[level-membership-for-neurosurgery-category]
Chapter 20 Cushing’s Disease
Pathophysiology
Cushing’s syndrome, the syndrome produced by chronic exposure to excess glucocorticoids, has several etiologies (Table 20-1). The most common cause of Cushing’s syndrome is iatrogenic—that is, prescribed glucocorticoids for patients with chronic obstructive pulmonary disease, autoimmune disorders, and transplant patients requiring chronic immunosuppression, among others. Excluding iatrogenic causes, the most common cause of Cushing’s syndrome is an ACTH-secreting pituitary adenoma, defined as CD, which affects 60% to 80% of patients with spontaneous (noniatrogenic) Cushing’s syndrome (Table 20-1).1 The other common causes of Cushing’s syndrome, which must be distinguished from CD, are adrenal tumors and adrenal cortical hyperplasia, and ectopic ACTH secretion from a nonpituitary tumor, most commonly small-cell–lung cancer or bronchial carcinoid. Secretion of ACTH by lung carcinoma is not rare, but the ACTH is usually inactive, termed “big ACTH.”2 A rare cause of Cushing’s syndrome is ectopic secretion of corticotropin-releasing hormone causing corticotroph hyperplasia.
| ACTH-Dependent | 85% |
| Cushing’s disease | 80–85% |
| Ectopic ACTH-secreting tumor | 15–20% |
| Ectopic CRH-secreting tumor | Rare (<1%) |
| ACTH-Independent | 15% |
| Adrenal adenoma | 7% |
| Adrenocortical carcinoma | 7% |
| Bilateral micronodular adrenocortical hyperplasia | Rare (1%) |
| Bilateral macronodular adrenocortical hyperplasia | Rare (<1%) |
Pathology
The typical pituitary adenoma causing CD is a microadenoma (<1 cm greatest diameter) that stains for ACTH by immunohistochemistry. Larger adenomas are occasionally seen and may even present with apoplexy. These tumors are monoclonal.3 They disrupt the normal acinar pattern of the gland most clearly seen when stained for reticulin. They are typically basophilic by hematoxylin and eosin staining, but this terminology is no longer used, as immunohistochemistry for ACTH is more specific. Occasionally Cushing’s syndrome is attributed to corticotroph hyperplasia. When no definite tumor is identified at surgery and partial or total hypophysectomy is performed, the specimen must be meticulously and thoroughly examined with serial slices at closely spaced intervals, because tumors as small as 1 mm diameter, or less, may cause endocrinopathy from excess ACTH secretion. Corticotroph hyperplasia, which is usually a diagnosis of exclusion (i.e., only after no tumor can be found in the specimen from pituitary surgery), should be considered only after a careful search of the gland has ruled out a discrete adenoma. In our experience, hyperplasia is exceedingly rare (only two suspected, but unproven, cases from over 1200 operations for CD). A unique cytoplasmic staining pattern, known as Crooke’s hyaline change, occurs in corticotrophs of the normal pituitary—cells from which ACTH production has been shut down by chronic exposure to hypercortisolemia. The “hyaline” is comprised of intracytoplasmic microfilaments, which do not stain for ACTH. Crooke’s changes may also be found in the cells of the adenoma itself.
Mechanism of Hypercortisolemia in ACTH-Secreting Pituitary Adenomas
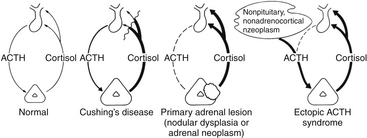
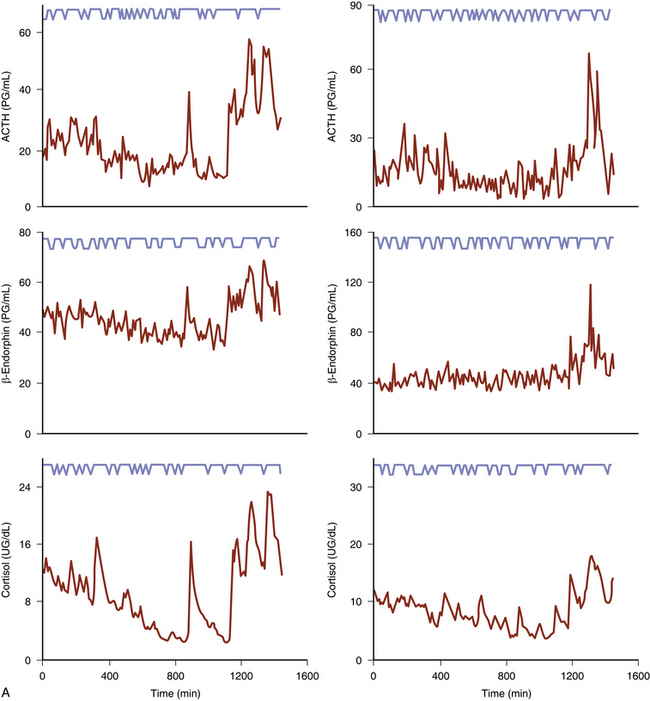
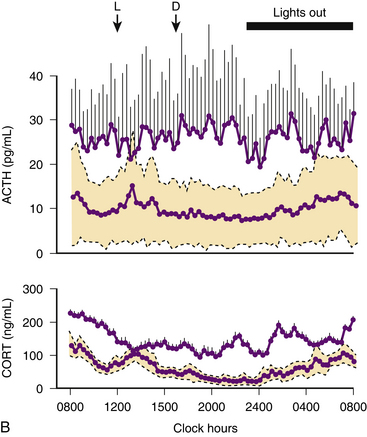
The basic endocrine disorder of the adenomas causing CD is that the sensitive negative feedback of cortisol on the production and release of ACTH is impaired (Fig. 20-1). However, ACTH-secreting pituitary adenomas are well-differentiated tumors derived from pituitary corticotrophs; thus, they typically retain negative feedback to glucocorticoids, it is just set at a higher threshold for suppression, as inhibition of ACTH release is retained in response to high doses of glucocorticoid. These well-differentiated tumor cells also retain their expression of CRH receptors and the cellular machinery necessary to respond to CRH. It is these features that underlie the typical diagnostic responses with provocative endocrine tests used for the differential diagnosis of Cushing’s syndrome (see below). Excessive production of ACTH leads to hyperplasia and overproduction of cortisol by the adrenal cortex, loss of normal diurnal plasma cortisol rhythm, and sustained hypercortisolemia. It is the excessive cortisol, rather than ACTH per se that causes the clinical manifestations of CD.
