[level-membership-for-neurology-category]
Chapter 57 Deficiency Diseases of the Nervous System
Malnutrition causes a wide spectrum of neurological disorders (Table 57.1). Despite socioeconomic advances, nutritional deficiency diseases such as kwashiorkor and marasmus are still endemic in many underdeveloped countries. The problem in Western countries is usually the result of dietary insufficiency from chronic alcoholism or malabsorption due to gastrointestinal (GI) diseases. The B vitamins (thiamine, pyridoxine, nicotinic acid, and vitamin B12), vitamin E, and perhaps folic acid are important for normal function of the nervous system. Emerging evidence also supports important roles of vitamin D and copper.
Table 57.1 Neurological Manifestations in Deficiency Diseases
| Neurological Manifestations | Associated Nutritional Deficiencies |
|---|---|
| Dementia, encephalopathy | Vitamin B12, nicotinic acid, thiamine, folate |
| Seizures | Pyridoxine |
| Myelopathy | Vitamin B12, vitamin E, folate, copper |
| Myopathy | Vitamin D, vitamin E |
| Peripheral neuropathy | Thiamine, vitamin B12, and many others |
| Optic neuropathy | Thiamine, vitamin B12, and many others |
Cobalamin (Vitamin B12)
Causes of Deficiency
The classic disease, pernicious anemia, is caused by defective intrinsic factor production by parietal cells, leading to malabsorption. These patients may have demonstrable circulating antibodies to parietal cells or lymphocytic infiltrations of the gastric mucosa, suggesting an underlying autoimmune disorder. Another common cause of malabsorption is food-cobalamin malabsorption (Dali-Youcef and Andres, 2009). Under some clinical settings, the normal digestive process fails to release cobalamin from food or intestinal transport protein. Cobalamin remains bound and cannot be absorbed even in the presence of available intrinsic factors. Predisposing factors include atrophic gastritis and hypochlorhydria, and malabsorption may be seen with Helicobacter pylori infection, gastrectomy or other gastric surgeries, intestinal bacterial overgrowth, and prolonged use of H2 antagonists, proton pump inhibitors, or biguanides (e.g., metformin). Patients with human immunodeficiency virus (HIV) are often observed to have a low serum cobalamin level, usually with normal homocysteine and methylmalonic acid. The significance of this association is unknown.
People who abuse nitrous oxide may develop a clinical syndrome of myeloneuropathy indistinguishable from that of cobalamin deficiency. The mechanism appears to be an interference with the cobalamin-dependent conversion of homocysteine to methionine. The other pathway, conversion of methylmalonyl co-A to succinyl co-A, is unaffected by nitrous oxide. Prolonged exposure to nitrous oxide is necessary to produce neurological symptoms in normal individuals. By contrast, patients who are already deficient in cobalamin may experience neurological deficits after only brief exposures during general anesthesia. Symptoms appear subacutely after surgery and resolve quickly with cobalamin treatment (Singer et al., 2008).
Laboratory Studies
Serum assays of vitamin B12 and cobalamin-dependent metabolites provide direct measures of cobalamin homeostasis, although there are important limitations (Solomon, 2005). Blood cobalamins are bound to two transport proteins, transcobalamin and haptocorrin. The cobalamin bound to transcobalamin, known as holotranscobalamin, is the fraction that is available to tissues, although it accounts for only 10% to 30% of the serum level measured by standard laboratory methods. Serum levels are influenced by conditions that affect the concentrations of these transport proteins. Myeloproliferative and hepatic disorders may raise the concentration of haptocorrin and cause a falsely normal serum level. A misleadingly high serum level also may result from the presence of an abnormal cobalamin-binding protein. In contrast, pregnancy and contraceptives may give falsely low measurements in the absence of deficiency. Folate deficiency also causes a falsely low cobalamin serum level that corrects after folate replacement. These confounding factors diminish the sensitivity and specificity of the commonly used assay of total serum cobalamin in the diagnosis of deficiency state. Although measurement of holotranscobalamin is better in theory, available data suggest that its diagnostic accuracy is approximately equivalent to that of total serum cobalamin (Miller et al., 2006).
