Chapter 9 Depression and Psychosis in Neurological Practice
The most widely recognized nomenclature used for discussion of mental disorders derives from the classification system developed for the Diagnostic and Statistical Manual of Mental Disorders (DSM). The American Psychiatric Association introduced the DSM in 1952 to facilitate psychiatric diagnosis through improved standardization of nomenclature. There have been consecutive revisions of this highly useful and relied-upon document since its inception, with the last revision being the DSM IV-TR in 2000 and a planned revision, the DSM V, scheduled for publication in 2013. Discussion about the potential secondary causes of depression and psychosis requires a familiarity with the most salient features of the primary psychiatric conditions. A brief outline of selected conditions derived from the DSM IV-TR is included in Boxes 9.1 and 9.2, which can be found at www.expertconsult.com, along with other content in this chapter marked “online only.”
Box 9.1 Diagnostic Features of Primary Psychiatric Disorders
Schizophrenia is a disorder that lasts for at least 6 months and includes at least 1 month of active symptoms (two or more of the following: delusions, hallucinations, disorganized speech, grossly disorganized or catatonic behavior, or negative symptoms).
Schizoaffective disorder is a disorder in which a mood episode and the active symptoms of schizophrenia occur together and were preceded or are followed by at least 2 weeks of delusions or hallucinations without prominent mood symptoms.
Major depressive disorder is characterized by one or more major depressive episodes (at least 2 weeks of depressed mood or loss of interest accompanied by at least four additional symptoms of depression). Additional symptoms of depression may include significant weight loss or loss of appetite, sleep dysfunction, psychomotor agitation or retardation, fatigue or loss of energy, feelings of worthlessness or guilt, cognitive problems, and suicidal ideational or thoughts of death.
A manic episode is defined by an abnormally and persistently elevated, expansive, or irritable mood persisting for at least 1 week (or less if hospitalization is required). At least three of the following symptoms must be present if the mood is elevated or expansive (four symptoms are required if the mood is irritable): inflated self esteem or grandiosity, decreased need for sleep, pressured speech, flight of ideas, distractibility, increased goal directed activities or psychomotor agitation, and excessive involvement in pleasurable activities with a high potential for painful consequences. Psychotic features may be present.
Bipolar I disorder is characterized by the presence of both manic and major depressive episodes or manic episodes alone.
Bipolar II is characterized by the presence of major depressive episodes alternating with episodes of hypomania.
Hypomania is characterized by an abnormally and persistently elevated, expansive, or irritable mood persisting for at least 4 days. Other criteria required for diagnosis are identical to that of a manic episode except that the symptoms are not so severe as to cause marked impairment in social or occupational functioning, hospitalization is not required, and no psychotic symptoms are present.
Box 9.2 Psychiatric Terms of Relevance to Neurologists
Abulia is the state of reduced impulse to act and think associated with indifference about consequences of action.
Affect is the examiner’s observation of the patient’s emotional state. Frequently used descriptive terms include:
Anxiety is the feeling of apprehension caused by anticipation of danger that may be internal or external.
Apathy is dulled emotional tone associated with detachment or indifference.
Comportment refers to self-regulation of behavior through complex mental processes that include insight, judgment, self-awareness, empathy, and social adaptation.
Compulsion is the uncontrollable impulse to perform an act repetitively.
Confusion is the inability to maintain a coherent stream of thought owing to impaired attention and vigilance. Secondary deficits in language, memory, and visual spatial skills are common.
Delusion is a false, unshakable conviction or judgment that is out of keeping with reality and with socially shared beliefs of the individual’s background and culture. It cannot be corrected with reasoning.
Depression is a sustained psychopathological feeling of sadness often accompanied by a variety of associated symptoms, particularly anxiety, agitation, feelings of worthlessness, suicidal ideation, abulia, psychomotor retardation, and various somatic symptoms and physiological dysfunctions and complaints that cause significant distress and impairment in social functioning.
Hallucination is a false sensory perception not associated with real external stimuli.
Mood is the emotional state experienced and described by the patient and observed by others.
Obsession is the pathological persistence of an irresistible thought or feeling that cannot be eliminated from consciousness by logical effort. It is associated with anxiety and rumination.
Paranoia is a descriptive term designating either morbid dominant ideas or delusions of self-reference concerning one or more of several themes, most commonly persecution, love, hate, envy, jealousy, honor, litigation, grandeur, or the supernatural.
Prosody is the melodic patterns of intonation in language that convey shades of meaning.
Psychosis is the inability or impaired ability to distinguish reality from hallucinations and/or delusions.
Thought process and content. Common descriptive terms include:
Principles of Differential Diagnosis
Emotional and cognitive processes are based on brain structure and physiology. Abnormal behavior can be attributable to the complex interplay of social influences, physical environment, and neural physiology. Psychosis, mania, depression, disinhibition, obsessive compulsive disorder (OCD), and anxiety all can occur as a result of neurological disease and can be indistinguishable from the idiopathic forms (Robinson and Travella, 1996). Neurological conditions must be considered in the differential diagnosis of any disorder with psychiatric symptoms.
Neuropsychiatric dysfunction can be correlated with altered functioning in anatomical regions. Any disease, toxin, drug, or process that affects a particular region can be expected to show changes in behavior mediated by the circuits within that region. The limbic system and the frontosubcortical circuits are most commonly involved in neuropsychiatric dysfunction. This neuroanatomical conceptual framework can provide useful information for localization and thus differential diagnosis. Klüver-Bucy syndrome, which consists of placidity, apathy, visual and auditory agnosia, hyperorality, and hypersexuality, occurs in processes that cause injury to the bilateral medial temporoamygdalar regions. A few of the most common causes of this syndrome include herpes encephalitis, traumatic brain injury (TBI), frontotemporal dementias (FTDs), and late-onset or severe Alzheimer disease (AD). Brain trauma, ischemic disease, demyelination, abscesses, or tumors, as well as degenerative dementias can also result in disinhibition. Damage to any portion of the circuit between the orbitofrontal cortex, ventral caudate nucleus, anterior globus pallidus, or mediodorsal thalamus can result in disinhibition (Tekin and Cummings, 2002).
Mood disorders, paranoia, disinhibition, and apathy derive from dysfunction in the limbic system and basal ganglia, which are phylogenetically more primitive (Mesulam, 2000). In some cases, the behavioral changes represent a response to the underlying disability; in others, behavioral abnormalities are part of the disease. For example, studies have shown that apathy in Parkinson disease (PD) is probably related to the underlying disease process, rather than being a psychological reaction to disability or to depression, and is closely associated with cognitive impairment (Kirsch-Darrow et al., 2006). Positron emission tomographic (PET) and single-photon emission computed tomographic (SPECT) studies suggest similar regions of abnormality in acquired forms of depression, mania, OCD, and psychosis, compared with their primary psychiatric presentations (Hirono et al., 1998; Rubinsztein et al., 2001; Saxena et al., 1998). Table 9.1 summarizes neuropsychiatric symptoms and their anatomical correlates. Additionally, the developmental phase during which a neurological illness occurs influences the frequency with which some neuropsychiatric syndromes are manifested. Adults with post-TBI sequelae tend to exhibit a higher rate of depression and anxiety. In contrast, post-TBI sequelae in children often involve attention deficits, hyperactivity, irritability, aggressiveness, and oppositional behavior (Geraldina et al., 2003). When temporal lobe epilepsy or Huntington disease (HD) begins in adolescence, a higher incidence of psychosis is noted than when their onset occurs later in life. Earlier onset of multiple sclerosis (MS) and stroke are associated with a higher incidence of depression (Rickards, 2005).
Table 9.1 Neuropsychiatric Symptoms and Corresponding Neuroanatomy
| Symptom | Neuroanatomical Region |
|---|---|
| Depression | Frontal lobes, left anterior frontal cortex, anterior cingulate gyrus, subgenu of the corpus callosum, basal ganglia, left caudate |
| Mania | Inferomedial and ventromedial frontal cortex, right inferomedial frontal cortex, anterior cingulate, caudate nucleus, thalamus, and temporothalamic projections |
| Apathy | Anterior cingulate gyrus, nucleus accumbens, globus pallidus, thalamus |
| OCD | Orbital or medial frontal cortex, caudate nucleus, globus pallidus |
| Disinhibition | Orbitofrontal cortex, hypothalamus, septum |
| Paraphilia | Mediotemporal cortex, hypothalamus, septum, rostral brainstem |
| Hallucinations | Unimodal association cortex, orbitofrontal cortex, paralimbic cortex, limbic cortex, striatum, thalamus, midbrain |
| Delusions | Orbitofrontal cortex, amygdala, striatum, thalamus |
| Psychosis | Frontal lobes, left temporal cortex |
OCD, Obsessive-compulsive disorder.
Patients with AD, PD, HD, and FTDs can develop multiple coexisting symptoms such as irritability, agitation, impulse-control disorders, apathy, depression, delusions, and psychosis that may be exacerbated by medications used to treat the underlying disorder (Table 9.2). For example, in patients with PD dopamine agonists such as pramipexole and ropinirole have been found to increase the risk of pathological gambling, compulsive shopping, hypersexuality, and other impulse-control disorders, sometimes referred to as dopamine dysregulation (Voon et al., 2006; Weintraub et al., 2006). Management outcome can be influenced by multiple factors. For instance, the complex relationship between behavioral changes and the caregiver’s ability to cope play a role in illness management and nursing home placement (de Vugt et al., 2005; Smith et al., 2001). Behavioral disturbances in patients with neurological illness have been related to the severity of caregiver distress (Kaufer et al., 1998).
Table 9.2 Neurological Disorders and Associated Prominent Behavioral Features
| Neurological Disorder | Associated Behavioral Disturbances |
|---|---|
| Alzheimer disease | Depression, irritability, anxiety, apathy, delusions, paranoia, psychosis |
| Lewy body dementia | Fluctuating confusion, hallucinations, delusions, depression, RBD |
| Vascular dementia | Depression, apathy, psychosis |
| Parkinson disease | Depression, anxiety drug-associated hallucinations and psychosis, RBD |
| FTD | Early impaired judgment, disinhibition, apathy, depression, delusions, psychosis |
| PSP | Disinhibition, apathy |
| TBI | Depression, disinhibition, apathy, irritability, psychosis uncommon |
| HD | Depression, irritability, delusions, mania, apathy, obsessive-compulsive disorder, psychosis |
| Corticobasal degeneration | Depression, irritability, RBD, alien hand syndrome |
| Epilepsy | Depression, psychosis |
| HIV infection | Apathy, depression, mania, psychosis |
| MS | Depression, irritability, anxiety, euphoria, psychosis |
| ALS | Depression, disinhibition, apathy, impaired judgment |
ALS, Amyotrophic lateral selerosis; FTD, frontotemporal dementia; HD, Huntington disease; HIV, human immunodeficiency virus; MS, multiple sclerosis; OCD, obsessive-compulsive disorder; PSP, progressive supranuclear palsy; RBD, rapid eye movement behavior disorder; TBI, traumatic brain injury.
Principles of Neuropsychiatric Evaluation
1. A normal neurological examination does not exclude neurological conditions. Lesions in the limbic, paralimbic, and prefrontal regions may manifest with cognitive-behavioral changes in the absence of elemental neurological abnormalities.
2. Normal routine laboratory testing, brain imaging, electroencephalography, and cerebral spinal fluid analysis do not necessarily exclude diseases of neurological origin.
3. New neurological complaints or behavioral changes should not be dismissed as being of psychiatric origin in a person with a preexisting psychiatric history.
4. The possibility of iatrogenically induced conditions such as lethargy with benzodiazepines, parkinsonism with neuroleptics, or hallucinations with dopaminergic medications must be taken into account. Medication side effects can significantly complicate the clinical history and physical examination in both the acute and long-term setting. Medication side effects can also potentially be harbingers of underlying pathology or progression of illness. Marked parkinsonism occurring after neuroleptic exposure can be a feature of PD and dementia with Lewy bodies (Aarsland et al., 2005) before the underlying neurodegenerative condition becomes clinically apparent. PD patients may develop hallucinations as a side effect of dopaminergic medications (Papapetropoulos and Mash, 2005).
5. Treatment of primary psychiatric and neurological behavioral disturbances share common principles. A response to therapy does not constitute evidence for a primary psychiatric condition.
The medical evaluation of affective illness and psychotic disorders must be individualized based on the patient’s family history, social environment, habits, risk factors, age, gender, clinical history, and examination findings. A careful review of the patient’s medical history and a general physical examination as well as a neurological examination (Murray and Price, 2008; Ovsiew et al., 2008) should be performed to assess for possible neurological and medical causes. The most basic evaluation should include vital signs (blood pressure, pulse, respirations, and temperature) and a laboratory evaluation that minimally includes a complete blood cell count (CBC); electrolyte panel; determination of serum levels for glucose, blood urea nitrogen (BUN), creatinine, calcium, total protein, and albumin; liver function assessment; thyroid function assessment; and additional laboratory testing as clinically indicated. Consideration should be given to checking the patient’s oxygen saturation on room air (especially in the elderly). Neurological abnormalities suggested by the clinical history or identified on examination, especially those attributable to the central nervous system (CNS), should prompt further evaluation for neurological and medical causes of psychiatric illness. A clear consensus has not been reached about when neuroimaging is indicated as part of the evaluation of new-onset depression in patients without focal neurological complaints and a normal neurological examination. This must be individualized based on clinical judgment. Treatment-resistant depression should prompt reassessment of the diagnosis and evaluation to rule out secondary causes of depressive illness. A careful history to rule out a primary sleep disorder such as sleep apnea should be considered in the evaluation of refractory depressive symptoms (Haba-Rubio, 2005) or cognitive complaints. When new-onset psychosis presents in the absence of identifiable infectious/inflammatory, metabolic, toxic, or other causes, we recommend that magnetic resonance imaging (MRI) of the brain be incorporated into the evaluation. In our experience, 5% to 10% of such patients have MRI abnormalities that identify potential neurological contributions (particularly in those 65 years of age and older). The MRI will help exclude lesions (e.g., demyelination, ischemic disease, neoplasm, congenital structural abnormalities, evidence of metabolic storage diseases) in limbic, paralimbic, and frontal regions, which may not be associated with neurological abnormalities on elemental examination (Walterfang et al., 2005). An electroencephalogram (EEG) should be considered to evaluate for complex partial seizures if there is a history of intermittent, discrete, or abrupt episodes of psychiatric dysfunction (e.g., confusion, spells of lost time, psychotic symptoms), stereotypy of hallucinations, automatisms (e.g., lip smacking, repetitive movements) associated with episodes of psychiatric dysfunction (or confusion), or a suspicion of encephalopathy (or delirium). Sensitivity of the EEG for detecting seizure activity is highest when the patient has experienced the specific symptoms while undergoing the study. Selected cases may require 24-hour or longer EEG monitoring to capture a clinical event to clarify whether a seizure disorder is present.