Clinical Manifestations
The typical patient with Cushing’s syndrome has truncal obesity with associated moon facies, enlarged dorsal fat pads (“buffalo hump”), and abdominal fat deposition with associated purple striae or “stretch marks” (Table 20-2). Hirsutism, especially noticeable on the face of women, is a common component, as are thin skin and easy bruisability, especially of the hands and forearms. Along with the outward appearance, mood or psychiatric disturbances are common, especially depression. A reversible form of brain atrophy is frequently displayed on imaging studies4 and may be a clue to the presence of Cushing’s syndrome in pediatric patients. Other signs include hypertension and hyperglycemia, often with frank diabetes mellitus. A hypercoagulable state has been described with Cushing’s syndrome, so prophylaxis for deep venous thrombosis in high-risk situations has been encouraged.5 Patients also may have complications related to immunosuppression, such as fungal or opportunistic infections. Spinal epidural lipomatosis may be symptomatic.6 Osteoporosis is common; related complications include vertebral compression fractures and susceptibility to traumatic long-bone fractures with minor trauma. Pediatric patients with CD stop growing linearly and gain weight, often producing morbid obesity.7 This “crossing of the weight and height curves” is so common in childhood Cushing’s syndrome that many consider it diagnostic of the condition. Affected children often appear cherubic. Symptoms caused by tumor growth and pressure on the optic nerves and chiasm are rare with CD, as the tumors are usually microadenomas.
| Fat Distribution | Skin Manifestations |
| Centripetal obesity | Purple striae |
| Moon facies | Plethora |
| “Buffalo hump” | Hirsutism |
| Supraclavicular fat pads | Acne |
| Epidural lipomatosis | Bruising |
| Musculoskeletal | Metabolic/Circulatory |
| Osteoporosis; fractures | Hypertension |
| Proximal muscle weakness Pituitary Dysfunction Amenorrhea Decreased libido, impotence Hypothyroidism Dwarfism (children) |
Glucose intolerance Hypokalemic alkalosis Mental Changes Irritability Psychosis |
Life expectancy is greatly foreshortened by untreated CD. If left untreated, most patients succumb early to complications of the disease (diabetes, hypertension, myocardial infarction, stroke, or complications associated with immunosuppression).8
Diagnosis
Establishing Hypercortisolism
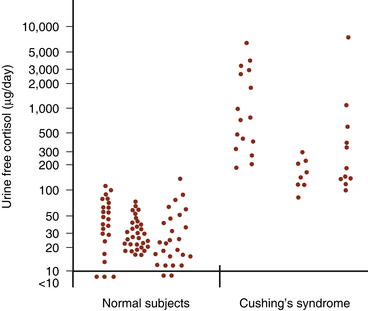
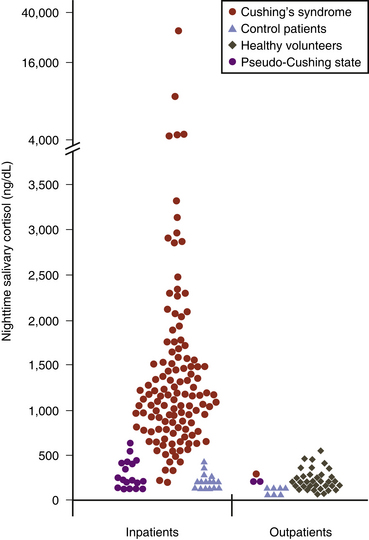
Urine-free cortisol (UFC) measurements are assayed using a variety of techniques. The normal upper levels vary with the technique used and with the laboratory performing the assay (Fig. 20-2). Hypercortisolism is associated with loss of normal diurnal variation in cortisol secretion, which is demonstrated by obtaining morning (8 to 9 a.m.) and evening (11 to 12 p.m.) plasma cortisol levels (Fig. 20-3). Salivary cortisol levels are also reliably used for this and are well suited for outpatient screening of adult and pediatric patients for hypercortisolism.9 A study of more than 140 patients demonstrated a sensitivity of 93% and a specificity of 100% using this test to determine the presence of Cushing’s syndrome (Fig. 20-4).10 Because of its simplicity, the overnight low-dose (1 mg) dexamethasone suppression test is commonly used to screen patients for hypercortisolism (Fig. 20-5). In persons with a normal hypothalamic-pituitary-adrenal axis, AM cortisol levels are suppressed by the overnight low-dose dexamethasone suppression test (1.0 mg given the night before a morning [7 to 8 a.m.] cortisol measurement). A morning plasma cortisol level greater than 1.8 μg/dl after the bedtime (11 p.m.) administration of 1 mg of dexamethasone detects most patients with Cushing’s syndrome and justifies further diagnostic evaluation.11
(Modified from Loriaux DL, Cutler GB Jr. Diseases of the adrenal glands. In: Kohler PO, ed. Clinical Endocrinology. New York: Wiley; 1986:167-238, with permission.)60
 , patient controls
, patient controls 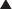 , pseudo-Cushing patients
, pseudo-Cushing patients  , and patients with Cushing’s syndrome
, and patients with Cushing’s syndrome  . Samples were collected at 12 midnight from inpatients and at bedtime (11 p.m. to 2 a.m.) from outpatients. To convert salivary cortisol nanograms per deciliter to nanomoles per liter, multiply by 0.0276.