Homocysteine and methylmalonic acid are precursors of cobalamin-dependent biochemical reactions. These metabolites accumulate during deficiency state. Measuring these metabolites is useful in settings of nitrous oxide abuse and in inherited metabolic disorders in which cobalamin-dependent pathways are impaired despite normal serum level. Homocysteine and methylmalonic acid assays are also useful when the cobalamin concentration is in the lower range of normal, between 200 and 350 pg/mL. Homocysteine level should be measured either at fasting or after an oral methionine load. The blood sample should be refrigerated immediately after collection because the level increases if whole blood is left at room temperature for several hours. Elevated levels of homocysteine and methylmalonic acid are not specific for cobalamin deficiency, as there are many other causes of increase in these metabolites (Box 57.1). In cobalamin-deficient patients, these levels typically normalize within 2 weeks of treatment.
Box 57.1 Causes of Elevated Serum Levels of Homocysteine and Methylmalonic Acid
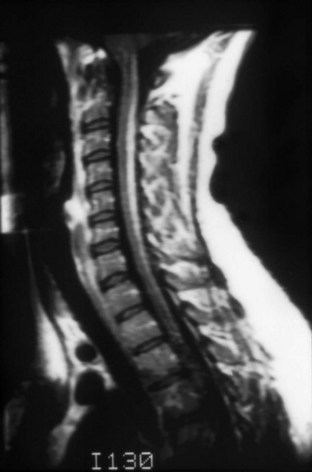
Because most patients present with clinical features suggesting a myelopathy or encephalopathy, imaging studies are necessary to exclude structural causes. Results of magnetic resonance imaging (MRI) may be normal, or T2-signal abnormalities may be seen in the lateral or posterior columns in patients with subacute combined degeneration (Kumar and Singh, 2009) (Fig. 57.1). Both gadolinium enhancement and spinal cord swelling have been described. Patients with encephalopathy or dementia often have multiple foci of T2 signal abnormalities in the deep white matter that may become confluent with disease progression. Radiographic improvement is seen within a few months after initiation of treatment. Nonspecific abnormalities of electroencephalography, as well as visual and somatosensory evoked responses, are present in most patients with neurological abnormalities. Nerve-conduction studies show small or absent rural nerve sensory potentials in approximately half of patients, providing evidence for an axonal polyneuropathy.
Pathology
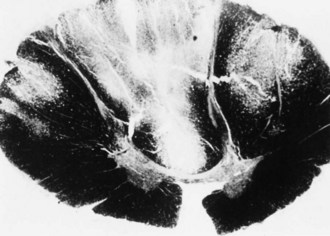
The term subacute combined degeneration of the spinal cord describes the pathological process seen in this disorder. Microscopically, spongiform changes and foci of myelin and axon destruction are seen in the white matter of the spinal cord. The most severely affected regions are the posterior columns at the cervical and upper thoracic levels (Fig. 57.2). Pathological changes also are seen commonly in the lateral columns, whereas the anterior columns are involved in only a small number of the advanced cases. The pathological findings of the peripheral nervous system are those of axonal degeneration, but in some cases there is evidence of demyelination. Involvement of the optic nerve and cerebral white matter also occurs.
Folate Deficiency and Homocysteine
Vitamin E
Like other fat-soluble compounds, vitamin E depends on the presence of pancreatic esterases and bile salts for its solubilization and absorption in the intestinal lumen. Neurological symptoms of deficiency occur most commonly in patients with fat malabsorption (Box 57.2). A reduced bile salt pool may be caused either by reduced hepatic excretion, as in congenital cholestasis, or by interruption of the enterohepatic reabsorption of bile, as in patients with extensive small-bowel resection. Pancreatic insufficiency contributes to malabsorption. Another setting is cystic fibrosis.
Box 57.2 Causes of Vitamin E Deficiency
A rare familial form of fat malabsorption is abetalipoproteinemia (Bassen-Kornzweig syndrome), a disorder in which impaired chylomicron and lipoprotein synthesis is partly responsible for the impaired fat absorption. In addition to a neurological syndrome similar to that seen in other vitamin E-deficient states, spiky red blood cells (acanthocytes) and retinal pigment changes are characteristic. Another hereditary cause of vitamin E deficiency may be a genetic defect in the assembly or secretion of chylomicrons, leading to a chylomicron retention disease that is demonstrable in the intestinal mucosa (Aguglia et al., 2000). A syndrome of ataxia with isolated vitamin E deficiency (AVED) occurs in patients without GI disease or generalized fat malabsorption. Mutations in the α-tocopherol transfer protein gene (TTPA) on chromosome 8q13 are responsible (Mariotti et al., 2004). This condition is inherited in an autosomal recessive manner. The defect appears to be impaired incorporation of the vitamin into hepatic lipoproteins that are necessary for delivery to tissues.
