Chapter 66 Dementias
Dementia Syndrome
Epidemiology
The exact incidence and prevalence of dementia from any cause remains unknown. The most common etiologies for the dementia syndrome are neurodegenerative disorders. Neurodegenerative dementias typically occur in late life with age; heretability; preexisting conditions, such as Parkinson disease; and vascular risk factors being the most important contributors. As the proportion of elderly individuals is rapidly increasing worldwide, dementia is becoming a highly significant global healthcare problem. Between 1997 and 2025, the elderly population—defined as persons 65 years of age and older—is projected to increase from 62.7 to 136.9 million in the Americas, from 17.7 to 37.9 million in Africa, from 112.5 to 169.8 million in Europe, from 60.5 to 166.7 million in Southeast Asia, and from 110.7 to 267.7 million in the Western Pacific Region of Asia (World Health Organization, 1998).
Recently the National Institutes of Health–funded Aging, Demographics, and Memory Study (ADAMS) published representative data of dementia prevalence in the United States (Langa et al., 2005; Plassman et al., 2007). One of ADAMS enrollment criteria was age of 70 years or older, and approximately a third of participants were older than 85 years. The study included a detailed cognitive battery (Langa et al., 2005). Diagnosis of dementia was based on Diagnostic and Statistical Manual of Mental Disorders (DSM)-III-R and DSM-IV criteria. Dementia prevalence increased from 5% among 71- to 79-year-olds to 37.4% in those 90 years and older (Plassman et al., 2007). In addition to older age, the authors reported that African American race and lower educational achievement independently contributed to increased odds of dementia syndrome of any etiology.
Diagnostic Criteria
In 2001, the Practice Parameters Subcommittee of the American Academy of Neurology (AAN) published a summary of evidence-based guidelines for early detection, diagnosis, and management of dementia (Knopman et al., 2001). These guidelines offered basic recommendations for diagnosis and both pharmacological and nonpharmacological management of dementia. The experts recommend the use of the American Psychiatric Association’s DSM criteria (the most recent being the DSM-IV) for establishing a diagnosis of dementia syndrome (Box 66.1). It should be noted, however, that despite the requirement for memory impairment in some forms of dementia, memory impairment is not an early symptom.
Box 66.1
DSM-IV Criteria for Dementia
 The development of multiple cognitive deficits that include memory impairment and at least one of the following:
The development of multiple cognitive deficits that include memory impairment and at least one of the following:









Adapted with permission from American Psychiatric Association, 1994. Diagnostic and Statistical Manual of Mental Disorders, fourth ed., American Psychiatric Association, Washington, DC.
Increased focus on the earliest stages of dementia has led to the recognition of a state called mild cognitive impairment (MCI). MCI was initially considered a transition state between normal aging and dementia of the Alzheimer type, but the concept quickly evolved to denote an intermediate state between normal cognitive aging and any type of dementia. Practice parameters on MCI from the Quality Standards Subcommittee of the AAN have been published (Petersen et al., 2001b). MCI is an increasingly important focus of research and clinical attention. Identifying early disease—especially Alzheimer disease (AD) and vascular cognitive impairment—and initiating therapeutic interventions to prevent or slow decline are the goals. However, the concept of MCI as a clinical entity has undergone refinement and multiple modifications (Dubois and Albert, 2004; Petersen et al., 2001a, 2006; Petersen and Morris, 2005; Winblad et al., 2004) and has most recently been challenged as an artificial construct (Dubois et al., 2007). MCI is now recognized as a nonspecific state that may progress to AD or a non-AD dementia, may remain stable, or may revert to normal cognition. Many studies now attempt to define MCI of the AD type as the harbinger of AD. Because the onset of functional decline is typically late in the course of the disease, the combination of characteristic cognitive features in the presence of positive disease biomarkers has been proposed as sufficient for the diagnosis of the disease in the early predementia stages. These new criteria were proposed for use in AD (Dubois et al., 2007), but it is very likely this idea will be adopted for other dementias in the near future.
General Approach to Dementia Diagnosis
The essential feature of dementia is the acquired and persistent compromise in multiple cognitive domains that is severe enough to interfere with everyday functioning (see Box 66.1). This definition stands in contrast to delirium or acute confusional states (ACSs), which are distinguished primarily by prominent deficits or fluctuations in attentional processing. Although dementia syndromes tend to be chronic, progressive, and irreversible, and ACSs tend to be acute to subacute, fluctuating, and reversible, these distinctions are more relative than absolute. Toxic, metabolic, or other systemic physiological disturbances are more likely to be reversible which is not the case for neurodegenerative or cerebrovascular dementia syndromes. On the other hand, dementia renders the patient more vulnerable to delirium, highlighting the need for a comprehensive evaluation of potentially reversible etiological disorders in the context of even well-established dementia syndrome. Careful evaluation of persons referred for dementia evaluation can identify treatable or reversible disorders in up to 20% of cases (Hejl et al., 2002).
History
A detailed comprehensive history of the presenting illness is the single most important part of the evaluation of cognitively impaired subjects. In general, the history should be obtained from both the patient and a reliable and knowledgeable informant, because cognitive deficits, lack of insight, and lapses in judgment actively interfere with accurate self-reporting. A comprehensive history should determine the initial manifestations, mode of onset, and course over time (Box 66.2).
Box 66.2 Historical Components of the Dementia Evaluation
Initial Manifestations
Impaired recent memory (repeats self, forgets what was heard or read, misplaces things)
Poor decision making, judgment, or problem solving; decreased organizational skills
Difficulty learning new tasks or performing routine tasks
Problems managing money (balancing checkbook, forgetting to pay bills)
Difficulties expressing self (word finding) or participating in conversation
Getting lost in familiar areas, forgetting known routes while driving
Change in personality (apathetic, disinhibited), mood (sad, irritable), or behavior (odd or bizarre)
Medical/Neurological Conditions
General medical conditions (hypothyroidism, hypertension, diabetes mellitus, heart disease)
Neurological conditions (transient ischemic attacks, strokes, seizures, syncope, head trauma)
Associated motor features (tremor, gait difficulties, speech/swallowing disturbance, ataxia)
Sleep disturbances (sleep apnea, insomnia, sleep-associated movement disorder)
Cognitive Assessment
Standardized assessments such as the Mini-Mental State Examination (MMSE) (Folstein et al., 1975) provide a brief focused survey of cognitive domains most often affected in AD. The MMSE is a crude assessment that offers very limited assessment of memory and executive functions, however. The Montreal Cognitive Assessment (MoCA) is another short focused test that can be easily adopted in the busy private neurologist’s practice (Nasreddine et al., 2005). In addition to the cognitive domains tapped into by the MMSE, the MoCA also includes drawing a cube, the clock-drawing test, and a shortened version of Trails B, a test of working memory, set shifting, and complex attention, as well as a more detailed verbal memory test. MoCA may be better suited for detection of cognitive changes in the predementia stage of AD and other types of dementia (Gagnon et al., 2010; Hoops et al., 2009; Nazem et al., 2009; Smith et al., 2007; Videnovic et al., 2010). Additional tests for attention, language, praxis, executive and visuomotor functioning, and abstract thinking should be used selectively at the bedside or during an office visit to augment the initial crude cognitive screen (Table 66.1). Finally, for more refined cognitive assessment, patients should be referred for an evaluation by a trained neuropsychologist. Assessment by the latter is especially informative in borderline cases when the physician is uncertain of the presence of cognitive decline beyond what is expected by aging alone or under special circumstances such as limited education.
Table 66.1 Clinical Cognitive Assessment* (Normal Ranges)
| Cognitive Feature | Means of Assessment |
|---|---|
| Working memory | Digit span forward (7 ± 2) |
| Complex attention (require manipulation of items in working memory) | Digit span backward (6 ± 2)Months in reverse order (15-20 seconds) |
| Orientation | Time, location, autobiographical data |
| Language | Confrontational naming (high- and low-frequency items) |
| Verbal fluency (e.g., animals, grocery items) (18 ± 6/1 minute) | |
| Repetition (sentences of varying length) | |
| Comprehension (yes-or-no questions; performing multistep tasks) | |
| Reading aloud/comprehension | |
| Sentence writing (spontaneous, to dictation) | |
| Visuospatial | Figure copying (two- and three-dimensional figures) |
| Visual scene analysis (describe whole and individual parts) | |
| Line bisection (place “X” in center of horizontal line) | |
| Clock drawing (draw clock face with hands set to “10 after 11”) | |
| Verbal recent memory | Word list (3-5 words): immediate/delayed/recognition recall |
| Paragraph recall | |
| Nonverbal recent memory | Figure copy recall |
| Remote memory | Historical events (family milestones, recent presidents) |
| Abstract conceptualization | Similarities (rose/tulip, poem/statue); proverb interpretation |
| Praxis | Have patient demonstrate saluting a flag, hammering a nail |
| Sequencing | Graphomotor sequencing (have patient draw a simple alternating pattern) |
| Luria gestures (sequentially alternating fist, side, palm) |
* Further details regarding administration and interpretation can be found in Hodges, J.R., 1994. Cognitive Assessment for Clinicians. Oxford University Press, Oxford.
Neuropsychiatric Assessment
Neuropsychiatric symptoms are exceedingly common among the cognitively impaired. Mood state (depressed, euphoric), vegetative status (eating, sleeping), changes in personality (apathetic, disinhibited), and alterations in perception (hallucinations) or thought (delusions) are major areas to be probed in the course of diagnostic assessment. The presence or absence of certain neuropsychiatric symptoms may guide the physician in the differential diagnosis. For instance, while apathy is exceedingly common among all neurodegenerative dementias, euphoria is only rarely seen in AD or DLB but common in frontal-variant frontotemporal dementia (fvFTD). Early hallucinations, delusions, and fluctuations, which can also predate the onset of cognitive decline, point to DLB. Attention to these and other neuropsychiatric concomitants of dementia is important because they represent treatable components of excess morbidity and often have a profound impact on caregiver burden and the need for institutionalization (Kaufer et al., 1998).
Although structured interviews for neuropsychiatric assessment in dementia have been developed, their use is restricted primarily to research settings. Recently a brief questionnaire version of the Neuropsychiatric Inventory (NPI) (Cummings, 1997) was designed for the practice setting. The Neuropsychiatric Inventory Questionnaire (NPIQ) (Kaufer et al., 2000) is relatively easy to administer and can provide a clinical screening examination for common neuropsychiatric manifestations of dementia and their associated impact on caregivers.
Laboratory Studies
An electroencephalogram (EEG) has not proved useful in the routine evaluation of AD (Knopman et al., 2001).
Neuroimaging
Based on evidence-based methodology, the AAN guidelines for diagnostic evaluation of dementia (Knopman et al., 2001) recommended the use of neuroimaging to screen patients with cognitive impairment. These recommendations resulted from the findings of one class II study showing that 5% of all patients with cognitive complaints harbored a causative nondegenerative lesion such as a slow-growing brain neoplasm (most commonly of the frontal lobes), subdural hematoma (SDH), or normal-pressure hydrocephalus (NPH) (Chui and Zhang, 1997). Inasmuch as the vast majority of MCI patients harbor neurodegenerative pathology, it is sensible to extend the guidelines for diagnostic imaging to that patient cohort as well. Justification for a structural imaging evaluation of the cognitively impaired individual is twofold: it can help detect a potentially treatable disorder in need of urgent targeted intervention, and it can identify vascular ischemic comorbidity that could either be the source of or a contributing factor to cognitive decline. Magnetic resonance imaging (MRI) is preferred, but in those instances where MRI technology is not available or when an MRI is contraindicated (for instance in patients in pacemakers), computed tomography (CT) should be used.
Neurodegenerative Dementias
Alzheimer Disease
AD was originally described in 1907 by the German psychiatrist and neuropathologist, Alois Alzheimer. The index case was a 51-year-old woman with paranoid delusions, progressive memory impairment, and subsequent progressive aphasia. At autopsy, Alzheimer noted brain atrophy, and as he applied the newly available silver stains, he uncovered the senile plaques (now termed neuritic or amyloid plaques) consisting of dystrophic neurites clustered around what subsequently was revealed to be a central amyloid core. He also described a second deposition, the neurofibrillary tangle, consisting of intraneuronal staining in a fibrillar pattern. In honor of Alois Alzheimer, Kraepelin subsequently named the condition Alzheimer’s disease. What initially was considered a presenile condition was later appreciated to be far more common in the elderly after 65 years of age, and with the age distinction removed, AD became the most common neurodegenerative disorder and one of the most common diseases of the aging population. It is now listed as the fifth most common cause of death (Alzheimer’s Association, 2010).
Diagnostic Criteria
The most commonly used and widely accepted criteria for dementia of the Alzheimer type are the National Institute of Aging and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria (McKhann et al., 1984). While these criteria are very useful and well operationalized for the dementia state of AD, the accumulating evidence of a long prodromal (or latent) AD state has challenged our former understanding of AD and resulted in incredibly insightful research into the MCI state and more recently into prodromal AD. The AD field is now rapidly moving toward early and presymptomatic AD diagnosis that will rely heavily on disease biomarkers as outlined in the Dubois criteria for prodromal AD (Dubois et al., 2007) (Box 66.3). As such, AD has moved from being a “diagnosis of exclusion,” post documenting normal findings on a battery of laboratory studies, to a specific diagnosis based on clinical course and a characteristic pattern (or evolving pattern) of neuropsychological deficits in the presence of disease-associated biomarkers.