. Samples were collected at 12 midnight from inpatients and at bedtime (11 p.m. to 2 a.m.) from outpatients. To convert salivary cortisol nanograms per deciliter to nanomoles per liter, multiply by 0.0276.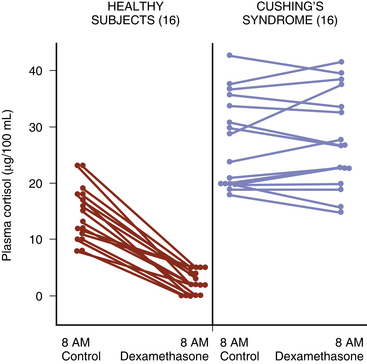
Differential Diagnosis of Hypercortisolism
Once excess cortisol production has been established, plasma ACTH levels are measured to distinguish between an ACTH-dependent and ACTH-independent etiology (Fig. 20-6; see also Fig. 20-1). With CD or ectopic ACTH secretion, the ACTH levels will be normal or elevated relative to the degree of glucocorticoid secretion. For this reason, these two entities are categorized as “ACTH-dependent” Cushing’s syndrome (see Table 20-1), in contrast to adrenal disease, in which plasma ACTH is low (<5 pg/ml) or undetectable (“ACTH-independent” Cushing’s syndrome, as the adrenal cortical cortisol secretion is autonomous).
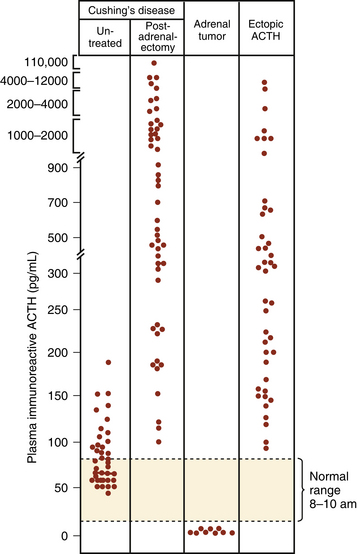
(From Besser GM, Edwards CRW. Cushing’s syndrome. Clin Endocrinol Metab. 1972;1:451-490, with permission.)64
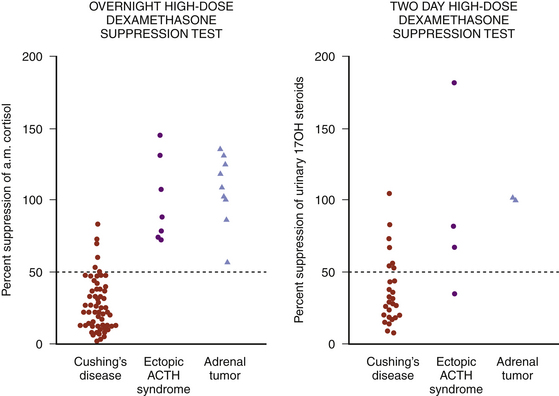
At one time, the presence of Cushing’s syndrome and then differentiating CD from adrenal disease or ectopic ACTH secretion were examined with the 6-day dexamethasone suppression test, as described by Liddle.12,13 The first 2 days of the test were used for measurement of basal cortisol secretion. In most patients with CD, cortisol secretion, an indirect measure of ACTH secretion by the pituitary gland or the tumor, is not suppressed during the 2 days of low-dose dexamethasone (0.5 mg every 6 hours for 48 hours), but 24-hour urinary cortisol secretion is suppressed to less than 10% of baseline values by 2 days of high-dose dexamethasone (2 mg every 6 hours for 48 hours). In contrast, high-dose dexamethasone fails to suppress cortisol secretion in most cases of ectopic ACTH secretion. Because of the difficulty in successfully completing this test today, it is now rarely used, but has been replaced with the high-dose overnight dexamethasone suppression test. For this test, 8 mg of dexamethasone is administered orally at 11 p.m. and a morning (7 to 8 a.m.) plasma cortisol measurement is obtained;14 for greatest diagnostic accuracy, suppression of morning serum cortisol of greater than 68% is required to assign a diagnosis of CD (Fig. 20-7).14,15
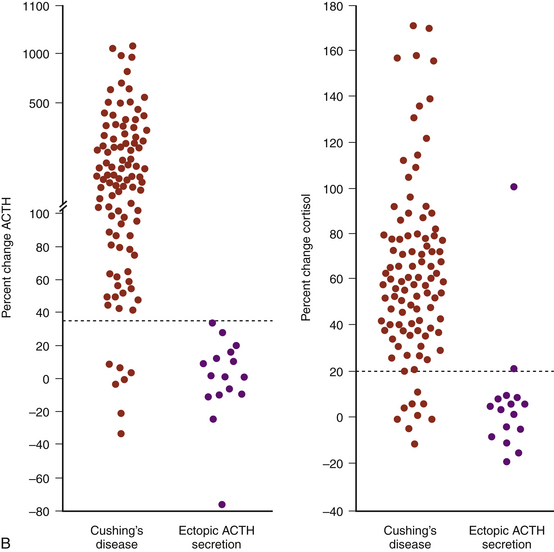
Another test available today to distinguish ectopic ACTH secretion from an ACTH-secreting pituitary tumor is the CRH stimulation test.16–18 Since they are well-differentiated tumors derived from pituitary corticotrophs, most ACTH-secreting pituitary adenomas retain receptors for, and response to, CRH, whereas most ectopic tumors, tumors that are not derived from pituitary tissue, do not express receptors for CRH and do not respond to it (Fig. 20-8). The sensitivity and specificity of the test are optimal using ≥35% for the maximum ACTH response from the 15- or 30-minute samples to indicate CD (see Fig. 20-8B).18
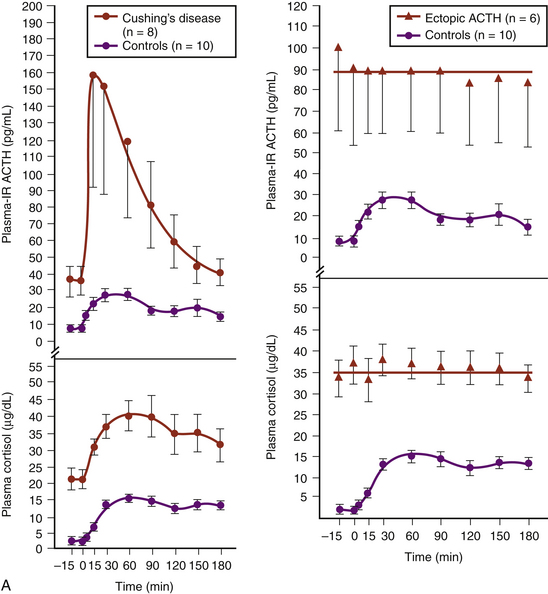
 , six patients with Cushing’s syndrome due to ectopic ACTH secretion
, six patients with Cushing’s syndrome due to ectopic ACTH secretion  , and 10 human controls
, and 10 human controls  . (Modified from Chrousos GP, Schulte HM, Oldfield EH, Gold PW, Cutler GB Jr, Loriaux DL. The corticotropin-releasing factor stimulation test. An aid in the evaluation of patients with Cushing’s syndrome. N Engl J Med. 1984;310:622-626.)B, Responses of plasma ACTH and cortisol to intravenous administration of ovine CRH in patients with CD and ectopic ACTH secretion. ACTH responses are expressed as the percent change in mean ACTH concentration 15 and 30 min after CRH from the basal value 1 and 5 min before the injection. Dashed line indicates a response of 35%. Cortisol responses are expressed as the percent change in mean cortisol concentration 30 and 45 min after CRH from the basal value 1 and 5 min before the injection. Dashed line indicates a response of 20%.