Cognitive-Behavioral Neuroanatomy
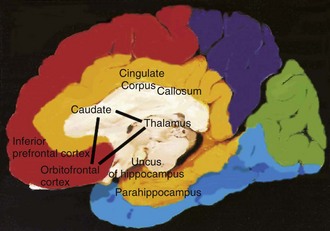
The cerebral cortex can be subdivided into five major functional subtypes: primary sensory-motor, unimodal association, heteromodal association, paralimbic, and limbic. The primary sensory areas are the point of entry for sensory information into the cortical circuitry. The primary motor cortex conveys complex motor programs to motor neurons in the brainstem and spinal cord. Processing of sensory information occurs as information moves from primary sensory areas to adjacent unimodal association areas. The unimodal and heteromodal cortices are involved in perceptual processing and motor planning. The complexity of processing increases as information is then transmitted to heteromodal association areas which receive input from more than one sensory modality. Examples of heteromodal association cortex include prefrontal cortex, posterior parietal cortex, parts of the lateral temporal cortex, and portions of the parahippocampal gyrus. These cortical regions have a six-layered architecture. Further cortical processing occurs in areas designated as paralimbic. These regions demonstrate a gradual transition of cortical architecture from the six-layered to the more primitive and simplified allocortex of limbic structures. The paralimbic regions consist of orbitofrontal cortex, insula, temporal pole, parahippocampal cortex, and cingulate cortex. Cognitive, emotional, and visceral inputs merge in these regions. The limbic subdivision is composed of the hippocampus, amygdala, substantia innominata, prepiriform olfactory cortex, and septal area (Figs. 9.1 and 9.2). These structures are to a great extent reciprocally interconnected with the hypothalamus. The limbic region is intimately involved with regulation of emotion, memory, motivation, autonomic, and endocrine function. The highest level of cognitive processing occurs in regions referred to as transmodal areas. These areas are composed of heteromodal, paralimbic, and limbic regions, which are collectively linked, in parallel, to other transmodal regions. Interconnections among transmodal areas (e.g., Wernicke area, posterior parietal cortex, hippocampal-enterorhinal complex) allow integration of distributed perceptual processing systems, resulting in perceptual recognition such as scenes and events becoming experiences and words taking on meaning (Mesulam, 2000).
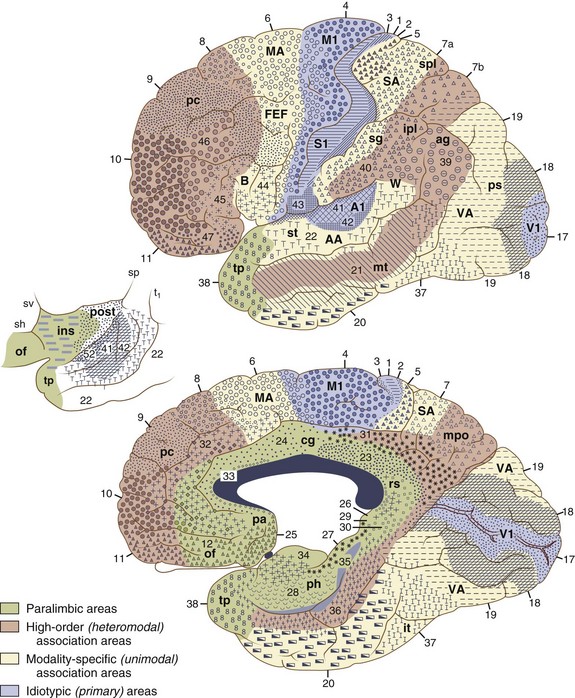
Cortical Networks
Five distinct cortical network regions govern various aspects of cognitive functioning: (1) the language network, which includes transmodal regions or “epicenters” in Broca and Wernicke areas; (2) spatial awareness, based in transmodal regions in the frontal eye fields and posterior parietal area; (3) the memory and emotional network, located in the hippocampal-enterorhinal region and amygdala; (4) the executive function–working memory network, based in transmodal regions in the lateral prefrontal cortex and possibly the inferior parietal cortices; and (5) the face-object recognition network, based in the temporopolar and midtemporal cortices (Mesulam, 1998).
The ability to empathize with another person’s psychological and physical circumstances is a foundation for social and moral behavior. The human mirror neuron system is now postulated to be involved in understanding the actions of others and the intentions behind the actions. It also may provide the basis for observational learning. The parietofrontal mirror system, which includes the parietal lobe and the premotor cortex plus the caudal part of the inferior frontal gyrus, is involved in recognition of voluntary behavior in other people, while the limbic mirror system, formed by the insula and the anterior mesial frontal cortex, is devoted to the recognition of affective behavior. Dysfunction of this system is postulated to underlie deficits in theory of mind and has been proposed as an explanation for the social deficits seen in autistic disorders (Cattaneo and Rizzolatti, 2009).
Frontosubcortical Networks
Five frontosubcortical circuits subserve cognition, behavior, and movement. Disruption of these networks at the cortical or subcortical level can be associated with similar neuropsychiatric symptoms. Each of these circuits shares the same components: (1) frontal cortex, (2) striatum (caudate, putamen, ventral striatum), (3) globus pallidus and substantia nigra, and (4) thalamus (which then projects back to frontal cortex) (Tekin and Cummings, 2002) (Fig. 9.3). Integrative connections also occur to and from other subcortical and distant cortical regions related to each circuit. Neurotransmitters such as dopamine (DA), glutamate, γ-aminobutyric acid (GABA), acetylcholine, norepinephrine, and serotonin are involved in various aspects of neural transmission and modulation in these circuits. The frontosubcortical networks are named according to their site of origin or function. Somatic motor function is subserved by the motor circuit originating in the supplementary motor area. Oculomotor function is governed by the oculomotor circuit originating in the frontal eye fields. Three of the five circuits are intimately involved in cognitive and behavioral changes: the dorsolateral prefrontal, the orbitofrontal, and the anterior cingulate circuits. Each circuit has both efferent and afferent connections with adjacent and distant cortical regions. The dorsolateral prefrontal circuit governs executive functions, including the ability to plan and maintain attention, problem solve, learn, retrieve remote memories, sequence the temporal order of events, shift cognitive and behavioral sets, and generate motor programs. Executive dysfunction is a principal component of subcortical dementias. Deficits identified in subcortical dementias include slowed information processing, memory retrieval deficits, mood and behavioral changes, gait disturbance, dysarthria, and other motor impairments. Vascular dementias, PD, and HD are a few examples of conditions that affect this circuit.
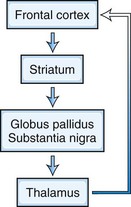
The orbitofrontal circuit connects frontal monitoring functions to the limbic system. This circuit governs appropriate responses to social cues, empathy, social judgment, and interpersonal sensitivity. It pairs thoughts, memories, and experiences with corresponding visceral and emotional states. This circuit is heavily involved in the process of decision making and evaluating the costs and benefits of specific behavioral responses to the environment. The medial orbitofrontal cortex (OFC) evaluates reward, whereas the lateral OFC monitors and decodes punishment as it pertains to motivating behavioral change. There is also an anterior-posterior gradient in which the reward value for more abstract and complex secondary reinforcing factors such as money are encoded in the anterior regions, and more concrete factors such as touch and taste are encoded in the posterior OFC areas. The posterior OFC is thought to have an important role in evaluating the emotional significance of stimuli (Barbas and Zikopoulos, 2007). Dysfunction in this circuit can lead to disinhibition, irritability, aggressive outbursts, inappropriate social responses, and impulsive decision making. Patients with OFC lesions show deficits in both the production and recognition of emotional expression from the face, voice, or gestures. Persons with bilateral OFC lesions may manifest “theory of mind” deficits. Theory of mind is a model of how a person understands and infers other people’s intentions, desires, mental states, and emotions (Bodden et al., 2010). Conditions that exhibit impairment in this circuit include schizophrenia (Bora et al., 2009), FTD (Adenzato et al., 2010), and HD. Other conditions that may affect this circuit include closed head trauma, rupture of anterior communicating aneurysms, and subfrontal meningiomas.
The anterior cingulate circuit includes the nucleus accumbens and has both afferent and efferent connections to the dorsolateral prefrontal cortex (DLPFC) and amygdala. It is involved in motivated behavior. Lesions in this circuit result in apathy, abulia, and akinetic mutism. There also is a reported decrease in the ability to understand new thoughts and participate in the creative thought process (Chow and Cummings, 1999; Mesulam, 2000). The medial prefrontal cortex is thought to play a significant role in generating emotions related to empathy, cognitive functions related to theory of mind, and the ability to recognize a moral dilemma (Robertson et al., 2007). The ventromedial frontal lobe evaluates the current relative value of stimuli helping to guide decision making by determining the goals toward which behavior is directed and through judging outcomes (Fellows, 2007). Some conditions that may affect this circuit include AD, FTD, PD, HD, head trauma, brain tumors, cerebral infarcts, and obstructive hydrocephalus.
Cerebrocerebellar Networks
The cerebellum is engaged in the regulation of cognition and emotion through a feed-forward and feedback loop. The cortex projects to pontine nuclei, which in turn project to the cerebellum. The cerebellum projects to the thalamus, which then projects back to the cortex. Cognitive processing tasks such as language, working memory, and spatial and executive tasks appear to activate the posterior cerebellar lobe. The posterior cerebellar vermis may function as a putative limbic cerebellum, modulating emotional processing (Stoodley and Schmahmann, 2010). Distractibility, executive and working memory problems, impaired judgment, reduced verbal fluency, disinhibition, irritability, anxiety, emotional lability or blunting, obsessive compulsive behaviors, depression, and psychosis have been reported in association with cerebellar pathology.
Biology of Psychosis
Among several etiological hypotheses for schizophrenia, the neurodevelopmental model is one of the most prominent. This model generally posits that schizophrenia results from processes that begin long before the onset of clinical symptoms and is caused by a combination of environmental and genetic factors (Murray and Lewis, 1987; Weinberger, 1987). Several postmortem and neuroimaging studies support this hypothesis with findings of brain developmental alterations such as agenesis of the corpus callosum, arachnoid cysts, and other abnormalities in a significant number of schizophrenic patients (Hallak et al., 2007; Kuloglu et al., 2008). Environmental factors are associated with an increased risk for schizophrenia. These factors include being a first-generation immigrant or the child of a first-generation immigrant, urban living, drug use, head injury, prenatal infection, maternal malnutrition, obstetrical complications during delivery, and winter birth (Tandon et al., 2008). Genetic risks are clearly present but not well understood. The majority of patients with schizophrenia lack a family history of the disorder. The population lifetime risk for schizophrenia is 1%, 10% for first-degree relatives, and 4% for second-degree relatives. There is an approximately 50% concordance rate for monozygotic twins, compared to approximately 15% for dizygotic twins. Advancing paternal age increases risk in a linear fashion, which is consistent with the hypothesis that de novo mutations contribute to the genetic risk for schizophrenia. It is most likely that many different genes make small but important contributions to susceptibility. The disease only manifests when these genes are combined or certain environmental factors are present. A number of susceptibility genes show association with schizophrenia: catechol-O-methyl-transferase, neuroregulin 1, dysbindin, disrupted in schizophrenia 1 (DISC1), metabotropic glutamate receptor type 3 gene and G27/G30 gene complex (Nöthen et al., 2010; Tandon et al., 2008). Research in twins and first-degree relatives of patients has shown that genes predisposing to schizophrenia and related disorders affect heritable traits related to the illness. Such traits include neurocognitive functioning, structural MRI brain volume measures, neurophysiological informational processing traits, and sensitivity to stress (van Os and Kapur, 2009). A small proportion of schizophrenia incidence may be explained by genomic structural variations known as copy number variants (CNVs). CNVs consist of inherited or de novo small duplications, deletions, or inversions in genes or regulatory regions. CNV deletions generally show higher penetrance (more severe phenotype) than duplications, and larger CNVs often have higher penetrance and/or more clinical features than smaller CNVs. These genomic structural variations contribute to normal variability, disease risk, and developmental anomalies, as well as act as a major mutational mechanism in evolution. The most common CNV disorder, 22q11.2 deletion syndrome (velocardiofacial syndrome), has an established association with schizophrenia. Individuals with 22q11.2 deletions have a 20-fold increased risk for schizophrenia and constitute about 0.9% to 1% of schizophrenia patients. When this syndrome is present, genetic counseling is helpful (Bassett and Chow, 2008).
A wide variety of neurological conditions, medications, and toxins are associated with psychosis. No consensus is available in the literature regarding the precise anatomical localization of various psychotic syndromes. Evidence from neurochemistry, cellular neuropathology, and neuroimaging studies support that schizophrenia is a brain disease, but there is no universally accepted theory regarding the specific nature of the brain dysfunction. The two best-known neurotransmitter models offered to explain the various manifestations of schizophrenia include the “dopamine hypothesis,” now in its third revision (Howes and Kapur, 2009), and the “glutamate hypothesis.” Schizophrenia has been associated with frontal lobe dysfunction and abnormal regulation of subcortical DA (Goldman-Rakic et al., 2004) and glutamate systems (Weinberger, 2005).
Functional imaging studies in persons with schizophrenia show decreased cerebral blood flow (CBF) in the DLPFC during specific cognitive tasks (Andreasen, 1996; Lehrer et al., 2005). Schizophrenic patients with prominent negative symptoms display reduced glucose utilization in the frontal lobes. Functional imaging studies suggest that disruption in distributed functional circuits is important in the development of schizophrenia. These functional circuit locations include the DLPFC, orbitofrontal cortex, mediofrontal cortex, anterior cingulate gyrus, thalamus, temporal lobe subregions, and the cerebellum (Schultz and Andreasen, 1999). Several conditions that may manifest psychosis (e.g., HD, PD, frontotemporal degenerations, stroke) are commonly associated with frontal and subcortical dysfunction. Dorsolateral and mediofrontal hypoperfusion on functional imaging has been demonstrated in a subset of AD patients with delusions (Hirono et al., 1998).
Biology of Depression
The connection between psychiatry and neurology is nowhere more evident than the remarkable comorbidity of psychiatric illness, especially depression, in many neurological disorders, with a 20% to 60% prevalence rate of depression in patients with stroke, neurodegenerative diseases, MS, headache, human immunodeficiency virus (HIV), TBI, epilepsy, chronic pain, obstructive sleep apnea, intracranial neoplasms, and motor neuron disease. Depression amplifies the physiological response to pain, while pain-related symptoms and limitations frequently lead to the emergence of depressive symptoms. In a community-based study, almost 50% of adolescents with chronic daily headaches had at least one psychiatric disorder, most commonly major depression and panic. Women with migraine who have major depression are twice as likely as those with migraine alone to report being sexually abused as a child. If the abuse continued past age 12, women with migraine were five times more likely to report depression (Tietjen et al., 2007). Despite the proliferation of antidepressant therapeutics, major depression is often a chronic and/or recurrent condition that remains difficult to treat. Up to 70% of patients taking antidepressants in a primary care setting may be poorly compliant, most often due to adverse side effects during both short and long-term therapy.
