Chapter 81 Defects in Metabolism of Carbohydrates
Carbohydrate synthesis and degradation provide the energy required for most metabolic processes. The important carbohydrates include 3 monosaccharides—glucose, galactose, and fructose—and a polysaccharide, glycogen. The relevant biochemical pathways of these carbohydrates are shown in Figure 81-1. Glucose is the principal substrate of energy metabolism. A continuous source of glucose from dietary intake, gluconeogenesis, and glycogenolysis of glycogen maintains normal blood glucose levels. Metabolism of glucose generates adenosine triphosphate (ATP) via glycolysis (conversion of glucose or glycogen to pyruvate), mitochondrial oxidative phosphorylation (conversion of pyruvate to carbon dioxide and water), or both. Dietary sources of glucose come from ingesting polysaccharides, primarily starch and disaccharides, including lactose, maltose, and sucrose. Oral intake of glucose is intermittent and unreliable. Glucose made de novo from amino acids, primarily alanine (gluconeogenesis), contributes to maintaining the euglycemic state, but this process requires time. The breakdown of hepatic glycogen provides the rapid release of glucose, which maintains a constant blood glucose concentration. Glycogen is also the primary stored energy source in muscle, providing glucose for muscle activity during exercise. Galactose and fructose are monosaccharides that provide fuel for cellular metabolism; their role is less significant than that of glucose. Galactose is derived from lactose (galactose + glucose), which is found in milk and milk products. Galactose is an important energy source in infants, but it is 1st metabolized to glucose. Galactose (exogenous or endogenously synthesized from glucose) is also an important component of certain glycolipids, glycoproteins, and glycosaminoglycans. The dietary sources of fructose are sucrose (fructose + glucose, sorbitol) and fructose itself, which is found in fruits, vegetables, and honey.
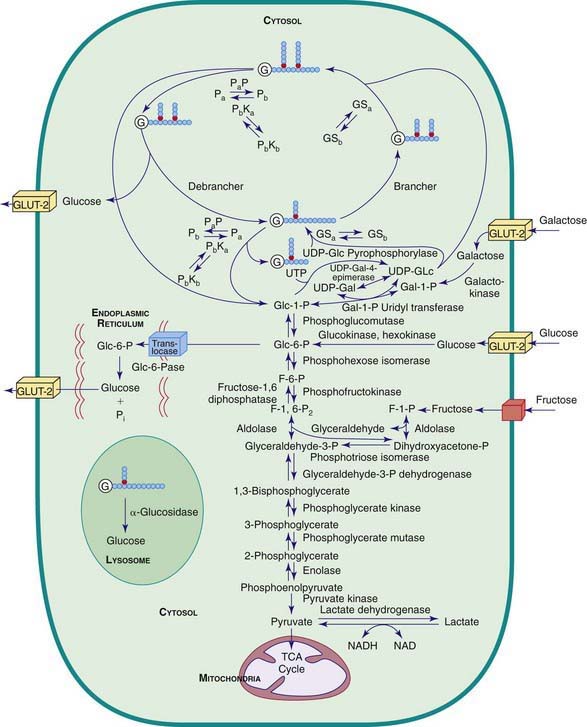
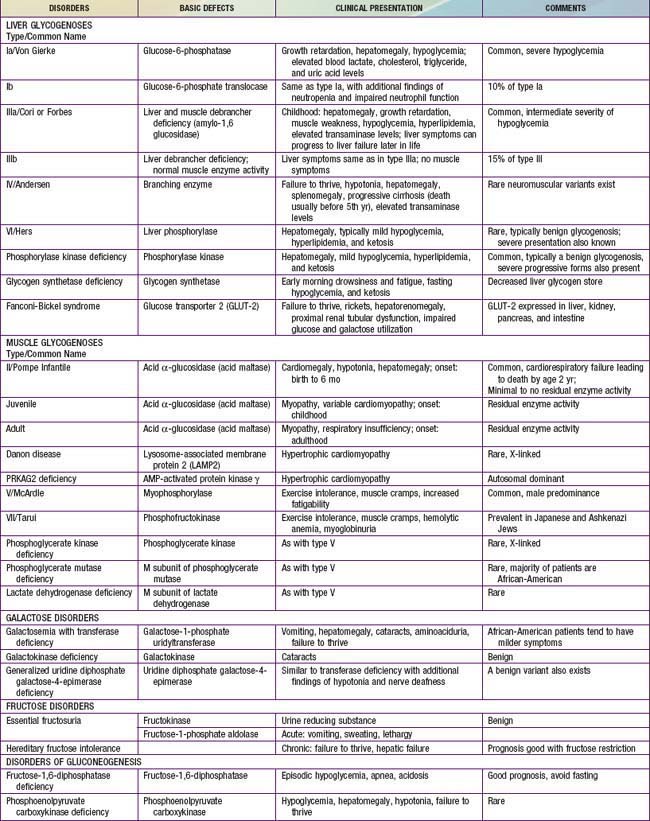
Defects in glycogen metabolism typically cause an accumulation of glycogen in the tissues, hence the name glycogen storage disease (Table 81-1). Defects in gluconeogenesis or the glycolytic pathway, including galactose and fructose metabolism, do not result in an accumulation of glycogen (see Table 81-1). The defects in pyruvate metabolism in the pathway of the conversion of pyruvate to carbon dioxide and water via mitochondrial oxidative phosphorylation are more often associated with lactic acidosis and some tissue glycogen accumulation.