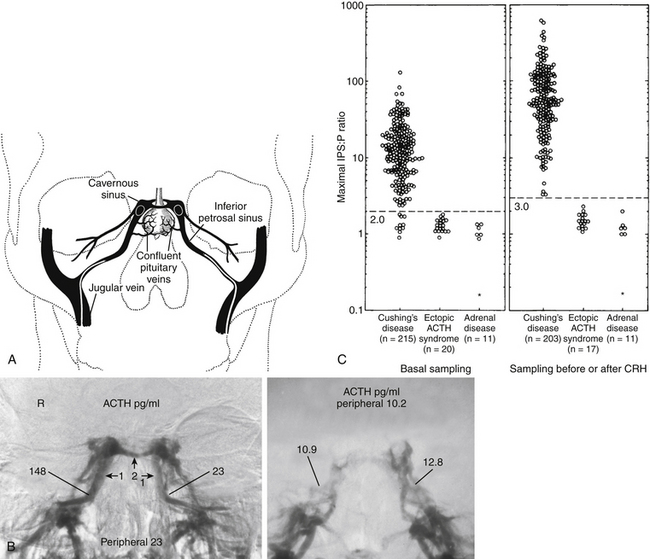
. (Modified from Chrousos GP, Schulte HM, Oldfield EH, Gold PW, Cutler GB Jr, Loriaux DL. The corticotropin-releasing factor stimulation test. An aid in the evaluation of patients with Cushing’s syndrome. N Engl J Med. 1984;310:622-626.)B, Responses of plasma ACTH and cortisol to intravenous administration of ovine CRH in patients with CD and ectopic ACTH secretion. ACTH responses are expressed as the percent change in mean ACTH concentration 15 and 30 min after CRH from the basal value 1 and 5 min before the injection. Dashed line indicates a response of 35%. Cortisol responses are expressed as the percent change in mean cortisol concentration 30 and 45 min after CRH from the basal value 1 and 5 min before the injection. Dashed line indicates a response of 20%.The test with the greatest diagnostic accuracy for the differential diagnosis of CD versus ectopic ACTH syndrome is bilateral simultaneous inferior petrosal sinus sampling performed with and without intravenous CRH administration.19 The test is performed by placement of catheters with their tips in the inferior petrosal sinuses and in a peripheral vein (Fig. 20-9A), and then obtaining serial, simultaneous samples for central and peripheral plasma ACTH concentrations at 2 and 0 minutes before and at 3, 5, and 10 minutes after intravenous CRH administration (1 μg/kg body weight). IPSS is only used in patients with confirmed hypercortisolism, as the test cannot discriminate between normal subjects and patients with CD. Further, since this is an invasive procedure with rare but serious associated risks,20 it is generally used in patients in whom the results of provocative endocrine testing to distinguish ectopic ACTH secretion from CD are conflicting or equivocal and in instances in which the sella MRI is negative. In this test the levels of ACTH in the primary venous drainage of the pituitary, the inferior petrosal sinuses, are compared to simultaneous ACTH measurements in the peripheral blood. A peak ratio of 2:1 petrosal:peripheral during baseline (before CRH) or 3:1 before or after CRH indicates a pituitary source of the excess ACTH, that is, CD.19 The sensitivity of the test is increased by sampling after administration of CRH, which, because it stimulates a pituitary adenoma to secrete ACTH, enhances the ACTH concentration differential between the central (inferior petrosal sinus) and the peripheral blood (Fig. 20-9B).19 It originally seemed that the procedure had a diagnostic accuracy 100%. However, reports of diagnostic errors, almost all false negative, have appeared, although in our experience the diagnostic accuracy approaches 100%. The accuracy of the test also relies upon successful placement of the catheter tips in the petrosal sinus bilaterally, since a false negative result may arise if the blood from both inferior petrosal sinuses cannot be catheterized successfully. Thus, venography is performed to ensure correct catheter placement and to evaluate the venous anatomy. A hypoplastic or anomalous inferior petrosal sinus in 0.8% of 501 patients was associated with false-negative results in patients with proven CD.21 It was briefly thought that comparison of petrosal sinus ACTH from the right to left sides would accurately indicate the side of the pituitary in which a small pituitary tumor was located, permitting a more focused search for it at surgery, or permitting removal of the half of the pituitary containing an adenoma that was too small to identify despite a thorough search of the gland during surgery.22 However, more experience with the technique for lateralization indicated that the lateralization accuracy is only about 70%, and even less so in pediatric patients,23 compromising its usefulness as a localizing measure during surgery.19