Box 66.3 Dubois Diagnostic Criteria for Alzheimer Disease
Probable Alzheimer Disease
Evidence satisfying the core diagnostic criteria (A) plus one or more supportive features (B-E)
 Gradual progressive change in memory function reported by patients or informants over more than 6 months
Gradual progressive change in memory function reported by patients or informants over more than 6 months





Exclusion Criteria











Epidemiology
AD is the most common cause of dementia worldwide. ADAMS recently published nationally representative data of dementia prevalence in the United States (Langa et al., 2005; Plassman et al., 2007). Diagnosis of dementia of the Alzheimer type was based on the NINCDS-ADRDA criteria. Overall dementia of the Alzheimer type accounted for 70% of dementia cases, ranging from 47% among those aged 71 to 75 years to 80% in the age 90+ group. The ADAMS data revealed that as of 2002, there were 3.4 million dementia cases in the United States (95% confidence interval [CI], 2.8-4.0), of whom 2.4 million (95% CI, 1.9-2.9) were due to AD. In addition to older age, the authors reported that African American race, lower educational achievement, and ApoE E4 genotype independently contributed to increased odds of dementia syndrome of any etiology (Plassman et al., 2007). In a following publication (Plassman et al., 2008) the ADAMS investigators reported that an estimated 22.2% (or 5.4 million Americans) 71 years or older have cognitive impairment in the absence of overt dementia. Of these, 2 million subjects were reported to have prodromal AD, defined as cognitive impairment without dementia but with a pattern of clinical symptoms or performance on neuropsychological testing suggestive of prodromal AD and no other medical or neuropsychiatric condition present to preclude an eventual diagnosis of AD. The annualized rate of progression to dementia of the Alzheimer type in prodromal AD subjects was substantially higher than the rate of progression to dementia among all cognitively impaired (17%-20% per year in prodromal AD versus 12% in cognitive impairment but no dementia from any cause). Among those who progressed to dementia, 83% were diagnosed with dementia of the Alzheimer type, 16.7% were diagnosed with vascular dementia, and 0.4% with dementia of undetermined cause (Plassman et al., 2008).
Based on our discussion thus far, it should not be surprising that advancing age is the most powerful risk factor for the development of AD. Other risk factors include the presence of one or more of the following: ApoE E4 allele in a given individual, lower education level, family history of AD, and cardiovascular risk factors (Bendlin et al., 2010; Rocchi et al., 2009; Steptoe et al., 2011). Prevalence but not incidence appears to be higher in women than in men, suggesting that susceptibility is similar, but duration of survival is greater in women. Midlife hypertension, elevated homocysteine, and elevated fat in the diet (the latter perhaps in combination with presence of an ApoE E4 allele) also are reported risk factors (Bendlin et al., 2010).
Clinical Presentation
AD is a progressive disorder of recent episodic memory, language, visuospatial function, and executive function associated with high frequency of neurobehavioral abnormalities at some point in the course. Onset of AD usually is in late life. If onset age is younger than 45 years, an autosomal dominant pedigree usually is involved. Most commonly, a PS1 mutation is the cause. In elderly individuals presenting with mild memory loss, the diagnosis of prodromal AD could be somewhat challenging owing to the presence of comorbid conditions that may alter cognition (e.g., congestive heart failure, cerebrovascular disease) or if medications for such conditions have anticholinergic side effects. Serial follow-up evaluations may be necessary to establish decline and a characteristic pattern of dementia. Nonetheless, if memory is not a prominent early complaint, other potential causes of dementia should be considered. In unselected series, diagnostic accuracy has been relatively low when tested against autopsy diagnosis. Using established diagnostic criteria for AD such as those of the NINCDS/ADRDA in conjunction with a standardized evaluation, clinical diagnostic accuracy of about 90% can be achieved (Knopman et al., 2001), accuracy approaching 90% can be anticipated, and few if any “treatable causes” are missed.
Memory Loss
A third type of memory, semantic memory (Tulving, 1987, 1992), is defined as our knowledge of facts about the world and is not associated with a spatiotemporal context of the learning event. For example, while we know that a tiger has stripes and that Washington DC is the capital of the United States, we do not recall the context in which we learned these pieces of information. Although episodic memory impairment is more prominent in AD, semantic memory is also impaired relatively early in the disease course. Some of the language impairment seen in AD relates to disintegration of semantic memory. A common test of semantic memory is the category fluency test, when the patient is asked to name as many items as possible from a given category such as animals or vegetables. Semantic memory can be reduced in the early stages of AD.
Aphasia, Apraxia, and Visuospatial Impairment
Language disturbance, especially verbal fluency and word finding, generally is an early feature of AD. In some patients, aphasia may be a prominent early feature, with more widespread cognitive disturbance occurring later. A helpful diagnostic aid in AD is that naming in semantic categories (e.g., animals) is more impaired than orthographically constrained tasks (e.g., words starting with a particular letter). This finding has been attributed to a breakdown in semantic memory. In practice, this means that patients with AD will produce more words beginning with a given letter (phonemic fluency task) than animals (semantic fluency task). This may be particularly useful in differentiating early AD from the effects of depression, in which the opposite pattern is seen. Of note, a reduction in category fluency also occurs with aging, so that in the “oldest old” (>80 years), it may be less informative in relation to the presence or absence of AD (Ravdin et al., 2003).
Atypical Alzheimer Disease Variants
The frontal variant of AD presents with prominent behavioral and/or personality changes in addition to short-term memory loss. These patients often are impatient, irritable, impulsive, and disinhibited. They show impairments on tests of frontal executive performance such as categorical verbal fluency and Trail-Making A (Chen et al., 1998; Johnson et al., 1999), as well as on response inhibition and set shifting (Chen et al., 1998) and show greater pathological involvement of the frontal neocortex (Johnson et al., 1999).
Posterior cortical atrophy (PCA) is a relatively rare AD variant. PCA patients present with prominent visuospatial dysfunction such as partial or full Balint syndrome (simultanagnosia, ocular apraxia and ocular ataxia), partial or full Gerstmann syndrome (acalculia, agraphia, right/left disorientation, finger agnosia), apperceptive visual agnosia, and environmental disorientation. In addition they frequently show visual-field deficits or constructional, dressing, and ideomotor apraxia. Relatively preserved memory and insight until later in the disease course is the norm (Mendez et al., 2002; Renner et al., 2004; Tang-Wai et al., 2004). The visual processing deficits can be related to both dorsal (“where”) and ventral (“what”) visual-stream impairment but tend to involve the former more prominently, consistent with the topography of the pathology. This form of AD is associated with profound parieto-occipital atrophy and higher AD pathology burden in the primary visual and secondary visual-association cortices (Renner et al., 2004; Tang-Wai et al., 2004). Other pathological conditions with this syndrome have been described, including the Heidenhain variant of Creutzfeldt-Jakob disease (CJD), DLB, corticobasal degeneration, and dementia lacking distinctive histological features (Renner et al., 2004; Tang-Wai et al., 2004).
Finally, the occasional AD patient may also present with early progressive language involvement (Kramer and Miller, 2000) or significant parkinsonian signs and symptoms (Cummings, 2000; Kurlan et al., 2000).
Neuropsychiatric Features
While dementia is classically defined by cognitive impairment, almost all AD patients exhibit a wide range of neuropsychiatric symptoms. Mood state (depressed, euphoric), vegetative status (eating, sleeping), changes in personality (apathetic, disinhibited), and alterations in perception (hallucinations) or thought (delusions) are the major areas to be probed in the course of diagnostic assessment. Although structured interviews for neuropsychiatric assessment in dementia have been developed, their use is restricted primarily to research settings. As noted earlier, a brief questionnaire version of the Neuropsychiatric Inventory (NPI) designed for practice settings—the Neuropsychiatric Inventory Questionnaire (NPIQ)—has been developed to provide a clinical screening examination for common neuropsychiatric manifestations of dementia and their associated impact on caregivers (Kaufer et al., 2000).
Neuropsychiatric assessment plays an important role in differential diagnosis and sometimes reveals the most pressing therapeutic needs. For instance, in the moderate to advanced stages of AD, patients may present with irritability and agitation to the point of violent verbal and physical outbursts, sundowning, hallucinations, and paranoid delusions may create a strain on the spouse, family, and caregivers. When prominent, these symptoms may have to be addressed first, even prior to initiation of anticholinergic medications or memantine. Behavioral symptoms once manifest tend to worsen over the course of the disease; however, for the individual patient, symptoms may fluctuate and may not be present at each clinical evaluation (Cummings, 2000).
Apathy can be found in 42% of those with mild, 80% of those with moderate, and 92% of those with advanced AD (Mega et al., 1996). It presents with loss of interest in previously enjoyed activities (e.g., hobbies, social outings, spending time with beloved relatives), aloofness, diminished spontaneity and emotional behavior, and reduced motivation. It is thought to reflect disruption of the connections within the frontosubcortical–anterior cingulate circuitry and their connections with other cortical regions. Apathy and depression commonly co-occur, but they are not synonymous with each other (Cummings, 2003). Apathy can be distinguished from primary depression on the basis of its neutral affect and the absence of vegetative signs.
Depression is very common in AD, occurring in 10% of mild, 40% to 60% of moderate, and 60% or more of severe AD patients (Mega et al., 1996). The symptoms are rarely severe enough to merit diagnosis of major depressive disorder; more often they represent minor depression/dysphoria. Risk factors for developing depression are familial or personal history of depressive disorder, female gender, and younger age (Lyketsos and Olin, 2002).
Agitation and irritability frequently co-occur. Agitation is more common in males, those with later onset of dementia, and those of more advanced age. It encompasses disruptive, aggressive, and/or resistive behaviors and is related to changes in frontal cortex on functional imaging studies and postmortem examination (Cummings, 2003). Common sources of resentment and irritability are the patient’s inability to successfully accomplish tasks that were accomplished with ease in the past, or a feeling of being mistreated or ignored.
AD patients may show a whole host of psychotic features such as hallucinations, delusions, or delusional misidentifications. These typically occur in the moderate to severe stages in AD, whereas in DLB they can occur early on and even be the first manifestation of the disorder. Most hallucinations are in the visual modality. Delusions also tend to favor the later stages of the disorder and occur in 30% to 50% of patients. Most common are delusions of infidelity, theft, and paranoia. Delusions often co-occur with aggression, anxiety, and aberrant motor behavior (Cummings, 2003).
Attention to the neuropsychiatric features of dementia is important because they represent treatable components of excess morbidity and often have a profound impact on caregiver burden and prompt institutionalization (Kaufer et al., 1998).
Laboratory Studies
The evidence-based guidelines of the AAN for diagnostic evaluation of dementia (Knopman et al., 2001) recommend routine screening for vitamin B12 deficiency and hypothyroidism. Other blood tests, such as screening for syphilis, are also justifiable if a clinical suspicion for neurosyphilis is present, either because of high-risk behavior or because of location in an endemic region.
Genetic testing for the ApoE genotype is not recommended on a routine basis. A large multicenter study demonstrated that the presence of the ApoE E4 allele increased the positive predictive value of diagnosing AD by only 4% over diagnoses made on clinical grounds alone (90% versus 94%) (Mayeux et al., 1998).
The AAN guidelines noted that CSF tests for β-amyloid, tau, and neuropil thread protein (AD7C-NTP) gave insufficient data, demonstrating values above and beyond the relatively high sensitivity and specificity of the clinical diagnosis of AD (Knopman et al., 2001). New studies assessing the diagnostic or predictive capabilities of β-amyloid and tau or phosphorylated tau (phosphotau) in the CSF, however, suggest the possibility that they may have a role to play in difficult cases, confirmation of diagnosis, or prediction of development of AD in patients in the predementia stages (Mattsson et al., 2009). Low CSF β-amyloid (Aβ1-42) coupled with elevated tau or phosphotau increases the likelihood that the patient has AD-type pathology (Mattsson et al., 2009; Sunderland et al., 2003). However, it should also be kept in mind that some pathologically confirmed AD patients have shown normal CSF Aβ1-42 and tau levels prior to death (Brunnstrom et al., 2010). In the predementia stages, a CSF pattern suggestive of AD relates to a very high probability of developing AD in the next 5 years (Hansson et al., 2006).
Genetics
Some but not all late-onset FAD pedigrees are associated with inheritance of the E4 allele of ApoE. In addition to the late-onset familial cases, ApoE also contributes to sporadic disease. Increased amyloid load and earlier age at onset are related to ApoE4 gene dosage (Mayeux et al., 1998). The apoE4 genotype is, however, only a risk factor, being neither sufficient nor necessary for disease development, and this is why it is not used routinely in clinical evaluations.
Neuroimaging
The AAN dementia practice parameter guidelines state that at least one unenhanced CT or MRI scan should be performed in patients with cognitive decline to rule out unexpected structural lesions and also to provide information about potential silent vascular injury (Knopman et al., 2001). MRI, with its improved resolution, allows better quantification of cerebral structures and better discrimination of normal from mildly affected patients with AD than is possible with CT. Noninvasive neuroimaging has greatly aided the accurate diagnosis of AD; structural lesions such as tumors, hydrocephalus, subdural hemorrhage, and strokes are identified easily. MRI and to a lesser extent CT aid in identification of vascular lesions that may be primary causes of dementia or contributory to cognitive decline in cases of mixed AD/vascular dementia.
