[level-membership-for-pediatrics-category]
Chapter 76 Cytogenetics
76.1 Methods of Chromosome Analysis
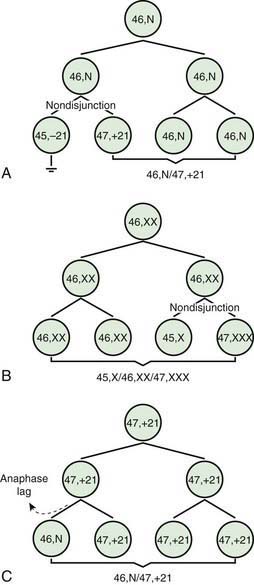
Two errors of cell division commonly occur during meiosis or mitosis, and either can result in an abnormal number of chromosomes. The 1st is nondisjunction, in which 2 chromosomes fail to separate during meiosis and thus migrate together into 1 of the new cells, producing 1 cell with 2 copies of the chromosome and another with no copy. The 2nd is anaphase lag, in which a chromatid or chromosome is lost during mitosis because it fails to move quickly enough during anaphase to become incorporated into 1 of the new daughter cells (Fig. 76-1).
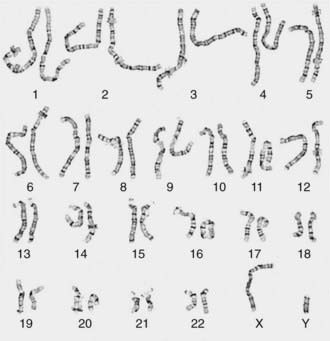
Other banding techniques such as Q-banding using quinacrine, reverse (R-banding) using acridine orange, and C-banding (constitutive heterchromatin) using barium hydroxide are available for use in certain circumstances but are losing ground to molecular technologies. Metaphase chromosome spreads are 1st evaluated microscopically, and then their images are photographed or captured by a video camera and stored on a computer to be later analyzed. Humans have 46 chromosomes or 23 pairs, which are classified as autosomes for chromosomes 1 to 22, and the sex chromosomes, often referred as sex complement: XX for females and XY for males. The homologous chromosomes from a metaphase spread can then be paired and arranged systematically to assemble a karyotype according to well-defined standard conventions like those established by International System for Human Cytogenetic Nomenclature (ISCN), with chromosome 1 being the largest and 22 the smallest. According to nomenclature, the description of the karyotype includes the total number of chromosomes followed by the sex chromosome constitution. A normal karyotype is 46,XX for females and 46,XY for males (Fig. 76-2). Abnormalities are noted after the sex chromosome complement.
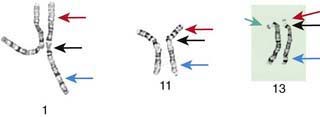
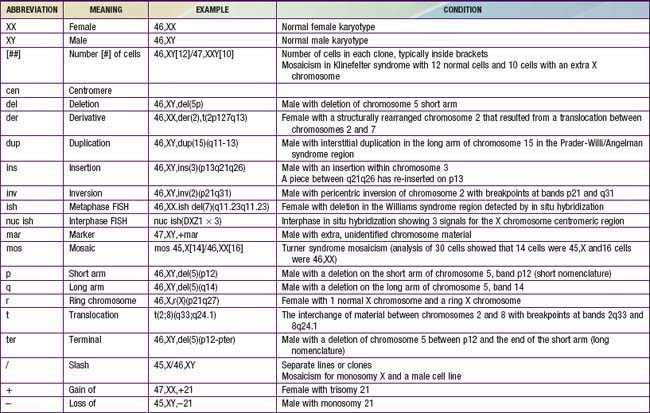
Although the internationally accepted system for human chromosome classification relies largely on the length and banding pattern of each chromosome, the position of the centromere relative to the ends of the chromosome also is a useful distinguishing feature (Fig. 76-3). The centromere divides the chromosome in 2, with the short arm designated as the p arm and the long arm designated as the q arm. A plus or minus sign before the number of a chromosome indicate that there is an extra or missing chromosome, respectively. Table 76-1 lists some of the abbreviations used for the descriptions of chromosomes and their abnormalities. A metaphase chromosome spread usually shows 450-550 bands. Prophase and prometaphase chromosomes are longer, are less condensed, and often show 550-850 bands. High-resolution analysis is useful for detecting subtle chromosome abnormalities that might otherwise go unrecognized.
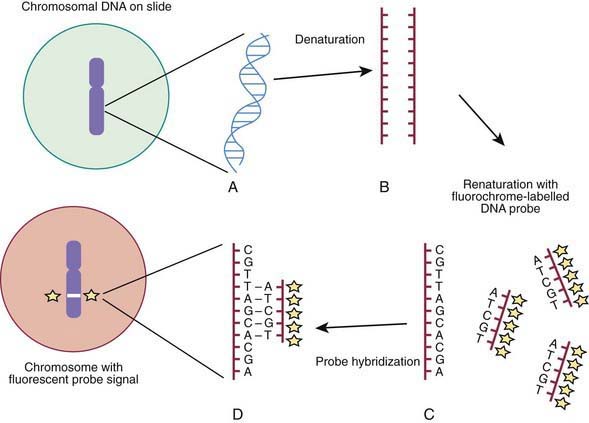
Molecular techniques such as fluorescence in situ hybridization (FISH) and comparative array genomic hybridization studies (conventional CGH and array CGH [aCGH]) have filled a significant void for the diagnosing cryptic chromosomal abnormalities. These techniques identify subtle abnormalities that are often below the resolution of standard cytogenetic studies. FISH is used to identify the presence, absence, or rearrangement of specific DNA segments and is performed with gene- or region-specific DNA probes. Several FISH probes are used in the clinical setting: unique sequence or single-copy probes, repetitive-sequence probes (alpha satellites in the pericentromeric regions), and multiple-copy probes (chromosome specific or painting). FISH involves using a unique known DNA sequence or probe labeled with a fluorescent dye that is complementary to the studied region of disease interest. The labeled probe is exposed to the DNA on a microscope slide, typically metaphase or interphase chromosomal DNA, that has been previously treated (denatured) to allow the DNA to become single stranded and to permit hybridization. When the probe pairs with its complementary DNA sequence, it can be then visualized by fluorescence microscopy (Fig. 76-4). In metaphase chromosome spreads, the exact chromosomal location of each probe copy can be documented and often the number of copies (deletions, duplications) of the DNA sequence as well, if they are not too close to each other; whereas in interphase cells, only the number of copies of a particular DNA segment can be determined. When the interrogated segments (as in genomic duplications) are close together, only interphase cells can accurately determine the presence of 2 or more copies or signals. In metaphase cells, some duplications might falsely appear as a single signal.
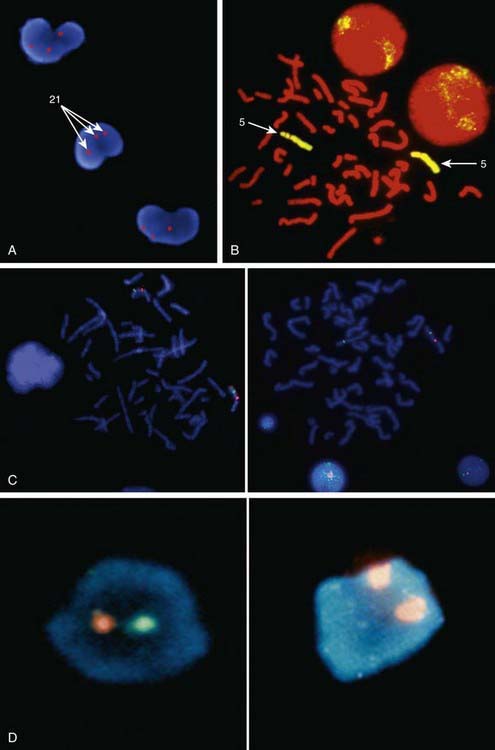
Metaphase and interphase FISH are particularly useful for detecting very small deletions that might escape notice with G-band analysis. In most cases the probe used for identification is used in conjunction with a control probe with a known location nearby the region studied. This allows correct identification of the hybridized signal to the right chromosome, and in some cases identification to the rearranged chromosome. With high-resolution chromosome analysis it is very difficult to recognize deletions of <5 million bp (5 Mb); FISH can reliably detect deletions as small as 50 to 200 kb of DNA. This has allowed the clinical characterization of a number of microdeletion syndromes. In addition to gene- or locus-specific probes, complex mixtures of DNA from a chromosome arm or an entire chromosome are available for fluorescence staining of large chromosome sections or entire chromosomes. The probe mixtures are referred to as chromosome paints (Fig. 76-5A and B). Other probes hybridize to repetitive sequences located to the pericentromeric regions. These probes are useful for the rapid identification of certain trisomies in interphase cells of blood smears, or even in the rapid analysis of prenatal samples from cells obtained through amniocentesis. Such probes are available for chromosomes 13, 18, and 21 and for the sex pair X and Y (Fig. 76-5C and D).