Efforts to link single genes to major depressive disorder (MDD) have been unsuccessful despite exhaustive mapping attempts. Consequently, behavioral geneticists have turned to the study of genetic polymorphisms in establishing a predisposition to depression and in shaping the response to environmental stressors. Perlis and coworkers (2007) found a strong association between variation at the CREB1 locus and anger expression in MDD. The most extensive studies in this field have focused on polymorphisms in the serotonin transporter (5-HTT) gene. Recent work has demonstrated that patients with a single nucleotide polymorphism on the long allele that entails an A-to-G transposition (LG) have low expression of 5-HT. The risk for major depression among hurricane survivors with either short or LG alleles was four to five times that of low-risk survivors. Although conflicting results have been reported with regard to these polymorphisms, a recent meta-analysis found that the 44bp Ins/Del short/long polymorphism was associated with MDD, whereas the VNTR intron 2 polymorphism was not. This study also reported significant associations for polymorphisms in the apolipoprotein E, guanine nucleotide binding protein, methylenetetrahydrofolate reductase, and dopamine transporter genes (López-León et al., 2008).
Behavioral genetics research based on diathesis-stress models of depression demonstrate that the risk of depression after a stressful event is enhanced in populations carrying genetic risk factors and is diminished in populations lacking such risk factors. A gene’s contribution to depression may be missed in studies that do not account for environmental interactions and may only be revealed when studied within the context of environmental stressors specifically mediated by that gene (Uher, 2008). Genotype-environment interactions are ubiquitous because genes not only impact the risk for depression by creating susceptibility to specific environmental stressors but also cause individuals to persistently place themselves in highly stressful environments.
The potential clinical relevance of neurogenesis in the adult mammalian brain represents the most recent major breakthrough in depression studies at the cellular neurobiological level. Imaging studies have demonstrated a 10% to 20% decrease in the hippocampal volume of human patients with chronic depression. Cell proliferation studies using 5-bromo-2′-deoxyuridine injection to label dividing cells show that antidepressants also lead to increased cell number in the mammalian hippocampus. This effect is seen with chronic but not acute treatment; the time course of the effect mirrors the known time course of the therapeutic action of antidepressants in humans (Czéh et al., 2001). Although a role for neurogenesis in the pathophysiology of depression appears to be a promising avenue of research, the relevance of animal studies described here remains controversial in the human (Reif et al., 2006).
Analysis at the systems level suggests that anterotemporal paralimbic and orbitofrontal regions are involved in mediating primary and acquired depression. Functional imaging studies of unmedicated patients with familial depression reveal increased CBF and glucose metabolism in the amygdala, orbital cortex, and medial thalamus and decreased CBF and glucose metabolism in the dorsomedial/dorsal anterolateral prefrontal cortex and anterior cingulate cortex (Charney and Manji, 2004). Damage to the prefrontal cortex from stroke or tumor, or to the striatum from degenerative diseases such as PD and HD, is associated with depression (Charney and Manji, 2004; Drevets, 2001). Functional imaging studies of subcortical disorders such as these reveal hypometabolism in paralimbic regions, including the anterotemporal cortex and anterior cingulate, which are correlated with depression seen in these patients (Ketter et al., 1996; Mayberg, 2003). Depression in PD, HD, and epilepsy has been correlated with reduced metabolic activity in the orbitofrontal cortex and caudatenucleus.
Mayberg (2003) proposed that primary depression is due to dysfunction in a network that includes two known pathways: the orbitofrontal–basal ganglia–thalamic circuit and the basotemporal limbic circuit that links the orbitofrontal cortex and the anterior temporal cortex by the uncinate fasciculus. Portions of this model are illustrated in Fig. 9.4. This has been expanded into a unifying depression circuit model that consists of four interconnected functional compartments. Each functional compartment consists of strongly interconnected anatomical structures upon which that compartment is dependent. Functional compartments are as follows: mood regulation (medial frontal, medial orbital-frontal and pregenu anterior cingulate cortex), mood monitoring (ventral striatum-caudate, amygdale, dorsomedial thalamus, midbrain-ventral tegmental area), interoception (subcallosal cingulated, ventral-anterior hippocampus, anterior insula, brain stem, hypothalamus) and exteroception (prefrontal, premotor, parietal, mid-cingulate and posterior cingulate cortices with dorsal-posterior hippocampus) (Mayberg, 2009).
Functional imaging studies of untreated depression have been extended to evaluate responses to pharmacological, cognitive-behavioral, and surgical treatments of depression. Clinical improvement after treatment with serotonin-specific reuptake inhibitors such as fluoxetine correlates with increased activity on PET in brainstem and dorsal cortical regions including the prefrontal, parietal, anterior, and posterior cingulate areas, and with decreased activity in limbic and striatal regions including the subgenual cingulate, hippocampus, insula, and pallidum (Mathew et al., 2003). These findings are consistent with the prevailing model for involvement of a limbic-cortical-striatal-pallidal-thalamic circuit in major depression. The same group has shown that imaging can be used to identify patterns of metabolic activity predictive of treatment response. Hypometabolism of the rostral anterior cingulate characterized patients who failed to respond to antidepressants, whereas hypermetabolism characterized responders. Dougherty and coworkers (2003) used PET to search for neuroimaging profiles that might predict clinical response to anterior cingulotomy in patients with treatment-refractory depression. Responders displayed elevated preoperative metabolism in the left prefrontal cortex and the left thalamus. A combination of functional imaging and pharmacogenomic technologies might allow subsets of treatment responders to be classified and predicted more precisely than with either technology alone. Goldapple and coinvestigators (2004) used PET to study the clinical response of cognitive-behavioral therapy in patients with unipolar depression and found increases in hippocampus and dorsal cingulate and decreases in dorsal, ventral, and medial frontal cortex. The authors speculate that the same limbic-cortical-striatal-pallidal-thalamic circuit is involved but that differences in the direction of metabolic changes may reflect different underlying mechanisms of action of cognitive-behavioral therapy (CBT) and selective serotonin reuptake inhibitors (SSRIs).
Clinical Symptoms and Signs Suggesting Neurological Disease
Many neurological conditions have associated psychiatric symptoms. Psychiatrists and neurologists need to be intimately acquainted with features of the clinical history and examination that indicate the need for further investigation. Box 9.3 outlines some key features that have historically suggested an underlying neurological condition. Box 9.4 (online only at www.expertconsult.com) reviews some key areas of the review of systems that can be helpful when assessing for neurological and medical causes of psychiatric symptoms. Table 9.3 (online only at www.expertconsult.com) reviews abnormalities in the elemental neurological examination associated with diseases that can exhibit significant neuropsychiatric features.
Box 9.3 Historical Features Suggesting Neurological Disease in Patients with Psychiatric Symptoms
Box 9.4 Review of Systems with Possible Neuropsychiatric Relevance and Related Neurological Conditions
ALS, Amyotrophic lateral sclerosis; CNS, central nervous system; MS, multiple sclerosis.
Table 9.3 Neurological Abnormalities Suggesting Diseases Associated with Psychiatric Symptoms
| Examination Abnormalities | Disease(s) or Underlying Etiology |
|---|---|
| Vital signs: | |
| Marked hypertension | Hypertensive encephalopathy, serotonin syndrome, neuroleptic malignant syndrome, preeclampsia |
| Tachypnea | Delirium due to systemic infection |
| Hypoventilation | Hypoxia, alcohol withdrawal, sedative intoxication |
| Behavior: | |
| Alien hand syndrome | Corticobasal ganglionic degeneration |
| Cranial nerves: | |
| Visual field deficit | Stroke, mass, MS, lupus |
| Pupils: | |
| Argyll Robertson | Neurosyphilis |
| Unilateral dilation | Brain herniation, porphyria |
| Horner syndrome | Stroke, carotid disease, demyelinating disease |
| Ophthalmoplegia: | |
| Vertical gaze palsy | PSP |
| Mixed | Wernicke-Korsakoff syndrome, chronic basilar meningitis |
| Cornea: Kayser-Fleischer rings | Wilson disease |
| Lens: cataracts | Chronic steroids, Down syndrome |
| Fundi: | |
| Papilledema | Intracranial mass lesion, MS |
| Optic pallor | MS, porphyria, Tay-Sachs |
| Extrapyramidal | Parkinson disease, DLB, HD, stroke, WD, numerous others |
| Cerebellar | Alcohol, hereditary degenerative ataxias, paraneoplastic, medication toxicity |
| Motor neuron | ALS, FTD with motor neuron disease |
| Peripheral nerve | Adrenomyeloneuropathy, metachromatic leukodystrophy, B12 deficiency, porphyria |
| Gait: | |
| Apraxia | Normal pressure hydrocephalus, frontal network dementias |
| Spasticity | Stroke, MS |
| Bradykinesia | Multiinfarct dementia, PD, PSP, DLB |
ALS, Amyotrophic lateral sclerosis; DLB, dementia with lewy bodies; HD, Huntington disease; MS, multiple sclerosis; PSP, progressive supranuclear palsy; WD, Wilson disease.
Psychiatric Manifestations of Neurological Disease
Virtually any process that affects the neuroanatomical circuits described earlier can result in behavioral changes and psychiatric symptoms at some point. Psychiatric symptoms may be striking and precede any neurological manifestation by years. Table 9.4 (online only at www.expertconsult.com) lists conditions that can be associated with psychosis or depression. Box 9.5 summarizes some key points from the preceding discussion. A general overview and discussion of a number of major categories of neurological and systemic conditions with prominent neuropsychiatric features follows. More detailed information regarding the evaluation, natural history, pathology, and specific treatment recommendations for these conditions is beyond the scope of this chapter.
Box 9.5 Key Points
1. Affective and psychotic disorders may occur as a result of neurological disease and be indistinguishable from the idiopathic forms.
2. Neuropsychiatric and cognitive dysfunction can be correlated with altered functioning in anatomical regions.
3. Cortical processing of sensory information proceeds from its point of entry through association areas with progressively more complex interconnections with other regions having sensory, memory, cognitive, emotional, and autonomic information, resulting ultimately in perceptual recognition and emotional meaning for experiences.
4. Frontosubcortical circuits are heavily involved in cognitive and behavioral functioning. Disruption of frontal circuits at the cortical or subcortical level by various processes can be associated with similar neuropsychiatric symptoms.
5. Features of the patient’s clinical history and examination can be suggestive of a medical or neurological cause of psychiatric symptoms.
6. Many medical and neurological conditions are associated with neuropsychiatric symptoms. Each condition may carry unique implications for prognosis, treatment, and long-term management.
Table 9.4 Selected Neurological and Systemic Causes of Depression and/or Psychosis
| Category | Disorders |
|---|---|
| Head trauma | Traumatic brain injury |
| Subdural hematoma | |
| Infectious | Lyme disease |
| Prion diseases | |
| Neurosyphilis | |
| Viral infections/encephalitides (HIV infection/encephalopathy, herpes encephalitis, cytomegalovirus, Epstein-Barr virus, etc.) | |
| Whipple disease | |
| Cerebral malaria | |
| Encephalitis | |
| Systemic infection | |
| Inflammatory | Systemic lupus erythematosus |
| Sjögren syndrome | |
| Temporal arteritis | |
| Hashimoto encephalopathy | |
| Sydenham chorea | |
| Sarcoidosis | |
| Neoplastic | Primary or secondary cerebral neoplasm |
| Systemic neoplasm | |
| Pancreatic cancer | |
| Paraneoplastic encephalitis | |
| Endocrine/acquired metabolic | Hepatic encephalopathy |
| Uremic encephalopathy | |
| Dialysis dementia | |
| Hypo/hyperparathyroidism | |
| Hypo/hyperthyroidism | |
| Addison disease/Cushing disease | |
| Postpartum | |
| Vitamin deficiency: B12, folate, niacin, vitamin C | |
| Gastric bypass associated nutritional deficiencies | |
| Hypoglycemia | |
| Vascular | Stroke |
| Multiinfarct dementia | |
| Central nervous system vasculitis | |
| Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) | |
| Degenerative | Alzheimer disease |
| Lewy body disease | |
| Frontotemporal dementias | |
| Parkinson disease | |
| Progressive supranuclear palsy | |
| Huntington disease | |
| Corticobasal ganglionic degeneration | |
| Multisystem atrophy/striatonigral degeneration/olivopontocerebellar atrophy | |
| Idiopathic basal ganglia calcifications/Fahr disease | |
| Demyelinating/dysmyelinating | Multiple sclerosis |
| Acute disseminated encephalomyelitis | |
| Adrenoleukodystrophy | |
| Metachromatic leukodystrophy | |
| Inherited metabolic | Wilson disease |
| Tay-Sachs disease | |
| Adult neuronal ceroid lipofuscinosis | |
| Niemann-Pick type C | |
| Acute intermittent porphyria | |
| Mitochondrial encephalopathy, lactic acidosis and stroke-like episodes | |
| Epilepsy | Ictal |
| Interictal | |
| Postictal | |
| Forced normalization | |
| Post epilepsy surgery | |
| Medications | Analgesics |
| Androgens | |
| Antiarrhythmics | |
| Anticonvulsants | |
| Anticholinergics | |
| Antibiotics | |
| Antihypertensives | |
| Antineoplastic agents | |
| Corticosteroids | |
| Dopamine agonists | |
| Oral contraceptives | |
| Sedatives/hypnotics | |
| Steroids | |
| Drugs of abuse | Alcohol |
| Amphetamines | |
| Cocaine | |
| Hallucinogens | |
| Marijuana | |
| MDMA (Ecstasy) | |
| Phencyclidine | |
| Drug withdrawal syndromes | Alcohol |
| Barbiturates | |
| Benzodiazepines | |
| Amphetamines | |
| Toxins | Heavy metals |
| Inhalants | |
| Other | Normal pressure hydrocephalus |
| Ionizing radiation exposure | |
| Decompression sickness |
Stroke and Cerebral Vascular Disease
Psychosis or psychotic features may present as a rare complication of a single stroke, but the prevalence of these features is not well established. Manifestations may include paranoia, delusions, ideas of reference, hallucinations, or psychosis. Paranoia and psychosis have been reported in association with left temporal strokes that result in Wernicke aphasia. Other regions producing similar neuropsychiatric symptoms include the right temporoparietal region and the caudate nuclei. Right hemispheric lesions may also be more associated with visual hallucinations and delusions. Reduplicative paramnesia and misidentifications syndromes such as Capgras syndrome and Fregoli syndrome have also been reported. Reduplicative paramnesia is a syndrome in which patients claim that they are simultaneously in two or more locations. It has been observed to occur in patients with combined lesions of frontal and right temporal lobe but has also been described as due to temporal-limbic-frontal dysfunction (Moser et al., 1998). Capgras syndrome is the false belief that someone familiar, usually a family member or close friend, has been replaced by an identical-appearing imposter. It has been proposed that this results from right temporal-limbic-frontal disconnection resulting in a disturbance in recognizing familiar people and places (Feinberg et al., 1999). In Fregoli syndrome, the patient believes a persecutor is able to take on a variety of faces, like an actor. Psychotic episodes can also be a manifestation of complex partial seizures secondary to stroke. Patients with poststroke psychosis are more prone to have comorbid epilepsy than poststroke patients without associated psychosis. Lesions or infarcts of the ventral midbrain can result in a syndrome characterized by well-formed and complex visual hallucinations referred to as peduncular hallucinosis. Obsessive-compulsive features have also been reported with strokes. These symptoms have been postulated to be due to dysfunction in the orbitofrontal-subcortical circuitry (Saxena et al., 1998).