81.1 Glycogen Storage Diseases
The disorders of glycogen metabolism, the glycogen storage diseases (GSDs), result from deficiencies of various enzymes or transport proteins in the pathways of glycogen metabolism (see Fig. 81-1). The glycogen found in these disorders is abnormal in quantity, quality, or both. GSDs are categorized by numeric type in accordance with the chronological order in which these enzymatic defects were identified. This numeric classification is still widely used, at least up to number VII. The GSDs can also be classified by organ involvement and clinical manifestations into liver and muscle glycogenoses (see Table 81-1).
Liver Glycogenoses
Type I Glycogen Storage Disease (Glucose-6-Phosphatase or Translocase Deficiency, Von Gierke Disease)
Treatment
Uncooked cornstarch acts as a slow-release form of glucose and can be introduced at a dose of 1.6 g/kg every 4 hr for infants <2 yr of age. The response of young infants is variable. As the child grows older, the cornstarch regimen can be changed to every 6 hr at a dose of 1.75-2.5 g/kg of body weight. New starch products, which are currently being developed, are thought to be longer acting, better tolerated, and more palatable. A short-term double-blind crossover pilot study comparing uncooked, physically modified cornstarch to traditional cornstarch showed that the majority of GSD patients treated with the new starch had better short-term metabolic control and longer duration of euglycemia; however, more extensive studies replicating these results are necessary at this time. Because fructose and galactose cannot be converted directly to glucose in GSD type I, these sugars are restricted in the diet. Sucrose (table sugar, cane sugar, other ingredients), fructose (fruit, juice, high fructose corn syrup), lactose (dairy foods), and sorbitol should be avoided or limited. Due to these dietary restrictions, vitamins and minerals such as calcium and vitamin D may be deficient and supplementation is required to prevent nutritional deficiencies. Dietary therapy improves hyperuricemia, hyperlipidemia, and renal function, slowing the development of renal failure. This therapy fails, however, to normalize blood uric acid and lipid levels completely in some individuals, despite good metabolic control, especially after puberty. The control of hyperuricemia can be further augmented by the use of allopurinol, a xanthine oxidase inhibitor. The hyperlipidemia can be reduced with lipid-lowering drugs such as HMG-CoA reductase inhibitors and fibrate (Chapter 80). Microalbuminuria, an early indicator of renal dysfunction in type I disease, is treated with angiotensin-converting enzyme (ACE) inhibitors. Citrate supplements can be beneficial for patients with hypocitraturia by preventing or ameliorating nephrocalcinosis and development of urinary calculi. Growth hormone should be used with extreme caution and limited to only those with a documented growth hormone deficiency. Even in those cases, there should be close monitoring of metabolic parameters and presence of adenomas.
Type III Glycogen Storage Disease (Debrancher Deficiency, Limit Dextrinosis)
Clinical Manifestations
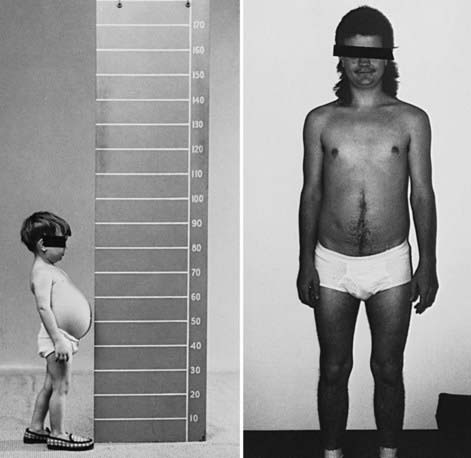
During infancy and childhood, the disease may be indistinguishable from type I GSD, because hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation are common (Fig. 81-2). Splenomegaly may be present, but the kidneys are not enlarged. Remarkably, hepatomegaly and hepatic symptoms in most patients with type III GSD improve with age and usually resolve after puberty. Progressive liver cirrhosis and failure can occur. Hepatocellular carcinoma has also been reported, more typically in patients with progressive liver cirrhosis. The frequency of adenomas in individuals with GSD III is far less, compared to GSD I. Furthermore, the relationship of hepatic adenomas and malignancy in GSD III is unclear. A single case of malignant transformation at the site of adenomas has been noted. In patients with muscular involvement (type IIIa), muscle weakness can present in childhood but can become severe after the 3rd or 4th decade of life, as evidenced by slowly progressive weakness and wasting. Because patients with GSD III can have decreased bone density, they are at an increased risk of potential fracture. Myopathy does not follow any particular pattern of involvement; both proximal and distal muscles are involved. Electromyography reveals a widespread myopathy; nerve conduction studies are often abnormal. Ventricular hypertrophy is a frequent finding, but overt cardiac dysfunction is rare. There have been some reports of life-threatening arrhythmia and need for heart transplant in some GSD III patients. Hepatic symptoms in some patients may be so mild that the diagnosis is not made until adulthood, when the patients show symptoms and signs of neuromuscular disease. The initial diagnosis has been confused with Charcot-Marie-Tooth disease. Polycystic ovaries are common; some patients can develop hirsutism, irregular menstrual cycles, and other features of polycystic ovarian syndrome. Fertility does not appear to be reduced; successful pregnancies in patients with GSD III have been reported.