Laboratory Studies
Other laboratory abnormalities, despite their nonspecific nature, help clarify the diagnosis. Stool fat is increased in many patients, and serum carotene concentration is often abnormally low, both reflecting a generalized state of fat malabsorption. CSF should be normal. Nerve conduction studies usually reveal a sensory polyneuropathy, although motor conduction abnormality and features of a demyelinating neuropathy have been reported rarely (Puri et al., 2005). Somatosensory and visual evoked responses are frequently abnormal, and there may be high signal lesions in the posterior columns on T2-weighted MRI.
Pellagra (Nicotinic Acid Deficiency)
The neurological syndrome of pellagra is not well defined. Reported cases, especially of patients with alcoholic pellagra, frequently are confounded by other coexisting central nervous system disorders. The primary early symptoms are neuropsychiatric (e.g., irritability, apathy, depressed mood, inattentiveness, memory loss) and may progress to stupor or coma. In addition to the confusional state, spasticity, Babinski sign, gegenhalten, and startle myoclonus may be prominent on neurological examination. Nonendemic pellagra occurs rarely in patients with alcoholism or malabsorption secondary to GI disease. The diagnosis of nonendemic pellagra can be made only on clinical grounds because there is no available method to make a blood niacin level determination, and diagnosis is frequently difficult because diarrhea and dermatological changes may be absent. Unexplained progressive encephalopathy in alcoholic patients not responsive to thiamine therapy (see Wernicke-Korsakoff Syndrome in this chapter) should raise the possibility of pellagra.
Vitamin B6 (Pyridoxine)
Another form of pyridoxine-responsive seizure occurs in infants with a hereditary dependency on pyridoxine due to a mutation of the antiquitin gene (Plecko et al., 2007). They develop symptoms despite a normal dietary supply of pyridoxine. In contrast to infants with dietary deficiency, most of these children manifest seizures earlier in life (within days of birth) and require much larger doses of pyridoxine (5-100 mg) to control their convulsions. Long-term administration of large amounts of pyridoxine is needed, generally in the range of 10 mg/day. Even after several years of successful treatment, seizures may reappear within days of pyridoxine withdrawal. This pyridoxine dependency should be considered in infants with undiagnosed seizures, especially in those with a poor response to anticonvulsants.
Wernicke-Korsakoff Syndrome
Wernicke encephalopathy is due to thiamine deficiency. The most common clinical setting for this disorder is chronic alcoholism. However, a large number of cases occur in other conditions, with the only prerequisite being a poor nutritional state, either from inadequate intake, malabsorption, or increased metabolic requirement (Box 57.3). Wernicke encephalopathy may be precipitated acutely in at-risk patients by IV glucose administration or carbohydrate loading. Although the classic triad of confusion, ophthalmoplegia, and gait ataxia is still relevant in defining Wernicke encephalopathy, all three elements are recognized in only about one-third of all patients. Thus, the disorder should be considered in the differential diagnosis of all patients with acute encephalopathy.
Laboratory Studies
Brain MRI is very helpful in acute Wernicke encephalopathy. MRI typically shows signal abnormalities on T2-weighted FLAIR (fluid-attenuated inversion recovery) and diffusion-weighted images, symmetrically distributed around the periaqueductal regions, tectal plates, medial thalami, and bilateral mammillary bodies. Other regions such as the cerebellar vermis, pons, medulla, dentate nuclei, cranial nerve nuclei, and basal ganglia are also at times affected. Some lesions may show contrast enhancement. Hemorrhages are rarely seen. There are differences in the distribution of lesions between alcoholic and nonalcoholic patients, but there is a considerable overlap between the two groups (Zuccoli et al., 2009). Brain computed tomography (CT) may be used when MRI is unavailable but is much less sensitive in demonstrating abnormalities. MRI signal abnormalities typically resolve completely with prompt treatment, but shrunken mammillary bodies may be seen as a late residual finding. The CSF is either normal or shows a mild elevation in protein. Serum thiamine level and erythrocyte transketolase activity may be depressed, and there may be an elevation of serum pyruvate.