Comparative genomic hybridization (CGH) is a molecular-based technique that involves differentially labeling the patient’s DNA with a fluorescent dye (green) and a normal reference DNA with another fluorescent dye (red) (Fig. 76-6). Equal amounts of the two-label DNA samples are mixed and then used as a painting probe for FISH with normal metaphase chromosomes. The ratio of green : red fluorescence is measured along each chromosome. Regions of amplification of the patient’s DNA display an excess of green fluorescence, and regions of loss show excess red fluorescence. If the patient’s and the control DNA are equally represented, the green : red ratio is 1 : 1 and the chromosomes appear yellow.
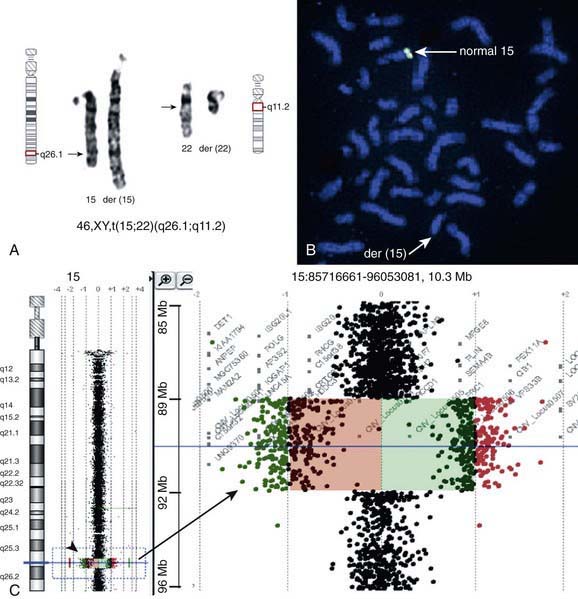
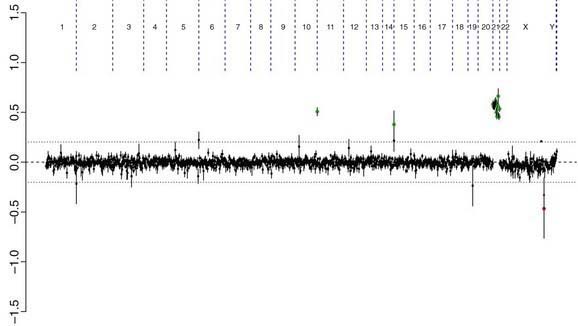
There are many copy number variations (CNVs) causing deletion or duplication in the human genome. Thus, most detected genetic abnormalities, unless associated with very well known clinical phenotypes, require parental investigations because a detected CNV that is inherited might turn out to be an incidental polymorphic variant. A de novo abnormality (i.e., 1 found only in the child and not the parents) is often more significant if it is associated with an abnormal phenotype and if it involves genes with important functions. aCGH is a very valuable technology alone or when combined with FISH and conventional chromosome studies (Fig. 76-7).
ACOG Practice Bulletin. Screening for fetal chromosomal abnormalities. Obstet Gynecol. 2007;109:217-227.
Bejjani BA, Saleki R, Ballif BC, et al. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet. 2005;134A:259-267.
Li MM, Andersson HC. Clinical application of microarray-based molecular cytogenetics: an emerging new era of genomic medicine. J Pediatr. 2009;155:311-317.
Saugier-Veber P, Girard-Lemaire F, Rudolf G, et al. Genetic compensation in a human genomic disorder. N Engl J Med. 2009;360:1211-1216.
76.2 Down Syndrome and Other Abnormalities of Chromosome Number
Aneuploidy and Polyploidy
The most common cause of aneuploidy is nondisjunction, the failure of chromosomes to disjoin normally during meiosis (see Fig. 76-1). Nondisjunction can occur during meiosis I or II or during mitosis. After meiotic nondisjunction, the resulting gamete either lacks a chromosome or has 2 copies instead of 1 normal copy, resulting in a monosomic or trisomic zygote, respectively.
FISH is a technique that can be used for rapid diagnosis in the prenatal detection of common fetal aneuploidies including chromosomes 13, 18, and 21 as well as sex chromosomes (see Fig. 76-5C and D). The most common numerical abnormalities in liveborn children include trisomy 21 (Down syndrome), trisomy 18 (Edwards syndrome), trisomy 13 (Patau syndrome), and sex chromosomal aneuploidies: Turner syndrome (usually 45,X), Klinefelter syndrome (47,XXY), 47,XXX, and 47,XYY, By far the most common type of trisomy in liveborn infants is trisomy 21 (47,XX,+21 or 47,XY,+21). Trisomy 18 and trisomy 13 are relatively less common and are associated with a characteristic set of congenital anomalies and severe mental retardation (Table 76-2). The occurrence of trisomy 21 and other trisomies increases with advanced maternal age (≥35 yr). Owing to this increased risk, women who are ≥35 yr at the time of delivery should be offered genetic counseling and prenatal diagnosis (including serum screening, ultrasonography, and amniocentesis or chorionic villus sampling; Chapter 90).
Table 76-2 CHROMOSOMAL TRISOMIES AND THEIR CLINICAL FINDINGS
| SYNDROME | INCIDENCE | CLINICAL MANIFESTATIONS |
|---|---|---|
| Trisomy 13, Patau syndrome | 1/10,000 births |
Cleft lip often midline; flexed fingers with postaxial polydactyly; ocular hypotelorism, bulbous nose; low-set, malformed ears; microcephaly; cerebral malformation, especially holoprosencephaly; microphthalmia, cardiac malformations; scalp defects; hypoplastic or absent ribs; visceral and genital anomalies
|
Down Syndrome
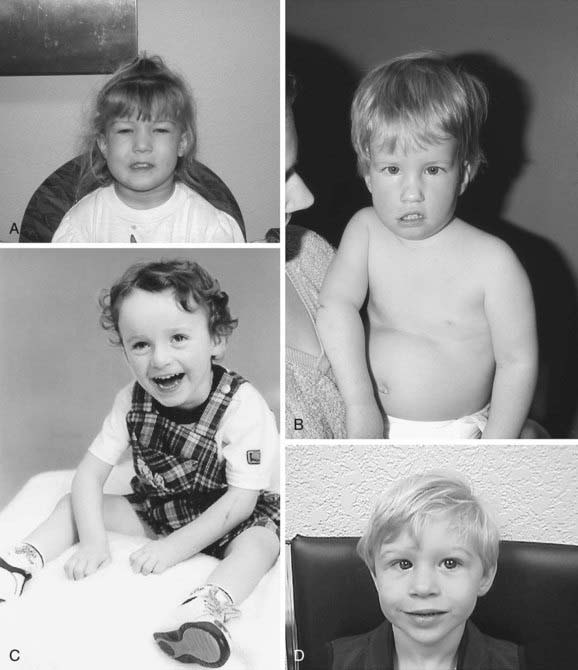
Trisomy 21 is the most common genetic cause of moderate mental retardation. The incidence of Down syndrome in live births is approximately 1 in 733; the incidence at conception is more than twice that rate; the difference is accounted by early pregnancy losses. In addition to cognitive impairment, Down syndrome is associated with congenital anomalies and characteristic dysmorphic features (Figs. 76-8 and 76-9; Table 76-3). Although there is variability in the clinical features, the constellation of phenotypic features is fairly consistent and permits clinical recognition of trisomy 21. Affected individuals are more prone to congenital heart defects (50%) such as atrioventricular septal defects, ventricular septal defects, isolated secundum atrial septal defects, patent ductus arteriosus, and tetralogy of Fallot. Congenital and acquired gastrointestinal anomalies and hypothyroidism are common (Table 76-4). Other abnormalities include megakaryoblastic leukemia, immune dysfunction, diabetes mellitus, and problems with hearing and vision (see Table 76-4). Alzheimer disease–like dementia is a known complication that occurs as early as the 4th decade and has an incidence 2-3 times higher than sporadic Alzheimer disease. Most males with Down syndrome are sterile, but some females have been able to reproduce, with a 50% chance of having trisomy 21 pregnancies. Two genes (DYRK1A, DSCR1) in the putative critical region of chromosome 21 may be targets for therapy.

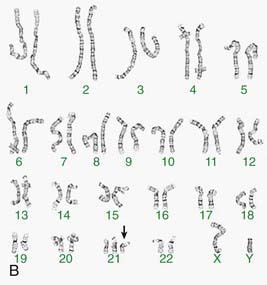
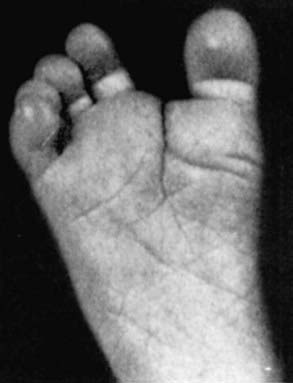
Figure 76-9 Prehensile foot in a 1-mo-old child.