Type IV Glycogen Storage Disease (Branching Enzyme Deficiency, Amylopectinosis, or Andersen Disease)
Glycogen Synthetase Deficiency (GSD 0)
Muscle Glycogenoses
Type II Glycogen Storage Disease (Lysosomal Acid α-1,4-Glucosidase Deficiency, Pompe Disease)
Treatment
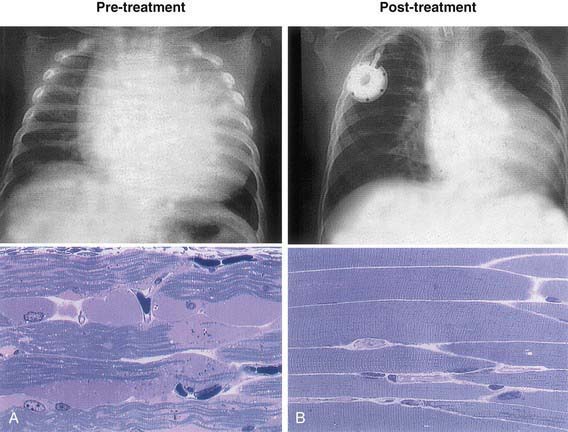
Treatment options were once limited to supportive or palliative care. Specific enzyme replacement therapy (ERT) with recombinant human acid α-glucosidase (alglucosidase alfa, [Myozyme]) is available for treatment of Pompe disease. Recombinant acid α-glucosidase is capable of preventing deterioration or reversing abnormal cardiac and skeletal muscle functions (Fig. 81-3). ERT should be initiated as soon as possible and preferably <6 mo of age; the dose is 20 mg/kg given every 2 wk. A high-protein diet and exercise therapy may also be beneficial. Nocturnal ventilatory support, when indicated, should be used. It has been shown to improve the quality of life and is particularly beneficial during a period of respiratory decompensation.
Type V Glycogen Storage Disease (Muscle Phosphorylase Deficiency, Mcardle Disease)
Type VII Glycogen Storage Disease (Muscle Phosphofructokinase Deficiency, Tarui Disease)
Akman HO, Sampayo JN, Ross FA, et al. Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the γ2-subunit of AMP-activated protein kinase. Pediatr Res. 2007;62:499-504.
Al-Hassnan ZN, Al Budhaim M, Al-Owain M, et al. Muscle phosphofructokinase deficiency with neonatal seizures and nonprogressive course. J Child Neurol. 2007;22:106-108.
Andersen ST, Haller RG, Vissing J. Effect of oral sucrose shortly before exercise on work capacity in McArdle disease. Arch Neurol. 2008;65:786-789.
Arad M, Maron BJ, Gorham JM, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352:362-372.
Beauchamp NJ, Dalton A, Ramaswami U, et al. Glycogen storage disease type IX: high variability in clinical phenotype. Mol Genet Metab. 2007;92:88-99.
Beauchamp NJ, Taybert J, Champion MP, et al. High frequency of missense mutations in glycogen storage disease type VI. J Inherit Metab Dis. 2007;30:722-734.
Beutler E. PGK deficiency. Br J Haematol. 2007;136:3-11.
Bhattacharya K, Orton RC, Qi X, et al. A novel starch for the treatment of glycogen storage diseases. J Inherit Metab Dis. 2007;30:350-357.
Chen LR, Chen CA, Chiu SN, et al. Reversal of cardiac dysfunction after enzyme replacement in patients with infantile-onset Pompe disease. J Pediatr. 2009;155:271-275.
Chien YH, Chiang SC, Zhang XK, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122:e39-45.
Franco LM, Krishnamurthy V, Bali D, et al. Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis. 2005;28:153-162.
Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alfa prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009;66:329-335.
Koeberl DD, Kishnani PS, Bali D, et al. Emerging therapies for glycogen storage disease type I. Trends Endocrinol Metab. 2009;20:252-258.
Koeberl DD, Kishnani PS, Chen YT. Glycogen storage disease types I and II: treatment updates. J Inherit Metab Dis. 2007;30:159-164.
Kollberg G, Tulinius M, Gilljam T, et al. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N Engl J Med. 2007;357:1507-1514.
Llerena JCJr, Horovitz DM, Marie SKN, et al. The Brazilian consensus on the management of Pompe disease. J Pediatr. 2009;155:S47-S56.
Lucchiari S, Pagliarani S, Salani S, et al. Hepatic and neuromuscular forms of glycogenosis type III: nine mutations in AGL. Hum Mutat. 2006;27:600-601.
Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210-219.
Pierre G, Chakupurakal G, McKiernan P, et al. Bone marrow transplantation in glycogen storage disease type 1b. J Pediatr. 2008;152:286-288.
Rommel O, Kley RA, Dekomien G, et al. Muscle pain in myophosphorylase deficiency (McArdle’s disease): the role of gender, genotype, and pain-related coping. Pain. 2006;124:295-304.
Spiegel R, Mahamid J, Orho-Melander M, et al. The variable clinical phenotype of liver glycogen synthase deficiency. J Pediatr Endocrinol Metab. 2007;20:1339-1342.
van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362(15):1396-1406.
81.2 Defects in Galactose Metabolism
Milk and dairy products contain lactose, the major dietary source of galactose. The metabolism of galactose produces fuel for cellular metabolism through its conversion to glucose-1-phosphate (see Table 81-1). Galactose also plays an important role in the formation of galactosides, which include glycoproteins, glycolipids, and glycosaminoglycans. Galactosemia denotes the elevated level of galactose in the blood and is found in 3 distinct inborn errors of galactose metabolism defective in 1 of the following enzymes: galactose-1-phosphate uridyl transferase, galactokinase, and uridine diphosphate galactose-4-epimerase. The term galactosemia, although adequate for the deficiencies in any of these disorders, generally designates the transferase deficiency.
Galactose-1-Phosphate Uridyl Transferase Deficiency Galactosemia
Barbouth DS, Velazquez DL, Konopka S, et al. Screening newborns for galactosemia using total body galactose oxidation to CO2 in expired air. Pediatr Res. 2007;62:720-724.
Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29:516-525.
Coman DJ, Murray DW, Byrne JC, et al. Galactosemia, a single gene disorder with epigenetic consequences. Pediatr Res. 2010;67(3):286-292.
Holton JB, Walter JH, Tyfield IA, et al. Galactosemia. In: Scriver CM, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular bases of inherited disease. ed 8. New York: McGraw-Hill; 2008:1553-1587.
Hughes J, Ryan S, Lambert D, et al. Outcomes of siblings with classical galactosemia. J Pediatr. 2009;154:721-726.
Noelmans L, Jacquemyn Y, De Naeyer S, et al. Pregnancy and galactosaemia. J Obstet Gynaecol. 2006;26:812-814.
Rubio-Gozalbo ME, Panis B, Zimmermann LJ, et al. The endocrine system in treated patients with classical galactosemia. Mol Genet Metab. 2006;89:316-322.
81.3 Defects in Fructose Metabolism
Two inborn errors are known in the specialized pathway of fructose metabolism: benign or essential fructosuria and hereditary fructose intolerance (HFI). Fructose-1,6-bisphosphatase deficiency, although strictly speaking not a defect of the specialized fructose pathway, is discussed in Chapter 81.4.
Deficiency of Fructokinase (Essential or Benign Fructosuria)
Fructokinase catalyzes the 1st step of metabolism of dietary fructose: conversion of fructose to fructose-1-phosphate (see Fig. 81-1). Without this enzyme, ingested fructose is not metabolized. Its level is increased in the blood, and it is excreted in urine because there is practically no renal threshold for fructose. Clinitest results reveal the urinary-reducing substance, which can be identified as fructose by chromatography.
81.4 Defects in Intermediary Carbohydrate Metabolism Associated with Lactic Acidosis
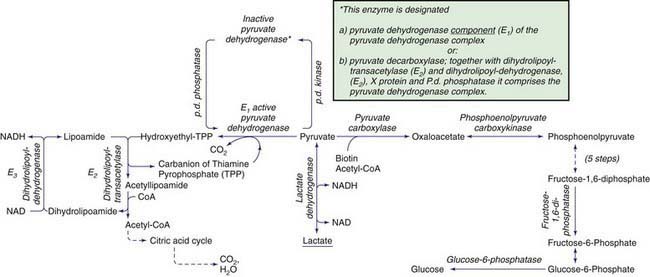
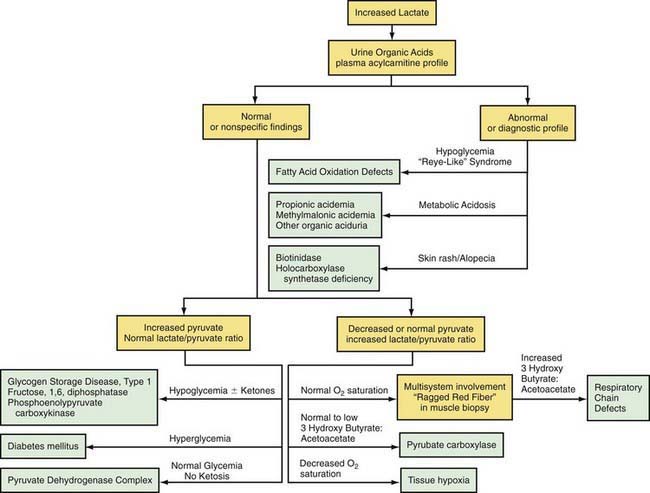
Lactic acidosis occurs with defects of carbohydrate metabolism that interfere with the conversion of pyruvate to glucose via the pathway of gluconeogenesis or to carbon dioxide and water via the mitochondrial enzymes of the Krebs cycle. Figure 81-4 depicts the relevant metabolic pathways. Type I GSD, fructose-1,6-diphosphatase deficiency, and phosphoenolpyruvate carboxylase deficiency are disorders of gluconeogenesis associated with lactic acidosis. Pyruvate dehydrogenase complex deficiency, respiratory chain defects, and pyruvate carboxylase deficiency are disorders in the pathway of pyruvate metabolism causing lactic acidosis. Lactic acidosis can also occur in defects of fatty acid oxidation, organic acidurias (Chapters 79.6, 79.10, and 80.1), or biotin utilization diseases. These disorders are easily distinguishable by the presence of abnormal acylcarnitine profiles, amino acids in the blood, and unusual organic acids in the urine. Blood lactate, pyruvate, and acylcarnitine profiles and the presence of these unusual urine organic acids should be determined in infants and children with unexplained acidosis, especially if there is an increase of anion gap.
Lactic acidosis unrelated to an enzymatic defect occurs in hypoxemia. In this case, as well as in defects in the respiratory chain, the serum pyruvate concentration may remain normal (<1.0 mg/dL with an increased lactate : pyruvate ratio), whereas pyruvate is usually increased when lactic acidosis results from an enzymatic defect in gluconeogenesis or pyruvate dehydrogenase complex (both lactate and pyruvate are increased and the ratio is normal). Lactate and pyruvate should be measured in the same blood specimen and on multiple blood specimens obtained when the patient is symptomatic because lactic acidosis can be intermittent. An algorithm for the differential diagnosis of lactic acidosis is shown in Figure 81-5.
Disorders of Gluconeogenesis
Deficiency of Glucose-6-Phosphatase (Type I Glycogen Storage Disease)
Type I GSD is the only glycogenosis associated with significant lactic acidosis. The chronic metabolic acidosis predisposes these patients to osteopenia; after prolonged fasting, the acidosis associated with hypoglycemia is a life-threatening condition (Chapter 81.1).