Consensus criteria for accurately diagnosing vascular cognitive impairments and dementia are lacking (Bowler and Gorelick, 2009; Wiederkehr et al., 2008). The vascular cognitive impairments can be conceptualized as being made up of three groups: vascular dementia, mixed vascular dementia and AD pathology, and vascular cognitive impairment not meeting criteria for dementia. These conditions may have variable contributions from mixed forms of small-vessel disease, large-vessel disease, and cardioembolic disease, which accounts for the clinical phenotypic heterogeneity. AD pathology is commonly found in association with cerebrovascular disease pathology, leading to uncertainty with respect to the relative contributions of each in some cases. A temporal relationship between a stroke and the onset of dementia or a stepwise progression of cognitive decline with evidence of cerebrovascular disease on examination and neuroimaging are considered most helpful. No specific neuroimaging profile exists that is diagnostic for pure cerebrovascular disease-related dementia. Vascular dementia may present with prominent cortical, subcortical, or mixed features. Cortical vascular dementia may manifest as unilateral sensorimotor dysfunction, abrupt onset of cognitive dysfunction and aphasia, and difficulties with planning, goal formation, organization, and abstraction. Subcortical vascular dementia often affects frontosubcortical circuitry, resulting in executive dysfunction, cognitive slowing, difficulties with abstraction, apathy, memory problems (recognition and cue recognition relatively intact), working memory impairment, and decreased ability to perform activities of daily living. Memory difficulties tend to be less severe than in AD. Limited data suggest that cholinesterase inhibitors are beneficial for treatment of vascular dementia, as demonstrated by improvements in cognition, global functioning, and performance of activities of daily living.
Infectious
Human Immunodeficiency Virus
Individuals infected with HIV can be affected by a variety of neuropsychiatric and neurological problems independent of opportunistic infections and neoplasms. These include cognitive impairment, behavioral changes, and sensorimotor disturbances. Psychiatrists and neurologists must anticipate a spectrum of psychiatric phenomena that can include depression, paranoia, delusions, hallucinations, psychosis, mania, irritability, and apathy. HIV-associated dementia (HAD) is the term given to the syndrome that presents with bradyphrenia, memory decline, executive dysfunction, impaired concentration, and apathy. These features are compatible with a subcortical dementia with prominent dysfunction in the frontal-basal ganglia circuitry (Woods et al., 2004). Minor cognitive motor disorder (MCMD) refers to a milder form of this syndrome that has become more common since the advent of highly active antiretroviral therapy (HAART). HAD may be the acquired immunodeficiency virus syndrome (AIDS)-defining illness in up to 10% of patients. It has been estimated to occur in 20% to 30% of untreated adults. HAART has reduced its frequency by approximately 50%, but the frequency of pathologically proven HIV encephalitis remains high.
Lifetime prevalence of depression in HIV-positive individuals is 22% to 45%, with depressed individuals demonstrating reduced compliance with antiretroviral therapy and increased HIV-related morbidity. Antidepressants have been efficacious in treating HAD (Himelhoch and Medoff, 2005). Psychostimulants may also be a helpful adjunct in treating HAD. Evidence suggests that HIV-infected patients with new-onset psychosis usually respond well to typical neuroleptic medications, but they are more sensitive to the side effects of these medications, particularly extrapyramidal symptoms and tardive dyskinesias. This sensitivity is thought to be due to HIV’s effect on the basal ganglia, resulting in a loss of dopaminergic neurons. When prescribing typical neuroleptics, caution is warranted owing to this sensitivity and the additional possible pharmacological interactions with antiretroviral medications. Atypical neuroleptics are favored.
Creutzfeldt-Jakob Disease
Prion diseases are a group of fatal degenerative disorders of the nervous system caused by a conformational change in the prion protein, a normal constituent of cell membranes. They are characterized by long incubation periods followed by relatively rapid neurological decline and death (Johnson, 2005). Creutzfeldt-Jakob disease (CJD) is the most common human prion disease but is rare, with an incidence of between 0.5 and 1.5 cases per million people per year. The sporadic form of the disease accounts for about 85% of cases, typically occurs later in life (mean age, 60 years), and manifests with a rapidly progressive course characterized by cerebellar ataxia, dementia, myoclonus, seizures, and psychiatric symptoms progressing to akinetic mutism and complete disability within months after disease onset. Psychiatric symptoms such as personality changes, anxiety, depression, paranoia, obsessive-compulsive features, and psychosis occur in about 80% of patients during the first 100 days of illness (Wall et al., 2005). About 60% present with symptoms compatible with a rapidly progressive dementia. The mean duration of the illness is 6 to 7 months.
The autosomal dominant familial form of CJD accounts for 10% to 15% of cases, and iatrogenically caused cases account for about 1%. New-variant CJD is a new form of acquired spongiform encephalopathy that emerged in 1994 in the United Kingdom. This form has been linked with consumption of infected animal products. Patients with the new variant have a different course characterized by younger age at onset (mean age, 29 years), prominent psychiatric and sensory symptoms, and a longer disease course. (Spencer and colleagues, 2002) reported that 63% demonstrated purely psychiatric symptoms at onset (dysphoria, anxiety, anhedonia), 15% had purely neurological symptoms, and 22% had features of both. Median duration of illness was 13 months, and by the time of death, prominent neurological and psychiatric manifestations were universal.
Neurosyphilis
A resurgence of neurosyphilis has accompanied the AIDS epidemic in the industrialized world. Neurosyphilis may occur in any stage of syphilis. Early neurosyphilis, seen in the first weeks to years of infection, is primarily a meningitic process in which the parenchyma is not typically involved. It can coexist with primary or secondary syphilis and be asymptomatic. Inadequate treatment of early syphilis and coinfection with HIV predispose to early neurosyphilis. Epidemiological studies in HIV-positive patients have documented increased HIV shedding associated with genital ulcers, suggesting that syphilis increases the susceptibility of infected persons to HIV acquisition and transmission (Golden et al., 2003). Symptomatic early neurosyphilis may present with meningitis, with or without cranial nerve involvement or ocular changes, meningovascular disease, or stroke. Late neurosyphilis affects the meninges, brain, or spinal cord parenchyma and usually occurs years to decades after primary infection. Manifestations of late neurosyphilis include tabes dorsalis, a rapidly progressive dementia with psychotic features, or general paresis, or both. Dementia as a symptom of neurosyphilis is unlikely to improve significantly with treatment, yet the course of the illness can be arrested. Presenting psychiatric symptoms of neurosyphilis can include personality changes, hostility, confusion, hallucinations, expansiveness, delusions, and dysphoria. Symptoms also reported in association with neurosyphilis include explosive temper, emotional lability, anhedonia, social withdrawal, decreased attention to personal affairs, unusual giddiness, histrionicity, hypersexuality, and mania. A significant incidence of depression has been associated with general paresis.
Metabolic and Toxic
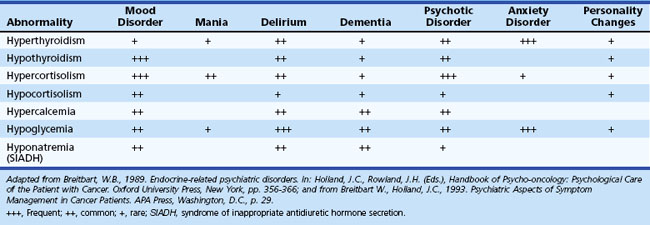
Essentially any metabolic derangement, if severe enough or combined with other conditions, can adversely affect behavior and cognition (Table 9.5). Metabolic disorders should remain within the differential diagnosis when evaluating patients with psychiatric symptoms.
Wilson Disease
Wilson disease (WD), also known as hepatolenticular degeneration, is an autosomal recessive disorder produced by a mutation on chromosome 13. The gene encodes a transport protein, the mutation of which causes abnormal deposition of copper in the liver, brain (especially the basal ganglia), and the cornea of the eyes. WD typically begins in childhood but in some cases has its onset as late as the fifth or sixth decade. About one-third of patients present with psychiatric symptoms, one-third present with neurological features, and one-third present with hepatic disease. Neurological manifestations are largely extrapyramidal, including chorea, tremor, and dystonia. Other symptoms include dysphagia, dysarthria, ataxia, gait disturbance, and a fixed (sardonic) smile. Seizures may also occur in a minority of patients. Potential neuropsychiatric symptoms are numerous, with at least half of patients manifesting symptoms early in the disease course. Personality and mood changes are the most common neuropsychiatric features, with depression occurring in approximately 30% of patients. Bipolar disorder occurs in about 20% of patients. Suicidal ideation is recognized in about 5% to 15%. WD patients can present with increased sensitivity to neuroleptics. Other symptoms include irritability, aggression, and psychosis. Cognitively, the profile is consistent with disturbance of frontosubcortical systems. Even long-term-treated WD patients develop psychiatric symptoms in about 70% of cases (Srinivas et al., 2008; Svetel et al., 2009).
Drug Abuse
Common neurological manifestations are broad and include the direct effects of intoxication, side effects, and withdrawal syndromes, as well as indirect effects. Direct effects can range from somnolence with sedatives to psychosis from hallucinogens. Side effects may be as severe as stroke or vasculitis from stimulant abuse. Withdrawal may be lethal as in the case of alcohol withdrawal and delirium tremens. Indirect effects can occur as a result of trauma, such as head injury, suffered while under the influence. Substance abuse has a high comorbidity with a variety of psychiatric conditions. Neuropsychiatric manifestations occur with abuse of all classes of drugs and are summarized in Box 9.6 (online only at www.expertconsult.com). The behavioral and cognitive manifestations of substance abuse may be transient but in a vulnerable subset of individuals may be chronic. Growing evidence suggests that drug use (e.g., 3,4-methylenedioxymethamphetamine [MDMA, “Ecstasy”]) may promote the development of chronic neuropsychiatric states such as depression and impaired cognition due to changes in structural and functional neuroanatomy (Montoya et al., 2002a). Although Cannabis use seems to be neither a sufficient nor a necessary cause of psychosis, it does confer an increased relative risk for developing schizophrenia later in life (Arseneault et al., 2004).
Autoimmune
Systemic Lupus Erythematosus
Neuropsychiatric symptoms are common, often episodic, and may occur in association with steroid treatment, which creates significant dilemmas in management. Depression and anxiety each occur in approximately 25% of SLE patients. Reports of the prevalence of overall mood disturbances range between 16% and 75%, and reports of anxiety disorders occur in 7% to 70%. Psychosis is more rare and tends to occur in the context of confusional states. Its overall prevalence has been reported to range from 5% to 8%. The incidence of psychotic symptoms in patients receiving prednisone doses between 60 mg and 100 mg/day is approximately 30%. These symptoms are reported to respond favorably to reduction in steroid dose and psychotropic management. Focal or generalized seizures may occur in the setting of active generalized SLE or as an isolated event. The prevalence of seizures ranges from 3% to 51%. Cognitive manifestations of SLE including temporary, fluctuating, or relatively stable characteristics eventually occur in up to 75% of patients; these manifestations range from mild attentional difficulties to dementia. In some patients, cognitive performance improves with resolution of any concurrent psychiatric disturbances. Cerebrovascular disease may underlie nonreversible cognitive dysfunction and when progressive may cause atrophy and multiinfarct dementia. Many patients with cognitive impairment have no demonstrable vascular lesions on neuroimaging. Cognitive impairment may manifest as subcortical features with deficits in processing speed, attention, learning and memory, conceptual reasoning, and cognitive flexibility. Reports of the prevalence of subclinical cognitive impairment range from 11% to 54% of patients. A number of brain-specific antibodies have been studied as potential diagnostic markers of psychosis associated with neuropsychiatric SLE (NPSLE), but none appear to be specific (Kimura et al., 2010). SLE patients identified as having a persistently positive immunoglobulin (Ig)G anticardiolipin antibody over a 5-year period have been demonstrated to have a greater reduction in psychomotor speed than antibody-negative SLE patients. Patients with a persistently elevated IgA anticardiolipin antibody level have been demonstrated to have poorer performance on tests of conceptual reasoning and executive function than antibody-negative SLE patients. Elevated IgG and IgA anticardiolipin antibody levels may be causative or a marker of long-term subtle deterioration in cognitive function in SLE patients. However, their role in routine evaluation and management remains controversial. Cerebrovascular disease is a well-known cause of neuropsychiatric dysfunction and is reported to occur in 5% to 18% of SLE patients.
Multiple Sclerosis
Cognitive impairment is found in approximately 40% of patients. Deficits have been described in working, semantic, and episodic memory as well as in the person’s ability to accurately assess his or her own memory function. Patients may also suffer from impaired attention, cognitive slowing, reduced verbal fluency, and difficulties with abstract reasoning and concept formation. Correlations between cognitive impairment and MRI location of lesions and indices of total lesion area have been noted. There is little data on the treatment of cognitive dysfunction in MS. The disease-modifying agent interferon beta-1a was noted to be associated with improvements in information-processing and problem-solving abilities over a 2-year longitudinal study. A small trial demonstrated an improvement in complex attention, concentration, and visual memory in a group of patients treated for 1 year with interferon beta-1b compared with controls (Barak and Achiron, 2002). Donepezil, 10 mg daily, has been reported to improve verbal learning and memory in some MS patients.
Neoplastic
Paraneoplastic syndromes represent remote nonmetastatic manifestations of malignancy. Neurological paraneoplastic syndromes are primarily immune-mediated disorders that may develop as a result of antigens shared between the nervous system and tumor cells. The most common primary malignancies that promote neurological paraneoplastic syndromes are ovarian and small-cell lung cancer (SCLC). These syndromes generally develop subacutely, often before the primary malignancy is identified, and may preferentially involve selected regions of the CNS. Typical sites of involvement include muscle, neuromuscular junction, peripheral nerve, cerebellum, and limbic structures. Limbic encephalitis, associated with SCLC and testicular cancer, produces a significant amnestic syndrome and neuropsychiatric symptoms including agitation, depression, personality changes, apathy, delusions, hallucinations, and psychosis. Complex partial and generalized seizures may also occur. Relevant markers include: (1) intracellular paraneoplastic antigens such as Hu, associated with SCLC, and Ta and Ma-2, associated with testicular cancer and (2) cell membrane antigens such as the N-methyl-d-aspartate receptor and voltage-gated potassium channels (Graus et al., 2010; Hoffmann et al., 2008). Paraneoplastic disorders are often progressive and refractory to therapy, although in some cases significant improvement follows tumor resection and immunotherapy. Significant neuropsychiatric sequelae can arise from the various chemotherapeutic and radiation therapies used for cancer treatment.