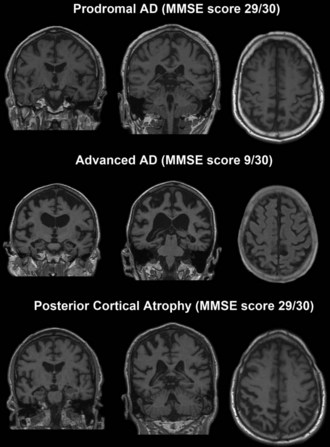
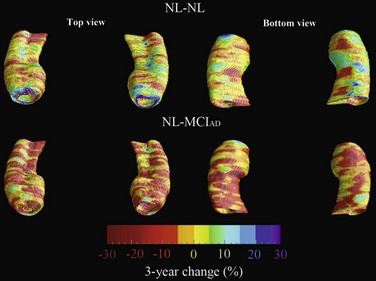
Mesial temporal atrophy including the entorhinal cortex, hippocampus, and amygdala are considered typical for the prodromal AD stages (Apostolova et al., 2006, 2009; Jack et al., 2004) (Fig. 66.1). In the dementia stage, global brain atrophy—more striking in the temporoparietal than in the frontal regions—and ventricular enlargement are also pronounced (Apostolova et al., 2007; Thompson et al., 2003) (Fig. 66.2). A gradient-echo sequence on MRI could reveal cortico-subcortical microhemorrhages suggestive of the presence of vascular amyloidosis.
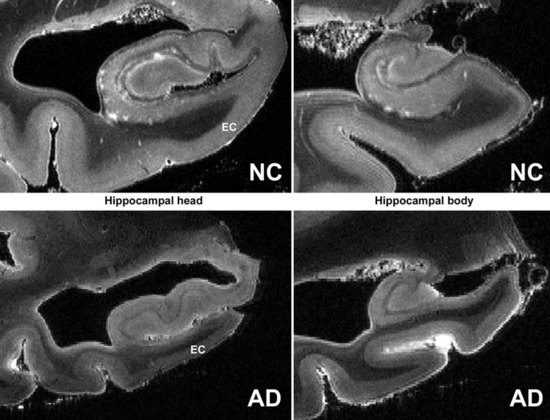
New and advanced methodologies provide unique opportunities to study the earliest changes in the hippocampal structure. Some studies have documented subtle atrophy present as early as 3 years prior to MCI and 6 years prior to the dementia stages of AD (Apostolova et al., 2010b) (Fig. 66.3). Imaging biomarkers are presently being developed as diagnostic and prognostic biomarkers as well as surrogate biomarkers for clinical trials, with hippocampal atrophy, the most validated structural biomarker, already being accepted as a biomarker criterion for AD presence in the prodromal AD stages (Dubois et al., 2007).
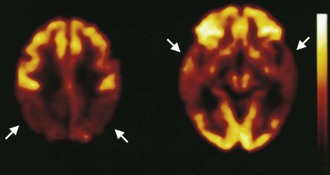
Functional brain imaging with single-photon emission computed tomography (SPECT) and positron emission tomography (PET) can identify disease-specific patterns such as temporoparietal abnormalities in AD, frontal or anterior temporal abnormalities in FTD, and temporo-parieto-occipital abnormalities in DLB (O’Brien, 2007) (Fig. 66.4). SPECT findings of blood-flow abnormalities in a temporoparietal distribution may aid in confirmation of AD, especially in cases without significant atrophy. PET using [18F]fluorodeoxyglucose (FDG), a measure of energy utilization in the brain that predominantly marks synaptic activity, also shows temporoparietal deficits, is more sensitive than SPECT, and can confirm the diagnosis (see Fig. 66.4). Although validation studies of these methods generally have shown high sensitivity, relatively lower specificity poses the risk of false-positive diagnoses. To date, the sensitivity and specificity of PET have not been assessed outside of research clinics (Silverman, 2004; Silverman et al., 2003). It is nevertheless intriguing that similar although less marked deficits in FDG-PET in the cingulate, temporal, and parietal cortices have been demonstrated in presymptomatic persons homozygous for ApoE E4 (Reiman et al., 1996). Further work to more rigorously validate these methods is necessary before they can become more widely applicable (Cummings et al., 2007; Thal et al., 2006).
The recent development of PET ligands for imaging proteins contained in amyloid plaques—Pittsburgh Compound B (PIB) (Klunk et al., 2004; Mathis et al., 2007)—or concomitantly labeling amyloid plaques and neurofibrillary tangles—[18F]-FDDNP (Small et al., 2006)—offers the prospect of a preclinical or early diagnostic biomarker for AD (Mintun et al., 2006). PIB uptake is seen in the same cortical regions that show diminished FDG-PET activity (Fig. 66.5), but PIB imaging offers the advantage of greater spatial resolution and greater effect sizes relative to conventional FDG-PET imaging (Ziolko et al., 2006).
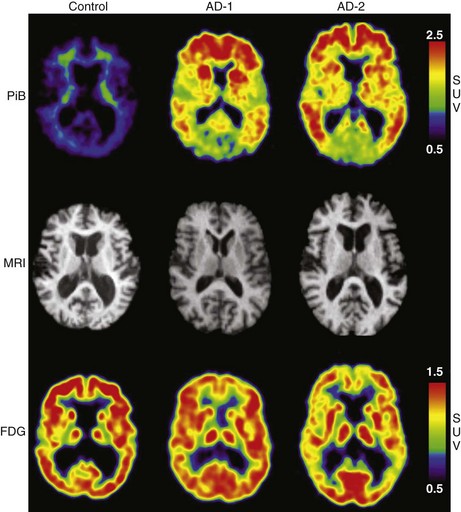
Amyloid PET imaging shows a bimodal uptake distribution in MCI, with some subjects showing AD-like and others normal control (NC)-like retention patterns (Kemppainen et al., 2007; Pike et al., 2007). Longitudinal amyloid imaging studies in the MCI and NC states has revealed that high PIB retention conveys substantially higher risk for progression from MCI to AD (Okello et al., 2009) and for cognitive decline in NC (Morris et al., 2009). PIB retention in amyloid-rich regions has also been documented in postmortem specimens (Ikonomovic et al., 2008; Thompson et al., 2009). Although PIB binding shows the expected correlation with cognitive function cross-sectionally (Jack et al., 2008, 2009; Mormino et al., 2009; Pike et al., 2007; Tolboom et al., 2009), longitudinal PIB studies have surprisingly uncovered that PIB retention levels off in the symptomatic stages, suggesting its greater usefulness as a preclinical rather than an overt clinical biomarker (Engler et al., 2006).
Pathology
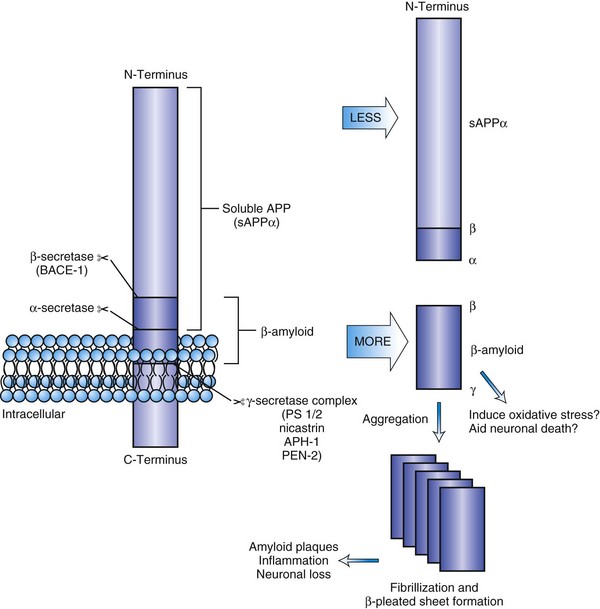
Amyloid protein isolated from plaque cores is predominantly a 40- to 42-amino-acid peptide (Aβ1-42 or Aβ1-40) derived from its larger precursor protein, APP. APP is a transmembrane protein expressed in both neural and non-neural tissue. The APP gene located on the long arm of chromosome 21 has been linked to autosomal dominant early-onset AD. Elevation of Aβ1-42 concentrations with increased synaptic activity suggests it plays a role in synaptic function (Cirrito et al., 2005). A number of metabolic pathways for the processing of APP are recognized, and a shift in metabolism to the β-peptide is thought to be fundamental to amyloid deposition (Selkoe and Schenk, 2003). In the primary metabolic pathway, α-secretase cleaves APP just above the surface of the membrane, resulting in a large-fragment, soluble APP (sAPPα). This cleavage occurs within the β-amyloid domain, precluding Aβ production (Fig. 66.6). The β-amyloid fragment, either Aβ1-42 or Aβ1-40, is cleaved by two different proteases, the β- and γ-secretases at the N- and C-termini, respectively. Monomers aggregate into an oligomeric structure that is neurotoxic. Fibrillization leads to insoluble amyloid that is less neurotoxic. Aβ1-42 has a greater propensity to fibril formation and is the predominant form in early plaques. Vascular amyloid is predominantly Aβ1-40. Posttranslational modification of the fragments may play a role in transition to neuritic plaques. In AD, an apparent partial shift to the amyloidogenic pathway may occur, or there may be impaired clearance, resulting in greater β-amyloid production and subsequent deposition of diffuse in the brain tissue. Basic research studies have shown that the monomeric Aβ1-42 can lower the threshold for neuron death in cell culture and induce oxidative stress. Thus, β- or γ-secretase inhibition is a proposed therapeutic strategy, as are immunotherapies or other strategies to decrease amyloid content in the brain (Fig. 66.7). Markers of inflammation such as α1-antichymotrypsin and other proteases and peptidases are found in neuritic plaques (NP), as is ApoE. ApoE is thought to play a role in amyloid removal. The ApoE E4 allele appears to be less effective in aiding the removal of β-amyloid from the brain, and this may be the reason that patients with AD who are ApoE4 carriers have more amyloid deposition in their brains and earlier onset of disease than non-ApoE E4 carriers.
NP, also called senile plaques, are found predominantly in the cerebral cortex, especially in the association areas and the hippocampus (Fig. 66.8). Amyloid deposition does not follow the topographic sequence for the development of NFTs; it is seen initially in the cortex, then in the hippocampus, and then in other regions (Arnold et al., 1991). NPs range in diameter between 25 and 200 mm and consist of abnormal nerve processes often referred to as dystrophic neuritis, processes of activated microglia and astrocytes, and a central core of β-amyloid (see Fig. 66.8, A). β-Amyloid is a 40- or 42-amino-acid peptide that is cleaved from the large APP molecule. Diffuse or immature plaques are not associated with dystrophic neurites or with fibrillar amyloid; immunostaining reveals the presence of the β-amyloid protein, predominantly Aβ-1-42. It is believed that diffuse plaques precede classical neuritic plaques and that progressive fibril formation by the monomeric Aβ-1-42 into oligomers leads to initiation of an inflammatory process that results in protease and peptidase deposition and neuropil destruction. Some authors refer to “burnt-out plaques” in which a prominent amyloid core is surrounded by a very thin rim of dystrophic neurites.
The primary component of NFTs is the microtubule-associated protein, tau. Tau protein is a cytoskeletal protein found predominantly in axons. In AD, tau is abnormally hyperphosphorylated and extensively cross-linked, forming an insoluble intracellular deposit (see Fig. 66.8, B). This altered form has reduced binding to microtubules, thereby disrupting the cytoskeleton and leading to interference with neuronal metabolism and subsequently to neuron death. Tau has six isoforms. These isofoms result from alternative splicing of the gene; three of the isoforms have three microtubule binding domain repeats (3R tau), andthree of the isoforms have four repeated sequences (4R). Variations seen in Western blots of tau isolated from brain in AD and Pick disease, corticobasal degeneration, and hereditary tauopathy indicate differential deposition of isoforms (Wszolek et al., 2006). Ubiquitin is frequently found in association with tangles, probably representing an attempt by the neuron to degrade the abnormal protein. Deposition of NFT appears to begin in the transentorhinal and entorhinal cortex, spreading from there to the hippocampus followed by the temporal neocortex and beyond (Delacourte et al., 1999). There is early laminar predilection is initially for layer 2 and then layer 5 of the entorhinal cortex followed by the CA1 and subicular region of the hippocampus, and the pyramidal cells of neocortical layers 3 and 5. Ultrastructurally, NFTs consist of paired helical filaments with an individual filament diameter of 10 nm wound in a double helix, with a total diameter of 200 nm and a periodicity of 160 nm. NFTs are not exclusive to AD and can be found in other conditions such as dementia pugilistica, prion disease, and Kufs disease. Neuropil threads (curly fibers) are also found and represent paired helical filament–containing neurites. Their number usually parallels the severity of neurofibrillary tangle formation.
Other histological AD features include granulovacuolar degeneration (see Fig. 66.8, C) and Hirano bodies (see Fig. 66.8, D). Granulovacuolar degeneration is typically seen in hippocampal and consists of clear, round, intracytoplasmic basophilic vesicles 4 to 5 mm in diameter that immunolabel for tubulin, ubiquitin, and neurofilament. Hirano bodies are eosinophilic rodlike structures located almost exclusively in the Sommer sector of the hippocampal pyramidal layer, adjacent to and occasionally in neuronal bodies. Hirano bodies contain actin and actin-associated proteins, tau, and a C-terminal fragment of APP. Mild cortical spongiosis that is distinct from the spongiform changes of CJD can be found in the association cortices, sparing the primary sensory and motor cortex in some cases.