Phosphoenolpyruvate Carboxykinase (PEPCK) Deficiency
PEPCK is a key enzyme in gluconeogenesis. It catalyzes the conversion of oxaloacetate to phosphoenolpyruvate (see Fig. 81-4). PEPCK deficiency is both a mitochondrial enzyme deficiency and a cytosolic enzyme deficiency, encoded by 2 distinct genes.
Disorders of Pyruvate Metabolism
Pyruvate is formed from glucose and other monosaccharides, from lactate, and from alanine. It is metabolized through 4 main enzyme systems: lactate dehydrogenase, alanine aminotransferase, pyruvate carboxylase, and pyruvate dehydrogenase complex. Deficiency of the M subunit of lactate dehydrogenase causes exercise intolerance and myoglobinuria (Chapter 81.1). Genetic deficiency of alanine aminotransferase has not been reported in humans.
Pyruvate Dehydrogenase Complex Deficiency
After entering the mitochondria, pyruvate is converted into acetyl coenzyme A (acetyl CoA) by the pyruvate dehydrogenase complex (PDHC), which catalyzes the oxidation of pyruvate to acetyl CoA, which then enters the tricarboxylic acid cycle for ATP production. The complex comprises 5 components: E1, an α-keto acid decarboxylase; E2, a dihydrolipoyl transacylase; E3, a dihydrolipoyl dehydrogenase; protein X, an extra lipoate-containing protein; and pyruvate dehydrogenase phosphatase. The most common is a defect in the E1 (see Fig. 81-4).
Deficiency of Pyruvate Carboxylase
Pyruvate carboxylase is a mitochondrial, biotin-containing enzyme essential in the process of gluconeogenesis; it catalyzes the conversion of pyruvate to oxaloacetate. The enzyme is also essential for Krebs cycle function as a provider of oxaloacetate and is involved in lipogenesis and formation of nonessential amino acids. Clinical manifestations of this deficiency have varied from neonatal severe lactic acidosis accompanied by hyperammonemia, citrullinemia, and hyperlysinemia (type B) to late-onset mild to moderate lactic acidosis and developmental delay (type A). In both types, patients who survived usually had severe psychomotor retardation with seizures, spasticity, and microcephaly. Some patients have pathologic changes in the brainstem and basal ganglia that resemble Leigh disease (see later). The clinical severity appears to correlate with the level of the residual enzyme activity. A “benign” form of PC deficiency characterized by recurrent attacks of lactic acidosis and mild neurologic deficits has also been described. Laboratory findings are characterized by elevated levels of blood lactate, pyruvate, alanine, and ketonuria. In the case of type B, blood ammonia, citrulline, and lysine levels are also elevated, which might suggest a primary defect of the urea cycle. The mechanism is likely caused by depletion of oxaloacetate, which leads to reduced levels of aspartate, a substrate for argininosuccinate synthetase in the urea cycle (Chapter 79.12).
Deficiency of Pyruvate Carboxylase Secondary to Deficiency of Holocarboxylase Synthetase or Biotinidase
Deficiency of either holocarboxylase synthetase (HCS) or biotinidase, which are enzymes of biotin metabolism, result in multiple carboxylase deficiency (pyruvate carboxylase and other biotin-requiring carboxylases and metabolic reactions) and in clinical manifestations associated with the respective deficiencies, as well as rash, lactic acidosis, and alopecia (Chapter 79.6). The course of HCS or biotinidase deficiency can be protracted, with intermittent exacerbation of chronic lactic acidosis, failure to thrive, seizures, and hypotonia leading to spasticity, lethargy, coma, and death. Auditory and optic nerve dysfunction can lead to deafness and blindness, respectively. Late-onset milder forms have also been reported. Laboratory findings include metabolic acidosis and abnormal organic acids in the urine. In HCS deficiency, biotin concentrations in plasma and urine are normal. Diagnosis can be made in skin fibroblasts or lymphocytes by assay for HCS activity, and in the case of biotinidase, in the serum by a screening blood spot.
Mitochondrial Respiratory Chain Defects (Oxidative Phosphorylation Disease)
The mitochondrial respiratory chain catalyzes the oxidation of fuel molecules and transfers the electrons to molecular oxygen with concomitant energy transduction into ATP (oxidative phosphorylation). The respiratory chain produces ATP from nicotinamide adenine dinucleotide (NADH) or FADH2 and includes 5 specific complexes (I: NADH–coenzyme Q reductase; II: succinate–coenzyme Q reductase; III: coenzyme QH2 cytochrome C reductase; IV: cytochrome C oxidase; V: ATP synthase). Each complex is composed of 4-35 individual proteins and, with the exception of complex II (which is encoded solely by nuclear genes), is encoded by nuclear or mitochondrial DNA (inherited only from the mother by mitochondrial inheritance). Defects in any of these complexes or assembly systems produce chronic lactic acidosis presumably due to a change of redox state with increased concentrations of NADH. In contrast to PDHC or pyruvate carboxylase deficiency, skeletal muscle and heart are usually involved in the respiratory chain disorders, and in muscle biopsy, “ragged red fibers” (indicating mitochondrial proliferation) are very suggestive when present (see Fig. 81-5). Because of the ubiquitous nature of oxidative phosphorylation, a defect of the mitochondrial respiratory chain accounts for a vast array of clinical manifestations and should be considered in patients in all age groups presenting with multisystem involvement. Some deficiencies resemble Leigh disease (see later), whereas others cause infantile myopathies such as MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes), MERRF (myoclonus epilepsy, with ragged red fibers), and Kearns-Sayre syndrome (external ophthalmoplegia, acidosis, retinal degeneration, heart block, myopathy, and high cerebrospinal fluid protein) (Table 81-2; Chapters 591.2 and 603.4). There is a higher incidence of psychiatric disorders in adults with a primary oxidative phosphorylation disease than in the general population. Diagnosis requires demonstration of abnormalities of oxidative phosphorylation enzyme complex activities in tissues or of mitochondrial DNA (mtDNA) or a nuclear gene coding form mitochondrial functions, or both (Fig. 81-6). Analysis of oxidative phosphorylation complexes I-IV from intact mitochondria isolated from fresh skeletal muscle is the most sensitive assay for mitochondrial disorders. Specific criteria may assist in making a diagnosis (Table 81-3). Clues to the diagnosis of mitochondrial diseases are noted in Table 81-4.