(From Wiedemann HR, Kunze J, Dibbern H: Atlas of clinical syndromes: a visual guide to diagnosis, ed 3, St Louis, 1989, Mosby.)
Table 76-3 CLINICAL FEATURES OF DOWN SYNDROME IN THE NEONATAL PERIOD
CENTRAL NERVOUS SYSTEM
CRANIOFACIAL
CARDIOVASCULAR
MUSCULOSKELETAL
GASTROINTESTINAL
CUTANEOUS
Cutis marmorta
Table 76-4 ADDITIONAL FEATURES OF DOWN SYNDROME THAT CAN DEVELOP OR BECOME SYMPTOMATIC WITH TIME
NEUROPSYCHIATRIC
SENSORY
CARDIOVASCULAR
MUSCULOSKELETAL
ENDOCRINE
HEMATOLOGIC
GASTROINTESTINAL
CUTANEOUS
Developmental delay is universal (Tables 76-5 and 76-6; Fig. 76-10). Cognitive impairment does not uniformly affect all areas of development. Social development is relatively spared, but children with Down syndrome have considerable difficulty using expressive language. Understanding these individual developmental strengths will maximize the educational process for children with Down syndrome. Persons with Down syndrome often benefit from programs aimed at stimulation, development, and education. These programs are most effective in addressing social skills that often appear advanced for the intellectual delay. Children with Down syndrome also benefit from anticipatory guidance, which establishes the protocol for screening, evaluation, and care for patients with genetic syndromes and chronic disorders (Table 76-7).
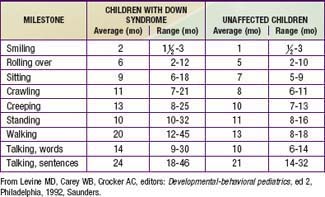
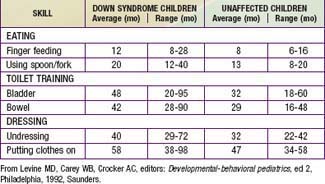
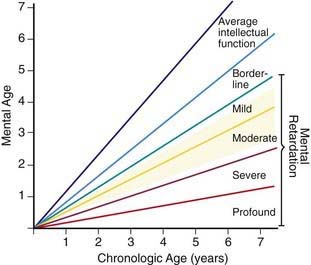
Figure 76-10 The area shaded in yellow denotes the range of intellectual function of the majority of children with Down syndrome.
(From Levine MD, Carey WB, Crocker AC, editors: Developmental-behavioral pediatrics, ed 2, Philadelphia, 1992, WB Saunders, p 226.)
Table 76-7 HEALTH SUPERVISION FOR CHILDREN WITH DOWN SYNDROME
| CONDITION | TIME TO SCREEN | COMMENT |
|---|---|---|
| Congenital heart disease |
IgA, immunoglobulin A; IgG, immunoglobulin G.
Extracted from Committee on Genetics: Health supervision for children with Down syndrome, Pediatrics 107:442–449, 2001; and Baum RA, Spader M, Nash PL, et al: Primary care of children and adolescents with Down syndrome: an update, Curr Prob Pediatr Adolesc Health Care 38:235–268, 2008.
In approximately 95% of the cases of Down syndrome there are 3 copies of chromosome 21. The origin of the supernumerary chromosome 21 is maternal in 97% of the cases as a result of errors in meiosis. The majority of these occur in maternal meiosis I (90%). Approximately 1% of persons with trisomy 21 are mosaics, with some cells having 46 chromosomes, and another 4% of have a translocation that involves chromosome 21. The majority of translocations in Down syndrome are fusions at the centromere between chromosomes 13, 14, 15, 21, and 22 known as Robertsonian translocations. The translocations can be de novo or inherited. Very rarely is Down syndrome diagnosed in a patient with only a part of the long arm of chromosome 21 in triplicate (partial trisomy). Isochromosomes and ring chromosomes are other rarer causes of trisomy 21. Down syndrome patients without a visible chromosome abnormality are the least common. It is not possible to distinguish the phenotypes of persons with full trisomy 21 and those with a translocation. Representative genes on chromosome 21 and their potential effects on development are noted in Table 76-8. Patients who are mosaic tend to have a milder phenotype.
Table 76-8 GENES LOCALIZED TO CHROMOSOME 21 THAT POSSIBLY AFFECT BRAIN DEVELOPMENT, NEURONAL LOSS, AND ALZHEIMER TYPE NEUROPATHOLOGY
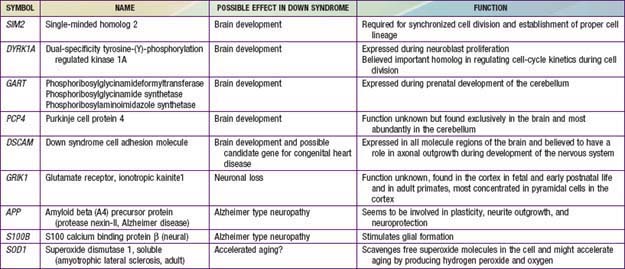
Tables 76-9 and 76-10 provide more information on other aneuploidies and partial autosomal aneuploidies (Figs. 76-11 to 76-14).
Table 76-9 OTHER RARE ANEUPLOIDIES AND PARTIAL AUTOSOMAL ANEUPLOIDIES
| DISORDER | KARYOTYPE | CLINICAL MANIFESTATIONS |
|---|---|---|
| Trisomy 8 | 47,XX/XY,+8 | |
| Trisomy 9 | 47,XX/XY,+9 | |
| Trisomy 16 | 47,XX/XY,+16 | The most commonly observed autosomal aneuploidy in spontaneous abortion; the recurrence risk is negligible |
| Tetrasomy 12p | 46,XX[12]/46,XX, +i(12p)[8] (mosaicism for an isochromosome 12p) | Known as Pallister-Killian syndrome. Sparse anterior scalp hair, eyebrows, and eyelashes, prominent forehead, chubby cheeks, long philtrum with thin upper lip and cupid-bow configuration, polydactyly, and streaks of hyper- and hypopigmentation |
Table 76-10 FINDINGS THAT MAY BE PRESENT IN TRISOMY 13 AND TRISOMY 18
| TRISOMY 13 | TRISOMY 18 |
|---|---|
| HEAD AND FACE | |
| CHEST | |
| EXTREMITIES | |
| GENERAL | |
VSD, ventricular septal defect; PDA, patent ductus arteriosus; ASD, atrial septal defect.
From Behrman RE, Kliegman RM: Nelson essentials of pediatrics, ed 4, Philadelphia, 2002, WB Saunders, p 142.

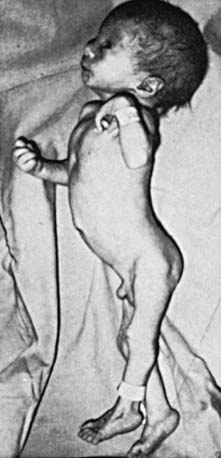
Figure 76-11 Facial appearance of a child with trisomy 13.
(From Wiedemann HR, Kunze J, Dibbern H: Atlas of clinical syndromes: a visual guide to diagnosis, ed 3, St Louis, 1989, Mosby.)
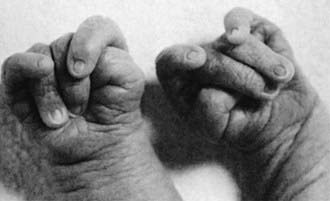
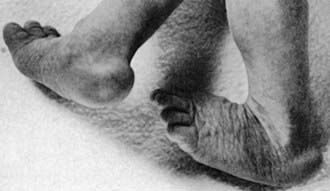
Figure 76-12 Trisomy 18: overlapping fingers and hypoplastic nails.
(From Wiedemann HR, Kunze J, Dibbern H: Atlas of clinical syndromes: a visual guide to diagnosis, ed 3, St Louis, 1989, Mosby.)
Alfirevic Z. Prenatal screening for Down’s syndrome. BMJ. 2009;338:421-422.
Baum RA, Nash PL, Foster JEA, et al. Primary care of children and adolescents with Down syndrome: an update. Curr Prob Pediatr Adolect Health Care. 2008;38:235-268.
Caine A, Maltby AE, Parkin CA, et al. Prenatal detection of Down’s syndrome by rapid aneuploidy testing for chromosomes 13, 18, and 21 by FISH or PCR without a full karyotype: a cytogenetic risk assessment. Lancet. 2005;366:123-128.
Carter M, McCaughey E, Annaz D, Hill CM. Sleep problems in a Down syndrome population. Arch Dis Child. 2009;94:308-310.
Chiu RWK, Akolekar R, Zheng YWL, et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ. 2011;342:217.