Imaging
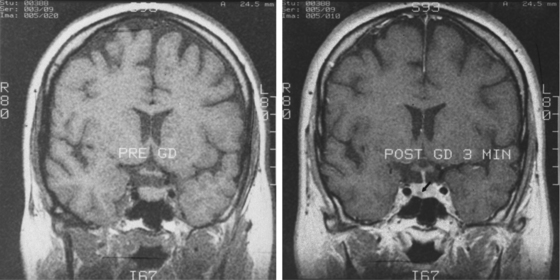
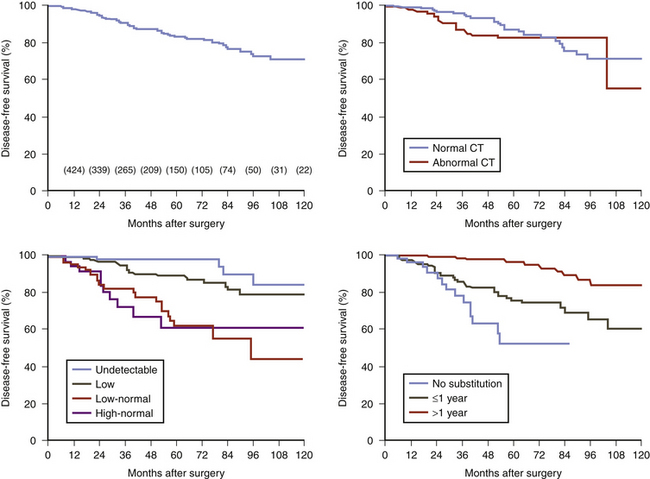
Sella magnetic resonance imaging (MRI) is the imaging procedure of choice for detecting and localizing the pituitary adenoma in patients with CD. MRI should be performed with and without contrast, as the adenomas typically have decreased enhancement compared to the normal gland (Fig. 20-10). The resolution of a 1.5-T magnet may reveal tumors as small as 3 mm in diameter. MRI provides other important anatomical information for the surgeon: aeration of the sphenoid, parasellar anatomy, location of the carotid arteries or coexisting aneurysms, extent of supra- or para-sellar extension of an adenoma, and ectopic parasellar tumors. Recently the spoiled gradient recalled acquisition (SPGR) technique, used with 1-mm nonoverlapping slices was shown to be more sensitive than the conventional spin echo approach (sensitivity 80% vs. 49%), a finding also true in pediatric CD.24,25 However, the incidence of false positives was also higher (4% vs. 2%). Since the sensitivity of the conventional MRI techniques (spin echo) for CD is only 50% to 75%,26–29 many patients have a negative MRI. Furthermore, MRI is not always available or possible, as in patients with an MRI incompatible cardiac pacemaker or a morbidly obese patient who cannot be accommodated by the scanner. In these patients, a sella computerized tomography image (CT) with and without contrast may demonstrate a tumor, but CT is less sensitive than MRI. Furthermore, since the specificity of MRI is not 100% (MRI abnormalities consistent with adenomas occur in the pituitary gland in 10% of normal volunteers30 and incidental adenomas are found in the pituitary gland in about 5% to 20% of subjects in unselected autopsy studies31,32), the results of MRI scanning must be confirmed by endocrinologic testing before surgery. In general, CD endocrine testing provides the diagnosis and CT localizes the adenoma within the pituitary and defines the anatomy in its vicinity.
Treatment
Surgery
However, many tumors that cause CD are small, frequently so small that they are not identified on the preoperative MRI. In these patients the first task for the surgeon is to locate the adenoma. A wide exposure of the pituitary gland permits visualization of the anterior lobe from one edge to the other. This requires removal of the bone of the anterior face of the sella until the medial portion of the cavernous sinus is visible bilaterally. The pseudocapsule of a microadenoma is almost always a grey-white or gray-yellow color. Because coagulation of the dura will discolor a circumscribed area of the pituitary surface, producing a local white region that can mimic an adenoma just beneath the site of dural coagulation, bipolar cautery of the dura is avoided. A wide cruciate dural opening extending to the lateral corners of the exposure, to the junction of the circular sinus with the cavernous sinus superiorly and to the medial wall of the cavernous sinus inferiorly, while avoiding entry into the capsule of the pituitary gland, allows visualization of most of the anterior surface of the anterior lobe. In many cases, an adenoma will be seen during careful inspection of the gland’s surface while using the operating microscope to look for a focal mass or discoloration on the surface of the gland. Localization of adenomas with intraoperative ultrasound, using a specially designed probe, has also proven useful in patients with negative MRI scans.33 When it is used, intraoperative ultrasound is obtained after the bone removal is complete, but before the dura is opened, and with a bloodless field. However, in our experience the most reliable clue to the identification of a microadenoma is visualization of its pseudocapsule. This is especially important for the small tumors that are buried in the gland.