Table 81-2 CLINICAL AND GENETIC HETEROGENEITY OF DISORDERS RELATED TO MUTATIONS IN MITOCHONDRIAL DNA (mtDNA)*
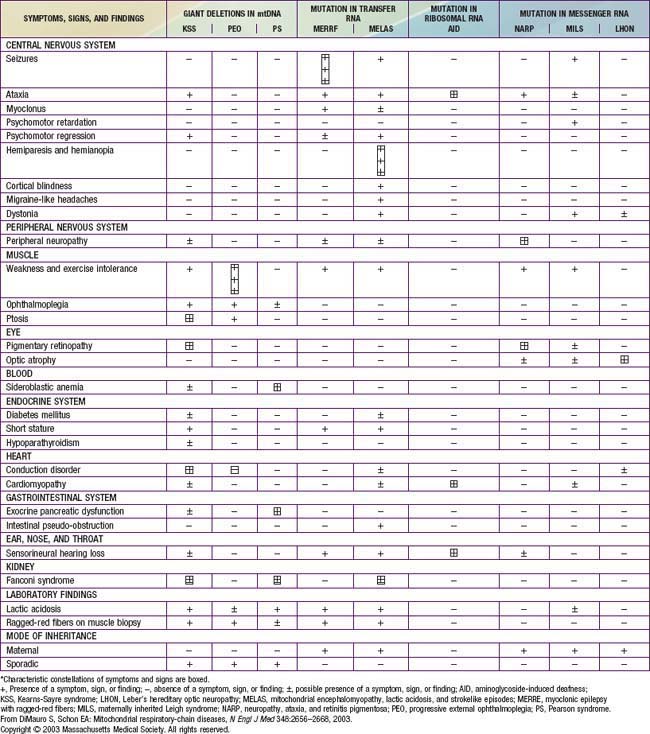
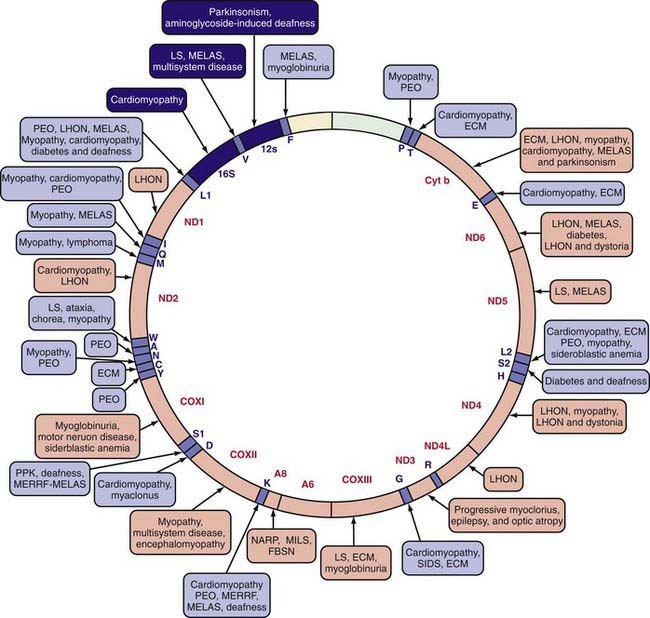
Table 81-3 MODIFIED WALKER CRITERIA APPLIED TO CHILDREN REFERRED FOR EVALUATION OF MITOCHONDRIAL DISEASE
| MAJOR CRITERIA | MINOR CRITERIA | |
|---|---|---|
| Clinical | Clinically complete RC encephalomyopathy* or a mitochondrial cytopathy defined as fulfilling 3 criteria† | Symptoms compatible with an RC defect‡ |
| Histology | >2% ragged red fibers (RRF) in skeletal muscle | Smaller numbers of RRF, SSAM, or widespread electron microscopy abnormalities of mitochondria |
| Enzymology | Cytochrome c oxidase–negative fibers or residual activity of an RC complex <20% in a tissue; <30% in a cell line, or <30% in 2 or more tissues | Antibody-based demonstration of an RC defect or residual activity of an RC complex 20-30% in a tissue, 30-40% in a cell line, or 30-40% in 2 or more tissues |
| Functional | Fibroblast ATP synthesis rates >3 SD below mean | Fibroblast ATP synthesis rates 2-3 SD below mean, or fibroblasts unable to grow in galactose media |
| Molecular | Nuclear or mtDNA mutation of undisputed pathogenicity | Nuclear or mtDNA mutation of probable pathogenicity |
| Metabolic | One or more metabolic indicators of impaired metabolic function |
ATP, adenosine triphosphate; RC, respiratory chain; SSAM, subsarcolemmal accumulation of mitochondria.