Cicero S, Bindra R, Rembouskos G, et al. Integrated ultrasound and biochemical screening for trisomy 21 using fetal nuchal translucency, absent fetal nasal bone, free β-hCG and PAPP-A at 11 to 14 weeks. Prenat Diagn. 2003;23:306-310.
Dennis J, Archer N, Ellis J, et al. Recognising heart disease in children with Down syndrome. Arch Dis Child Educ Pract Ed. 2010;95:98-104.
Dykens EM. Psychiatric and behavioral disorders in persons with Down syndrome. Mental Ret Dev Dis Res Rev. 2007;13:272-278.
Einfeld SL, Brown R. Down syndrome—new prospects for an ancient disorder. JAMA. 2010;303:2525-2526.
Garrison MM, Jeffries H, Christakis DA. Risk of death for children with Down syndrome and sepsis. J Pediatr. 2005;147:748-752.
Gibson PA, Newton RW, Selby K, et al. Longitudinal study of thyroid function in Down’s syndrome in the first two decades. Arch Dis Child. 2005;90:574-578.
Irving C, Basu A, Richmond S, et al. Twenty-year trends in prevalence and survival of Down syndrome. Eur J Hum Genet. 2008;16:1336-1340.
Juj H, Emery H. The arthropathy of Down syndrome: an undiagnosed and under-recognized condition. J Pediatr. 2009;154:234-238.
Malone FD, Canick JA, Ball RH, et al. First-trimester or second-trimester screening, or both, for Down’s syndrome. N Engl J Med. 2005;353:2001-2010.
McDowell KM, Craven DI. Pulmonary complications of Down syndrome during childhood. J Pediatr. 2011;158:319-325.
Morris JK, Alberman E. Trends in Down’s syndrome live births and antenatal diagnosis in England and Wales from 1989 to 2008: analysis of data from the national Down syndrome cytogenetic register. BMJ. 2009;339:b3794.
Papageorgiou EA, Karagrigoriou A, Tsaliki E, et al. Fetal-specific DNA methylation ratio permits noninvasive prenatal diagnosis of trisomy 21. Nat Med. Mar 6, 2011. epub, doi: 10.1038/nm.2312
Rasmussen SA, Whitehead N, Collier SA, et al. Setting a public health research agenda for Down syndrome: summary of a meeting sponsored by the CDC and the national Down syndrome society. Am J Med Genet. 2008;46A:2998-3010.
Reynolds T. Giving antioxidants to infants with Down’s syndrome. BMJ. 2008;336:568-570.
Shin M, Besser LM, Kucik JE, et al. Prevalence of Down syndrome among children and adolescents in 10 regions of the United States. Pediatrics. 2009;124:1565-1571.
Shott SR, Amin R, Chini B, et al. Obstructive sleep apnea: should all children with Down syndrome be tested? Arch Otolaryngol Head Surg. 2006;132:432-436.
Webb D, Roberts I, Vyas P. Haematology of Down syndrome. Arch Dis Child. 2007;92:F503-F507.
Weijerman ME, Van Furth M, Noordegraaf AV, et al. Prevalence, neonatal characteristics, and first-year mortality of Down syndrome: a national study. J Pediatr. 2008;152:15-19.
Wouters J, Weijerman ME, Van Forth AM, et al. Prospective human leukocyte antigen, endomysium immunoglobulin A antibodies, and transglutaminase antibodies testing for celiac disease in children with Down syndrome. J Pediatr. 2009;154:239-242.
76.3 Abnormalities of Chromosome Structure
Translocations
Translocations, which involve the transfer of material from 1 chromosome to another, occur with a frequency of 1/500 liveborn human infants. They may be inherited from a carrier parent or appear de novo, with no other affected family member. Translocations are commonly reciprocal or Robertsonian, involving 2 chromosomes (Fig. 76-15).
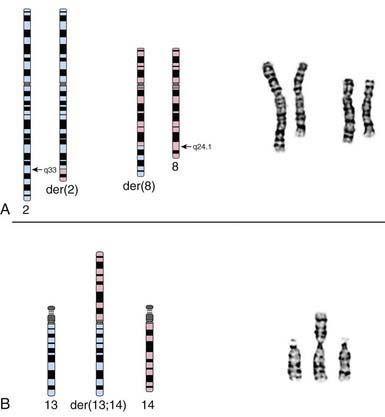
Deletions and Duplications
A carrier of a deletion is monosomic for the genetic information of the missing segment. Deletions are usually associated with mental retardation and malformations. The most commonly observed deletions in routine chromosome preparations include 1p-, 4p-, 5p-, 9p-, 11p-, 13q-, 18p-, 18q-, and 21q- (Table 76-11 and Fig. 76-16), all distal or terminal deletions of the short or the long arms of chromosomes. Deletions may be observed in routine chromosome preparations, and deletions and translocations larger than 5-10 Mb are usually visible microscopically.
Table 76-11 COMMON DELETIONS AND THEIR CLINICAL MANIFESTATIONS
| DELETION | CLINICAL ABNORMALITIES |
|---|---|
| 4p- | Wolf-Hirschhorn syndrome. The main features are a typical “Greek helmet” facies secondary to ocular hypertelorism, prominent glabella, and frontal bossing; microcephaly, dolichocephaly, hypoplasia of the orbits, ptosis, strabismus, nystagmus, bilateral epicanthic folds, cleft lip and palate, beaked nose with prominent bridge, hypospadias, cardiac malformations, and mental retardation. |
| 5p- | Cri-du-chat syndrome. The main features are hypotonia, short stature, characteristic shrill cry in the first few weeks of life (cat like cry), microcephaly with protruding metopic suture, hypertelorism, bilateral epicanthic folds, high arched palate, wide and flat nasal bridge, and mental retardation. |
| 9p- | The main features are craniofacial dysmorphology with trigonocephaly, slanted palpebral fissures, discrete exophthalmos secondary to supraorbital hypoplasia, arched eyebrows, flat and wide nasal bridge, short neck with low hairline, genital anomalies, long fingers and toes with extra flexion creases, cardiac malformations, and mental retardation. |
| 13q- | The main features are low birthweight, failure to thrive, microcephaly, and severe mental retardation. Facial features include high wide nasal bridge, hypertelorism, ptosis, micrognathia. Ocular malformations are common (retinoblastoma). The hands have hypoplastic or absent thumbs and syndactyly. |
| 18p- | A few patients (15%) are severely affected and have cephalic and ocular malformations: holoprosencephaly, cleft lip and palate, ptosis, epicanthal folds, and varying degrees of mental retardation. Most (80%) have only minor malformations and mild mental retardation. |
| 18q- | Growth deficiency, hypotonia with “froglike” position with the legs flexed, externally rotated, and in hyperabduction. The face is characteristic with depressed midface and apparent protrusion of the mandible, deep-set eyes, short upper lip, everted lower lip (“carplike” mouth); antihelix of the ears is very prominent; varying degrees of mental retardation and belligerent personality. Myelination abnormalities in the CNS. |
High-resolution banding techniques, FISH, and molecular studies like aCGH can reveal deletions that are too small to be seen in ordinary or routine chromosome spreads (see Fig. 76-7). Microdeletions involve loss of small chromosome regions, the largest of which are detectable only with prophase chromosome studies and/or molecular methods. For submicroscopic deletions, the missing piece can only be detected using molecular methodologies such as FISH or DNA-based studies like aCGH. The presence of extra genetic material from the same chromosome is referred to as duplication. Duplications can also be sporadic or result from abnormal segregation in translocation or inversion carriers.
Microdeletions and microduplications usually involve regions that include several genes, so that the affected individuals can have a distinctive phenotype depending on the number of genes involved. When such a deletion involves more than a single gene, the condition is referred to as a contiguous gene deletion syndrome (Table 76-12). With the advent of clinically available aCGH, a large number of duplications, most of them microduplications, have been uncovered. Most of those microduplication syndromes are the reciprocal duplications of the known deletions or microdeletion counterparts and have distinctive clinical features (Table 76-13).