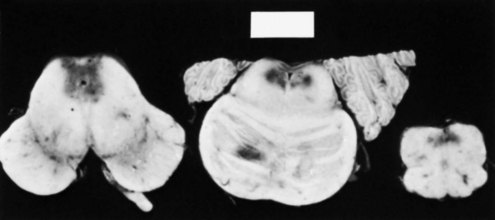
Pathology
The pathological process depends on the age of the lesions. Macroscopically, varying degrees of congestion, petechial hemorrhages, shrinkage, and discoloration are present (Fig. 57.3). Glial proliferation and myelin pallor characterize the more chronic lesions. The regions affected are the same as those observed to be involved on MRI. The frequency of Wernicke encephalopathy as estimated from various autopsy studies is approximately 0.8% to 2.8%, a figure far greater than that expected from clinical studies. Only 20% of the autopsy cases in one series were diagnosed during life. This is unfortunate because Wernicke encephalopathy is preventable and treatable. The underrecognition may result from an overemphasis on alcoholism as a cause (see Table 57.4) or a misconception that all three elements of the clinical triad are needed for a diagnosis. Wernicke encephalopathy occurring under other settings may be mistaken for encephalopathy of uremia, dialysis, sepsis, or other systemic diseases.
Treatment
If left untreated, Wernicke encephalopathy is progressive. The mortality, even with thiamine treatment, was 10% to 20% in the early studies. With treatment, the majority of ocular signs resolve within hours, although a fine horizontal nystagmus persists in approximately 60% of patients. Apathy and lethargy improve over days or weeks. The gait disturbance resolves much more slowly, and in over one-third of the cases, gait may be abnormal even months after treatment. As the global confusional state recedes, some patients are left with the Korsakoff syndrome. The treatment of Korsakoff syndrome is usually limited to social support. Many patients require at least some form of supervision, either at home or in a chronic care facility. There are anecdotal reports of success treating the memory loss with acetylcholinesterase inhibitors or memantine, but controlled studies in small number of patients did not show a consistent benefit (Luykx et al., 2008).
Other Diseases Associated with Alcoholism
Despite the increased risk of malnutrition, only Wernicke-Korsakoff syndrome and rare cases of pellagra in alcoholics are clearly linked to nutritional deficiency. The pathogenesis of other neurological disorders is less clear (Box 57.4), though many have postulated a direct toxic effect of alcohol on both the central and peripheral nervous systems. For instance, neuropathy sometimes develops in alcohol abusers with normal nutritional status. The pattern of nerve fiber loss in these patients appears to be different from that in beriberi neuropathy from thiamine deficiency, thus suggesting a different pathological mechanism (Koike et al., 2003).
Alcohol-Withdrawal Syndromes
Benzodiazepines and supportive care are the mainstays in the treatment of a severe alcohol-withdrawal state (Korsten and O’Connor, 2003). A fast-acting benzodiazepine such as diazepam, lorazepam, or oxazepam should be given via the IV route. They are effective in controlling the agitation and sympathetic hyperactivity as well as any withdrawal seizures. This should be accompanied by aggressive support with IV fluids, nutritional supplementation (see Wernicke-Korsakoff Syndrome, earlier), treatment of coexisting complications, and close monitoring of vital signs, fluid status, and electrolytes. Less proven agents such as β-adrenergic antagonists, clonidine, and carbamazepine may also be used as adjunctive measures in controlling alcohol-withdrawal symptoms. The improvement of treatment has reduced the mortality rate of delirium tremens from over 30% at the beginning of the 20th century to the current rate of no more than 5%.
Alcoholic Neuropathy
Clinical Features
Alcoholic neuropathy is a mixed sensory and motor disorder that affects large- and small-diameter nerve fibers to varying degrees (Zambelis et al., 2005). Symptom onset is insidious, beginning in the feet and progressing proximally and symmetrically. Paresthesia is the most common presenting complaint. Many patients also complain of pain, either an aching discomfort in the calves or a burning sensation over the soles. Dysesthesia may be so severe that a light touch or gentle rubbing over the skin is intensely unpleasant. Interestingly, pain is more often a problem in those with milder neuropathy. On examination, both deep and superficial sensations are affected. Ankle tendon reflexes and sometimes knee reflexes are lost. Weakness and wasting are limited to the distal feet in mild cases but can involve the distal upper extremities in more severe cases. Rarely there may be vagus or recurrent laryngeal nerve involvement, with prominent hoarseness and weakness of voice. Both alcohol neurotoxicity and thiamine deficiency likely play important roles in alcoholic neuropathy. One study (Koike et al., 2003) suggests that pure alcoholic neuropathy without thiamine deficiency is more likely to be painful and has less motor involvement than that associated with concomitant thiamine deficiency.