* Leigh disease, Alpers disease, LIMD, Pearson syndrome, Kearns-Sayre syndrome, MELAS, MERRF, NARP, MNGIE, and LHON.
† (1) Unexplained combination of multisystemic symptoms that is essentially pathognomonic for an RC disorder, (2) a progressive clinical course with episodes of exacerbation or a family history strongly indicative of an mtDNA mutation, and (3) other possible metabolic or nonmetabolic disorders have been excluded by appropriate testing.
‡ Added pediatric features: stillbirth associated with a paucity of intrauterine movement, neonatal death or collapse, movement disorder, severe failure to thrive, neonatal hypotonia, and neonatal hypertonia as minor clinical criteria.
From Scaglia F, Towbin JA, Craigen WJ, et al: Clinical spectrum, morbidity and mortality in 113 pediatric patients with mitochondrial disease, Pediatrics 114:925–931, 2004.
Table 81-4 CLUES TO THE DIAGNOSIS OF MITOCHONDRIAL DISEASE
NEUROLOGIC
CARDIOVASCULAR
OPHTHALMOLOGIC
GASTROENTEROLOGIC
OTHER
From Haas RH, Parikh S, Falk MJ, et al: Mitochondrial disease: a practical approach for primary care physicians, Pediatrics 120:1326–1333, 2007, Table 1, p 1327.
Leigh Disease (Subacute Necrotizing Encephalomyelopathy)
Leigh disease is a heterogenous neurologic disease that remains a neuropathologic description characterized by demyelination, gliosis, necrosis, relative neuronal sparing, and capillary proliferation in specific brain regions. In decreasing order of severity, the affected areas are the basal ganglia, brainstem cerebellum, and cerebral cortex (Chapter 591). The classic presentation is of an infant who presents with central hypotonia, developmental regression or arrest, and signs of brainstem or basal ganglia involvement. The clinical presentation is highly variable. Diagnosis is usually confirmed by radiologic or pathologic evidence of symmetric lesions affecting the basal ganglia, brainstem, and subthalamic nuclei. Patients with Leigh disease have defects in several enzyme complexes. Dysfunction in cytochrome C oxidase (complex IV) is the most commonly reported defect, followed by NADH-coenzyme Q reductase (complex I), PDHC, and pyruvate carboxylase. Mutations in the nuclear SURF1 gene, which encodes a factor involved in the biogenesis of cytochrome C oxidase and mitochondrial DNA mutations in the ATPase 6 coding region, are common molecular findings in patients with Leigh disease. Patients with Leigh disease frequently present with developmental delay, seizures, altered consciousness, failure to thrive, pericardial effusion, and dilated cardiomyopathy. The prognosis for Leigh syndrome is poor. In a study of 14 cases, there were 7 fatalities before the age of 1.5 yr.
Copeland WC. Inherited mitochondrial diseases of DNA replication. Annu Rev Med. 2008;59:131-146.
Debray FG, Lambert M, Chevalier I, et al. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics. 2007;119:722-733.
DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91-123.
Koene S, Kozicz TL, Rodenburg RJ, et al. Major depression in adolescent children consecutively diagnosed with mitochondrial disorder. J Affect Disord. 2009;114:327-332.
Garcoa-Cazorla A, De Lonlay P, Nassogne MC, et al. Long-term follow-up of neonatal mitochondrial cytopathies: a study of 57 patients. Pediatrics. 2005;116:1170-1177.
Genc GA, Sivri-Kalkanoglu HS, Dursun A, et al. Audiologic findings in children with biotinidase deficiency in Turkey. Int J Pediatr Otorhinolaryngol. 2007;71:333-339.
Haas RH, Parikh S, Falk MJ, et al. Mitochondrial disease: a practical approach for primary care physicians. Pediatrics. 2007;120:1326-1333.
Huntsman RJ, Sinclair DB, Bhargava R, et al. Atypical presentations of Leigh syndrome: a case series and review. Pediatr Neurol. 2005;32:334-340.
Lee HF, Tsai CR, Chi CS, et al. Leigh syndrome: clinical and neuroimaging follow up. Pediatr Neurol. 2009;40:88-93.
Lee NC, Dimmock D, Hwu WL, et al. Simultaneous detection of mitochondrial DNA depletion and single-exon deletion in the deoxyguanosine gene using array-based comparative genomic hybridization. Arch Dis Child. 2009;94:55-58.
Montini G, Malaventura C, Saliati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med. 2008;358:2849-2850.
Pithukpakorn M. Disorders of pyruvate metabolism and the tricarboxylic acid cycle. Mol Genet Metab. 2005;85:243-246.
Sarzi E, Bourdon A, Chretien D, et al. Mitochondrial DNA depletion is a prevalent cause of multiple respiratory chain deficiency in childhood. J Pediatr. 2007;105:531-534.
Scheers I, Bachy V, Stephenne X, et al. Risk of hepatocellular carcinoma in liver mitochondrial respiratory chain disorders. J Pediatr. 2005;146:414-417.
Schon EA, DiMauro S. Mitochondrial mutations: genotype to phenotype. Novartis Found Symp. 2007;287:214-225.
Shanske S, Coku J, Lu J, et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol. 2008;65:368-372.
Shoffner JM. Oxidative phosphorylation diseases. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The online metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2008.
Stacpoole PW, Gilbert LR, Neiberger RE, et al. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 2008;121:e1223-1228.
Stacpoole PW, Kerr DS, Barnes C, et al. Controlled clinical trial of dichloro-acetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519-1531.