Table 76-12 MICRODELETION AND CONTIGUOUS GENE SYNDROMES AND THEIR CLINICAL MANIFESTATIONS
| DELETION | SYNDROME | CLINICAL MANIFESTATIONS |
|---|---|---|
| 1p36 | 1p deletion | Growth retardation, dysmorphic features with midface hypoplasia, straight thin eyebrows, pointy chin, sensorineural hearing loss, progressive cardiomyopathy, hypothyroidism, seizures, mental retardation |
| 5q35 | Sotos (50% are deletions of NSD1 gene in Asians but only 6% in whites) | Overgrowth, macrocephaly, prominent forehead, prominence of extra-axial fluid spaces on brain imaging, large hands and feet, hypotonia, clumsiness, mental disabilities |
| 6p25 | Axenfeld-Rieger | Axenfeld-Rieger malformation, hearing loss, congenital heart defects, dental anomalies, developmental delays, facial dysmorphism |
| 7q11.23 | Williams | Round face with full cheeks and lips, long philtrum, stellate pattern in iris, strabismus, supravalvular aortic stenosis and other cardiac malformations, varying degrees of mental retardation, friendly personality |
| 8p11 | 8p11 | Kallman syndrome 2 (hypogonadotropic hypogonadism and anosmia), spherocytosis (deletions of ankyrin 1), multiple congenital anomalies, mental retardation |
| 8q24.1-q24.13 | Langer-Giedion or trichorhinophalangeal type II | Sparse hair, multiple cone-shaped epiphyses, multiple cartilaginous exostoses, bulbous nasal tip, thickened alar cartilage, upturned nares, prominent philtrum, large protruding ears, mild mental retardation |
| 9q22 | Gorlin | Multiple basal cell carcinomas, odontogenic keratocysts, palmoplantar pits, calcification falx cerebri |
| 9q34 | 9q34 deletion | Distinct face with synophrys, anteverted nares, tented upper lip, protruding tongue, midface hypoplasia, conotruncal heart defects, mental retardation |
| 10p12-p13 | DiGeorge 2 | Many of the DiGeorge 1 and velocardiofacial 1 features (conotruncal defects, immunodeficiency, hypoparathyroidism, dysmorphic features) |
| 11p11.2 | Potocki-Shaffer | Multiple exostoses, parietal foramina, craniosynostosis, facial dysmorphism, syndactyly, mental retardation |
| 11p13 | WAGR | Hypernephroma (Wilms tumor), aniridia, male genital hypoplasia of varying degrees, gonadoblastoma, long face, upward slanting palpebral fissures, ptosis, beaked nose, low-set poorly formed auricles, mental retardation |
| 11q24.1-11qter | Jacobsen | Mental and growth retardation, cardiac and digit anomalies, thrombocytopenia |
| 15q11-q13 (pat) | Prader-Willi | Severe hypotonia and feeding difficulties at birth, voracious appetite and obesity in infancy, short stature (responsive to growth hormone), small hands and feet, hypogonadism, mental retardation |
| 15q11-q13 (mat) | Angelman | Hypotonia, feeding difficulties, GE reflux, fair hair and skin, midface hypoplasia, prognathism, seizures, tremors, ataxia, sleep disturbances, inappropriate laughter, poor or absent speech, severe mental retardation |
| 16p13.3 | Rubinstein-Taybi | Microcephaly, ptosis, beaked nose with low-lying philtrum, broad thumbs and large toes, mental retardation |
| 17p11.2 | Smith-Magenis | Brachycephaly, midfacial hypoplasia, prognathism, myopia, cleft palate, short stature, severe behavioral problems, mental retardation |
| 17p13.3 | Miller-Dieker | Microcephaly, lissencephaly, pachygyria, narrow forehead, hypoplastic male external genitals, growth retardation, seizures, profound mental retardation |
| 20p12 | Alagille syndrome | Bile duct paucity with cholestasis; heart defects, particularly pulmonary artery stenosis; ocular abnormalities (posterior embryotoxon); skeletal defects such as butterfly vertebrae; long nose |
| 22q11.2 | Velocardiofacial-DiGeorge syndrome | Conotruncal cardiac anomalies, cleft palate, velopharyngeal incompetence, hypoplasia or agenesis of the thymus and parathyroid glands, hypocalcemia, hypoplasia of auricle, learning disabilities, psychiatric disorders |
| 22q13.3 deletion | Hypotonia, developmental delay, normal or accelerated growth, severe expressive language deficits, autistic behavior | |
| Xp21.2-p21.3 | Duchenne muscular dystrophy, retinitis pigmentosa, adrenal hypoplasia, mental retardation, glycerol kinase deficiency | |
| Xp22.2-p22.3 | Ichthyosis, Kallman syndrome, mental retardation, chondrodysplasia punctata | |
| Xp22.3 | Microphthalmia with linear defects (MLS) | Microphthalmia, linear skin defects, poikiloderma, congenital heart defects, seizures, mental retardation |
pat, paternal; mat, maternal; GE, gastroesophageal.
Table 76-13 MICRODUPLICATIONS AND THEIR CLINICAL MANIFESTATIONS
| DUPLICATION CHROMOSOME REGION | DISEASE REGION | CLINICAL FEATURES |
|---|---|---|
| 1q21.1 | Macrocephaly, DD, learning disabilities | |
| 3q29 | Mild to moderate MR, microcephaly | |
| 7q11.23 | Williams syndrome | DD and severe expressive language disorder, autistic features, subtle dysmorphisms |
| 15q13.3 | Prader-Willi/Angelman syndrome region | DD, MR, autistic features in duplications of maternal origin |
| 15q24 | Growth retardation, DD, microcephaly, digital anomalies, hypospadias, connective tissue abnormalities | |
| 16p11.2 | FTT, severe DD, short stature, GH deficiency, dysmorphic features | |
| 17p11.2 | Potocki-Lupski syndrome | Hypotonia, cardiovascular anomalies, FTT, DD, verbal apraxia, autism, anxiety |
| 17q21.31 | Severe DD, microcephaly, short and broad digits, dysmorphic features | |
| 22q11.2 | Velocardiofacial-DiGeorge syndrome | Cardiovascular defects, velopharyngeal insufficiency |
| Xq28 | MECP2 gene region (Rett syndrome) | In males: infantile hypotonia, immune deficiency, dysmorphic features, DD, speech delay, autistic behavior, regression in childhood |
DD, developmental delay; MR, mental retardation; FTT, failure to thrive; GH, growth hormone.
Battaglia A, Hoyme HE, Dallapiccola B, et al. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121:404-410.
Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353-2365.
Fillion M, Deal C, Van Vliet G. Retrospective study of the potential benefits and adverse events during growth hormone treatment in children with Prader-Willi syndrome. J Pediatr. 2009;154:230-233.
Gicquel C, Rossignol S, Cabrol S, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. 2005;37:1003-1007.
Mefford H, Shapr A, Baker C, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685-1698.
Peters J. Prader-Willi and snoRNAs. Nat Genet. 2008;40:688-689.
Rappold GA, Shanske A, Saenger P. All shook up by SHOX deficiency. J Pediatr. 2005;147:422-424.
Robin NH, Shprintzen RJ. Defining the clinical spectrum of deletion 22q11.2. J Pediatr. 2005;147:90-96.
Sahoo T, del Gaudio D, German JR, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719-721.
Sharp AJ, Mefford HC, Li K, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322-328.
Stafler P, Wallis C, et al. Prader-Willi syndrome: who can have growth hormone? Arch Dis Child. 2008;93:341-345.
Vissers LELM, van Ravenswaaji CMA, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955-957.
Walter S, Sandig K, Hinkel GK, et al. Subtelomere FISH in 50 children with mental retardation and minor anomalies, identified by a checklist, selects 10 rearrangements including a de novo balanced translocation of chromosomes 17p13.3 and 20q13.33. Am J Med Genet. 2004;128A:364-373.
Youings S, Ellis K, Ennis S, et al. A study of reciprocal translocations and inversions detected by light microscopy with special reference to origin, segregation, and recurrent abnormalities. Am J Med Genet. 2004;126A:46-60.
76.4 Sex Chromosome Aneuploidy
About 1/400 males and 1/650 females have some form of sex chromosome abnormality. Considered together, sex chromosome abnormalities are the most common chromosome abnormalities seen in liveborn infants, children, and adults. Sex chromosome abnormalities can be either structural or numerical and can be present in all cells or in a mosaic form. Those affected with these abnormalities might have few or no physical or developmental problems (Table 76-14).
| DISORDER | KARYOTYPE | APPROXIMATE INCIDENCE |
|---|---|---|
| Klinefelter syndrome | 47,XXY | 1/575-1/1,000 males |
| 48,XXXY | 1/50,000-1/80,000 male births | |
| Other (48,XXYY; 49,XXXYY; mosaics) | ||
| XYY syndrome | 47,XYY | 1/800-1,000 males |
| Other X or Y chromosome abnormalities | 1/1,500 males | |
| XX males | 46,XX | 1/20,000 males |
| Turner syndrome | 45,X | 1/2,500-1/5,000 females |
| Variants and mosaics | ||
| Trisomy X | 47,XXX | 1/1,000 females |
| 48,XXXX and 49,XXXXX | Rare | |
| Other X chromosome abnormalities | 1/3,000 females | |
| XY females | 46,XY | 1/20,000 females |
Turner Syndrome
Turner syndrome is a condition characterized by complete or partial monosomy of the X chromosome and defined by a combination of phenotypic features (Table 76-15). Half of the patients with Turner syndrome have a 45,X chromosome complement. The other half exhibits mosaicism and varied structural abnormalities of the X or Y chromosome. Maternal age is not a predisposing factor for children with 45,X. Turner syndrome occurs in approximately 1/5,000 female live births. In 75% of patients, the lost sex chromosome is of paternal origin (whether an X or a Y). 45,X is 1 of the chromosome abnormalities most often associated with spontaneous abortion. It has been estimated that 95-99% of 45,X conceptions are miscarried.