Cell loss preferentially involves the large neurons of the deeper layers of the cortex. Dysfunction of the cholinergic system in early stages (before loss of the synthetic enzyme ChAT) is suggested by the loss of basal forebrain neurons and receptors for the cholinergic neurotrophin nerve growth factor, as well as by the sensitivity of patients with AD to anticholinergics. Studies of patients with end-stage AD in the 1970s and 1980s uncovered extensive neuron loss in the nucleus basalis of Meynert and other cholinergic basal forebrain nuclei. These structures are the origin of the cholinergic projection to cerebral cortex and other forebrain regions. This loss was found to associate with loss of choline acetyltransferase (ChAT) in the projection fields of these nuclei. More recent work, however, revealed that (1) the loss of ChAT does not occur until much later in AD’s course (DeKosky et al., 2002), and (2) that cholinergic cells in the cholinergic basal forebrain neurons are preserved until very late in the course (Mufson et al., 2002). Neuron loss occurs to a lesser extent within the locus ceruleus and the nucleus raphe. As these studies were performed in late-stage cases, it is possible that these neurons, too, are preserved longer than was earlier thought.
Treatment
Acetylcholinesterase Inhibitors
Acetylcholinesterase inhibitors (AChEIs) are the first line of pharmacological treatment in AD. Treatment of AD with AChEIs was judged to be an accepted standard of care in the AAN practice guidelines for dementia (Doody et al., 2001). The following three agents are now available in the United States, Europe, and many other parts of the world: donepezil (Aricept), rivastigmine (Exelon), and galantamine (Reminyl). All three medications are effective and as shown in double-blind placebo-controlled trials can potentially stimulate or stabilize performance. Initial studies were 3 to 6 months in duration; double-blind placebo-controlled trials of up to 1 year (donepezil) show maintenance of medication effects, and open-label extension studies suggest continued efficacy for several years. Sudden improvement in cognitive function occurs in a small percentage of subjects. Slowing of decline or stabilization over time, difficult to determine in a brief exposure to the drug, also occurs with these medications. Gastrointestinal cholinergic-mediated side effects are commonly seen with AChEIs. Side effects are more common during the dose escalation phase of treatment; emphasizing this to patients and caregivers will enhance compliance. Gastrointestinal effects can be reduced by giving these drugs after meals or slowing the dose escalation rate. Beneficial effects of these agents also have been shown on neurobehavioral symptoms, notably apathy and visual hallucinations, and on stabilizing activities of daily living functions or slowing their decline. If therapy with one agent appears ineffective or causes intolerable side effects, another AChEI should be tried. Because of the nature of the neurochemical action of these medications, withdrawal effects are possible on switching drugs, so prolonged washout periods should be avoided.
Glutamate Receptor Modulators
The abnormalities in glutamate metabolism and receptors noted postmortem in brains of patients who had AD led to hypotheses that glutamate metabolic dysregulation leads to excitotoxicity and neuronal death in AD (Rogawski and Wenk, 2003). Memantine, an N-methyl-d-aspartate (NMDA) antagonist that had been available in Germany since the 1980s, was assessed as solitary therapy in two pivotal trials in moderate to severe AD (MMSE score of 5 to 15) in the United States (Reisberg et al., 2003) or as an add-on to ongoing AChEI therapy (Tariot et al., 2004). Both studies showed a significant benefit at the end of the trial in cognition and function, and memantine was approved for use in moderate to severe AD. No specific side effects were identified in the pivotal trials; side effects were significantly more frequent than in the placebo groups.
Other Cognitive-Enhancing or Disease-Modifying Drugs
With the vast increase in knowledge of the disease mechanisms operative in AD, many medications aimed at slowing the disease process and ameliorating cognitive decline are under development or evaluation. These include drugs currently approved for other indications, new medications, and complementary or alternative medications. Most proposed or unregulated preparations have been suggested by virtue of their potential ability to interfere with the known pathobiological cascades in AD, notably oxidative stress and inflammation. Vitamin E, an antioxidant, in a dose of 1000 IU twice daily had some effect on delaying time to nursing home placement, decline from moderate to severe AD, or death (Sano et al., 1997). However, recent data suggesting that high doses of vitamin E may cause cardiac toxicity, coupled with the failure of high-dose vitamin E to slow conversion of MCI to AD (Petersen et al., 2005) have resulted in abandonment of vitamin E as a treatment. Other free radical inhibitors and antioxidants are used, but without support from prospective data.
Estrogen or estrogen-progesterone replacement therapy was shown in several retrospective studies to delay or prevent emergence of AD, but two studies, one large and one small, showed that they have no effect on disease progression (Henderson et al., 2000; Mulnard et al., 2000). The Women’s Health Initiative Memory Study (WHIMS) showed no effect of these agents on incidence of either MCI or AD; in fact, a slight increase in dementia incidence was noted (Shumaker et al., 2003).
The use of Ginkgo biloba as a treatment for AD has now been credibly disputed by two separate studies. A carefully performed double-blind placebo-controlled 6-month-long study of ginkgo showed no effect at 6 months (Schneider et al., 2000). Another randomized double-blind placebo-controlled 7-year-long clinical trial with 3069 community-dwelling participants aged 72 to 96 demonstrated no benefit of gingko on cognitive performance in normal cognition or MCI (Snitz et al., 2009).
Latrepirdine (Dimebon) attracted interest after showing potential beneficial effects in AD animal models, a phase II human trial, and a phase III double-blind trial with mild- to moderate-stage patients (Doody et al., 2008). A second phase III monotherapy trial showed no benefit. An ongoing study is assessing the utility of latrepirdine in patients receiving donepezil.
Medications for Behavioral and Neuropsychiatric Symptoms
Agitation or disruptive behavior may require a neuroleptic. The newer atypical antipsychotic medications (quetiapine, risperidone, olanzapine) have been recommended in low doses with careful titration, allowing time for the medications to exert their effects. The Clinical Antipsychotic Trials of Intervention Effectiveness (Schneider et al., 2006), however, indicated that within the limitations inherent in attempts to do a carefully controlled outpatient trial on agitation, side effects, and limited efficacy, the atypical antipsychotics did not show clear advantages over the older antipsychotic in the study. Traditional neuroleptics are more likely to produce extrapyramidal symptoms, which may worsen cognitive function. The newer antipsychotic agents are associated with increased mortality in the elderly and have had a “black box” warning label added by the U.S. Food and Drug Administration (FDA). The older antipsychotics, however, also carry an increased risk of death (Schneeweiss et al., 2007). Thus, judicious use of all of these medications, with frequent reassessment of the need for such a drug (at least every month or two), is appropriate.
Future Therapies
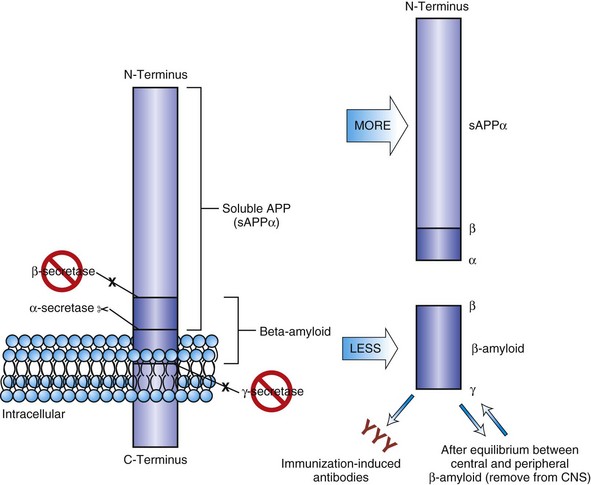
Interruption of the amyloid cascade also has a number of strategies, including β-secretase inhibitors, γ-secretase inhibitors or modifiers, or α-secretase enhancers (see Fig. 66.7). Compounds that would insert themselves on the polymerizing β-amyloid, the so-called plaque busters, conceivably could aid in slowing β-amyloid accumulation. Accelerated removal of β-amyloid once it is formed, either by antibody infusion (passive immunization) or active immunization to produce endogenous antibodies, is effective in transgenic mouse models. Initial active immunization trials in humans were interrupted by neuroinflammation and encephalitis in a subset of the subjects, probably due to T-cell sensitization. Autopsy studies in several immunized individuals from that study showed the expected neuroinflammatory changes (Orgogozo et al., 2003), as well as a remarkable amyloid clearance in several areas of the cortex, suggesting that—as in mice—the amyloid in humans can be removed (Masliah et al., 2005; Nicoll et al., 2003). Future studies will determine the feasibility of removing amyloid safely, whether it results in slowing of cognitive decline, and whether it has any effects on NFT formation.
Cerebral Amyloid Angiopathy
β-Amyloid deposits can be seen in the cerebral blood vessels of all patients with AD (see Fig. 66.8, E). Severity of involvement varies, ranging from the presence of small amounts of amyloid to major deposits that distort artery architecture, causing cortical microinfarcts and microaneurysm with or without cerebral hemorrhages. Familial cases of AD and cerebral amyloid angiopathy (CAA) are associated with multiple hemorrhages in the brain, spinal cord, and leptomeninges. Although these hemorrhages could be clinically silent, patients may present with progressive dementia and spastic paraparesis. MRI using a gradient-echo sequence can show multiple small hemorrhages in the brain and spinal cord. The most common forms of familial CAA without AD manifest with spontaneous lobar hemorrhages rather than dementia (i.e., Icelandic and Dutch cerebral amyloid angiopathy). Lobar hemorrhage in late life in nonfamilial cases also is suggestive of CAA (Table 66.2). This diagnosis is suspect when the hemorrhage is “lobar,” located at the cortical gray/white-matter junction, and not in the typical distribution of hypertensive hemorrhages (i.e., basal ganglia, brainstem, or cerebellum). AD can be accompanied by severe amyloid angiopathy.
Table 66.2 Boston Criteria for Diagnosis of Cerebral Amyloid Angiopathy–Related Hemorrhage
| Definite CAA | Full postmortem examination demonstrating CAA |
| Absence of other diagnostic lesions | |
| Probable CAA with supporting pathology in specimen | Clinical data and histopathological specimen (evacuated hematoma or biopsy) demonstrating some degree of CAA |
| Absence of other diagnostic lesions | |
| Probable CAA | Clinical data and MRI or CT demonstrating multiple hemorrhages restricted to lobar, cortical, or corticosubcortical regions (cerebellar hemorrhage allowed) in individuals age ≥55 years |
| Possible CAA | Clinical data and MRI or CT demonstrating single lobar, cortical, or corticosubcortical region hemorrhage in individuals age ≥55 years |
CAA, Cerebral amyloid angiopathy; CT, Computed tomography; MRI, magnetic resonance imaging.
Adapted from Knudsen, K.A., Rosand, J., Karluk, D., et al., 2001. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston Criteria. Neurology 56, 537-539.
More recently, an inflammatory presentation of amyloid angiopathy has been described. This syndrome is associated with subacute cognitive decline, seizures, and extensive white-matter abnormalities and microhemorrhages on imaging studies. Histologically, this presentation was found to be associated with giant-cell inflammatory response. The CAA-related inflammation appears to be relatively responsive to corticosteroids and cyclophosphamide (Eng et al., 2004).
Frontotemporal Dementia Spectrum
The frontotemporal dementias (FTDs) are a group of neurodegenerative dementias of varied etiology in which the frontal or temporal lobes, or both, are affected out of proportion to the rest of the brain, with variable degrees of subcortical pathology and degeneration. The first pathologically described case of FTD consistent with Pick disease was published in 1892 by the German neurologist, Arnold Pick. The clinical syndrome of frontal lobe degeneration of non-Alzheimer type with onset typically between the ages of 50 and 60 years and featuring insidious personality change, disinhibition, and subsequent gradual loss of speech output was described by Gustafson and Brun in the early 1990s (Gustafson et al., 1990). The FTD spectrum is a clinically and pathologically inhomogeneous group. Several distinct clinical phenotypes have been described: one behavioral variant, frontal-variant FTD (fvFTD); two language variants, primary progressive aphasia (PPA) and semantic dementia (SD); and one variant with associated motor neuron disease (MND), FTD-MND. Pathologically, FTDs have been attributed to a variety of molecular defects with new ones still being described. Population estimates of the incidence of FTD are not available, but most series estimate that it makes up 10% to 15% of the neurodegenerative dementias in clinical autopsy series.
Diagnostic Criteria
The most recently proposed criteria (McKhann et al., 2001) developed for the FTD spectrum in general are presented in Box 66.4. These are relatively well suited for clinical practice. In the research environment, however, the most widely accepted criteria for the FTD spectrum disorders are the Neary criteria (Neary et al., 1998). These are presented in Boxes 66.5, 66.6, and 66.7 (also see Clinical Presentation section).
Box 66.4
Recently Proposed General Criteria for the Frontotemporal Dementia Spectrum Disorders
1. Development of behavioral or cognitive deficit manifested by either:
2. The behavioral or language deficits cause significant impairment in social and/or occupational functioning and represent a significant decline from a previous level of functioning
3. The course of progression is gradual and continuous
4. The behavioral or language deficits are not due to other neurologic, systemic, psychiatric or substance abuse condition
5. The behavioral or language deficits are not due to delirium or depression
From McKhann, G.M., Albert, M.S., Grossman, M., et al., 2001. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58, 1803-1809.
Box 66.5
Neary Criteria for Frontal-Variant Frontotemporal Dementia
Adapted from Neary, D., Snowden, J.S., Gustafson, L., et al., 1998. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546-1554.
Box 66.6
Neary Criteria for Primary Progressive Aphasia
2. Supportive Diagnostic Features
Adapted from Neary, D., Snowden, J.S., Gustafson, L., et al., 1998. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546-1554.