Rodriquez MC, MacDonald JR, Mahoney DJ, et al. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007;35:235-242.
Wani AA, Ahanger SH, Bapat SA, et al. Analysis of mitochondrial DNA sequences in childhood encephalomyopathies reveals new disease-associated variants. PLoS One. 2007;2:e942.
81.5 Defects in Pentose Metabolism
Wamelink MM, Smith DE, Jakobs C, et al. Analysis of polyols in urine by liquid chromatography-tandem mass spectrometry: a useful tool for recognition of inborn errors affecting polyol metabolism. J Inherit Metab Dis. 2005;28:951-963.
Wamelink MM, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis. 2008;31:703-717.
Verhoeven NM, Wallot M, Huck JH, et al. A newborn with severe liver failure, cardiomyopathy and transalodase deficiency. J Inherit Metab Dis. 2005;28:169-179.
81.6 Disorders of Glycoprotein Degradation and Structure
Congenital Disorders of Glycosylation (CDGs)
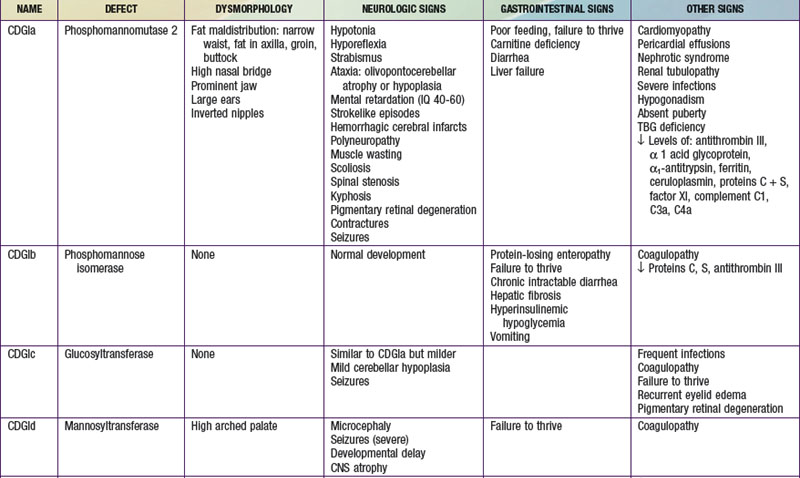
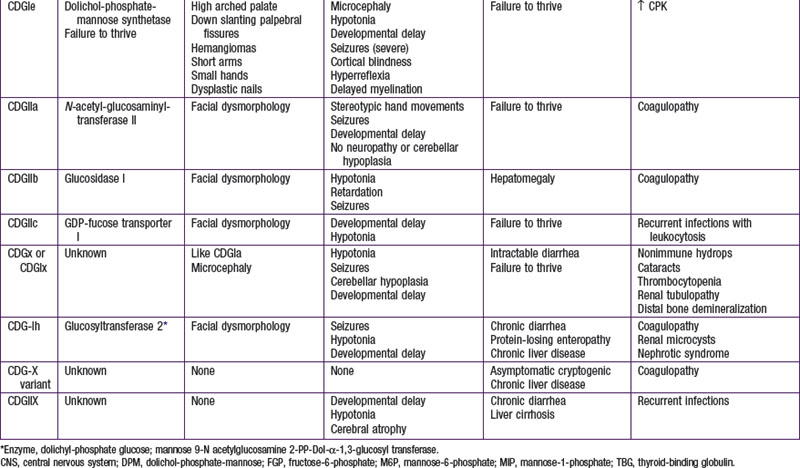
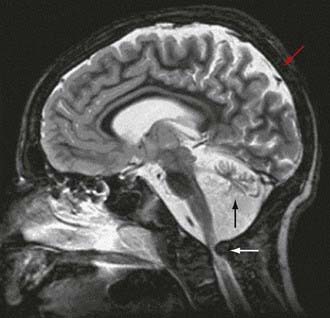
To date, more than 30 CDG subtypes have been identified (Table 81-5). In general most CDG disorders are multisystemic and present with variable involvement of the central nervous system (most often hypotonia and ataxia), abnormal fat distribution, ocular movement defects, coagulation abnormalities, gastrointestinal symptoms including protein-losing enteropathy, retinitis pigmentosa, hormonal abnormalities, and in some cases dysmorphic features. CDG Type Ia, which results from mutations in the gene that encodes phosphomannomutase, is the most common form. The most consistent clinical features of this disorder include variable degrees of psychomotor retardation, subcutaneous fat pads and inverted nipples. Frequent neurologic findings in infancy include cerebellar atrophy (Fig. 81-7), hypotonia, weakness, hyperreflexia, and strokelike episodes.
Collins AE, Ferriero DM. The expanding spectrum of congenital disorders of glycosylation. J Pediatr. 2005;147:728-730.
Denecke J, Kranz C, Von Kleist-Retzow JC, et al. Congenital disorder of glycosylation type 1d: clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatr Res. 2005;58:248-253.
Dinopoulos A, Mohaned I, Jones B, et al. Radiologic and neurophysiologic aspects of stroke-like episodes in children with congenital disorder of glycosylation type Ia. Pediatrics. 2007;119:e768-e772.
Leroy JG. Congenital disorders of N-glycosylation including diseases associated with O- as well as N-glycosylation defects. Pediatr Res. 2006;60:643-656.
Vodopiutz J, Bodamer OA. Congenital disorders of glycosylation—a challenging group of IEMs. J Inherit Metab Dis. 2008;31:267-269.