Clinical findings in the newborns can include small size for gestational age, webbing of the neck, protruding ears, and lymphedema of the hands and feet, although many newborns are phenotypically normal (Fig. 76-17). Older children and adults have short stature and exhibit variable dysmorphic features. Congenital heart defects (40%) and structural renal anomalies (60%) are common. The most common heart defects are bicuspid aortic valves, coarctation of the aorta, aortic stenosis, and mitral valve prolapse. The gonads are generally streaks of fibrous tissue (gonadal dysgenesis). There is primary amenorrhea and lack of secondary sex characters. These children should receive regular endocrinologic testing (Chapter 580). Most patients tend to be of normal intelligence, but mental retardation is seen in 6% of affected children. They are also at increased risk for behavioral problems and deficiencies in spatial and motor perception. Guidelines for health supervision for children with Turner syndrome are published by the American Academy of Pediatrics (AAP).
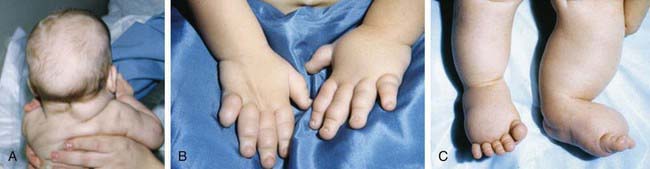
Figure 76-17 Redundant nuchal skin (A) and puffiness of the hands (B) and feet (C) in Turner syndrome.
(From Sybert VP, McCauley E: Turner’s syndrome, N Engl J Med 351:1227–1238, 2004. Copyright © 2004 Massachusetts Medical Society. All rights reserved.)
Noonan syndrome shares many clinical features with Turner syndrome, although it is an autosomal dominant disorder resulting from mutations in several genes that are involved in the RAS-MAPK (mitogen activated protein kinase) pathway. The most common of these is PTPN11 (50%), which encodes a nonreceptor tyrosine kinase (SHP-2) on chromosome 12q24.1. Other genes include SOS1 in 10-15%, RAF1 in 3-8%, and KRAS in 5%. Features common to Noonan syndrome include short stature, low posterior hairline, shield chest, congenital heart disease, and a short or webbed neck (Table 76-16). In contrast to Turner syndrome, Noonan syndrome affects both sexes and has a different pattern of congenital heart disease typically involving right-sided lesions.
Klinefelter Syndrome
Persons with Klinefelter syndrome are phenotypically male; this syndrome is the most common cause of hypogonadism and infertility in males and the most common sex chromosome aneuploidy in humans (Chapter 577). Eighty percent of children with Klinefelter syndrome have a male karyotype with an extra chromosome X-47,XXY; the remaining 20% have multiple sex chromosome aneuploidies (48,XXXY; 48,XXYY; 49,XXXXY), mosaicism (46,XY/47,XXY), or structurally abnormal X chromosomes. The greater the aneuploidy, the more severe the mental impairment and dysmorphism. Early studies showed that the birth prevalence is approximately 1/1,000 males. The current prevalence of 47,XXY appears to have increased to approximately 1/580 liveborn boys; the reasons for this are still unknown. Errors in paternal nondisjunction in meiosis I account for half of the cases.
Lanfranco F, Kamischke A, Zitzmann M, et al. Klinefelter’s syndrome. Lancet. 2004;364:273-283.
Lin AE. Focus on the heart and aorta in Turner syndrome. J Pediatr. 2007;150:572-574.
Massa G, Verlinde F, De Schepper J, et al. Trends in age at diagnosis of Turner syndrome. Arch Dis Child. 2005;90:267-268.
Mazzanti L, Cicognani A, Baldazzi L, et al. Gonadoblastoma in Turner syndrome and Y-chromosome–derived material. Am J Med Genet. 2005;135A:150-154.
Roberts AE, Araki T, Swanson KD, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70-74.
Schubbert S, Zenker M, Rowe SL, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331-336.
Sybert VP, McCauley E. Turner’s syndrome. N Engl J Med. 2004;351:1227-1238.
Tartaglia M, Pennacchio LA, Zhao C, et al. Gain-of-function SOSI mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75-79.
Zeger MPD, Zinn AR, Lahlou N, et al. Effect of ascertainment and genetic features on the phenotype of Klinefelter syndrome. J Pediatr. 2008;152:716-722.
Zenker M, Gernot Buheitel G, Rauch R, et al. Genotype-phenotype correlations in Noonan syndrome. J Pediatr. 2004;144:368-374.
76.5 Fragile Chromosome Sites
Fragile sites are regions of chromosomes that show a tendency for separation, breakage, or attenuation under particular growth conditions. They appear as a gap in the staining. At least 120 chromosomal loci, many of them heritable, have been identified as fragile sites in the human genome (see Table 75-1).
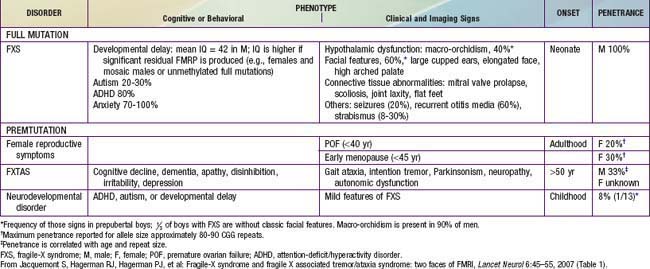
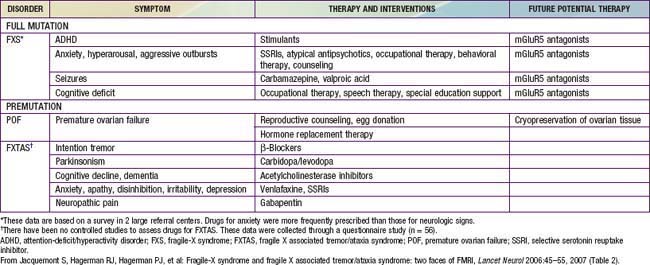
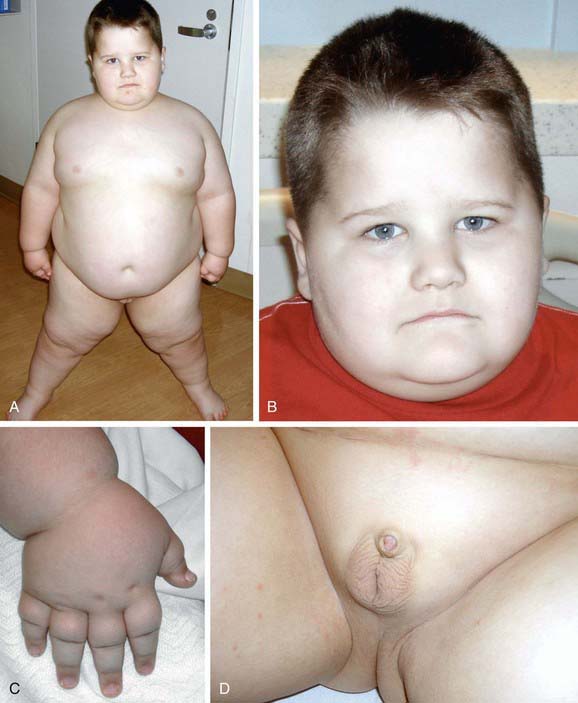
The main clinical manifestations of fragile X syndrome in affected males are mental retardation, autistic behavior, macro-orchidism, and characteristic facial features (Table 76-17). The macro-orchidism may not be evident until puberty. The facial features, which include a long face, large ears, and a prominent square jaw, become more obvious with age. Females affected with fragile X show varying degrees of mental retardation and/or learning disabilities. Diagnosis of fragile X is possible by DNA testing that shows an expansion of a triplet DNA repeat inside the FMR1 gene on the X chromosome. The expansion involves an area of the gene that contains a variable number of trinucleotide (CGG) repeats. The larger the triplet repeat expansion, the more significant the mental retardation. In cases where the expansion is large, females can also manifest different degrees of mental retardation. Therapy of the diverse neuropsychiatric manifestations associated with fragile X syndrome is noted in Table 76-18. Inhibitors of the metabolic glutamate receptor (overexpressed in fragile X) are undergoing clinical trials.
Dobkin C, Radu G, Ding XH, et al. Fragile X prenatal analyses show full mutation females at high risk for mosaic Turner syndrome: fragile X leads to chromosome loss. Am J Med Genet. 2009;149A:2152-2157.