When the location of an adenoma is identified by inspection of the surface of the gland, the pituitary capsule is sharply incised just beyond the visible margin of the tumor pseudocapsule, which is a margin of compressed normal gland surrounding the adenoma (Fig. 20-11). If the adenoma is not identified by examining the pituitary capsule anteriorly and inspecting the lateral surfaces of the anterior lobe, a series of incisions is made vertically in the gland at intervals of 1.5 to 2 mm, each carried deeper in increments until either the pseudocapsule of the tumor is identified or the juncture of the anterior and posterior lobes is reached (Fig. 20-12). By identification of the interface between the pseudocapsule of the adenoma and the surrounding normal gland, the adenoma is discretely dissected from the normal pituitary, ideally without entering the soft body of the adenoma. In this way, total resection is commonly performed and lasting remission is achieved.34 If the tumor breaches the capsule of the pituitary gland, careful inspection of the contiguous dura is performed to ensure that dural invasion by the tumor is not overlooked. This is particularly important laterally along the medial wall of the cavernous sinus, where even small tumors may spread into the adjacent dura and cavernous sinus.
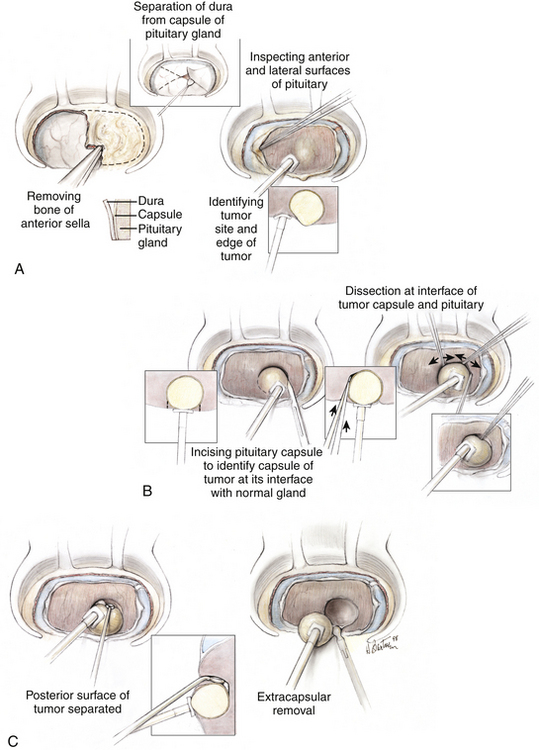
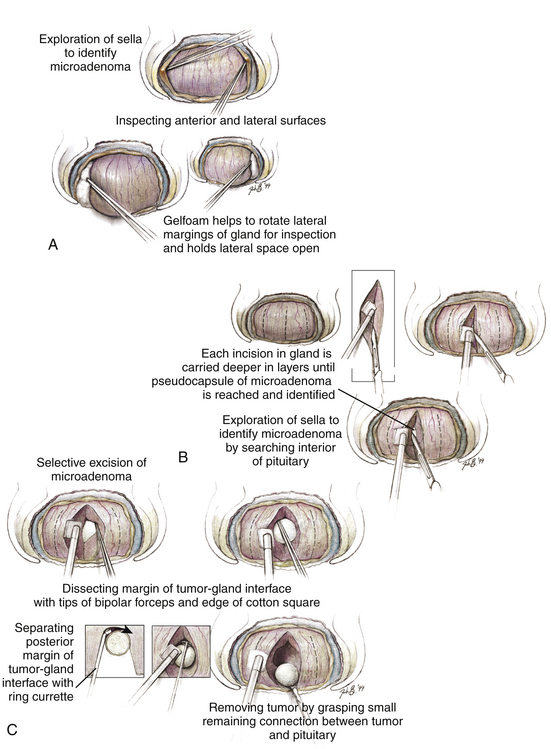
FIGURE 20-12 Exploration of the pituitary gland in patients with an endocrine-active adenoma whose imaging studies appear normal. The goal of the exploration is to find and identify the distinct encapsulated margin of the adenoma. Success depends on beginning with a widely exposed, bloodless surgical field (A). Coagulation of the anterior and inferior sella dura is avoided during the exposure and dural opening because it would produce a white area on the surface of the underlying gland that may falsely suggest the site of the adenoma. After widely opening the dura, which provides exposure of an intact pituitary capsule covering the anterior surface of the gland, and after exposure of the extreme lateral margins of the anterior surface, the surfaces are carefully inspected for regions of focal discoloration. The adenoma usually appears to be gray-blue or yellow-white, and can be identified against the background of the anterior lobe surface whose color is orange-pink. B, If no tumor is seen, the lateral surfaces of the anterior lobe are then inspected. For this the lateral dural wall of the sella (the medial wall of the cavernous sinus) is separated from the pituitary capsule by gently passing a disc dissector between these two layers from top to bottom. The space produced provides room for dissection of the interface between the lateral pituitary capsule and the dural wall with the closed tips of a fine-tipped bipolar forceps (B). Dissection is initially superficial and then progresses in stages to deeper levels until the posterior sella has been reached. After these two tissues have been separated, small pieces of Gelfoam are packed into the intervening space to rotate the lateral surface anteriorly and to gently displace it medially into the surgeon’s direct view. After both lateral surfaces are exposed and examined in this fashion, the inferior surface of the gland is separated from the dura. (This is the site at which these two layers—the surface of the pituitary and the dura mater—are most tenaciously attached to each other.) If a tumor can be identified from inspection of the surface, it is removed as described in the text and the legend to Fig. 20-11. If no tumor is identified on inspection of the superficial gland, a series of vertical incisions is then made (B), each of which begins 1 to 2 mm below the superior edge of the pituitary exposure and is directed downward, initially to a depth of only approximately 1 mm, and then in stages deeper through the anterior lobe until either the intermediate lobe or the glistening white anterior surface of the posterior lobe is reached. Because the pituitary blood supply and the delivery of hypothalamic trophic factors to the pituitary is oriented vertically, vertical incisions should be less likely to cause an infarction in a portion of the pituitary or to isolate the gland from its hypothalamic regulation. A distinct tissue capsule is the primary object of the search, with the intent to identify the margin of the tumor before entering it and spilling its contents, by using the surgical capsule of the adenoma (B and C). When the surgical capsule of the adenoma is identified, the tumor is removed using dissection along the interface of the adenoma and the normal gland, as described in Fig. 20-11 (A and B). In cases of tumors lying in the posterior portion of the anterior lobe, a 2-mm-wide slice of the anterior lobe may be removed to provide space for dissection and removal of the deep microadenoma (not shown).