Box 66.7
Neary Criteria for Semantic Dementia
Adapted from Neary, D., Snowden, J.S., Gustafson, L., et al., 1998. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546-1554.
Epidemiology
The FTD spectrum disorders usually affect persons between 45 and 65 years of age, except for SD, where the typical age of onset is 70 years. The prevalence of FTD in those between 45 and 64 years of age is 15/10,000 (Ratnavalli et al., 2002). Approximately half of the cases are familial.
Clinical Presentation
Consideration of a diagnosis of FTD follows elimination of possible systemic causes, other CNS cognitive or behavioral degenerative disorders (e.g., Huntington disease, AD) and substance abuse, which because of the lack of social concern in the patient, frequently is entertained as a cause by family, friends, or physicians (see Box 66.4). Absence of widespread knowledge of features and symptoms of FTD probably keeps it from being diagnosed more frequently.
The classification of FTDs is exceedingly controversial owing to large gaps in our understanding of the cellular and molecular pathophysiology and their associations with distinct clinical phenotypes (Geser et al., 2010). In this chapter, we will first describe the clinical phenotypes, and later on (see Genetics and Pathology sections) we will talk about the distinct molecular and neuropathological subtypes.
Frontal-Variant Frontotemporal Dementia
The classic clinical variant of FTD is fvFTD, also known as behavioral-variant FTD, primary progressive behavioral disorder, and behavioral disorder and dysexecutive syndrome. Behavioral characteristics vary along a spectrum depending on the part of the frontal lobes affected. Some patients are disinhibited, overactive, and restless and show prominent orbitofrontal involvement. Others with more widespread frontal involvement are apathetic and lack motivation and initiative. In general, fvFTD is associated with insidious early personal conduct and personality changes. Patients are frequently socially inappropriate, tactless, and disinhibited or aloof and socially isolated. FvFTD patients show impaired judgment and readily disobey socially accepted norms and boundaries. These are frequently described as exceedingly self-centered, shallow, lacking empathy and insight, emotionally labile or withdrawn, or impulsive. They pay less attention to their work or home responsibilities. Their behavior can be purposeless and lacking in goals. Obsessions and compulsions are common and may take the form of hoarding, unusual collecting, consumption of the same food item (especially sweets), and even consumption of inedible items. Patients experience difficulty in organizing or sequencing, without evidence of agnosia or ideomotor dyspraxia. Detailed neuropsychological evaluation reveals impairment of frontal executive abilities such as abstract thinking, decision making, planning, sequencing and set shifting, and diminished free verbal recall, with characteristic sparing of visual memory (Hodges and Miller, 2001). Box 66.5 lists the Neary criteria for fvFTD.
Primary Progressive Aphasia
Nonfluent PPA (also known as progressive nonfluent aphasia or PNFA) is characterized by early prominent language impairment with dysfluent, effortful, and agrammatical language output in the context of preserved language comprehension until late in the disease course (see Box 66.6). Anomia and phonemic paraphasic errors are pronounced early on. Speech output requires manifest effort and is nonfluent and hesitant with phonemic and semantic errors. Writing often is similarly affected, although dissociations in the degree of speech versus written impairment occasionally may be noted. Word finding and repetition are impaired, and spelling is poor. Early on, comprehension is preserved, as is insight into the disorder and nonlanguage cognitive skills including memory, allowing some patients to maintain employment and productivity for years after symptom onset. Speech output becomes progressively constrained, and dyspraxia ensues, impairing communication. As the disease advances, comprehension eventually deteriorates, but patients may retain some understanding even without the ability to speak or communicate with gestures. Social skills are preserved early in the course, but frontal symptoms may occur later. Behavioral disturbances are mild, and PPA patients are typically socially competent until late in the disease.
Some investigators now also identify the fluent form of PPA with greater early impairment in comprehension and preservation of articulation and words per utterance (Mesulam, 2003). Other investigators consider any fluent form of progressive aphasia to be SD, regardless of the presence of agnosia. It is unclear whether such subdivisions have meaning with regard to etiology and histopathology. To some extent, these forms differ by the prominence of pathology along the perisylvian fissure, with changes in the nonfluent form located more anteriorly along the axis. In almost all cases, anomia is a prominent initial symptom.
Semantic Dementia
SD (also known as semantic aphasia and temporal-variant FTD) presents with characteristic language impairment resulting from semantic knowledge loss (i.e., loss of conceptual knowledge and word meaning) (see Box 66.7). As a result, the language output in SD, while being fluent, well articulated, and grammatically correct, shows impoverished content and semantic supraordinate paraphasias (e.g., naming a lion an “animal” or a flower a “plant”). Further semantic loss leads to frequent substitutions of many nouns with “it” or “thing” to the point that the message the individual is trying to convey is completely lost. Impaired comprehension for both spoken and written language is also characteristic. Fluency belies profound anomia and word comprehension deficits, most evident on neuropsychological testing, which usually is necessary to diagnose and evaluate the patient over time. SD is characterized by a unique type of dyslexia called surface dyslexia, where irregular words such as “colonel,” “pint” or “yacht” are read and written with spelling-to-sound correspondence. At the same time, ability to repeat is preserved, and reading and writing are fluent.
Unlike individuals with PPA, SD patients frequently show frontally mediated behavioral abnormalities similar to those seen in FTD (Bozeat et al., 2000; Hodges and Miller, 2001). Although the dramatic disinhibition of FTD typically is absent, other frontal behaviors such as obsessions, compulsions, preoccupations, and behavioral stereotypy may appear. Ultimately, both PPA and SD patients progress to mutism or use of only a few words.
Frontotemporal Dementia with Motor Neuron Disease
FTD-MND patients may have behavioral or aphasic FTD presentation with additional amyotrophic lateral sclerosis (ALS)-like motor neuron symptoms such as fasciculations, muscle weakness, hyperreflexia, spasticity, and upgoing plantar reflexes. Additionally, they may have parkinsonian signs. Swallowing difficulties and choking may emerge, necessitating alternative methods of feeding and airway protection. On the other hand, frontal lobe deficits frequently can be found with detailed testing in ALS. It is estimated that FTD develops in 10% of patients with ALS, suggesting that these two disorders may be along the clinicopathological spectrum of a similar underlying neurodegenerative phenomenon (Chen-Plotkin et al., 2010).
Differentiating Frontotemporal Dementia from Alzheimer Disease
Although FTD patients sometimes complain of “diminished memory function” this usually is due to poor attention or a dysexecutive syndrome. Careful testing usually reveals relatively intact memory function. Obviously, memory testing is limited in patients with significant language or semantic impairment, but notably, recognition memory is spared, in contrast with AD (Graham et al., 1997). Neuropsychological testing, especially of recent memory function, may give normal results early in the course. Executive dysfunction can manifest as problems in set shifting, sequencing, abstract thinking, fluency, or confrontation naming. In contradistinction to those with AD, patients with FTD often show greater difficulty in naming words starting with a particular letter (e.g., words beginning with f) than in identifying members of a category (e.g., animals).
Genetics
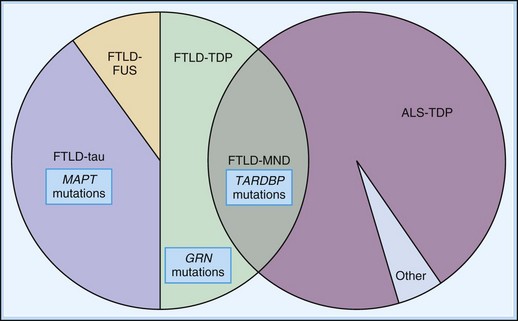
The genetic underpinnings of FTD are exceedingly complex and evolving concepts (Fig. 66.9). The discovery of genetic mutations in the tau (MAPT) gene on chromosome 17 led to increased focus on tau mutations or other metabolic abnormalities in the tau gene in FTD. A consensus conference on FTD with parkinsonism reviewed data for 13 families with linkage of their clinical symptoms to chromosomal locus 17q21-22 (Foster et al., 1996). A host of tau abnormalities have been described since the initial FTDP17 mutation was described. It has been determined that MAPT mutations cause between 5% and 15% of FTD cases, with age of onset ranging between 25 and 65 years of age. Some cases may also present as progressive supranuclear palsy and corticobasal degeneration (van Swieten and Heutink, 2008).
Tau protein, a microtubule binding protein found in neurons and glia, is needed for stabilization and polymerization of microtubules. The region of the tau molecule that binds to microtubules has repeat sequences of 31 or 32 amino acids. Six isoforms of tau in the adult human brain result from alternative splicing of the gene; three of the isoforms have three microtubule binding domain repeats (3R tau), and three of the isoforms have four repeated sequences (4R). Mutations in the MAPT gene associated with FTDP17 may occur in either exons or introns and may alter the production of either 3R or 4R tau, or alter the ability of tau to bind to microtubules. Most genotypic alterations do not correlate well with clinical phenotypes. However, overproduction of the 4R isoforms (associated with mutations that affect the splicing of exon 10) appears to associate with parkinsonian symptoms, while mutations that do not affect exon 10 splicing of exon 10 are associated more frequently with dementia-prominent forms. Ghetti and colleagues (2003) have summarized the current knowledge of the relationship of the mutations and their associated clinical syndromes in a recent review paper.
In 2006, some cases previously attributed to FTDP17, with conspicuous absence of tau pathology, were shown to be actually due to mutations in the progranulin gene (GRN) located on chromosome 17 (Chen-Plotkin et al., 2010; van Swieten and Heutink, 2008). The age at onset in these cases can be highly variable, ranging from 39 to 89 years (van Swieten and Heutink, 2008). 70% to 90% of patients with GRN mutations have a positive family history of dementia with parkinsonism. With more than 50 GRN gene mutations described to date, there is considerable phenotypical heterogeneity with GRN mutations. While described in FTD, GRN mutations have been reported to also cause AD, and corticobasal degeneration. Plasma and CSF levels of progranulin have been found to be reduced nearly fourfold in both affected and unaffected subjects with PGRN mutations Low (75% reduction) plasma progranulin levels may be used as a screening tool for PGRN mutations (Finch et al., 2009).
FTD with TAR-binding DNA protein (TDP-43) inclusions is the most common pathological variant in the FTD spectrum. TARDBP mutations have been strongly associated with familial forms of ALS (Geser et al., 2010). Yet familial FTD-MND cases without mutations in the TARDBP gene on chromosome 1 that encodes TDP-43 have been described suggesting that other genetic causes of FTD-MND exist. (Gijselinck et al., 2009). Recently one group reported a single base substitution in the TARDBP gene in an elderly woman diagnosed with the apathetic subtype of fvFTD without MND (Borroni et al., 2009). Cases with GRN mutations characteristically present with ubiquitin-positive TDP-43 positive inclusions (Chen-Plotkin et al., 2010).
Recent studies also have identified mutations in the valosin gene on chromosome 9 and the CHMP2B gene on chromosome 3 associated with FTD. The FTD cases related to valosin mutations are very rare and may include Paget disease and inclusion body myositis as part of the clinical manifestations. FTD cases with CHMP2B mutations are similarly rare and can be associated with MND (van Swieten and Heutink, 2008).
Neuroimaging
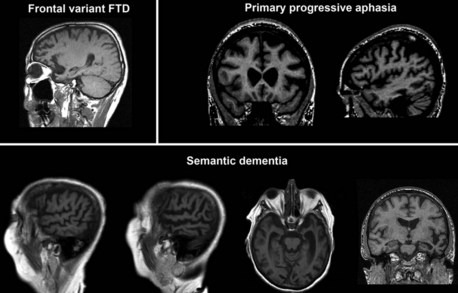
MRI (or CT) scans in fvFTD frequently show frontal and temporal cortical atrophy (Fig. 66.10), which are often times asymmetrical. Nonfluent PPA usually presents with left perisylvian atrophy, particularly in the inferior frontal cortex, while fluent PPA tends to be associated with more inferior, middle, and polar temporal lobe involvement (Mesulam, 2003). SD presents with bilateral anterior temporal lobe involvement. Greater right temporal lobe disease is associated with prosopagnosia, while progressive limb apraxia is associated with frontal premotor and parietal pathology.
Pathology
Histopathology
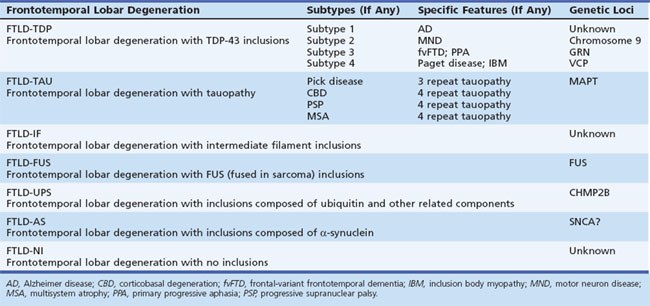
There is a growing trend to classify the FTD spectrum disorders based on their histopathological appearance. While it is true the underlying histology does not appear to relate closely to either the clinical syndrome (fvFTD versus PPA, for instance) or the presence of a positive family history, it is the histopathological defect that is most closely related to the underlying pathophysiology. Recognizing the great need for systematic nomenclature for pathological classification, an expert panel recently suggested a protein-based nomenclature as the most simple, consistent, transparent, and capable of accommodating future discoveries (Mackenzie et al., 2009) (Table 66.3).