Degenerative
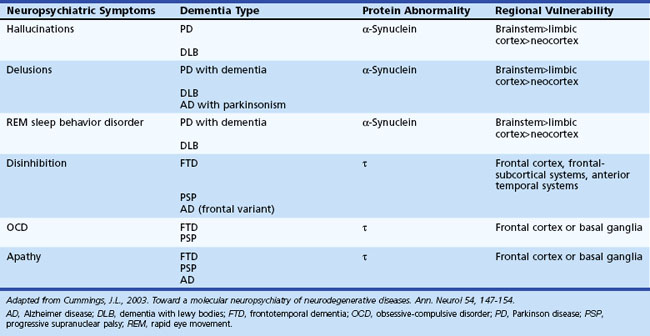
Neuropsychiatric symptoms are common in most degenerative disorders that produce significant dementia. The individual presentations of such symptoms are related to a number of factors specific to the disease: location of lesion burden, rate of progression of disease, and factors specific to the individual (e.g., premorbid personality, education level, psychiatric history, social support system, and coping skills). Neurodegenerative diseases are increasingly recognized as involving abnormalities of protein metabolism. About 70% of dementias in the elderly and more than 90% of neurodegenerative dementias can be linked to abnormalities of three proteins: β-amyloid, α-synuclein, and τ. Disorders of protein metabolism have associated neuroanatomical regions of vulnerable cell populations that are related to the clinical manifestations. AD, for example, has associated disorders of β-amyloid, τ, and α-synuclein metabolism that involve specific anatomical regions. PD, dementia with Lewy bodies (DLB), and multisystem atrophies are synucleinopathies. α-Synuclein is the main component of Lewy bodies, which are a major histological marker seen in PD and DLB. In these disorders, Lewy bodies may be found in the substantia nigra, locus ceruleus, nucleus basalis, limbic system, and transitional and neocortex. Frontotemporal dementia, progressive supranuclear palsy (PSP), and corticobasal ganglionic degeneration implicate abnormal τ metabolism in their pathogenesis. Tauopathies are associated with selective involvement of the frontal and temporal cortex and frontosubcortical circuitry. Table 9.6 lists associated regions of vulnerability and neuropsychiatric symptoms.
Table 9.6 Correlations of Neuropsychiatric Symptoms to Regions of Vulnerability in Neurodegenerative Disorders
Alzheimer Disease and Mild Cognitive Impairment
Neuropsychiatric symptoms of AD may include paranoia, agitation, aggression, delusions, hallucinations, anxiety, apathy, social withdrawal, reduced speech output, reduction or alteration of long-standing family relationships, and loss of sense of humor. A review of 100 cases of autopsy-proven AD demonstrated that 74% of patients had behavioral symptoms detected at the time of the initial evaluation. Symptoms included apathy (51%), hallucinations (25%), delusions (20%), depressed mood (6.6%), verbal aggression (36.8%), and physical aggression (17%). The presence of behavioral symptoms at the initial evaluation was associated with greater functional impairment not directly related to their cognitive impairments. Depressive symptoms, dysphoria, or major depression eventually occur in approximately half of patients. Psychosis has been reported to occur in 30% to 50% of patients at some time during the course of the illness, more commonly in the later stages. Mania occurs in less than 5%. Behavioral changes have been shown to be problematic and to precipitate earlier nursing home placement. Social comportment has been viewed as being relatively spared in AD, but subtle personality changes occur in nearly every individual over time. Significant impairment in the ability to recognize facial expressions of emotion and an inability to repeat, comprehend, and discriminate affective elements of language have been reported. It has been hypothesized that 15% of AD patients may have a frontal variant wherein they present with difficulties attributable to frontal lobe circuitry rather than an amnestic syndrome. Impairments in driving ability (Dawson et al., 2009) and decision-making abilities such as medical decision making (Okonkwo et al., 2008) and financial management (Marson et al., 2009) may be present even in early AD.
Atypical antipsychotic drugs are widely used to treat psychosis, aggression, and agitation in patients with AD. Their benefits are uncertain, and concerns about safety have emerged. Adverse effects may offset advantages in the efficacy of atypical antipsychotic drugs for the treatment of psychosis, aggression, or agitation in AD patients. Limited evidence suggests that electroconvulsive therapy (ECT) may be effective for management of agitation (Sutor and Rasmussen, 2008).
The concept of mild cognitive impairment (MCI) was developed to characterize a population of individuals exhibiting symptoms that are between normal age-related cognitive decline and dementia. These patients have a very slight degree of functional impairment and minimal decline from their prior level of functioning and therefore do not meet criteria for dementia. MCI was initially defined as a condition of memory impairment beyond what was expected for age, in the absence of impairments in other domains of cognitive functioning such as working memory, executive function, language, and visual-spatial ability. This concept has since evolved and now includes a total of four subtypes of impairment that are not of sufficient severity to warrant the diagnosis of dementia. The second type of MCI, called amnestic multiple domain, is associated with memory impairment plus impairment in one or more other cognitive domains. The third subtype is called nonamnestic single domain, and the fourth is known as nonamnestic multiple domain MCI. In many cases, the natural history of these subtypes leads to different endpoint conditions. Combining the clinical syndrome with the presumed cause may allow for reliable prediction of outcome of the MCI syndrome. When associated with only memory impairment, MCI may represent normal aging or depression or progress to AD. Amnestic MCI–multiple domains has a higher association with depression or progression to AD or vascular dementia. Nonamnestic single-domain MCI may have a higher likelihood of progression to frontal temporal dementia. Nonamnestic multiple-domain MCI may have a higher likelihood of progression to Lewy body dementia or vascular dementia (Petersen and Negash, 2008).
In 2008, it was estimated that more than 5 million people in the United States older than age 71 had MCI. The prevalence of MCI among persons younger than age 75 has been estimated to be 19% and for those older than 85 years, 29%. Almost a third of these individuals have amnestic MCI which may to progress to AD at a rate of 10% to 15% per year. The conversion rate of amnestic MCI to dementia over a 6-year period may be as high as 80%. Neuropsychiatric symptoms are common in persons with MCI. Depression occurs in 20%, apathy in 15%, and irritability in 15%. Increased levels of agitation and aggression are also present. Almost half of MCI patients demonstrate one of these neuropsychiatric symptoms coincident with the onset of cognitive impairment. Impaired awareness of memory dysfunction may also be present to a degree comparable to that found in persons with early AD. Evidence suggests that persons with MCI have an increased risk of motor vehicle accidents when risk factors such as a having a history of driving citations, crashes, reduced driving mileage, situational avoidance, or aggression or impulsivity are present. Difficulties with medical decision making have also been identified in some individuals with MCI (Okonkwo et al., 2008).
Idiopathic Parkinson Disease
Neuropsychiatric manifestations of PD are common. Depression is the most common psychiatric symptom, with a reported prevalence of 25% to 50%. Establishing the diagnosis of depression is complicated by the presence of comorbid confounding symptoms including dementia, facial masking, bradykinesia, apathy, and hypophonia. Menza et al. (2009) conducted a placebo-controlled trial in PD patients with depression and found that nortriptyline was efficacious, but paroxetine was not. Psychosis is also particularly prevalent and generally related to dopaminergic agents. The onset of motor impairment almost always precedes that of psychosis. Hallucinations, usually fleeting and nocturnal, are typically visual and occur in 30% of treated patients. Auditory and olfactory hallucinations, however, are rare. Visual hallucinations are associated with impaired cognition, use of anticholinergic medications, and impaired vision. In contrast to the hallucinations associated with DLB, patients with PD generally have at least partial insight into the nature of their hallucinations. Delusions occur less commonly and are often persecutory in nature. Management is complicated by neuroleptic sensitivity to both typical and atypical agents. Typical neuroleptics should be avoided. Novel atypical neuroleptics with potentially more favorable pharmacological properties, such as quetiapine and clozapine, may have theoretical advantages over other agents for treating PD. Evidence suggests that clozapine is effective, quetiapine may be effective, and olanzepine is not effective. Impulse-control disorders including pathological gambling, binge-eating, compulsive sexual behavior and buying are associated with dopamine agonist treatment in PD (Weintraub et al., 2010).
Huntington Disease
Huntington Disease is a degenerative disorder of autosomal dominant inheritance resulting from an expanded trinucleotide (cytosine-adenine-guanine [CAG]) repeat on chromosome 4. Symptoms typically develop during the fourth or fifth decade, initially manifesting with neurological features, psychiatric features, or both. Neurologically, patients often demonstrate generalized chorea, motor impersistence, and oculomotor dysfunction. In the juvenile form, the Westphal variant, early parkinsonian features are prominent, as are seizures, ataxia, and myoclonus. Significant cognitive impairment is inevitable and is often present early in the disease. Features of a subcortical dementia are present with involvement of frontosubcortical circuits. Common features include cognitive slowing, memory retrieval deficits, attentional difficulties, and executive dysfunction. Patients often lack awareness of their chorea and their cognitive and emotional deficits. Psychiatric features such as personality changes, apathy, irritability, and depression are common. Depression may be exacerbated by tetrabenazine used for the treatment of chorea, since this drug is a dopamine-depleting agent. Psychosis may occur in up to 25% of patients with HD. Anxiety and obsessive tendencies also occur (Phillips et al., 2008).
Epilepsy
There has been speculation that PIP may sometimes be caused by complex partial (limbic) status (Elliott et al., 2009). When this is thought to be the case, acute therapy with antiepileptic medications would be advised, possibly in conjunction with antipsychotic medication. Risk factors for PIP include a cluster of seizures, insomnia within 1 week of onset of PIP (particularly within 1-3 days), epilepsy of more than 10 years’ duration, generalized tonic-clonic seizures or secondarily generalized complex partial seizures, prior episodes of PIP, prior psychiatric hospitalizations or a history of psychosis, bilateral independent seizure foci (particularly temporal), history of TBI or encephalitis, and low intellectual function. PIP is usually short-lived, lasting several days to weeks, but chronic psychosis may develop after recurrent episodes or even after a single episode. Research data are lacking for treatment of PIP, and recommendations are based on expert opinion. Recommendations include vigilant monitoring of patients with risk factors for PIP after a cluster of seizures, ensuring that there is not ongoing seizure activity, early implementation of antipsychotic medications (preferably atypical agents) after the emergence of symptoms, consideration of treatment after a cluster of seizures in patients with a history of PIP, and consideration of treatment with the emergence of sleeplessness, which can be a harbinger of PIP. PIP can respond to ECT, but it is rarely necessary to utilize this resource.
Treatment of epilepsy-related psychosis is complicated by the propensity of antipsychotics to cause paroxysmal EEG abnormalities (Centorrino et al., 2002) and induce seizures. EEG changes occur in the nonepileptic population treated with antipsychotics but in most circumstances are of little consequence. Studies defining the effects of neuroleptics on the EEG of persons with epilepsy are lacking. The potential for increasing seizures has led to some anxiety about the use of antipsychotics in individuals with epilepsy. In most circumstances, the risk of increasing seizures is considered low, but formal studies investigating the efficacy of antipsychotic medications for treating epilepsy-related psychosis and the risks for precipitating seizures are lacking. The specific causes and characteristics of a given epilepsy have to be considered carefully, as these may increase risk. Seizure potential is generally dose related, so high-dose therapy should be avoided. Careful monitoring of anticonvulsants is advised. The lowest possible effective dose should be used, medications selected carefully, and psychiatric polypharmacy avoided if possible, since this may increase the risk of seizures. Seizure risk is particularly increased with use of clozapine and chlorpromazine. Potential problems with antipsychotic treatment in persons with epilepsy include pharmacokinetic interactions due to common metabolism with P450 isoenzymes, as well as side effects (sedation, weight gain, hyperlipidemia, decreased glycemic control, hematotoxicity, and hepatotoxicity).
Traumatic Brain Injury
The pathological correlates of moderate to severe TBI are numerous and particular to the types and mechanisms of injuries suffered. Various types of pathology, which are often found in combinations, include penetrating wounds, depressed skull fractures, diffuse axonal injury (DAI), petechial hemorrhages, contusions, lacerations, hematomas (epidural, subdural, and intraparenchymal), subarachnoid hemorrhage, edema, herniation, and focal or diffuse hypoxic ischemic injury. Many of these specific types of injuries have their own prognosis and time course of recovery. Concussion or mild TBI occurs most frequently in young adults. There is no consensus about what clinical findings constitute mild TBI, with many practitioners advocating that loss of consciousness is not an absolute requirement. Others differ on the required duration of loss of consciousness, with ranges from 20 minutes to any event lasting less than 1 hour. Any traumatic process associated with a generalized alteration in cerebral function, including amnesia (retrograde or anterograde) or alteration in consciousness at the time of the accident, may be associated with brain injury. Persons who sustain a mild TBI often complain of a number of emotional/behavioral, cognitive, and physical symptoms, which can persist for months to years after the injury, and rarely may be permanent. Such symptoms can include anxiety, depression, irritability, mood lability, cognitive slowing, judgment problems, difficulty concentrating, memory problems, fatigue, sensitivity to noise, dizziness, and headaches. Postconcussive symptoms occur after moderate and severe TBI as well. It is estimated that 80% to 90% of persons sustaining a mild TBI make a favorable recovery. When symptoms persist, the patient is said to suffer from a postconcussive syndrome. The overall prevalence for postconcussive symptoms, self-limited and persistent at 3 months after injury, ranges from approximately 25% to 85%. Well-controlled research data are not available on optimal pharmacological management or rehabilitation strategies for post-TBI neuropsychiatric and cognitive difficulties. Limited evidence supports the effectiveness of methylphenidate for enhancing attention, processing speed, and memory function. Other medications such as d-amphetamine, amantadine, donepezil, levodopa, and bromocriptine may also have some benefit for treating symptoms that include attentional difficulties, cognitive slowing, poor initiation, aspects of poor memory, fatigue, or motor deficits. Cognitive rehabilitation may be helpful for management of attention and executive difficulties, as well as improving communication skills (Cicerone et al., 2009). Evidence-based reviews generally support holistic rehabilitation programs that support community reintegration, awareness of deficits, regulation of behavior and affect, improved physical and social function, and effective communication (Cernich et al., 2010). Psychiatric disorders occur at high rates in TBI patients, with criteria for axis I disorders (as defined by the DSM-IV-TR) being met in 50% to 80% of patients in a community sample of patients with a mixed level of severity. Axis II disorders were identified in 25% to 65% of patients (Price, 2004; Warriner and Velikonja, 2006). Axis I disorders include major mood and anxiety disorders, schizophrenia, and other psychoses; axis II disorders include major personality disorders and other maladaptive personality features. Psychiatric symptoms have been observed to occur immediately after injury up to decades later. It is likely that a complex interplay of factors results in the particular cognitive and psychiatric manifestations in a given individual. These factors include the nature and severity of the neurological injury, premorbid personality and cognition, preexisting psychiatric illness, substance abuse history, family psychiatric history, educational level, occupational status, coping strategies, age at injury, stressors, support systems, and the possibility of psychological or financial gains.
