age and patient’s condition into account. Children can have difficulty in swallowing solid dosage forms and for this reason many oral preparations are prepared as pleasantly flavoured syrups or mixtures. Adults generally prefer solid dosage forms, primarily because of their convenience. However, alternative liquid preparations are usually available for those unable to take tablets and capsules.
Interest has grown in the design of drug-containing formulations which deliver drugs to specific ‘targets’ in the body, for example the use of liposomes and nanoparticles, as well as providing drugs over longer periods of time at controlled rates. Alternative technologies for preparing particles with required properties – crystal engineering – provide new opportunities. Supercritical fluid processing using carbon dioxide as a solvent or anti-solvent is one such method, allowing fine tuning of crystal properties and particle design and fabrication. Undoubtedly, these new technologies and others, as well as sophisticated formulations, will be required to deal with the advent of gene therapy and the need to deliver such labile macromolecules to specific targets and cells in the body. Interest is also likely to be directed to individual patient requirements such as age, weight and physiological and metabolic factors, features which can influence drug absorption and bioavailability, and the increasing application of diagnostic agents will play a key role in this area.
Other areas of innovation in formulation science responding to drug regulatory agency requirements in applications for marketing authorization of medicines are emerging, such as the concepts of ‘computational pharmaceutics’. This topic incorporates i) the use of in-silico procedures to predict drug substance properties, and ii) decision making and optimization tools, such as experimental design, artificial intelligence and neural computing. All of these can facilitate faster and rational design of formulations and manufacturing processes.
Summary
This chapter has demonstrated that the formulation of drugs into dosage forms requires the interpretation and application of a wide range of information and knowledge from several study areas. Whilst the physical and chemical properties of drugs and additives need to be understood, the factors influencing drug absorption and the requirements of the disease to be treated also have to be taken into account when identifying potential delivery routes. The formulation and associated preparation of dosage forms demand the highest standards with careful examination, analysis and evaluation of wide-ranging information by pharmaceutical scientists to achieve the objective of creating high-quality, safe and efficacious dosage forms.
Bibliography
1. Blagden N, de Matas M, Gavan PT, York P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rate. Advanced Drug Delivery Reviews. 2007;59:617–630.
2. British Pharmacopoeial Commission. British Pharmacopoeia. London: Stationery Office; 2013.
3. Byrn SR, Pfeiffer RR, Stowell JG. Solid State Chemistry of Drugs. 2nd edn West Lafayette: SSCI; 1999.
4. Colbourn E, Rowe RC. Neural computing and formulation optimization. In: Swarbrick J, Boylan J, eds. Encyclopedia of Pharmaceutical Technology. New York: Marcel Dekker; 2005.
5. Florence AT, Attwood D. Physicochemical Principles of Pharmacy. 5th edn London: Pharmaceutical Press, Royal Pharmaceutical Society; 2011.
6. Shekunov BYu, York P. Crystallisation processes in pharmaceutical technology and drug delivery design. Journal of Crystal Growth. 2000;211:122–136.
7. Sweetman SC, ed. Martindale: The Complete Drug Reference. 37th edn London: Pharmaceutical Press, Royal Pharmaceutical Society; 2011.
8. Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharmaceutical Research. 2005;22:11–23.