Marchiafava-Bignami Disease
The neurological presentation is variable. The most common are an acute confusional state or a dementing syndrome. Patients may present with a variable combination of psychomotor slowing, behavioral changes, incontinence, dysarthria, and spasticity. Seizures, hemiparesis, and coma are sometimes seen. Pathologically, there is selective involvement of the central portion of the corpus callosum; the dorsal and ventral regions are spared or affected to a lesser degree. There also may be symmetrical involvement of other white-matter tracts. MRI is valuable and shows increased T2 and FLAIR signals along with restricted diffusion in the body of the corpus callosum, sometimes with extension into the genu or the splenium (Menegon et al., 2005). Abnormalities may also be seen in the subcortical white matter and cerebellar peduncles. Thinning of the corpus callosum is seen commonly in alcoholics without symptoms of Marchiafava-Bignami disease. It is unclear what causes the overt disease in susceptible individuals.
Vitamin D
Derangement in vitamin D and calcium metabolism may be caused by a diversity of systemic conditions including dietary insufficiency, inadequate sunlight exposure, immobility, anticonvulsant use, malabsorption, hypophosphatemia, and hyperparathyroidism. A common assay is the serum level of 25-hydroxyvitamin D. Levels below 10 ng/mL indicate deficiency, and levels between 10 and 20 ng/mL suggest some degree of insufficiency. Recent interest in the role of vitamin D arise from three lines of observation. First, around 50% of the elderly population may have insufficient amounts of vitamin D, most likely from a lack of adequate dietary intake and exposure to sunlight. Second, vitamin D has potentially diverse effects in the nervous system through its action on inflammatory cytokines, neurotrophins, and calcium-binding proteins. Third, low levels of 25-hydroxyvitamin D have been associated with a number of neurological diseases including multiple sclerosis, Parkinson disease, stroke, and cognitive decline (Miller, 2010). However, the observed associations do not prove a causative role of vitamin D deficiency in these neurological disorders. Whether vitamin D supplementation has any beneficial impact remains to be seen.
The best-documented syndrome attributable to overt vitamin D deficiency is a myopathy characterized by proximal weakness (Al-Said et al., 2009). Progressive weakness develops over many months. Weakness leads to difficulty in going upstairs and rising from a chair. When severe, some patients are wheelchair dependent. Diffuse bone pain, muscle pain, or back pain are common. Stretch reflexes and sensation are normal. Some patients may already have a diagnosis of osteomalacia. Serum creatine kinase level is usually normal or only mildly elevated. Serum alkaline phosphatase is abnormally high, and calcium and phosphorus may be normal or mildly decreased. Electromyography typically shows short-duration low-amplitude and polyphasic motor unit potentials without spontaneous activities; these features are similar to those of other metabolic myopathies. Nonspecific type II muscle fiber atrophy is seen on biopsy.
Miscellaneous Deficiency Diseases
Complications after Bariatric Surgery
The most commonly reported neurological complication is peripheral neuropathy. Most of these patients have a sensorimotor polyneuropathy. Mononeuropathies and plexopathies are less frequent. Wernicke encephalopathy is the most common central nervous system disorder. Other less common complications include optic neuropathy, myelopathy, and myopathy (Koffman et al., 2006). Some patients may not become symptomatic until many years after surgery. All patients should have long-term medical follow-up, dietary counseling, and periodic laboratory evaluations. They should all take dietary supplements on an indefinite basis. Supplements should include multivitamins, folic acid, iron, calcium, and additional oral vitamin B12 supplements.
Copper Deficiency
Deficiency of copper classically leads to a myelopathy or myeloneuropathy characterized by sensory ataxia and gait difficulty. The syndrome may be difficult to distinguish from the subacute combined degeneration of vitamin B12 deficiency. Common symptoms are lower-limb paresthesias and gait difficulty. The syndrome is characterized by sensory loss and sensory ataxia. Examination shows prominent proprioceptive impairment, less severe sensory loss to other modalities, brisk reflexes, and variable presence of Babinski sign (Kumar, 2006). Diminished ankle reflex and evidence of axonal loss on nerve conduction studies, indicating the presence of a sensorimotor polyneuropathy, are common. Other observers have described unusual clinical features such as cognitive dysfunction, optic neuropathy, sensory ganglionopathy, and asymmetrical weakness from a lower motor neuron syndrome.