(From Oldfield EH, Vortmeyer AO. Development of a histological pseudocapsule and its use as a surgical capsule in the excision of pituitary tumors. J Neurosurg. 2006;104:7-19.)
ACTH-producing tumors may also be found in the posterior lobe,35 so careful inspection of the gland back to the neurohypophysis should be preformed when no tumor is encountered anteriorly.
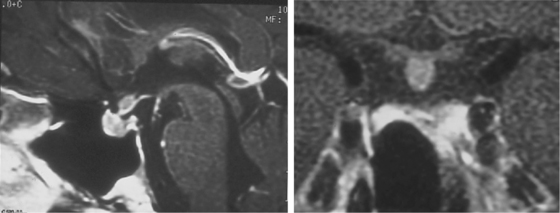
In the rare instance when the ACTH-secreting adenoma is located outside of the adenohypophysis, such as in a suprasellar (Fig. 20-13) or parasellar location,36 special techniques are required. For tumors of the pituitary stalk in the suprasellar cistern, an extended trans-sphenoidal approach may be used.37 With this technique, during the standard trans-septal approach, bone removal is extended rostrally over the planum sphenoidale. The dura anterior to the gland is opened in the midline to expose the suprasellar cistern (Fig. 20-14). Bleeding from the opened circular sinus is stopped by packing it with small pieces of Gelfoam or with bipolar coagulation. Direct visual access of the pituitary stalk is thus provided.
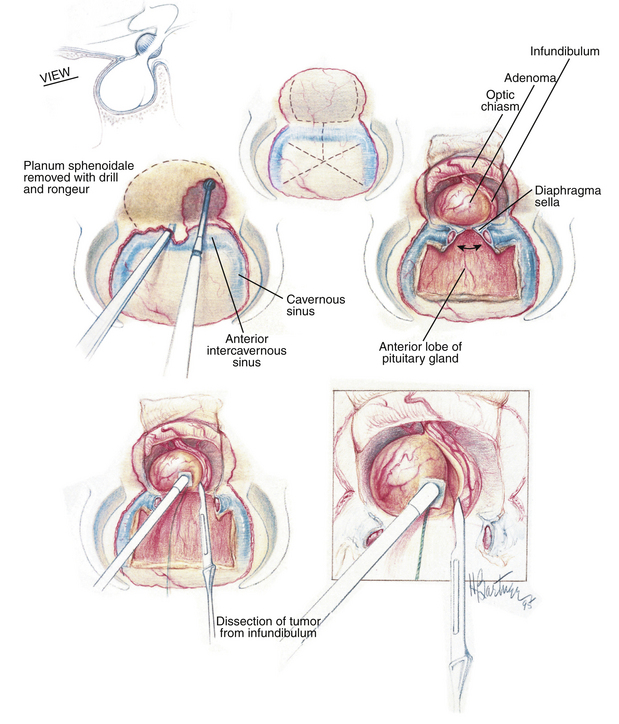
Postoperative Assessment
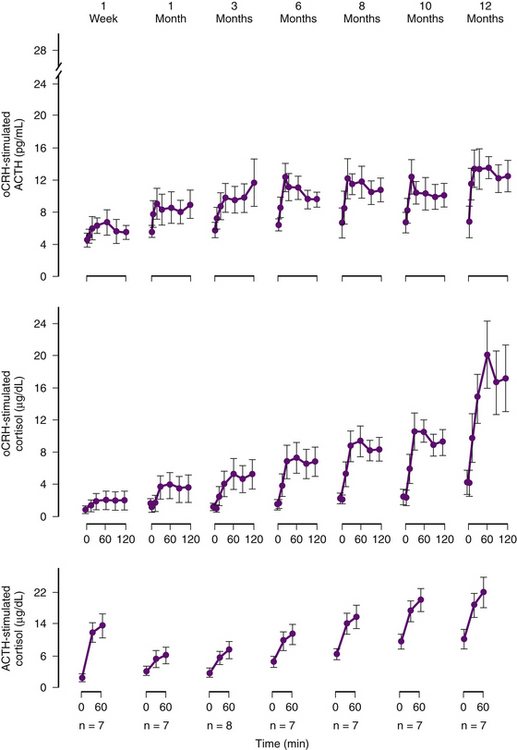
The chronic hypercortisolism associated with CD suppresses the hypothalamic CRH-producing cells and the secretion of ACTH by the normal corticotrophs, but does not fully suppress ACTH secretion by the tumor. In most patients who are cured by surgery it takes several months for the hypothalamic-pituitary-adrenal axis to recover and the patient suffers from hypocortisolism and requires glucocorticoid replacement therapy for the 6 to 24 months required for recovery of the hypothalamic neuron, the normal pituitary corticotroph, and the adrenal cortex and return of normal endogenous circadian cortisol secretion (Fig. 20-15). Therefore, complete removal of the abnormal source of ACTH should produce hypocortisolism in the immediate postoperative interval and for several months.38 Thus, in almost all instances of successful surgery for CD, the patient will be hypocortisolemic (a morning cortisol of less than 5 μg/dl and 24-hour urine-free cortisol secretion of less than 20 μg). Because of this, postoperative cortisol replacement must be provided. There are several protocols for postoperative cortisol replacement therapy. One approach used in our practices is to administer 0.5 mg of dexamethasone every 6 hours for 36 hours. The dexamethasone is then discontinued and daily AM cortisol and 24-hour urine cortisol are obtained for 3 consecutive days. This permits immediate determination if surgery has been successful in eliminating hypercortisolism and provides the potential for immediate early reoperation, if indicated, which is successful in many patients.39 Others use postoperative low-dose dexamethasone suppression testing as follows: on the first postoperative day, the patient receives 50 mg of hydrocortisone intravenously in the morning and 25 mg in the evening. On the second day, they are given 50 mg of hydrocortisone in the morning and 1 mg of dexamethasone in the evening at 10 p.m. Blood is withdrawn from the patient at 8 a.m. on the third postoperative morning and a fasting serum cortisol level is measured. With this approach patients with a morning plasma cortisol greater than 2 μg/dl will have a high likelihood of having recurrent disease: 100% of patients in the report by Arnott et al.,40 and in the larger experience of Chen et al.41 On the other hand, 93% of patients with an AM cortisol value of 2 μg/dl or less still had sustained remission of CD at 5 years (Fig. 20-16).41 Many authors prefer either to provide cortisol replacement therapy and to test the patient’s cortisol secretion several weeks after hospital discharge, or to provide no cortisol replacement and sample serum cortisol levels every 6 hours after surgery.42–44 Most cured patients have serum cortisol drop below 2 μg/dl within the first 48 hours after surgery.