Post-TBI depression occurs in up to 60% of patients, with comorbid anxiety and aggressive behavior being common. Both right and left hemispheric lesions have been implicated. SSRIs are most commonly prescribed and may be helpful for management of depression, irritability, agitation, and aggression. CBT may decrease depression, anxiety, and anger and improve problem-solving skills (Silver et al., 2009). TBI-associated hypomania and mania have also been observed, although at much lower frequencies. Psychosis in association with TBI has a reported incidence ranging from 0.7% to 20%. Reliable incidence and prevalence information is unavailable. An increased risk of developing chronic psychosis has been observed in individuals suffering severe diffuse brain injury involving the temporal and frontal lobes. Patients undergoing evaluation for potential TBI-related psychosis need to be carefully distinguished from those with preexisting psychotic symptoms and schizophrenia. The mean delay to the onset of psychotic symptoms after injury has been reported to be about 4 years (Guerreiro et al., 2009). The latency of the injury to the onset of symptoms has been reported to range from 2 days to 48 years. Delusions occur in more than 75%, and hallucinations occur in almost 50% of patients. Approximately 70% of affected individuals were noted to have abnormal findings on EEG. Neuropsychological testing demonstrated abnormalities in almost 90%. Psychosis in the majority of patients eventually improves with antipsychotics.
Depression-Related Cognitive Impairment
Converging evidence suggests that late-onset depressive symptoms may be both a prodrome of and an independent risk factor for cognitive decline as seen in AD and vascular dementia (Saczynski et al., 2010). Late-onset depression is also a risk factor for MCI (Dotson et al., 2010). Four possible mechanisms may underlie the association between depression and dementia/MCI. First, depression may cause cognitive impairment. For example, depression produces excessive release of glucocorticoids which may lead to hippocampal damage. Second, depression may be an emotional reaction on the part of the patient to the onset of dementia. Third, an underlying neurodegenerative process may cause both the depression and the dementia. Fourth, there may be a synergistic interaction between depression and a neurodegenerative process that produces dementia. Although a causal relationship between depression and dementia is speculative at this time, future studies may distinguish between these four possible mechanisms (Geda, 2010).
Catatonia
Catatonia, once felt to be rare, has been reported to occur among psychiatric inpatients with a prevalence ranging from 7% to 30%. Up to 20% of catatonia in psychiatric inpatients is associated with mania, and 5% to 15% is associated with schizophrenia. In general, catatonia is characterized by motor abnormalities that occur in association with changes in thought, mood, and vigilance. The specific manifestations vary and commonly include mutism, stupor, stereotypies, mannerisms, diminished motor function (including waxy flexibility or rigidity), staring, negativism, automatic obedience, echopraxia, and echolalia. Stereotypies are purposeless repetitions of sounds, words, phrases, or movements. Unexplained foreign accents, whispered or robotic speech, and tiptoe walking have also been observed. There are two principal forms of catatonia: a hypokinetic retarded-stuporous variety and a hyperkinetic excited-delirious variety. Patients with the excited form can present with impulsive or combative behavior that may be difficult to distinguish from mania. If untreated, catatonia may progress to a malignant state marked by fever, hyperexcitability, and autonomic instability, which after several days can be followed by exhaustion, dehydration, coma, cardiac arrest, and death. Although the majority of catatonic patients have an underlying affective (most often mania) or psychotic disorder, some 10% to 20% have significant medical or neurological conditions that contribute to their catatonic state. Stroke, demyelinating disease, encephalitis, head trauma, medications, and CNS malignancy are individually associated with catatonia. Medical disorders that can result in catatonia include heat stroke, autoimmune disease, uremia, hyperthyroidism, diabetic ketoacidosis, porphyria, and Cushing disease. Catatonia has been reported in association with use of illicit recreational drugs, antipsychotics, and opiates, as well as withdrawal from benzodiazepines and dopaminergic drugs. Important considerations in the differential diagnosis include neuroleptic malignant syndrome, serotonin syndrome, and nonconvulsive status epilepticus. Treatment with IV benzodiazepines, IV sodium amobarbital, or ECT can result in dramatic improvement. Bilateral ECT is more effective than unilateral in patients who are febrile, delirious, or do not respond to benzodiazepines (Fink and Taylor, 2009).
Treatment Modalities
Clinicians face a wide array of antidepressant drug options (Table 9.7). The most commonly prescribed drugs are the second-generation antidepressants: SSRIs, serotonin and norepinephrine reuptake inhibitors (SNRIs), and bupropion. First-generation antidepressants (tricyclic antidepressants [TCAs] and monoamine oxidase inhibitors [MAOIs]) offer similar effectiveness, but with more toxicity. Generally, TCAs are avoided because of considerable dry mouth, constipation, and dizziness. TCAs are relatively contraindicated in patients with coronary artery disease, congestive heart failure, and arrhythmias. They are also potentially fatal in overdose. MAOIs are also used infrequently, even by psychiatrists, because of the many dietary restrictions and the potential for hypertensive crisis. The selegiline patch (20-mg formulation) is a U.S. Food and Drug Administration (FDA)-approved MAOI that does not require dietary tyramine restrictions. Antidepressant selection is based on tolerability, safety, evidence of effectiveness in the patient or a first-degree relative, and cost. The goal of treatment is complete remission of symptoms and return to normal functioning. About 50% of patients achieve full remission with antidepressant therapy, while the other half achieves partial remission or are nonresponders. For the first episode, antidepressant treatment may take 1 to several months until remission is achieved, and medication should be continued for another 4 to 9 months. Some clinicians advocate treatment for at least 1 year to maintain remission for a full annual cycle of holidays and anniversaries. For patients older than 70 years who respond to an SSRI, consider treating for 2 years to prevent recurrence. Increasing the dose of the current medication or changing medications is often necessary. For a partial response, the dose of the initial agent should be maximized as tolerated before switching to another medication or adding a second drug. When a partial response continues, the clinician can refer for psychotherapy, change antidepressants, or augment treatment with bupropion, mirtazapine, or a nontraditional agent. Compared with withdrawing one drug and initiating another, combination therapy offers faster effects and avoidance of withdrawal symptoms when stopping the first agent. Combinations of MAOIs and either SSRIs or TCAs are not recommended because of an increased risk for serotonin syndrome (with confusion, nausea, autonomic instability, and hyperreflexia). Adding adjunctive atypical antipsychotics, psychostimulants, and thyroid hormone remains controversial. Antipsychotics added to SSRIs for treatment-resistant depression show some benefit but also carry significant risks, so their use should be limited to psychiatrists (Shelton and Papakostas, 2008). A Cochrane review of monotherapy treatment with psychostimulants (dexamphetamine, methylphenidate, methyl amphetamine, and pemoline) for moderate to severe depression found short-term improvement in depression symptoms and fatigue (Candy et al., 2008). A second review of 19 controlled trials on adults older than 65 years supported this recommendation for methylphenidate (Hardy, 2009).
| Agent, Daily Dose* | Benefits/Selected Side Effects |
|---|---|
| First-Generation Antidepressants: | As a class: dry mouth, dizziness, nausea, sedation, anticholinergic effects, orthostatic hypotension. Contraindicated with MAOIs. Do not use with prolonged QT interval. Use with caution in patients with cardiovascular disease or predisposition to urinary retention or narrow-angle glaucoma. Follow ECGs and orthostatic blood pressure changes. |
| Amitriptyline, 25-300 mg | May aid with sleep and treat neuropathic pain and migraines. Highly sedating and anticholinergic. |
| Clomipramine, 25-250 mg | Possibly useful in comorbid anxiety, panic disorders, and OCD. |
| Desipramine, 25-300 mg | |
| Imipramine, 25-300 mg | |
| Protriptyline, 15-60 mg | |
| MAOIs: | As a class: hypertensive crisis, orthostatic hypotension, insomnia, agitation, sedation, weight change, dry mouth, urinary hesitancy, and sexual dysfunction. Special dietary restrictions except for selegiline patch. Potential severe drug-drug interactions. |
| Phenelzine, 45-60 mg | |
| Tranylcypromine, 30-50 mg | |
| Selegiline transdermal patch, 6-12 mg/day | |
| Second-Generation Antidepressants: | Nausea, diarrhea, decreased appetite, nervousness, insomnia, somnolence, sweating, impaired sexual function; hyponatremia in the elderly.† Contraindicated with MAOIs. Potential for drug interactions with drugs metabolized in liver. Risk/benefit analysis needed in pregnancy. |
| Bupropion, 200-300 mg | NE/DA reuptake inhibitor. Less weight gain and fewer sexual side effects than other agents. |
| (75-225mg) | Lowers seizure threshold. Relatively contraindicated in patients with history of seizures, family history of seizures, or head trauma. |
| Citalopram, 20-40 mg (10-40 mg) | SSRI |
| Escitalopram, 5-20 mg (5-10 mg) | SSRI; similar to citalopram. |
| Duloxetine, 30-120 mg | SNRI; may be effective in comorbid pain and depression‡ |
| Fluoxetine, 20-40 mg (5-40 mg) | SSRI; long half-life mitigates effects of missed doses. Withdrawal symptoms rare. |
| Mirtazapine, 15-45 mg (7.5-30 mg) | ARA; increased appetite and somnolence. Use caution with renal impairment. Avoid concomitant benzodiazepines and alcohol. |
| Paroxetine, 20-40 mg (5-40 mg) | SSRI; more weight gain and sexual adverse events. Withdrawal syndrome not uncommon. |
| Sertraline, 50-150 mg (25-150 mg) | SSRI |
| Trazodone, 50-400 mg (50-225 mg) | SRA/A; less effective in doses <300 mg. Somnolence, rare priapism in young men. Useful in low doses (50-100 mg) as a sleeping aid. |
| Venlafaxine, 75-300 mg (50-225 mg) | SNRI; higher incidence of nausea, vomiting, dry mouth, sexual side effects, and hypertension. Occasional hypertensive urgencies. |
ARA, α2-Receptor antagonist; ECG, electrocardiogram; MAOI, monoamine oxidase inhibitor; NDRI, norepinephrine and dopamine reuptake inhibitor; OCD, obsessive compulsive disorder; SNRI, serotonin and norepinephrine reuptake inhibitor; SRA/A, serotonin receptor antagonists/agonists; SSRI, selective serotonin reuptake inhibitor.
* Dose range for geriatric patients is in parentheses.
† Wright, S.K., Schroeter, S., 2008. Hyponatremia as a complication of selective serotonin reuptake inhibitors. J Am Acad Nurse Pract 20, 47–51.
‡ Kroenke, K., Krebs, E.E., Bair, M.J., 2009. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry 31, 206–19.
Studies are conflicting about the effectiveness of adding thyroid hormone (triiodothyronine [T3] and levothyroxine [T4]) to antidepressants. More research is needed before these therapies can be recommended. Augmentation with other nontraditional agents has also shown mixed results: omega-3 fatty acids added to sertraline in patients with coronary heart disease did not improve depressive outcomes (Carney et al., 2009). For seasonal depression, light therapy, 6000 to 10,000 lux for 30 to 90 minutes each morning may be helpful (Golden et al., 2005). Yoga, exercise (Mead et al., 2009), self-help books, and relaxation therapy (Morgan and Jorm, 2008) may also be useful. Specific types of side effects are more common with particular drugs and should guide choice of medications (see Table 9.7). Sexual side effects of SSRIs include decreased libido or interest (men and women), anorgasmia (women), and delayed ejaculation (men). To address these side effects, consider pretreatment counseling, switching to a drug with a different mechanism of action (e.g., bupropion, mirtazapine), or using sildenafil for SSRI-associated erectile dysfunction if there are no contraindications. Switching to bupropion can reduce undesired weight gain. Agitation or excessive activation may occur with fluoxetine and warrants a switch to a different SSRI.
Treatment for schizophrenia is most successful when antipsychotic medications are combined with psychological and social supports. These supports may include CBT, vocational interventions, and the use of multidisciplinary mental health professional teams who work with the patient and caregivers inside and outside the hospital to insure health and social care (van Os and Kapur, 2009). CBT may improve coping and reduce distress and affective symptoms associated with psychotic symptoms. Despite these interventions, about one-third may remain symptomatic. All antipsychotics share varying degrees of striatal D2 receptor blockade. The first-generation antipsychotics (e.g., haloperidol, perphenazine, chlorpromazine) possess equivalent efficacy but differ in potency and side effects. All potentially produce extrapyramidal symptoms (EPS), tardive dyskinesia, and hyperprolactinemia. The second-generation antipsychotics (e.g., clozapine, risperidone, olanzapine, quetiapine), also known as atypical antipsychotics, generally produce fewer EPS, less risk of tardive dyskinesia, and less hyperprolactinemia. Table 9.8 lists selected antipsychotic side effects. Some atypical antipsychotics such as clozapine and olanzapine have been associated with weight gain, impairment of glucose metabolism, and dyslipidemia. Clozapine has greater efficacy than first-generation drugs but requires regular blood test monitoring throughout the course of treatment owing to an increased risk for agranulocytosis (1%-4%). Risperidone is known for its association with hyperprolactinemia. The primary symptom targets for antipsychotics are psychotic symptoms, agitation, and negative symptoms (e.g., apathy, social withdrawal, diminished affect). Response times for psychotic symptoms may range from responding within hours of administration to several weeks of administration. Maximal response may require months. Some patients are nonresponders. Agitation responds well to most antipsychotics, but negative symptoms generally respond only modestly.