Finding a low serum copper level, along with low serum ceruloplasmin and low urinary copper excretion, helps establish the diagnosis of copper deficiency. There may be associated hematological abnormalities such as anemia (microcytic, normocytic, or macrocytic) and neutropenia. Abnormal T2 signals on MRI may be seen along the posterior column of the spinal cord. Interpretation of copper and ceruloplasmin levels may be complicated. Ceruloplasmin is the chief carrier protein for copper, and its serum level generally parallels that of copper. Ceruloplasmin is also an acute-phase reactant, and its concentration is increased in pregnancy, liver disease, and various infectious and inflammatory disorders. Thus, serum copper level in a deficiency state may be falsely normal under some conditions. Low levels of ceruloplasmin and low serum copper are seen in Wilson disease (see Chapter 71). In such patients, urinary copper excretion is typically elevated (>100 µg over 24 hours).
The most common cause is impaired absorption of dietary copper after gastric surgeries, including bariatric surgery. GI disorders predisposing to malabsorption, such as sprue, celiac disease and bacterial overgrowth, are also risk factors. Excessive dietary consumption of zinc and iron may impair the absorption of copper. Some cases have been reported in the setting of parenteral zinc overload from renal dialysis. Menkes disease is a form of congenital copper deficiency and is due to an inherited disorder of intestinal copper absorption. Clioquinol, an antibiotic with the property of being a copper-zinc chelator, may rarely be responsible. Even in cases of malabsorption, dietary supplementation of 2 to 6 mg of copper salt per day is usually sufficient to reverse a deficiency state. Intravenous infusion may be used if needed. Replenishment appears to halt progression of disease but with little neurological improvement (Kelkar et al., 2008).
Aguglia U., Annesi G., Pasquinelli G., et al. Vitamin E deficiency due to chylomicron retention disease in Marinesco-Sjogren syndrome. Ann Neurol. 2000;47:260-264.
Al-Said Y., Al-Rached H., Al-Qahtani H., Jan M. Severe proximal myopathy with remarkable recovery after vitamin D treatment. Can J Neurol Sci. 2009;36:336-339.
Dali-Youcef N., Andres E. An update on cobalamin deficiency in adults. Quart J Med. 2009;102:17-28.
Kelkar P., Chang S., Muley S.A. Response to oral supplementation in copper deficiency myeloneuropathy. J Clin Neuromusc Dis. 2008;10:1-3.
Koffman B.M., Greenfield L.J., Ali I.I., Pirzada N.A. Neurologic complications after surgery for obesity. Muscle Nerve. 2006;33:166-176.
Koike H., Iijima M., Sugiura M., et al. Alcoholic neuropathy is clinicopathologically distinct from thiamine-deficiency neuropathy. Ann Neurol. 2003;54:19-29.
Korsten T., O’Connor P. Management of drug and alcohol withdrawal. N Engl J Med. 2003;348:1786-1795.
Kumar A., Singh A.K. Teaching NeuroImage: Inverted V sign in subacute combined degeneration of spinal cord. Neurology. 2009;72:e4.
Kumar N. Copper deficiency myelopathy (human swayback). Mayo Clin Proc. 2006;81:1371-1384.
Luykx H.J., Dorresteijn L.D., Haffmans P.M., et al. Rivastigmine in Wernicke-Korsakoff’s syndrome: five patients with rivastigmine showed no more improvement than five patients without rivastigmine. Alcohol Alcohol. 2008;43:70-72.
Mariotti C., Gellera C., Rimoldi M., et al. Ataxia with isolated vitamin E deficiency: neurological phenotype, clinical follow-up and novel mutations in TTPA gene in Italian families. Neurol Sci. 2004;25:130-137.
Menegon P., Sibon I., Pachai C., et al. Marchiafava-Bignami disease: diffusion-weighted MRI in corpus callosum and cortical lesions. Neurology. 2005;65:475-477.
Miller J.W. Vitamin D and cognitive function in older adults: are we concerned about vitamin D-mentia? Neurology. 2010;74:13-15.
Miller J.W., Garrod M.G., Rockwood A.L., et al. Measurement of total vitamin B12 and holotranscobalamin, singly and in combination, in screening for metabolic vitamin B12 deficiency. Clin Chem. 2006;52:278-285.
Plecko B., Paul K., Paschke E., et al. Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat. 2007;28:19-26.
Puri V., Chaudhry N., Tatke M., et al. Isolated vitamin E deficiency with demyelinating neuropathy. Muscle Nerve. 2005;32:230-235.