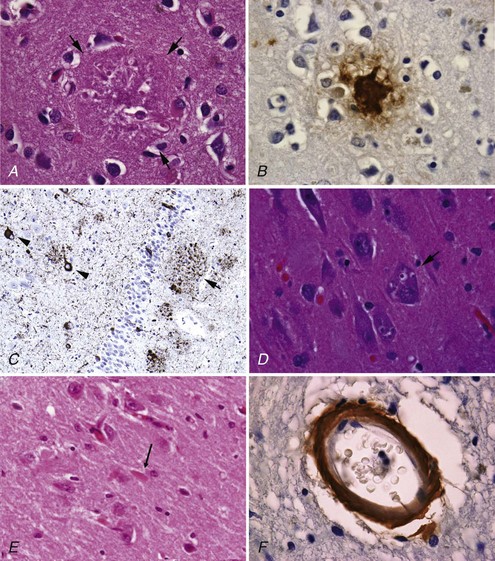
The tau-negative FTLD spectrum consists of several histopathological subtypes. FTLD-TDP is the subtype characterized by inclusions staining for TDP-43 (Fig. 66.11, A-B). The majority of tau-negative ubiquitin-positive cases appear to be associated with TDP-43 aggregation (Chen-Plotkin et al., 2010; Geser et al., 2010). TDP-43 is a nuclear protein of uncertain function. In FTLD cases, TDP-43 is hyperphosphorylated, undergoes ubiquitination, and is cleaved to generate C-terminal fragments. TDP-43 aggregates in the cytoplasm and disappears from the nucleus. Of importance, the inclusions occur in the neurons of the affected regions that correspond well with the clinical symptoms. FTLD-TDP commonly presents as FTD-MND. Similar inclusions are also found in lower motor neurons in patients with ALS. Patients found to have TDP-43-positive inclusions can present anywhere along a clinical spectrum from pure ALS to pure FTD. The fact that ALS and FTD-MND feature TDP-43 inclusions as a primary pathological finding suggests that these two disorders share a similar neurodegenerative pathobiology and belong to the spectrum of TDP-43 proteinopathies (Chen-Plotkin et al., 2010; Geser et al., 2010). Three subtypes with distinct clinicopathological correspondence have been described: (1) cases with neuronal TDP-43 inclusions and glial inclusions but few or absent neurites and predilection for frontal cortex, usually present with FTD-MND; (2) cases with thick dystrophic neurites without a discrete cortical laminar predilection that tend to present with striking temporal atrophy and SD features; and (3) cases with superficial cortical spongiosis and predominantly neuronal pleomorphic intracytoplasmic TDP-43 and thin immunoreactive neurites in the pyramidal layer of the hippocampus inclusions, usually present with the fvFTD subtype but can also manifest as nonfluent PPA with or without MND (Dickson, 2009). Presence of neuronal intracytoplasmic inclusions correlates with short survival (Geser et al., 2010).
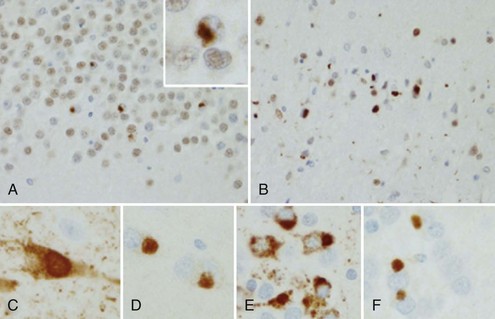
(Adapted with permission from Dickson, D.W., 2009. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol 3, 1–23.)
A few of the FTDs with ubiquitin-positive inclusions have been identified as neuronal intermediate filament inclusion disease (Cairns et al., 2004). In these cases, the ubiquitin-positive inclusions immunostain for neurofilament and α-internexin. Such cases are now proposed to be classified as FTLD-intermediate filament (FTLD-IF), whereas the formerly known dementia lacking distinctive neuropathology (DLDH) cases are now classified as FTLD-no inclusions (FTLD-ni). Cases with basophilic inclusions of unidentified biochemistry are now referred to as basophilic inclusion body disease (BIBD) (Mackenzie et al., 2009).
In addition to characteristic inclusions, FTLD can also show microvacuolar or spongiform changes and gliosis with or without swollen neurons. Classic Pick disease, which was the first FTLD disorder described by Arnold Pick in 1892, is characterized histologically by severe astrocytic gliosis, ballooned cells, and intraneuronal inclusions (see Fig. 66.11, C-F). In other cases the large cortical neurons, primarily in laminae III and V, are lost, and spongiform change and microvacuolation are seen in lamina II. Occasionally the neuronal loss is accompanied by diffuse gliosis but with little or no spongiform change or microvacuolation. Swollen neurons or inclusions that are both tau- and ubiquitin-positive are present in some cases. The limbic system and striatum show more severe damage.
Parkinsonian Dementias
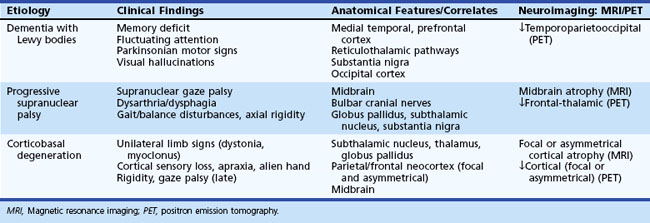
Parkinsonian dementia syndromes encompass a broad range of etiological disorders that share a heterogeneous combination of cognitive and motor features (Table 66.4). In general, the disorders in this group have an insidious onset and gradual progression. A positive family history of a similar clinical syndrome implicates an autosomal dominant or familial variant. The type, timing, and topography of parkinsonian motor signs are important for differential diagnosis, as are associated cognitive, neuropsychiatric, or other specific clinical manifestations. Although most degenerative disorders in this category are more prevalent with advancing age, others, particularly the metabolic disorders, tend to occur earlier in life. The clinical and anatomical features of selected degenerative parkinsonian dementias are illustrated in Table 66.5.
Table 66.4 Classification and Salient Features of Parkinsonian Dementia Syndromes
| Etiology/Syndrome | Distinguishing Features |
|---|---|
| DEGENERATIVE (SPORADIC) | |
| Parkinson disease with dementia (PDD) | Parkinsonism initially, later onset of dementia |
| Dementia with Lewy bodies (DLB) | Recurrent visual hallucinations, fluctuating cognition, variable parkinsonian signs |
| Progressive supranuclear palsy (PSP) | Balance and bulbar dysfunction, vertical gaze palsy |
| Corticobasal degeneration (CBD) | Asymmetric limb signs (apraxia, myoclonus) |
| Multisystem atrophy (MSA) | “Parkinson-plus” syndromes |
| Cerebellar type | Brainstem/cerebellar atrophy, ocular dysmotility |
| Parkinsonian type | Motor parkinsonism, dysautonomia |
| DEGENERATIVE (FAMILIAL) | |
| Huntington disease (HD) | Autosomal dominant; chorea, athetosis, personality changes |
| Neuroacanthocytosis | Autosomal dominant; HD mimic, acanthocytic red blood cells |
| Machado-Joseph disease | Autosomal dominant; ataxia and dysarthria, cerebellar atrophy |
| Progressive subcortical gliosis | Autosomal dominant; white matter gliosis, frontal atrophy |
| Familial frontotemporal dementia (FTDP-17) | Autosomal dominant; chromosome 17–linked, frontal atrophy |
| SECONDARY PARKINSONIAN SYNDROMES | |
| Drug-induced encephalopathy/parkinsonism | Relevant drug exposure (neuroleptic, antiemetic) |
| Vascular parkinsonism | Multiple subcortical infarcts; may mimic PSP |
| Normal-pressure hydrocephalus | Prominent gait disturbance (magnetic), urinary incontinence, subcortical dementia |
| Whipple disease | May mimic PSP; cerebrospinal fluid pleocytosis, gastrointestinal symptoms prominent |
| Dementia pugilistica | Repetitive head trauma ± cavum septum pellucidum on MRI |
| INHERITED METABOLIC DISORDERS | |
| Wilson disease | Autosomal recessive; early onset, impaired copper clearance |
| Neurodegeneration with brain iron accumulation (Hallervorden-Spatz disease) | Familial and sporadic disorders with variable age of onset and subcortical iron deposits |
| Idiopathic basal ganglia calcification | Autosomal dominant and recessive; subcortical calcium deposits |
| Parkinson disease with dementia (PDD) | Parkinsonism initially, later onset of dementia |
Classification
Parkinsonian dementia syndromes encompass a broad range of etiological disorders that share a heterogeneous combination of cognitive and motor features (see Table 66.4). The two main etiological categories are primary degenerative syndromes, representing disorders that typically are either sporadic or inherited, and secondary parkinsonian syndromes, attributable to a variety of cerebral insults including drug-induced cognitive-motor syndromes. An intermediate (and less common) category of disorders that feature extrapyramidal dysfunction and associated cognitive alterations includes systemic metabolic derangements involving the excess accumulation of heavy metals or minerals in basal ganglia structures.
Differentiating Features
In PDD, PSP, and MSA, parkinsonian motor signs typically dominate the initial presentation, whereas neuropsychiatric and cognitive symptoms are often less prominent early on. DLB and CBD have more variable initial presentations with respect to motor, cognitive, and neuropsychiatric features. Cognitive disturbances may be a presenting feature of DLB and CBD, although relatively circumscribed cortical cognitive deficits (i.e., agnosia or aphasia) are more characteristic in the latter. Typical parkinsonian signs are seen in PDD, while atypical ones are characteristic for DLB, PSP, and MSA (Box 66.8). The motor characteristics of DLB range from typical to atypical parkinsonism and may be either symmetrical or asymmetrical. PDD and CBD characteristically feature asymmetrical motor signs, whereas PSP and MSA feature axial symmetrical motor dysfunction.
Box 66.8 Motor Profiles of Parkinsonian Dementia Syndromes
Typical parkinsonian signs (e.g., Parkinson disease with dementia): resting tremor, limb rigidity, bradykinesia
Atypical parkinsonian signs (e.g., progressive supranuclear palsy, corticobasal degeneration): intention tremor, axial rigidity, bulbar or balance disturbances, vertical gaze paresis, myoclonus
Parkinsonian + other motor signs (e.g., multisystem atrophies): pyramidal, cerebellar signs
Synucleinopathies
Dementia with Lewy Bodies
Diagnostic Criteria
Parkinson disease (PD), PDD, and DLB are neurodegenerative disorders from the Lewy body spectrum disorders. DLB achieved consensus recognition as a distinct clinicopathological entity in 1996 (McKeith et al., 1996). Our conceptualization of it continues to evolve (McKeith et al., 1999, 2000b, 2005). From a nosological standpoint, DLB is conceived as being distinct from AD and PD, but substantial clinical and pathological overlap with both AD and PD is recognized, making the relationship among these disorders controversial. In the recently revised diagnostic criteria (McKeith et al., 2005) (Table 66.6), two additional clinical features have been designated as “suggestive”: rapid eye movement (REM) sleep behavior disorder and neuroleptic sensitivity. Either of these two features can count as a core feature if at least one core feature is present.
Table 66.6 Consensus Diagnostic Criteria for Dementia with Lewy Bodies
| Symptom/Sign | Cardinal Manifestations | Frequency |
|---|---|---|
| Dementia | Attentional, frontal-executive, and visuospatial deficits, often worse than in Alzheimer disease; short-term (episodic) memory relatively better than in Alzheimer disease | 100% |
| CORE FEATURES | ||
| Fluctuating cognition | Variable timing of altered level of attention or arousal; distinct from sundowning | 60%-80% |
| Visual hallucinations | Recurrent; typically involve animate subjects; variable degree of insight; reminiscent of anticholinergic delirium | 50%-75% |
| Parkinsonian motor signs | Spontaneous; rigidity and bradykinesia most common; intention tremor more common than resting tremor | 80%-90% |
| SUGGESTIVE FEATURES | ||
| REM sleep behavior disorder | Loss of atonia during REM sleep; individuals appear to act out dreams; may be combative or violent | 25%-50% |
| Neuroleptic sensitivity | Severe cognitive and motor adverse reaction to antipsychotic agents; may increase mortality | 50% |
| Decreased tracer uptake | ||
CT, Computed tomography; EEG, electroencephalogram; MIBG, metaiodobenzylguanidine; MRI, magnetic resonance imaging; PET, positron emission tomography; REM, rapid eye movement; SPECT, single-photon emission computed tomography.
Modified from McKeith, I.G., Dickson, D.W., Lowe, J., et al., 2005. Dementia with Lewy bodies: diagnosis and management: third report of the DLB Consortium. Neurology 65, 1863-1872.
Epidemiology
DLB is found at autopsy in approximately 20% of late-onset dementias (alone or in combination with AD). In one European study, probable DLB was diagnosed in 15.8% and possible DLB in 4.1% of all dementia cases (Aarsland et al., 2008b). The estimated population-based prevalence of DLB is between 10 and 200/10,000 (Brayne et al., 2006). The mean age of onset is 75 years, with a range of 50 to 80 years. There is a slight male predominance (Geser et al., 2005). Older age at onset, fluctuating cognition and hallucinations at disease onset, and associated AD pathology herald shorter survival (Jellinger et al., 2007).
Clinical Presentation
Although the dementia syndrome of DLB is similar enough to AD that many patients will warrant both diagnoses, marked variability in the clinical presentation is characteristic. The initial manifestations of DLB may be cognitive deficits or any of the core features of DLB alone or in conjunction with a dementia syndrome (see Table 66.6).