Electroconvulsive Therapy
MDD is the most common indication for ECT. The mechanisms by which this procedure alleviates depressive symptoms are not fully understood. Remission rates of 70% to 90% have been reported in clinical trials of ECT for MDD (Popeo, 2009). It is also an effective treatment for bipolar disorder but may uncommonly precipitate hypomania or mania. Suicidal thoughts respond favorably to ECT and are an indication for early transition from drug therapy. ECT is not routinely used for treatment of schizophrenia, but when combined with antipsychotic medications, it may result in improvement in 80% of drug-resistant chronic schizophrenia patients. Patients with mania also respond favorably to ECT. There are few absolute contraindications to ECT, but cardiac conditions may worsen and should be addressed. Conditions such as vascular aneurysms and aortic stenosis should preferably be repaired prior to ECT, but persons with such conditions have been reported to tolerate the procedure. Those with properly functioning cardiac pacemakers generally tolerate ECT well. Case reports of ECT performed on individuals with a recent cerebral infarction suggest a low complication rate and a favorable response to treatment. ECT has been successfully used in persons with mental retardation, MS, HD, arteriovenous malformations, and hydrocephalus. Patients with depression and PD may experience improvement of mood and motor symptoms with ECT. Some research supports the effectiveness of ECT for treatment of the core motor symptoms of PD (Popeo and Kellner, 2009). There is no evidence of structural brain damage due to ECT. Posttreatment memory difficulty (anterograde amnesia) is usually experienced during the course of ECT treatments but normally resolves within one month after the last treatment. Retrograde amnesia is more prominent for the events closer to the time of ECT treatment. Posttreatment confusion is variable and may be associated with bilateral electrode placement, high stimulus intensity, prolonged seizure activity, and inadequate oxygenation. There is controversy about whether unilateral electrode placement for ECT is as effective as bilateral placement. Several studies have shown equal efficacy so long as unilateral ECT is performed with a stimulus intensity well above seizure threshold (Lisanby, 2007). Studies also indicate a lower incidence of cognitive side effects with right unilateral electrode placement and electrical brief pulse waveform stimulus. There is uncertainty about the efficacy of ultra-brief pulse stimulus, but preliminary evidence suggests it is associated with a significant reduction in memory-related side effects (Peterchev et al., 2010).
Vagus Nerve Stimulation
The FDA approved vagus nerve stimulation (VNS) for treatment-refractory depression (TRD) in July 2005. Consensus criteria are unavailable for what constitutes TRD. Interest in VNS as a treatment for depression arose when it was noticed that persons being treated with VNS for treatment-resistant epilepsy (which is associated with an increased prevalence of depression) experienced improvements in their mood. Long-term studies of VNS for use as an adjunct to medications and therapy showed that it was well tolerated and resulted in a successful response in one-half of patients and complete remission in one-third (Andrade et al., 2010). The mechanism of VNS’s effects are not completely understood. It is believed that input information from the vagus nerve projects to the solitary tract nucleus and follows an ascending pathway to modulate various structures such as the amygdala, dorsal raphe, locus coeruleus, and the ventromedial prefrontal cortex that produce its effects on mood. The solitary tract nucleus also communicates with the parabrachial nucleus (PBN), cerebellum, and periaquaductal gray matter. The PBN communicates with other regions implicated in the pathophysiology of depression such as the hypothalamus, thalamus, amygdala, and nucleus of the stria terminalis. Only the left vagus nerve is used for VNS, because the right vagus nerve has parasympathetic branches to the heart. Aside from standard surgical risks, the most common side effects are voice alteration (54%-60% of patients), cough, neck pain, paresthesia, and dyspnea. These side effects typically decrease over time. It is recommended that VNS be used as an adjunctive treatment to medications and psychotherapy. Right unilateral ECT has been well tolerated in persons with VNS (Sharma et al., 2009).
Repetitive Transcranial Magnetic Stimulation
Repetitive transcranial magnetic stimulation (rTMS) is an emerging and recently FDA-approved noninvasive, well-tolerated treatment modality for MDD in adults. It is being studied for potential therapeutic applications in OCD, posttraumatic stress disorder, and auditory hallucinations in schizophrenia. The rTMS procedure uses a pulsed magnetic field introduced on the scalp surface to generate focal electrical stimulation of the cortical surface; it does not require anesthesia, does not produce a seizure, and can be administered in the office setting. The mechanism of action for treatment of depression is not well understood. The rTMS field can be pulsed at different frequencies to produce excitatory or inhibitory effects on cortical neurons. Frequencies of less than or equal to 1 Hz (slow rTMS) are believed to have mostly inhibitory neuronal effects by means of preferentially activating GABA-ergic interneurons in the cortex; this may result in transsynaptic depression. Use of rTMS frequencies greater than 1 Hz (fast rTMS) are believed to have mostly glutamatergic or excitatory neuronal effects (Kim et al., 2009). In depression, the target area is the left DLPFC. Left DLPFC high-frequency rTMS has shown effectiveness in almost one-third of pharmacotherapy-resistant MDD patients (Andrade et al., 2010). Headaches and site application pain are the most common side effects; the risk for seizure is estimated at less than 1 per 10,000 rTMS sessions. Monotherapy with rTMS is associated with few adverse effects but significant antidepressant effects for unipolar depressed patients who do not respond to medications or who cannot tolerate them (George et al., 2010).
Psychiatric Neurosurgery or Psychosurgery
Neurosurgical procedures are not commonly used for treatment of neuropsychiatric symptoms but should be considered for selected conditions when patients have failed combined pharmacotherapy and psychotherapy. Psychosurgery, both ablative and by deep brain stimulation, is still experimental and should be performed at institutions supporting psychosurgery research, having multidisciplinary involvement, and appropriate follow-up. In the middle of the 20th century, procedures such as frontal lobotomy were performed without defined indications or an understanding of the limbic system. This resulted in severe adverse events, even death. During the latter half of the 20th century, smaller stereotactically targeted lesions were used, resulting in benefits for patients, useful research data, and a precipitous decline in adverse events. Currently used ablative procedures include anterior cingulotomy, subcaudate tractotomy, limbic leucotomy (combined anterior cingulotomy and subcaudate tractotomy), and anterior capsulotomy. Carefully selected patients with intractable mood and anxiety disorders have experienced response rates ranging from 30% to 70%. Postoperative side effects are mostly transient and include headache, nausea, and edema. More serious adverse events include infection, urinary dysfunction, seizures, cognitive deficits, and cerebral infarct or hemorrhage (Andrade, 2010).
Deep brain stimulation involves the placement of electrodes into targeted deep brain regions so electrical stimulation can be delivered. Its advantages over ablative procedures are its adjustability (by manipulation of stimulation parameters) and reversibility. Published brain targets include the subcallosal cingulate gyrus (Hamani et al., 2009), nucleus accumbens, ventral internal capsule/ventral striatum, inferior thalamic peduncle, and lateral habenula. Response rates for MDD range from 35% to 50% (Blomstedt et al., 2010). Targets for OCD include the anterior limb of the internal capsule, ventral striatum, nucleus accumbens, or subthalamic nucleus (Denys et al., 2010). Transient hypomania may occur with deep brain stimulation for OCD. Response rates for OCD range from 20% to 75% (Haynes and Mallet, 2010). Nine brain regions have been targeted for treatment of Gilles de la Tourette syndrome, with in most cases some diminution of tics (Hariz and Robertson, 2010; Porta et al., 2009). The effects on other neuropsychiatric comorbidities are uncertain.
Treatment Principles
The unique features of each condition discussed should be carefully taken into account when developing a treatment plan. Transient, progressive, or static impairments in abilities such as driving, medical decision making, and management of finances may be present. Increased vigilance when monitoring patients for these impairments may improve care and allow for earlier interventions to protect the welfare of the patient, their family, and society. Patients with underlying neurological conditions tend to be more susceptible to the adverse reactions of psychotropic medications, particularly to extrapyramidal and cognitive side effects. These adverse reactions tend to be minimized with initiation of medications at low doses and use of gentle titration. When clinically indicated, atypical antipsychotics are often preferred over typical agents because of their fewer adverse side effects, but longitudinal studies are needed to better confirm this impression (Lieberman et al., 2005; Tarsy and Baldessarini, 2006). Further options to consider for treatment of refractory primary depression and other carefully selected psychiatric conditions include ECT, VNS (Elger et al., 2000; Groves and Brown, 2005; Marangell et al., 2002; Rush et al., 2000), transcranial magnetic stimulation (Rosa et al., 2006), deep brain stimulation (Skidmore et al., 2006), or stereotactic ablative surgery (Dougherty et al., 2002, 2003; Montoya et al., 2002b; Price et al., 2001). There is currently little evidence to guide the optimal treatment approach for patients with neurological disease and comorbid psychiatric symptoms.
In conclusion, advances in neuroscience have improved our understanding of the neural substrates of cognition and emotional behavior. The traditional boundaries between neurology and psychiatry have become obsolete. The future of psychiatric and neurological care, training, and research will inevitably require effective collaboration between both disciplines (Cunningham et al., 2006; Price et al., 2000).
Aarsland D., Perry R., Larsen J.P., et al. Neuroleptic sensitivity in Parkinson’s disease and parkinsonian dementias. J Clin Psychiatry. 2005;66:633-637.
Adenzato M., Cavallo M., Enrici I. Theory of mind ability in the behavioural variant of frontotemporal dementia: an analysis of the neural, cognitive, and social levels. Neuropsychologia. 2010;48:2-12.
Andrade P., Noblesse L.H., Temel Y., et al. Neurostimulatory and ablative treatment options in major depressive disorder: a systematic review. Acta Neurochir (Wien). 2010;152:565-577.
Andreasen N.C. Linking mind and brain in the study of mental illnesses: a project for a scientific psychopathology. Science. 1996;275:1586-1593.
Arseneault L., Cannon M.C., Witton J.W., et al. Causal association between cannabis and psychosis: examination of the evidence. Br J Psychiatry. 2004;184:110-117.
Barak Y., Achiron A. Effect of interferon-beta-1b on cognitive functions in multiple sclerosis. Eur Neurol. 2002;47:11-14.
Barbas H., Zikopoulos B. The prefrontal cortex and flexible behavior. Neuroscientist. 2007;13:532-545.
Bassett A.S., Chow E.W. Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep. 2008;10:148-157.
Blomstedt P., Sjöberg R.L., Hansson M., et al. Deep brain stimulation in the treatment of depression. Acta Psychiatr Scand. 2010:1-8. doi: 10.1111/j.1600-0447.2010.01625.x
Bodden M.E., Dodel R., Kalbe E. Theory of mind in Parkinson’s disease and related basal ganglia disorders: a systematic review. Mov Disord. 2010;25:13-27.
Bora E., Yucel M., Pantelis C. Theory of mind impairment in schizophrenia: meta-analysis. Schizophr Res. 2009;109:1-9.
Bowler J.V., Gorelick P.B. Advances in vascular cognitive impairment. Stroke. 2009;40:e315-318.
Candy M., Jones L., Williams R., et al. Psychostimulants for depression. Cochrane Database Syst Rev. 2008;16:CD006722.
Carney R.M., Freedland K.E., Rubin E.H., et al. Omega-3 augmentation of sertraline in treatment of depression in patients with coronary heart disease: a randomized controlled trial. JAMA. 2009;21:1651-1657.
Cattaneo L., Rizzolatti G. The mirror neuron system. Arch Neurol. 2009;66:557-560.
Centorrino F., Price B.H., Tuttle M., et al. EEG abnormalities during treatment with typical and atypical antipsychotics. Am J Psychiatry. 2002;159:109-115.
Cernich A.N., Kurtz S.M., Mordecai K.L., et al. Cognitive rehabilitation in traumatic brain injury. Curr Treat Options Neurol. 2010;12:412-423.
Charney D.S., Manji H.K. Life stress, genes, and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci STKE. 2004:re5.
Chow T.W., Cummings J.L. Frontal-subcortical circuits. In: Miller B., Cummings J.L. The Human Frontal Lobes. New York: Guilford Publications; 1999:3-26.
Cicerone K.D., Azulay J., Trott C. Methodological quality of research on cognitive rehabilitation after traumatic brain injury. Arch Phys Med Rehabil. 2009;90:S52-S59.
Cummings J.L. Toward a molecular neuropsychiatry of neurodegenerative diseases. Ann Neurol. 2003;54:147-154.
Cunningham M.G., Goldstein M., Katz D., et al. Coalescence of psychiatry, neurology, and neuropsychology: from theory to practice. Harv Rev Psychiatry. 2006;14:127-140.
Czéh B., Michaelis T., Watanabe T., et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci U S A. 2001;98:12796-12801.
Dawson J.D., Anderson S.W., Uc E.Y., et al. Predictors of driving safety in early Alzheimer disease. Neurology. 2009;72:521-527.
Denys D., Mantione M., Figee M., et al. Deep brain stimulation of the nucleus accumbens for treatment-refractory obsessive-compulsive disorder. Arch Gen Psychiatry. 2010;67:1061-1068.
de Vugt M.E., Stevens F., Aalten P., et al. A prospective study of the effects of behavioral symptoms on the institutionalization of patients with dementia. Int Psychogeriatr. 2005;17:577-589.
Dotson V.M., Beydoun M.A., Zonderman A.B. Recurrent depressive symptoms and the incidence of dementia and mild cognitive impairment. Neurology. 2010;75:27-34.
Dougherty D.D., Baer L., Cosgrove G.R., et al. Prospective long-term follow-up of 44 patients who received cingulotomy for treatment-refractory obsessive-compulsive disorder. Am J Psychiatry. 2002;159:269-275.
Dougherty D.D., Weiss A.P., Cosgrove G.R., et al. Cerebral metabolic correlates as potential predictors of response to anterior cingulotomy for treatment of major depression. J Neurosurg. 2003;99:1010-1017.
Drevets W.C. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol. 2001;11:240-249.
Elger G., Hoppe C., Falkai P., et al. Vagus nerve stimulation is associated with mood improvements in epilepsy patients. Epilepsy Res. 2000;42:203-210.
Elliott B., Joyce E., Shorvon S. Delusions, illusions and hallucinations in epilepsy: 2. Complex phenomena and psychosis. Epilepsy Res. 2009;85:172-186.
Feinberg T.E., Eaton L.A., Roane D.M., et al. Multiple Fregoli delusions after brain injury. Cortex. 1999;35:373-387.
Fellows L.K. Advances in understanding ventromedial prefrontal function: the accountant joins the executive. Neurology. 2007;27:991-995.
Fink M., Taylor M.A. The catatonia syndrome: forgotten but not gone. Arch Gen Psychiatry. 2009:1173-1177.
Geda Y.E. Blowing hot and cold over depression and cognitive impairment. Neurology. 2010;75:12-14.
Geraldina P., Mariarosaria L., Annarita A., et al. Neuropsychiatric sequelae in TBI: a comparison across different age groups. Brain Injury. 2003;17:835-846.
George M.S., Lisanby S.H., Avery D., et al. Daily left prefrontal transcranial magnetic stimulation therapy for major depressive disorder: a sham-controlled randomized trial. Arch Gen Psychiatry. 2010;67:507-516.
Goldapple K., Segal Z., Garson C., et al. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61:34-41.
Golden M.R., Marra C.M., Holmes K.K. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
Golden R.N., Gaynes B.N., Ekstrom R.D., et al. The efficacy of light therapy in the treatment of mood disorders: a review and meta-analysis of the evidence. Am J Psychiatry. 2005;162:656-662.
Goldman-Rakic P.S., Castner S.A., Svensson T.H., et al. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology (Berl). 2004;174:3-16.