Singer M.A., Lazaridis C., Nations S.P., Wolfe G.I. Reversible nitrous oxide-induced myeloneuropathy with pernicious anemia: case report and literature review. Muscle Nerve. 2008;37:125-129.
Solomon L.R. Cobalamin-responsive disorders in the ambulatory care setting: unreliability of cobalamin, methylmalonic acid and homocysteine testing. Blood. 2005;105:978-985.
Zambelis T., Karandreas N., Tzavellas E., et al. Large and small fiber neuropathy in chronic alcohol-dependent subjects. J Peripher Nerv Syst. 2005;10:375-381.
Zuccoli G., Santa Cruz D., Bertolini M., et al. MR imaging findings in 56 patients with Wernicke encephalopathy: nonalcoholics may differ from alcoholics. Am J Neuroradiol. 2009;30:171-176.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 57 Deficiency Diseases of the Nervous System
Malnutrition causes a wide spectrum of neurological disorders (Table 57.1). Despite socioeconomic advances, nutritional deficiency diseases such as kwashiorkor and marasmus are still endemic in many underdeveloped countries. The problem in Western countries is usually the result of dietary insufficiency from chronic alcoholism or malabsorption due to gastrointestinal (GI) diseases. The B vitamins (thiamine, pyridoxine, nicotinic acid, and vitamin B12), vitamin E, and perhaps folic acid are important for normal function of the nervous system. Emerging evidence also supports important roles of vitamin D and copper.
Table 57.1 Neurological Manifestations in Deficiency Diseases
| Neurological Manifestations | Associated Nutritional Deficiencies |
|---|---|
| Dementia, encephalopathy | Vitamin B12, nicotinic acid, thiamine, folate |
| Seizures | Pyridoxine |
| Myelopathy | Vitamin B12, vitamin E, folate, copper |
| Myopathy | Vitamin D, vitamin E |
| Peripheral neuropathy | Thiamine, vitamin B12, and many others |
| Optic neuropathy | Thiamine, vitamin B12, and many others |
Cobalamin (Vitamin B12)
Causes of Deficiency
The classic disease, pernicious anemia, is caused by defective intrinsic factor production by parietal cells, leading to malabsorption. These patients may have demonstrable circulating antibodies to parietal cells or lymphocytic infiltrations of the gastric mucosa, suggesting an underlying autoimmune disorder. Another common cause of malabsorption is food-cobalamin malabsorption (Dali-Youcef and Andres, 2009). Under some clinical settings, the normal digestive process fails to release cobalamin from food or intestinal transport protein. Cobalamin remains bound and cannot be absorbed even in the presence of available intrinsic factors. Predisposing factors include atrophic gastritis and hypochlorhydria, and malabsorption may be seen with Helicobacter pylori infection, gastrectomy or other gastric surgeries, intestinal bacterial overgrowth, and prolonged use of H2 antagonists, proton pump inhibitors, or biguanides (e.g., metformin). Patients with human immunodeficiency virus (HIV) are often observed to have a low serum cobalamin level, usually with normal homocysteine and methylmalonic acid. The significance of this association is unknown.
People who abuse nitrous oxide may develop a clinical syndrome of myeloneuropathy indistinguishable from that of cobalamin deficiency. The mechanism appears to be an interference with the cobalamin-dependent conversion of homocysteine to methionine. The other pathway, conversion of methylmalonyl co-A to succinyl co-A, is unaffected by nitrous oxide. Prolonged exposure to nitrous oxide is necessary to produce neurological symptoms in normal individuals. By contrast, patients who are already deficient in cobalamin may experience neurological deficits after only brief exposures during general anesthesia. Symptoms appear subacutely after surgery and resolve quickly with cobalamin treatment (Singer et al., 2008).
Laboratory Studies
Serum assays of vitamin B12 and cobalamin-dependent metabolites provide direct measures of cobalamin homeostasis, although there are important limitations (Solomon, 2005). Blood cobalamins are bound to two transport proteins, transcobalamin and haptocorrin. The cobalamin bound to transcobalamin, known as holotranscobalamin, is the fraction that is available to tissues, although it accounts for only 10% to 30% of the serum level measured by standard laboratory methods. Serum levels are influenced by conditions that affect the concentrations of these transport proteins. Myeloproliferative and hepatic disorders may raise the concentration of haptocorrin and cause a falsely normal serum level. A misleadingly high serum level also may result from the presence of an abnormal cobalamin-binding protein. In contrast, pregnancy and contraceptives may give falsely low measurements in the absence of deficiency. Folate deficiency also causes a falsely low cobalamin serum level that corrects after folate replacement. These confounding factors diminish the sensitivity and specificity of the commonly used assay of total serum cobalamin in the diagnosis of deficiency state. Although measurement of holotranscobalamin is better in theory, available data suggest that its diagnostic accuracy is approximately equivalent to that of total serum cobalamin (Miller et al., 2006).