Fluctuating cognition is observed in up to 90% (Geser et al., 2005) and should be routinely solicited from the informant. Fluctuations in attention, arousal, or cognitive performance can lead to high variability in cognitive performance on formal testing.
DLB patients frequently display REM sleep behavior disorder (RBD) with loss of physiological skeletal muscle tone during REM cycles and dream enactment. RBD can be an early prodrome of DLB symptom predating dementia or extrapyramidal feature onset by decades. The positive predictive value for DLB in the demented patient with parkinsonism and RBD is 92% (Boeve et al., 2001). RBD is also common in other α-synucleinopathies including PD, PDD, and MSA. In addition to RBD, DLB patients also suffer from decreased sleep efficiency, increased upper airway resistance, obstructive sleep apnea/hypopnea, and periodic limb movements of sleep (Boeve et al., 2003).
Dysautonomia is usually seen later in the disease course, although some cases with dysautonomia at initial presentation have been reported, as well as frequent falls, dysphagia, and urinary incontinence. A rapidly progressive course with aphasia, dyspraxia, and severe visuospatial disturbances can be seen. Some DLB patients progress to end stages and death within 1 to 2 years (Geser et al., 2005).
Laboratory Studies
There are no specific blood or CSF tests for DLB to date. Recently, increased levels of CSF total tau have been associated with shorter survival (Bostrom et al., 2009). Increase in the oxidized α-helical form of Aβ1-40 (Aβ1-40*) yielded a diagnostic sensitivity and specificity of 81% and 71% in differentiating DLB and PDD from AD (Bibl et al., 2006). Research efforts toward the development of reliable fluid biomarkers for DLB are underway.
Genetics
The genetics of DLB has been poorly understood. Familial DLB in a Belgian kindred has been recently mapped to chromosome 2q35-q36 (Meeus et al., 2010). DLB has been also associated with duplications in the α-synuclein (SNCA) gene on chromosome 4 (Kasuga et al., 2010) and the leucine-rich repeat kinase 2 (LRRK2) gene on chromosome 12 (Qing et al., 2009). It has been suggested that oxidative stress up-regulates both SNCA and LRRK2 expression in DLB (Qing et al., 2009). Mutations in the glucocerebrosidase (GBA) gene on chromosome 1 have been associated with both DLB and PD (Mata et al., 2008).
Neuroimaging
Structural neuroimaging (MRI or CT) can reveal relatively nonspecific generalized brain atrophy with typically modest hippocampal involvement in DLB (Fig. 66.12). Several research groups have demonstrated diffuse temporal, parietal, and frontal cortical atrophy (Ballmaier et al., 2004; Beyer et al., 2007b; Burton et al., 2002) as well as atrophic changes of the dorsal midbrain, hypothalamus, and substantia innominata (Whitwell et al., 2007).
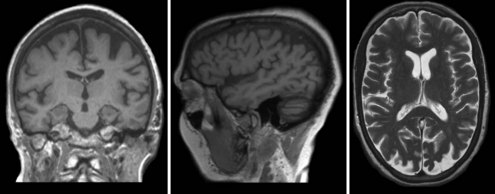
Functional brain imaging offers the prospect of aiding the clinical diagnosis of DLB. Occipital lobe hypoperfusion on SPECT or PET (Fig. 66.13) differentiates DLB from AD with a sensitivity of 65% and specificity of 87% (Lobotesis et al., 2001). A PET study of autopsy-confirmed AD and DLB subjects also demonstrated that the presence of occipital hypometabolism differentiated DLB from AD with a sensitivity of 90% and specificity of 87% (Minoshima et al., 2001). Unfortunately occipital hypometabolism is not always present in DLB patients hence its diagnostic contribution is somewhat limited.
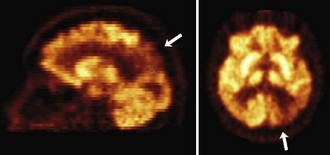
The most established functional neuroimaging biomarker in DLB is dopamine transporter imaging. [123I]-FP-CIT SPECT was reported to show 78% sensitivity and 90% specificity for differentiating DLB from non-DLB dementia (McKeith et al., 2007). Evidence of dopaminergic abnormalities in the basal ganglia has been accepted as a suggestive feature for DLB in the revised McKeith criteria (McKeith et al., 2005).
Myocardial iodine-131-meta-iodobenzylguanidine (MIBG) scintigraphy is another imaging modality that may help with DLB diagnosis. Recently Estorch et al. reported sensitivity of 94% and a specificity of 96% of MIBG scanning in predicting DLB diagnosis in MCI subjects (Estorch et al., 2008).
Pathology
Whereas PD is characterized by Lewy bodies primarily affecting the midbrain substantia nigra and other brainstem nuclei (e.g., locus ceruleus, raphe nuclei), the additional presence of Lewy bodies and Lewy neurites in limbic (e.g., hippocampus, amygdala), paralimbic (e.g., anterior cingulate), and neocortical regions is the neuropathological hallmark of DLB. Cortical Lewy bodies have a less distinctive microscopic appearance than brainstem Lewy bodies. α-Synuclein protein is a primary constituent of both. Lewy neurites are dystrophic curvilinear or dot-like processes with highest density in limbic cortex, amygdala, and CA2/3 sectors of the hippocampus (Dickson et al., 2009) (Fig. 66.14).
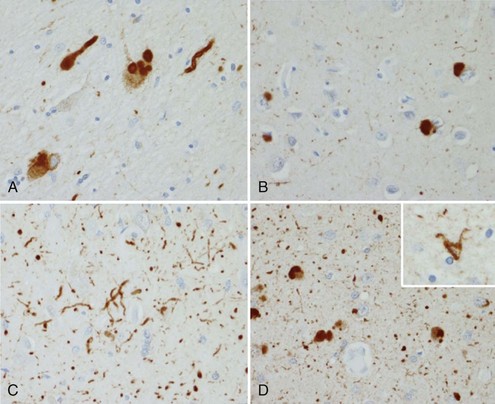
Treatment
Acetylcholinesterase Inhibitor Drugs
Preliminary clinical observations led to the suggestion that patients with DLB may be generally more responsive to AChEI therapy than patients with AD. At autopsy patients with DLB have a greater degree of neocortical cholinergic deficit fewer NFTs, and perhaps lesser neuronal loss relative to patients with AD. Given the proportionately greater degree of functional neurochemical to structural pathology in DLB, one would expect generally more favorable therapeutic substrate for cholinergic therapy. Case reports and open-label studies in DLB suggested that the core symptoms of hallucinations and mental status fluctuations might be responsive to AChEI treatment. The only controlled treatment study of any kind in DLB published to date, a multicenter study of rivastigmine (Exelon), showed no overall worsening of motor function in 59 treated subjects (McKeith et al., 2000a). During a 20-week treatment period, subjects receiving rivastigmine (6 to 12 mg/day) showed statistically significant differences relative to placebo in the total NPI score and a four-item derivative Lewy body cluster composed of delusions, hallucinations, apathy, and depression subscores. Subjects receiving rivastigmine also performed better on timed attention-demanding cognitive tasks.
Antipsychotic Agents
Quetiapine (Seroquel) is a relatively new atypical antipsychotic agent with potential benefit for treating psychosis in parkinsonian syndromes. A 12-week open-label trial of quetiapine in 151 persons with PD and other late-life psychotic disorders showed significant improvement in overall neuropsychiatric symptom burden and global disease severity rating at a median dose of 100 mg/day (McManus et al., 1999). Somnolence, dizziness, and postural hypotension were the most common side effects reported, with extrapyramidal side effects occurring in only 6% of subjects. The dose range of quetiapine varies widely, from 12.5 to 400 mg/day, based on individual differences in sensitivity and tolerability, with excess sedation being the most common dose-limiting side effect.
The increased risk of mortality in dementia associated with the use of three of the novel antipsychotics—risperidone, olanzapine, and quetiapine—to treat agitation in AD (Schneider et al., 2006) resulted in a “black box” warning for several of these agents.
Parkinson Disease Dementia
Idiopathic PD, the most common neurodegenerative movement disorder, results from a defect in the metabolism of α-synuclein. In addition to the classic motor features of rigidity, bradykinesia, tremor, and postural abnormalities, individuals with PD frequently display a host of non-motor features. Progressive cognitive impairment and behavioral problems are commonly seen in PD patients even early in the disease course (Janvin et al., 2006a, 2006b; Muslimovic et al., 2005).
Epidemiology
PD affects 1 in 100 people over the age of 60 (Galvin, 2006; Poewe and Wenning, 1998). PD incidence and prevalence increase with age and are greater in males. According to a recent meta-analysis, the worldwide incidence and prevalence of PD are 1.6/1000 and 9.5/1000, respectively, among the elderly age 65 years and older (Hirtz et al., 2007).
Up to 90% of PD patients develop overt dementia (PDD) (Buter et al., 2008). Cognitive impairment in PD is perhaps one of the most understudied non-motor syndromes. The onset of cognitive decline in relation to initial PD presentation is highly variable (Aarsland et al., 2007), suggesting individually predetermined neuronal vulnerability, with the additional contribution of environmental factors and comorbidities.
Clinical Presentation
PD manifests with four cardinal symptoms: tremor, rigidity, bradykinesia, and postural instability. Progressive cognitive impairment and behavioral problems are commonly seen in PD patients even early in the disease course (Foltynie et al., 2004). Individuals with PD are at two- to sixfold increased risk for developing PDD relative to elderly normal controls (Breteler et al., 1995; Rajput et al., 1987). Both overt dementia (Aarsland et al., 2003) and a predementia state (i.e., PDMCI) (Janvin et al., 2003) have been described in PD.
PDD is characterized by pronounced slowing of cognitive and motor skills, episodic memory deficits, and executive, attentional, and visuospatial dysfunction (Aarsland et al., 2008a; Bronnick et al., 2007; Camicioli and Fisher, 2004; Galvin, 2006). Behavioral disturbances are common in PDD. Visual hallucinations and RBD are frequently seen and have been shown to correlate with executive dysfunction and attention deficits (Barnes and Boubert, 2008).
PDMCI is associated with impaired visuospatial and executive function, but recently, deficits in episodic memory have also been reported (Aarsland et al., 2009; Dubois and Pillon, 1997; Foltynie et al., 2004; Janvin et al., 2003; Muslimovic et al., 2005). PD patients who show cognitive decline consistent with PDMCI are five times more likely to receive a diagnosis of dementia over the next 4 years (Janvin et al., 2006a). There is some evidence that tests of attention and executive function may predict conversion from PDMCI to PDD (Janvin et al., 2006a; Williams-Gray et al., 2007), but these data need to be expanded upon and replicated. Other risk factors for future development of dementia include increasing age, greater motor disability, significant bradykinesia, presence of postural instability and significant gait disorder, as well as early hallucinations (Aarsland et al., 2001, 2003; Alves et al., 2006; Mayeux et al., 1992).
Genetics
In the last decade, several causative genetic mutations for both early- and late-onset PD have been identified. Mutations in the synuclein (SNCA or PARK1) gene on chromosome 4, the parkin (PARK2) gene on chromosome 12, the DJ-1 (PARK7) gene on chromosome1, or the leucine-rich repeat kinase 2 (LRRK2 or PARK8) gene on chromosome 12 can cause both early- and late-onset PD (Klein et al., 2009). Recently the glucosidase (GBA) gene on chromosome 1, coding the enzyme deficient in Gaucher disease, has gained a lot of attention. PD patients are in fact five times more likely to carry mutations in GBA relative to the normal population. GBA mutations are now received as the most common genetic cause of PD (Velayati et al., 2010).
Neuroimaging
To date, structural imaging has not been regarded as diagnostically useful in PD and PDD. The role of structural MRI in PD is confined to ruling out other etiologies such as basal ganglia stroke lesions, diffuse white-matter ischemic changes, midbrain atrophy, or strikingly unilateral frontoparietal atrophy that could suggest PSP or CBD, respectively. However, several recent studies have documented that structural differences can be seen in PD and especially in PDD. There seems to be widespread cortical atrophy of the limbic, temporal, parietal, frontal, and occipital regions in PDD relative to normal controls and nondemented PD subjects (Beyer et al., 2007a; Burton et al., 2004; Hwang et al., 2010). PDMCI subjects were also reported to show frontal and temporal atrophy (Beyer et al., 2007a; Meyer et al., 2007). Other structural changes associated with cognitive decline in PD are ventricular enlargement and caudate atrophy (Apostolova et al., 2010a; Meyer et al., 2007).
An FDG-PET pattern similar to the one seen in AD, with hypometabolism affecting the posterior cingulate, parietal, and temporal cortices, has been reported in PDD (Brooks, 2010). Striatal [123I]-FP-CIT SPECT binding is reduced to a similar degree in PD, PDD, and DLB versus AD. (O’Brien et al., 2004).
Multiple pathological substrates may contribute to the cognitive and neuropsychiatric features of PDD. These include neurotransmitter deficiencies in dopamine (substantia nigra and ventral tegmental area), norepinephrine (locus ceruleus), and serotonin (raphe nuclei), as well as Lewy bodies in cortical and subcortical regions. Bohnen et al. conducted PET imaging with a radiolabeled acetylcoline analogue and found cholinegric deficits of 70% or higher in the cerebral cortex in PDD. Nondemented patients with PD also demonstrated marked cholinergic deficits relative to both control subjects and patients with AD (Bohnen et al., 2003). Whether marked cholinergic deficit is a prognostic sign of impending dementia in PD remains to be determined.