Hagerman RJ, Berry-Kravis E, Kaufman WE, et al. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123:378-390.
Jacquemont S, Curie A, des Portes V, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3:64ra1.
Jacquemont S, Hagerman RJ, Hagerman PJ, et al. Fragile-X syndrome and fragile X associated tremor/ataxia syndrome: two faces of FMRI. Lancet Neurol. 2007;6:45-55.
Kuehn BM. Scientists find promising therapies for fragile X and Down syndrome. JAMA. 2011;305:344-346.
Paribello C, Tao L, Folino A, et al. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010;10:91.
Saul RA, Friez M, Eaves K, et al. Fragile X syndrome detection in newborns—pilot study. Genet Med. 2008;10:714-719.
76.6 Mosaicism
Carlos A. Bacino and Brendan Lee
Mosaicism describes an individual or tissue that contains ≥2 different cell lines typically derived from a single zygote and the result of mitotic nondisjunction (see Fig. 76-1). Study of placental tissue from chorionic villus samples collected at or before the 10th wk of gestation has shown that 2% or more of all conceptions are mosaic for a chromosome abnormality. With the exception of chromosomes 13, 18, and 21, complete autosomal trisomies are usually nonviable; the presence of a normal cell line might allow these other trisomic conceptions to survive to term. Depending on the point at which the new cell line arises during early embryogenesis, mosaicism may be present in some tissues but not in others. Germline mosaicism, which refers to the presence of mosaicism in the germ cells of the gonad, may be associated with an increased risk for recurrence of an affected child whether the germ cells are affected with a chromosomal abnormality or specific gene mutation.
Hypomelanosis of Ito
Hypomelanosis of Ito is characterized by unilateral or bilateral macular hypo- or hyperpigmented whorls, streaks, and patches (Chapter 645). Sometimes these pigmentary defects follow the lines of Blaschko. Hair and tooth anomalies are common. Abnormalities of the eyes, musculoskeletal system (growth asymmetry, syndactyly, polydactyly, clinodactyly), and central nervous system (microcephaly, seizures, mental retardation) may also be present. Patients with hypomelanosis of Ito might have 2 genetically distinct cell lines. The mosaic chromosome anomalies that have been observed involve both autosomes and sex chromosomes and have been demonstrated in about 50% of patients. The mosaicism might not be visible in lymphocyte-derived chromosome studies; it is more likely to be found when chromosomes are analyzed from skin fibroblasts. The distinct cell lines might not always be due to observable chromosomal anomalies but might result from single gene mutations or other mechanisms.
76.8 Uniparental Disomy and Imprinting
Carlos A. Bacino and Brendan Lee
Uniparental Disomy
Uniparental disomy (UPD) occurs when both chromosomes of a pair or areas from 1 chromosome in any individual have been inherited from a single parent. UPD can be of 2 types: uniparental isodisomy or uniparental heterodisomy. Uniparental isodisomy means that both chromosomes or chromosomal regions are identical (typically the result of monosomy rescue by duplication). Uniparental heterodisomy means that the 2 chromosomes are different members of a pair, both of which were still inherited from 1 parent. This results from a trisomy that is later reduced to disomy, leaving 2 copies from 1 parent. The phenotypic result of UPD varies according to the chromosome involved, the parent who contributed the chromosomes, and whether it is isodisomy or heterodisomy. Three types of phenotypic effects are seen in UPD: those related to imprinted genes (i.e., the absence of a gene that is normally expressed only when inherited from a parent of a specific sex), those related to the uncovering of autosomal recessive disorders, and those related to a vestigial aneuploidy producing mosaicism (Chapter 75).
UPD for chromosome 15 is seen in some cases of Prader–Willi syndrome and Angelman syndrome. In Prader-Willi syndrome, about 25-29% of cases have maternal UPD (missing the paternal chromosome 15). In Angelman syndrome, paternal UPD of chromosome 15 is rarer and is observed in approximately 5% of the cases (missing the maternal chromosome 15). The phenotype for Prader-Willi syndrome (Fig. 76-18) and Angelman syndrome in cases of UPD is thought to result from the lack of the functional contribution from a particular parent of chromosome 15. In Prader-Willi syndrome the paternal contribution is missing, and the maternal contribution is missing in Angelman syndrome. Prader-Willi may be due to paternal deficiency of HB11-85 snoRNAs (small nucleolar RNAs). These findings suggest that there are differences in function of certain regions of chromosome 15, depending on whether it is inherited from the mother or from the father.
Imprinting
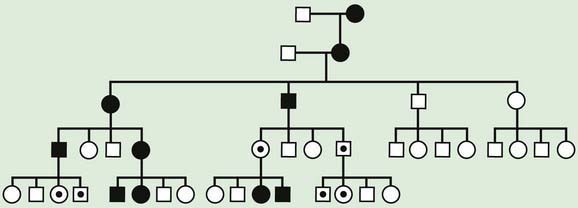
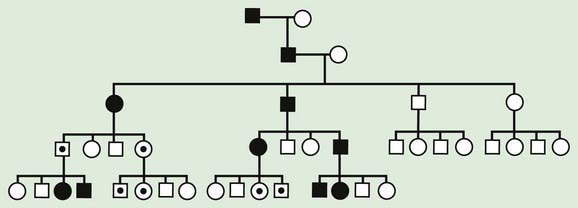
Traditional genetics has for many years suggested that most genes are equally expressed when inherited from maternal vs. paternal lineages. The only exception to this rule were genes on the X chromosome that are subject to inactivation, and the immunoglobulin genes subject to allelic exclusion, a phenomenon that results in monoallelic expression of a particular immunoglobulin chain by switching on and off expression of parental alleles. Genomic imprinting occurs when the phenotypic expression of a gene depends on the parent of origin for certain genes or in some cases entire chromosome regions. Whether the genetic material is expressed or not depends on the sex of the parent from whom it was derived. Genomic imprinting can be suspected in some cases on the basis of a pedigree. In these pedigrees, the disease is always transmitted from 1 sex and could be passed on silently for several generations by the opposite sex (Figs. 76-19 and 76-20). Imprinting probably occurs in many different parts of the human genome and is thought to be particularly important in gene expression related to development, growth, cancer, and even behavior.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 76 Cytogenetics
76.1 Methods of Chromosome Analysis
Two errors of cell division commonly occur during meiosis or mitosis, and either can result in an abnormal number of chromosomes. The 1st is nondisjunction, in which 2 chromosomes fail to separate during meiosis and thus migrate together into 1 of the new cells, producing 1 cell with 2 copies of the chromosome and another with no copy. The 2nd is anaphase lag, in which a chromatid or chromosome is lost during mitosis because it fails to move quickly enough during anaphase to become incorporated into 1 of the new daughter cells (Fig. 76-1).
Other banding techniques such as Q-banding using quinacrine, reverse (R-banding) using acridine orange, and C-banding (constitutive heterchromatin) using barium hydroxide are available for use in certain circumstances but are losing ground to molecular technologies. Metaphase chromosome spreads are 1st evaluated microscopically, and then their images are photographed or captured by a video camera and stored on a computer to be later analyzed. Humans have 46 chromosomes or 23 pairs, which are classified as autosomes for chromosomes 1 to 22, and the sex chromosomes, often referred as sex complement: XX for females and XY for males. The homologous chromosomes from a metaphase spread can then be paired and arranged systematically to assemble a karyotype according to well-defined standard conventions like those established by International System for Human Cytogenetic Nomenclature (ISCN), with chromosome 1 being the largest and 22 the smallest. According to nomenclature, the description of the karyotype includes the total number of chromosomes followed by the sex chromosome constitution. A normal karyotype is 46,XX for females and 46,XY for males (Fig. 76-2). Abnormalities are noted after the sex chromosome complement.
Although the internationally accepted system for human chromosome classification relies largely on the length and banding pattern of each chromosome, the position of the centromere relative to the ends of the chromosome also is a useful distinguishing feature (Fig. 76-3). The centromere divides the chromosome in 2, with the short arm designated as the p arm and the long arm designated as the q arm. A plus or minus sign before the number of a chromosome indicate that there is an extra or missing chromosome, respectively. Table 76-1 lists some of the abbreviations used for the descriptions of chromosomes and their abnormalities. A metaphase chromosome spread usually shows 450-550 bands. Prophase and prometaphase chromosomes are longer, are less condensed, and often show 550-850 bands. High-resolution analysis is useful for detecting subtle chromosome abnormalities that might otherwise go unrecognized.