Homocysteine and methylmalonic acid are precursors of cobalamin-dependent biochemical reactions. These metabolites accumulate during deficiency state. Measuring these metabolites is useful in settings of nitrous oxide abuse and in inherited metabolic disorders in which cobalamin-dependent pathways are impaired despite normal serum level. Homocysteine and methylmalonic acid assays are also useful when the cobalamin concentration is in the lower range of normal, between 200 and 350 pg/mL. Homocysteine level should be measured either at fasting or after an oral methionine load. The blood sample should be refrigerated immediately after collection because the level increases if whole blood is left at room temperature for several hours. Elevated levels of homocysteine and methylmalonic acid are not specific for cobalamin deficiency, as there are many other causes of increase in these metabolites (Box 57.1). In cobalamin-deficient patients, these levels typically normalize within 2 weeks of treatment.
Box 57.1 Causes of Elevated Serum Levels of Homocysteine and Methylmalonic Acid
Because most patients present with clinical features suggesting a myelopathy or encephalopathy, imaging studies are necessary to exclude structural causes. Results of magnetic resonance imaging (MRI) may be normal, or T2-signal abnormalities may be seen in the lateral or posterior columns in patients with subacute combined degeneration (Kumar and Singh, 2009) (Fig. 57.1). Both gadolinium enhancement and spinal cord swelling have been described. Patients with encephalopathy or dementia often have multiple foci of T2 signal abnormalities in the deep white matter that may become confluent with disease progression. Radiographic improvement is seen within a few months after initiation of treatment. Nonspecific abnormalities of electroencephalography, as well as visual and somatosensory evoked responses, are present in most patients with neurological abnormalities. Nerve-conduction studies show small or absent rural nerve sensory potentials in approximately half of patients, providing evidence for an axonal polyneuropathy.
Pathology
The term subacute combined degeneration of the spinal cord describes the pathological process seen in this disorder. Microscopically, spongiform changes and foci of myelin and axon destruction are seen in the white matter of the spinal cord. The most severely affected regions are the posterior columns at the cervical and upper thoracic levels (Fig. 57.2). Pathological changes also are seen commonly in the lateral columns, whereas the anterior columns are involved in only a small number of the advanced cases. The pathological findings of the peripheral nervous system are those of axonal degeneration, but in some cases there is evidence of demyelination. Involvement of the optic nerve and cerebral white matter also occurs.
Folate Deficiency and Homocysteine
Vitamin E
Like other fat-soluble compounds, vitamin E depends on the presence of pancreatic esterases and bile salts for its solubilization and absorption in the intestinal lumen. Neurological symptoms of deficiency occur most commonly in patients with fat malabsorption (Box 57.2). A reduced bile salt pool may be caused either by reduced hepatic excretion, as in congenital cholestasis, or by interruption of the enterohepatic reabsorption of bile, as in patients with extensive small-bowel resection. Pancreatic insufficiency contributes to malabsorption. Another setting is cystic fibrosis.
Box 57.2 Causes of Vitamin E Deficiency
A rare familial form of fat malabsorption is abetalipoproteinemia (Bassen-Kornzweig syndrome), a disorder in which impaired chylomicron and lipoprotein synthesis is partly responsible for the impaired fat absorption. In addition to a neurological syndrome similar to that seen in other vitamin E-deficient states, spiky red blood cells (acanthocytes) and retinal pigment changes are characteristic. Another hereditary cause of vitamin E deficiency may be a genetic defect in the assembly or secretion of chylomicrons, leading to a chylomicron retention disease that is demonstrable in the intestinal mucosa (Aguglia et al., 2000). A syndrome of ataxia with isolated vitamin E deficiency (AVED) occurs in patients without GI disease or generalized fat malabsorption. Mutations in the α-tocopherol transfer protein gene (TTPA) on chromosome 8q13 are responsible (Mariotti et al., 2004
[/not-level-membership-for-neurology-category]