Graus F., Saiz A., Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol. 2010;257:509-517.
Groves D.A., Brown V.J. Vagal nerve stimulation: a review of its applications and potential mechanisms that mediate its clinical effects. Neurosci Biobehav Rev. 2005;29:493-500.
Guerreiro D.F., Navarro R., Silva M., et al. Psychosis secondary to traumatic brain injury. Brain Inj. 2009;23:358-361.
Haba-Rubio J. Psychiatric aspects of organic sleep disorders. Dialogue Clin Neurosci. 2005;7:335-346.
Hallak J.E., Crippa J.A., Pinto J.P., et al. Total agenesis of the corpus callosum in a patient with childhood-onset schizophrenia. Arq Neuropsiquiatr. 2007;65:1216-1219.
Hamani C., Mayberg H., Snyder B., et al. Deep brain stimulation of the subcallosal cingulate gyrus for depression: anatomical location of active contacts in clinical responders and a suggested guideline for targeting. J Neurosurg. 2009;111:1209-1215.
Hardy S.E. Methylphenidate for the treatment of depressive symptoms, including fatigue and apathy, in medically ill older adults and terminally ill adults. Am J Geriatr Pharmacother. 2009;7:34-59.
Hariz M.I., Robertson M.M. Gilles de la Tourette syndrome and deep brain stimulation. Eur J Neurosci. 2010;32:1128-1134.
Haynes W.I., Mallet L. High-frequency stimulation of deep brain structures in obsessive-compulsive disorder: the search for a valid circuit. Eur J Neurosci. 2010;32:1118-1127.
Himelhoch S., Medoff D.R. Efficacy of antidepressant medication among HIV-positive individuals with depression: a systematic review and meta-analysis. AIDS Patient Care STDS. 2005;19:813-822.
Hirono N., Mori E., Ishii K., et al. Alteration of regional cerebral glucose utilization with delusions in Alzheimer’s disease. J Neuropsychiatry Clin Neurosci. 1998;10:433-439.
Hoffmann L.A., Jarius S., Pellkofer H.L., et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J Neurol Neurosurg Psychiatry. 2008;79:767-773.
Howes O.D., Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bull. 2009;35:549-562.
Johnson R.T. Prion diseases. Lancet Neurol. 2005;4:635-642.
Kaufer D.I., Cummings J.L., Christine D., et al. Assessing the impact of neuropsychiatric symptoms in Alzheimer’s disease: the Neuropsychiatric Inventory Caregiver Distress Scale. J Am Geriatr Soc. 1998;46:210-215.
Ketter T.A., George M.S., Kimbrell T.A., et al. Functional brain imaging, limbic function, and affective disorders. Neuroscientist. 1996;2:55-65.
Kim D.R., Pesiridou A., O’Reardon J.P. Transcranial magnetic stimulation in the treatment of psychiatric disorders. Curr Psychiatry Rep. 2009;11:447-452.
Kimura A., Kanoh Y., Sakurai T., et al. Antibodies in patients with neuropsychiatric systemic lupus erythematosus. Neurology. 2010;74:1372-1379.
Kirsch-Darrow L., Fernandez H.F., Marsiske M., et al. Dissociating apathy and depression in Parkinson disease. Neurology. 2006;67:33-38.
Kuloglu M., Caykoylu A., Yilmaz E., et al. A left temporal lobe arachnoid cyst in a patient with schizophrenia-like psychosis: a case report, Prog. Neuropsychopharmacol. Biol Psychiatry. 2008;32:1353-1354.
Lehrer D.S., Christian B.T., Mantil J., et al. Thalamic and prefrontal FDG uptake in never medicated patients with schizophrenia. Am J Psychiatry. 2005;162:931-938.
Lieberman J.A., Stroup T.S., McEvoy J.P., et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209-1223.
Lisanby S.H. Electroconvulsive therapy for depression. N Engl J Med. 2007;8:1939-1945.
López-León S., Janssens A.C., González-Zuloeta Ladd A.M., et al. Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry. 2008;13:772-785.
Marangell L.B., Rush A.J., George M.S., et al. Vagus nerve stimulation (VNS) for major depressive episodes: one-year outcomes. Biol Psychiatr. 2002;51:280-287.
Marson D.C., Martin R.C., Wadley V., et al. Clinical interview assessment of financial capacity in older adults with mild cognitive impairment and Alzheimer’s disease. J Am Geriatr Soc. 2009;57:806-814.
Mathew S.J., Shungu D.C., Mao X., et al. A magnetic resonance spectroscopic imaging study of adult nonhuman primates exposed to early-life stressors. Biol Psychiatry. 2003;54:727-735.
Mayberg H.S. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull. 2003;65:193-207.
Mayberg H.S. Targeted electrode-based modulation of neural circuits for depression. J Clin Invest. 2009;119:717-725.
Mead, G.E., Morley, W., Campbell, P., et al., 2009. Exercise for depression. Cochrane Database Syst Rev 8, CD004366.
Menza M., Dobkin R.D., Marin H., et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology. 2009;72:886-892.
Mesulam M.M. From sensation to cognition. Brain. 1998;121:1013-1052.
Mesulam M.M. Behavioral neuroanatomy. Large scale networks, association cortex, frontal syndromes, the limbic system and hemisphere specializations. In: Mesulam M.M., editor. Principles of Behavioral and Cognitive Neurology. New York: Oxford University Press; 2000:1-120.
Montoya A.G., Sorrentino R., Lukas S.E., et al. Long-term neuropsychiatric consequences of “ecstasy” (MDMA): a review. Harv Rev Psychiatry. 2002;10:212-220.
Montoya A., Weiss A.P., Price B.H., et al. Magnetic resonance imaging-guided stereotactic limbic leukotomy for treatment of intractable psychiatric disease. Neurosurgery. 2002;50:1043-1049. discussion 1049-1052
Morgan A.J., Jorm A.F. Self-help interventions for depressive disorders and depressive symptoms: a systematic review. Ann Gen Psychiatry. 2008;19:13.
Moser D.J., Cohen R.A., Malloy P.F., et al. Reduplicative paramnesia: longitudinal neurobehavioral and neuroimaging analysis. J Geriatr Psychiatry Neurol. 1998;11:174-180.
Murray E.D., Price B.H. The Neurological Examination. Stern T.A., Rosenbaum J.F., Fava M., Rauch S., Biederman J. Comprehensive Clinical Psychiatry, first ed, Philadelphia: Mosby/Elsevier, 2008.
Murray R.M., Lewis S.W. Is schizophrenia a neurodevelopmental disorder? BMJ. 1987;295:681-682.
Nöthen M.M., Nieratschker V., Cichon S., et al. New findings in the genetics of major psychoses. Dialogues Clin Neurosci. 2010;12:85-93.
Okonkwo O.C., Griffith H.R., Copeland J.N., et al. Medical decision-making capacity in mild cognitive impairment: a 3-year longitudinal study. Neurology. 2008;71:1474-1480.
Ovsiew F., Murray E.D., Price B.H. Neuropsychiatric approach to the psychiatric inpatient. Ovsiew F., Munich R. Principles of Inpatient Psychiatry, first ed, Baltimore: Lippincott Williams and Wilkins, 2008.
Papapetropoulos S., Mash D.C. Psychotic symptoms in Parkinson’s disease. From description to etiology. J Neurol. 2005;252:753-764.
Perlis R.H., Purcell S., Fagerness J., et al. Clinical and genetic dissection of anger expression and CREB1 polymorphisms in major depressive disorder. Biol Psychiatry. 2007;62:536-540.
Peterchev A.V., Rosa M.A., Deng Z.D. Electroconvulsive therapy stimulus parameters: rethinking dosage. J ECT. 2010;26:159-174.
Petersen R.C., Negash S. Mild cognitive impairment: an overview. CNS Spectr. 2008;13:45-53.
Phillips W., Shannon K.M., Barker R.A. The current clinical management of Huntington’s disease. Mov Disord. 2008;23:1491-1504.
Popeo D.M. Electroconvulsive therapy for depressive episodes: A brief review. Geriatrics. 2009;64:9-12.
Popeo D., Kellner C.H. ECT for Parkinson’s disease. Med Hypotheses. 2009;73:468-469.
Porta M., Brambilla A., Cavanna A.E., et al. Thalamic deep brain stimulation for treatment-refractory Tourette syndrome: two-year outcome. Neurology. 2009;73:1375-1380.
Price B.H. Psychiatric disorders following traumatic brain injury. Rev Neurol Dis. 2004;1:107-110.
Price B.H., Adams R.D., Coyle J.T. Neurology and psychiatry: closing the great divide. Neurology. 2000;54:8-14.
Price B.H., Baral I., Cosgrove G.R., et al. Improvement in severe self-mutilation following limbic leucotomy: a series of 5 consecutive cases. J Clin Psychiatry. 2001;62:925-932.
Saczynski J.S., Beiser A., Seshadri S., et al. Depressive symptoms and risk of dementia: The Framingham Heart Study. Neurology. 2010;75:35-41.
Reif A., Fritzen S., Finger M., et al. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006;11:514-522.
Rickards H. Depression in neurological disorders: Parkinson’s disease, multiple sclerosis, and stroke. J Neurol Neurosurg Psychiatry. 2005;76(Suppl. 1):i48-i52.
Robertson D., Snarey J., Ousley O., et al. The neural processing of moral sensitivity to issues of justice and care. Neuropsychologia. 2007;2:755-766.
Robinson R.G., Travella J.I. Neuropsychiatry of mood disorders. In: Fogel B.S., Schiffer R.B., Rao S.M. Neuropsychiatry. Baltimore: Williams & Wilkins; 1996:287-305.
Rosa M.A., Gattaz W.F., Pascual-Leone A., et al. Comparison of repetitive transcranial magnetic stimulation and electroconvulsive therapy in unipolar non-psychotic refractory depression: a randomized, single-blind study. Int J Neuropsychopharmacol. 2006;21:1-10.
Rubinsztein J.S., Fletcher P.C., Rogers R.D., et al. Decision-making in mania: a PET study. Brain. 2001;124:2550-2563.
Rush A.J., George M.S., Sackeim H.A., et al. Vagus nerve stimulation (VNS) for treatment-resistant depressions: a multicenter study. Biol Psychiatr. 2000;47:276-286.
Saxena S., Brody A.L., Schwartz J.M., et al. Neuroimaging and frontal-subcortical circuitry in obsessive-compulsive disorder. Br J Psychiatry. 1998;35(suppl):26-37.
Schultz S.K., Andreasen N.C. Schizophrenia. Lancet. 1999;24:1425-1430.
Sharma A., Chaturvedi R., Sharma A., et al. Electroconvulsive therapy in patients with vagus nerve stimulation. J ECT. 2009;25:141-143.
Shelton R.C., Papakostas G.I. Augmentation of antidepressants with atypical antipsychotics for treatment-resistant major depressive disorder. Acta Psychiatr Scand. 2008;117:253-259.
Silver J.M., McAllister T.W., Arciniegas D.B. Depression and cognitive complaints following mild traumatic brain injury. Am J Psychiatry. 2009;166:653-661.
Skidmore F.M., Rodriguez R.L., Fernandez H.H., et al. Lessons learned in deep brain stimulation for movement and neuropsychiatric disorders. CNS Spectr. 2006;11:521-536.
Smith G.E., O’Brien P.C., Ivnik R.J., et al. Prospective analysis of risk factors for nursing home placement of dementia patients. Neurology. 2001;57:1467-1473.
Spencer M.D., Knight R.S.G., Will R.G. First hundred cases of variant Creutzfeldt-Jakob disease: retrospective case note review of early psychiatric and neurological features. BMJ. 2002;324:1479-1482.
Srinivas K., Sinha S., Taly A.B., et al. Dominant psychiatric manifestations in Wilson’s disease: A diagnostic and therapeutic challenge!. J Neurological Sci. 2008;266:104-108.
Stoodley C.J., Schmahmann J.D. Evidence for topographic organization in the cerebellum of motor control versus cognitive and affective processing. Cortex. 2010;46:831-844.
Sutor B., Rasmussen K.G. Electroconvulsive therapy for agitation in Alzheimer disease: a case series. J ECT. 2008;24:239-241.
Svetel M., Potrebic A., Pekmezovic T., et al. Neuropsychiatric aspects of treated Wilson’s disease. Parkinsonism Relat Disord. 2009;15:772-775.
Tandon R., Keshavan M.S., Nasrallah H.A. Schizophrenia, “just the facts” what we know in 2008. 2. Epidemiology and etiology. Schizophr Res. 2008;102:1-18.
Tarsy D., Baldessarini R.J. Epidemiology of tardive dyskinesia: is risk declining with modern antipsychotics? Mov Disord. 2006;21:589-598.
Tekin S., Cummings J.L. Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002;53:647-654.
Tietjen G.E., Brandes J.L., Digre K.B., et al. History of childhood maltreatment is associated with comorbid depression in women with migraine. Neurology. 2007;4:959-968.
Uher R. The implications of gene-environment interactions in depression: will cause inform cure? Mol Psychiatry. 2008;13:1070-1078.
van Os J., Kapur S. Schizophrenia. Lancet. 2009;22:635-645.
Voon V., Hassan K., Zurowski M., et al. Prospective prevalence of pathologic gambling and medication association in Parkinson disease. Neurology. 2006;66:1750-1752.
Wall C.A., Rummans T.A., Aksamit A.J., et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17:489-495.
Walterfang M., Wood S.J., Velakoulis D., et al. Diseases of white matter and schizophrenia-like psychosis. Aust N Z J Psychiatry. 2005;39:746-756.
Warriner E.M., Velikonja D. Psychiatric disturbances after traumatic brain injury: neurobehavioral and personality changes. Curr Psychiatry Rep. 2006;8:73-80.
Weinberger D.R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660-669.
Weinberger D.R. Genetic mechanisms of psychosis: in vivo and postmortem genomics. Clin Ther. 2005;27(Suppl. A):S8-S15.
Weintraub D., Koester J., Potenza M.N., et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol. 2010;67:589-595.
Weintraub D., Siderowf A.D., Potenza M.N., et al. Association of dopamine agonist use with impulse control disorders in Parkinson disease. Arch Neurol. 2006;63:969-973.
Wiederkehr S., Simard M., Fortin C., et al. Comparability of the Clinical Diagnostic Criteria for Vascular Dementia: A Critical Review. Part I. J Neuropsychiatry Clin Neurosci. 2008;20:150-161.
Wiederkehr S., Simard M., Fortin C., et al. Comparability of the clinical diagnostic criteria for vascular dementia: a critical review. Part II. J Neuropsychiatry Clin Neurosci. 2008;20:162-177.
Woods S.P., Conover E., Rippeth J.D., et al. Qualitative aspects of verbal fluency in HIV-associated dementia: a deficit in rule-guided lexical-semantic search processes? Neuropsychologia. 2004;42:801-809.