Pathology
The pathology underlying cognitive deficits in PD is not yet completely understood but seems to include both neurotransmitter deficits (dopamine, serotonin, acetylcholine, noradrenalin), structural changes, and cortical Lewy body (LB) deposits (Braak et al., 2005; Emre, 2003). The pathological changes in PDMCI and PDD consist of progressive degeneration of the dopaminergic and other neurotransmitter systems and dysfunction of multiple neuronal circuits (Perry et al., 1991). On gross macroscopic inspection, the brain frequently shows mild to moderate atrophy and ventriculomegaly. Brainstem dissection reveals the pathognomonic macroscopic signs of PD: pallor of the substantia nigra and locus coeruleus. Histopathologically, the neurodegenerative changes of PD consist of abundant LB, Lewy neurites, and neuronal loss in the midbrain, substantia nigra, and locus coeruleus, with associated depletion of pigmented neurons (Braak et al., 2005; Emre, 2003). In addition, widespread argyrophilic α-synuclein-positive tau-negative glial inclusions can be seen diffusely in the cerebral cortex, basal ganglia, brainstem, cerebellum, and even in the spinal cord (Jellinger and Mizuno, 2003).
Postmortem investigations in PDD have demonstrated several neuropathological findings: cortical LB pathology, degenerative changes in critical cortico-subcortical circuits, and coexistent AD pathology (Jellinger and Mizuno, 2003). Individuals with PD who develop dementia late in the disease course tend to show diffuse LB but only modest AD pathology (Apaydin et al., 2002). Brains of PDD subjects show significantly higher densities of LB and Lewy neurites in the entorhinal cortex as well as the CA2-3 hippocampal subregions and the amygdala relative to brains of nondemented PD subjects (Bertrand et al., 2004).
The extent of contribution of AD pathology to PDD has proven difficult to understand. Concomitant AD pathology (i.e., neuritic plaques, NFTs) is seen in over 50% of PDD subjects on postmortem examination (Jellinger, 2001) and contributes to cognitive decline (Harding and Halliday, 2001). Nondemented PD (PDND) typically exhibits AD changes corresponding to Braak staging of IV or less (Braak and Braak, 1991), whereas PDD subjects usually harbor advanced Braak stage cortical pathology (V-VI) (Delacourte et al., 1999). AD pathology in PDD has been associated with much shorter survival (Jellinger et al., 2002).
Cortico-subcortical circuit degeneration in PDD manifests in reduced prefrontal choline acetyltransferase activity, dopaminergic system hypofunction, and D1 receptor decline in the caudate nuclei. These changes are seen even in the absence of AD pathology (Bruck et al., 2001; Mattila et al., 2001). Additionally, the involvement of the noradrenergic and serotoninergic systems has been implicated for the development of cognitive and behavioral changes in PD (Gerlach et al., 2001; Jellinger, 1999). The relationship between cholinergic depletion, PDD, and visual hallucinations is particularly intriguing. Recent research has shown that there is more severe cholinergic depletion in PDD compared to AD, which appears to be related to cognitive impairment (Bohnen et al., 2003, 2006). In addition, PD patients with hallucinations were found to respond better to cholinesterase inhibitors than those without (Burn et al., 2006).
Treatment
Treatment with the AChEI, rivastigmine, should be attempted in PDD in light of a recent multicenter placebo-controlled study of rivastigmine in PDD that showed a positive effect of treatment in cognitive, neuropsychiatric (especially hallucinations), and functional domains without significant exacerbation of parkinsonian motor features (Emre et al., 2004). Findings from this study led to formal approval of rivastigmine for treatment of PDD in 2006 by the FDA.
Multisystem Atrophy
Multisystem atrophy refers to a degenerative parkinsonian disorder with variable associated features including autonomic, cerebellar, and pyramidal tract dysfunction. The prevalence of MSA has been estimated to be 4.4 cases per 100,000, making it slightly less common than PSP (Schrag et al., 1999). Three clinical variants of MSA were initially recognized: (1) striatonigral degeneration, (2) Shy-Drager syndrome, and (3) olivopontocerebellar atrophy (OPCA). All share a common pathological substrate of α-synuclein–containing glial cytoplasmic inclusions, which are distributed variably in the cortex, subcortical regions, cerebellum, spinal cord, and dorsal root ganglia. Newer consensus diagnostic criteria for MSA (Gilman et al., 1998) have suggested two main types based on the predominant clinical feature: (1) MSA-parkinsonian (MSA-P) and (2) MSA-cerebellar (MSA-C). Accordingly, previously described cases of striatonigral degeneration and Shy-Drager syndrome are now classified as MSA-P, and sporadic OPCA conforms to MSA-C. Of importance, however, most cases of MSA have some degree of parkinsonism and autonomic dysfunction, and about half will have either cerebellar or pyramidal tract dysfunction. RBD is associated with MSA, just as with PD and DLB, suggesting that this disorder may be a common manifestation of synucleinopathies.
MSA can show several characteristic features on MRI. These include atrophy and hyperintensity of the putamen, slit-like hyperintensity of the posterolateral margin of the putamen, brainstem atrophy, hyperintensity of the middle cerebellar peduncles, and cruciform hyperintensity of the pons (the so-called hot cross bun sign) (Fig. 66.15). As expected MSA-C also shows significant cerebellar atrophy.
Pathologically MSA is characterized by extensive oligodendroglial α-synuclein pathology known as glial cytoplasmic inclusions. Dystrophic neurites can be found in the putamen, inferior olive, and brainstem nuclei (Fig. 66.16).
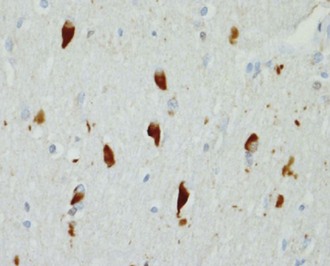
Tauopathies
Corticobasal Degeneration
MRI frequently helps with the differential diagnosis in CBD. CBD patients usually show asymmetrical frontoparietal atrophy affecting the motor and sensory cortices, which is more severe on the side contralateral to the affected limb. High T1 signal intensity in the subthalamic nucleus, midbrain atrophy, and T2 striatal hypointensity have also been reported (Sitburana and Ondo, 2009; Tokumaru et al., 2009). As expected, functional imaging with PET and SPECT show strikingly asymmetrical hypoactivity contralateral to the affected side (Fig. 66.17).
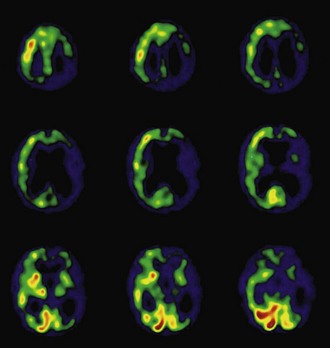
The pathological hallmarks of CBD are astrocytic gliosis, most prominent in the superficial cortical layers and at the grey/white junction, and swollen achromatic neurons (neuronal achromasia) that are distributed asymmetrically in discrete (frontal or parietal) cortical areas and in subcortical regions (Fig. 66.18). Several types of intraneuronal tau-immunoreactive inclusions are seen in the cortex, thalamus, basal ganglia, substantia nigra, and locus coeruleus. These are commonly seen as densely packed, small, globose NFT-like inclusions and dispersed filamentous inclusions. Tau-immunoreactive cell processes, oligodendroglial coiled bodies, and astrocytic plaque-like lesions are characteristic for CBD.
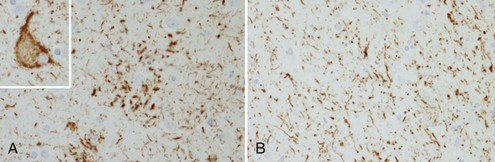
(Adapted with permission from Dickson, D.W., 2009. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol 3, 1–23.)
Parkinsonian motor signs in CBD show a modest response to l-dopa, with approximately 25% of patients who received this treatment showing some benefit (Kompoliti et al., 1998). Clonazepam may be useful for treating myoclonus and dystonia in some patients. Limited clinical experience with AChEI therapy in CBD generally has been unrewarding.
Progressive Supranuclear Palsy
PSP (Steele-Richardson-Olszewski syndrome) is distinguished clinically from PD by the absence of a rest tremor, greater axial (neck and trunk) than limb rigidity, the presence of severe dysarthria and dysphagia as a result of the supranuclear palsy, a greater degree of gait and balance impairment, and limitation in voluntary downward gaze that can be overcome with the doll’s eyes head maneuver (“supranuclear” gaze palsy). Prevalence estimates of PSP based on community studies are as high as 6.4 per 100,000 (Schrag et al., 1999). Clinicopathological studies often find this entity misdiagnosed as PD. Standardized clinical criteria for diagnosing PSP, derived from symptoms in histopathologically proven cases, have been proposed (Box 66.9). High sensitivity and specificity for these criteria have been demonstrated in an independent clinical sample with autopsy-confirmed diagnosis (Lopez et al., 1999).
Box 66.9
National Institute of Neurological Disorders and Stroke Clinical Criteria for Progressive Supranuclear Palsy
Adapted from Litvan, I., Agid, Y., Calne, D., et al., 1996. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP International Workshop. Neurology 44, 1-9.
Gross atrophy of the midbrain tegmentum may be evident on MRI (Boxer et al., 2006; Oba et al., 2005). Additional features suggestive of PSP include enlargement of the third ventricle and signal increase in the midbrain and inferior olives. Mild to moderate frontal and temporal cortical atrophy can be seen in PSP, never quite reaching the severity of cortical atrophy seen in CBD (Fig. 66.19). Recently, using susceptibility-weighted MRI, one research group reported that PSP could be distinguished from MSA and idiopathic PD based on hypointensity of the red nucleus and putamen, respectively (Gupta et al., 2010).
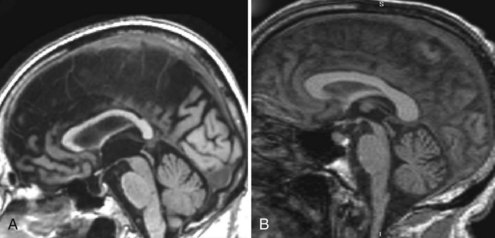
The histopathological hallmark of PSP are globose NFTs composed of aggregated 4R tau protein that accumulate in prefrontal cortex and a number of subcortical regions, principally the globus pallidus, substantia nigra, and subthalamic nucleus (Dickson, 2009) (Fig. 66.20). Tau-reactive tangles also occur in glial cells. Reactive gliosis and neuronal loss are variable features. The localization of tau pathology in PSP corresponds well to the clinical phenotype. Severe brainstem involvement is seen in the akinetic type, while dementia and CBD-like presentation and/or speech apraxia are commonly associated with cortical tau pathology. Characteristic features are the tufted astrocytes usually found in motor cortex and striatum, the abundance of thread-like processes, and oligodendroglial coiled bodies. This is distinct from CBD, where threads but not oligodendroglial coiled bodies are seen (Dickson, 2009).
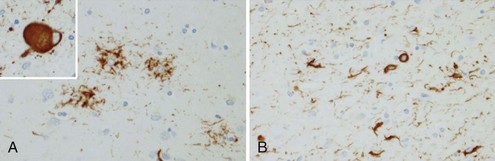
(Adapted with permission from Dickson, D.W., 2009. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol 3, 1–23.)
Symptomatic pharmacological therapy of PSP has had very limited success. Although occasional modest and transient improvement in motor function has been observed with l-dopa and other dopaminergic agonists, adverse effects (i.e., orthostatic hypotension, psychosis) often outweigh the benefits. Similar observations with the AChEI, donepezil, have been reported. In a placebo-controlled double-blind study involving 21 patients with PSP, modest improvement in memory test scores was offset by deterioration in functional mobility (Litvan et al., 2001). Depression, anxiety, and pseudobulbar affect may respond to SSRI agents, which generally are well tolerated.
Metabolic Parkinsonian Disorders
Wilson Disease
Kayser-Fleischer rings composed of copper-containing granules in the periphery of the cornea almost invariably are present in cases of WD with neurological signs and are visualized reliably by slit-lamp examination. MRI abnormalities typically are evident on T2-weighted images—bilateral symmetrical signal hyperintensities in the basal ganglia, midbrain, pons, and thalamus (Fig. 66.21).
Neurodegeneration with Brain Iron Accumulation
Neurodegeneration with brain iron accumulation (NBIA) was initially described in 1922 as an autosomal recessive childhood-onset parkinsonian dementia associated with rusty brown discoloration of the medial globus pallidus and substantia nigra pars reticulata at autopsy. A variety of atypical forms have been included under the rubric of NBIA and referred to as NBIA syndrome. These tend to present as later-onset dementia, dystonia, and parkinsonism and have associated basal ganglia iron deposition and neuroaxonal dystrophy. Virtually all patients with classic NBIA and approximately 33% to 50% of those with later-onset neuroaxonal dystrophy, or NBIA syndrome, have a mutation in the gene encoding pantothenate kinase 2 (PANK2) located on chromosome 20 (Hayflick, 2003). Compared with affected persons with the late-onset syndrome who test negative for PANK2 mutation, patients with late-onset syndrome and a mutation at the PANK2 locus can exhibit more severe speech and psychiatric disturbances. Persons with a PANK2 mutation, whether associated with early-onset classic NBIA or a later-onset syndrome of NBIA, can exhibit the “eye-of-the-tiger” sign (central hyperintensity surrounded by an area of hypointensity) in the globus pallidus on T2-weighted MRI. There is currently no available therapy for NBIA; management is largely symptomatic and supportive.