Molecular techniques such as fluorescence in situ hybridization (FISH) and comparative array genomic hybridization studies (conventional CGH and array CGH [aCGH]) have filled a significant void for the diagnosing cryptic chromosomal abnormalities. These techniques identify subtle abnormalities that are often below the resolution of standard cytogenetic studies. FISH is used to identify the presence, absence, or rearrangement of specific DNA segments and is performed with gene- or region-specific DNA probes. Several FISH probes are used in the clinical setting: unique sequence or single-copy probes, repetitive-sequence probes (alpha satellites in the pericentromeric regions), and multiple-copy probes (chromosome specific or painting). FISH involves using a unique known DNA sequence or probe labeled with a fluorescent dye that is complementary to the studied region of disease interest. The labeled probe is exposed to the DNA on a microscope slide, typically metaphase or interphase chromosomal DNA, that has been previously treated (denatured) to allow the DNA to become single stranded and to permit hybridization. When the probe pairs with its complementary DNA sequence, it can be then visualized by fluorescence microscopy (Fig. 76-4). In metaphase chromosome spreads, the exact chromosomal location of each probe copy can be documented and often the number of copies (deletions, duplications) of the DNA sequence as well, if they are not too close to each other; whereas in interphase cells, only the number of copies of a particular DNA segment can be determined. When the interrogated segments (as in genomic duplications) are close together, only interphase cells can accurately determine the presence of 2 or more copies or signals. In metaphase cells, some duplications might falsely appear as a single signal.
Metaphase and interphase FISH are particularly useful for detecting very small deletions that might escape notice with G-band analysis. In most cases the probe used for identification is used in conjunction with a control probe with a known location nearby the region studied. This allows correct identification of the hybridized signal to the right chromosome, and in some cases identification to the rearranged chromosome. With high-resolution chromosome analysis it is very difficult to recognize deletions of <5 million bp (5 Mb); FISH can reliably detect deletions as small as 50 to 200 kb of DNA. This has allowed the clinical characterization of a number of microdeletion syndromes. In addition to gene- or locus-specific probes, complex mixtures of DNA from a chromosome arm or an entire chromosome are available for fluorescence staining of large chromosome sections or entire chromosomes. The probe mixtures are referred to as chromosome paints (Fig. 76-5A and B). Other probes hybridize to repetitive sequences located to the pericentromeric regions. These probes are useful for the rapid identification of certain trisomies in interphase cells of blood smears, or even in the rapid analysis of prenatal samples from cells obtained through amniocentesis. Such probes are available for chromosomes 13, 18, and 21 and for the sex pair X and Y (Fig. 76-5C and D).
Comparative genomic hybridization (CGH) is a molecular-based technique that involves differentially labeling the patient’s DNA with a fluorescent dye (green) and a normal reference DNA with another fluorescent dye (red) (Fig. 76-6). Equal amounts of the two-label DNA samples are mixed and then used as a painting probe for FISH with normal metaphase chromosomes. The ratio of green : red fluorescence is measured along each chromosome. Regions of amplification of the patient’s DNA display an excess of green fluorescence, and regions of loss show excess red fluorescence. If the patient’s and the control DNA are equally represented, the green : red ratio is 1 : 1 and the chromosomes appear yellow.
There are many copy number variations (CNVs) causing deletion or duplication in the human genome. Thus, most detected genetic abnormalities, unless associated with very well known clinical phenotypes, require parental investigations because a detected CNV that is inherited might turn out to be an incidental polymorphic variant. A de novo abnormality (i.e., 1 found only in the child and not the parents) is often more significant if it is associated with an abnormal phenotype and if it involves genes with important functions. aCGH is a very valuable technology alone or when combined with FISH and conventional chromosome studies (Fig. 76-7).
ACOG Practice Bulletin. Screening for fetal chromosomal abnormalities. Obstet Gynecol. 2007;109:217-227.
Bejjani BA, Saleki R, Ballif BC, et al. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet. 2005;134A:259-267.
Li MM, Andersson HC. Clinical application of microarray-based molecular cytogenetics: an emerging new era of genomic medicine. J Pediatr. 2009;155:311-317.
Saugier-Veber P, Girard-Lemaire F, Rudolf G, et al. Genetic compensation in a human genomic disorder. N Engl J Med. 2009;360:1211-1216.
76.2 Down Syndrome and Other Abnormalities of Chromosome Number
Aneuploidy and Polyploidy
The most common cause of aneuploidy is nondisjunction, the failure of chromosomes to disjoin normally during meiosis (see Fig. 76-1). Nondisjunction can occur during meiosis I or II or during mitosis. After meiotic nondisjunction, the resulting gamete either lacks a chromosome or has 2 copies instead of 1 normal copy, resulting in a monosomic or trisomic zygote, respectively.
FISH is a technique that can be used for rapid diagnosis in the prenatal detection of common fetal aneuploidies including chromosomes 13, 18, and 21 as well as sex chromosomes (see Fig. 76-5C and D). The most common numerical abnormalities in liveborn children include trisomy 21 (Down syndrome), trisomy 18 (Edwards syndrome), trisomy 13 (Patau syndrome), and sex chromosomal aneuploidies: Turner syndrome (usually 45,X), Klinefelter syndrome (47,XXY), 47,XXX, and 47,XYY, By far the most common type of trisomy in liveborn infants is trisomy 21 (47,XX,+21 or 47,XY,+21). Trisomy 18 and trisomy 13 are relatively less common and are associated with a characteristic set of congenital anomalies and severe mental retardation (Table 76-2). The occurrence of trisomy 21 and other trisomies increases with advanced maternal age (≥35 yr). Owing to this increased risk, women who are ≥35 yr at the time of delivery should be offered genetic counseling and prenatal diagnosis (including serum screening, ultrasonography, and amniocentesis or chorionic villus sampling; Chapter 90).
Table 76-2 CHROMOSOMAL TRISOMIES AND THEIR CLINICAL FINDINGS
| SYNDROME | INCIDENCE | CLINICAL MANIFESTATIONS |
|---|---|---|
| Trisomy 13, Patau syndrome | 1/10,000 births |
Cleft lip often midline; flexed fingers with postaxial polydactyly; ocular hypotelorism, bulbous nose; low-set, malformed ears; microcephaly; cerebral malformation, especially holoprosencephaly; microphthalmia, cardiac malformations; scalp defects; hypoplastic or absent ribs; visceral and genital anomalies
|
Down Syndrome
Trisomy 21 is the most common genetic cause of moderate mental retardation. The incidence of Down syndrome in live births is approximately 1 in 733; the incidence at conception is more than twice that rate; the difference is accounted by early pregnancy losses. In addition to cognitive impairment, Down syndrome is associated with congenital anomalies and characteristic dysmorphic features (Figs. 76-8 and 76-9; Table 76-3). Although there is variability in the clinical features, the constellation of phenotypic features is fairly consistent and permits clinical recognition of trisomy 21. Affected individuals are more prone to congenital heart defects (50%) such as atrioventricular septal defects, ventricular septal defects, isolated secundum atrial septal defects, patent ductus arteriosus, and tetralogy of Fallot. Congenital and acquired gastrointestinal anomalies and hypothyroidism are common (Table 76-4). Other abnormalities include megakaryoblastic leukemia, immune dysfunction, diabetes mellitus, and problems with hearing and vision (see Table 76-4). Alzheimer disease–like dementia is a known complication that occurs as early as the 4th decade and has an incidence 2-3 times higher than sporadic Alzheimer disease. Most males with Down syndrome are sterile, but some females have been able to reproduce, with a 50% chance of having trisomy 21 pregnancies. Two genes (DYRK1A, DSCR1) in the putative critical region of chromosome 21 may be targets for therapy.
Figure 76-9 Prehensile foot in a 1-mo-old child.
(From Wiedemann HR, Kunze J, Dibbern H: Atlas of clinical syndromes: a visual guide to diagnosis, ed 3, St Louis, 1989, Mosby.)
Table 76-3 CLINICAL FEATURES OF DOWN SYNDROME IN THE NEONATAL PERIOD
CENTRAL NERVOUS SYSTEM
CRANIOFACIAL
CARDIOVASCULAR
MUSCULOSKELETAL
GASTROINTESTINAL
CUTANEOUS
Cutis marmorta
Table 76-4 ADDITIONAL FEATURES OF DOWN SYNDROME THAT CAN DEVELOP OR BECOME SYMPTOMATIC WITH TIME
NEUROPSYCHIATRIC
SENSORY
CARDIOVASCULAR
MUSCULOSKELETAL
ENDOCRINE
HEMATOLOGIC
GASTROINTESTINAL
CUTANEOUS
Developmental delay is universal (Tables 76-5 and 76-6; Fig. 76-10). Cognitive impairment does not uniformly affect all areas of development. Social development is relatively spared, but children with Down syndrome have considerable difficulty using expressive language. Understanding these individual developmental strengths will maximize the educational process for children with Down syndrome. Persons with Down syndrome often benefit from programs aimed at stimulation, development, and education. These programs are most effective in addressing social skills that often appear advanced for the intellectual delay. Children with Down syndrome also benefit from anticipatory guidance, which establishes the protocol for screening, evaluation, and care for patients with genetic syndromes and chronic disorders (Table 76-7).