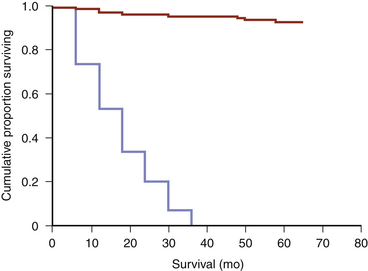
In patients with eucortisolism immediately or within a few weeks of surgery, clinical remission is still likely, though CD is more likely to recur than in patients with postoperative hypocortisolism (Fig. 20-17).45,46
Early Repeat Surgery
If results of cortisol measurement shortly after surgery indicate persistent hypercortisolism, immediate repeat surgery should be considered in certain circumstances.39 Early reoperation (within 1 to 6 weeks) can induce remission in the majority of patients, although with a risk of hypopituitarism. Surgical or pathologic confirmation of an ACTH-positive adenoma that is incompletely resected during the initial pituitary exploration is the most significant predictor of success with early re-exploration, in terms of both remission and avoidance of hypopituitarism. Patients who have undergone limited exploration of the pituitary and selective excision of an area that at surgery appears to be, but proves not to have been, a corticotroph adenoma, also are likely to benefit from repeat surgery. On the other hand, patients who undergo an extensive initial exploration and partial resection of the anterior lobe are unlikely to benefit from a repeat operation if there is no ACTH-positive adenoma in the excised specimen.39 With appropriate patient selection early repeat surgery deserves consideration, especially when prompt control of hypercortisolism is required.
Surgical Management of Recurrent Cushing’s Disease
Patients who have remission of hypercortisolism following surgery occasionally develop recurrent CD. What should be done for the patient with recurrent CD after previous surgery? In a report detailing the results of trans-sphenoidal surgery in 31 patients who had previously undergone a trans-sphenoidal operation and two patients who had had previous pituitary irradiation only, in 24 (73%) of the 33 patients, remission of hypercortisolism was achieved by surgery.47 Although CT identified an adenoma in only three of the 33 patients, in 20 patients a discrete adenoma was identified at pituitary exploration. The incidence of remission of hypercortisolism was greatest if an adenoma was identified at surgery and the patient received selective adenomectomy (19, or 95% of 20 patients), if there was evidence at surgery or by preoperative imaging that the previous surgical exposure of the pituitary was incomplete (seven, or 78% of nine patients), if an adenoma was seen on preoperative imaging (three of three patients), or if the patient had had prior pituitary irradiation without surgery (two of two patients). In contrast, only five (42%) of 12 patients who received subtotal or total hypophysectomy had remission of hypercortisolism. Surgically induced hypopituitarism occurred in six (50%) of these 12 patients, but in only one (5%) of the 20 patients who underwent selective adenomectomy. Three (13%) of the 24 patients who were in remission from hypercortisolism following repeat surgery developed recurrent hypercortisolism 10 to 47 months postoperatively.
More recent reports have established that the recurrent adenoma is always at, or immediately contiguous to, the site of the adenoma at the original surgery.48,49 Furthermore, the recurrent tumor usually is invading the dura, usually the cavernous sinus wall contiguous to the former location of the adenoma. In the Dickerman and Oldfield series, at repeat surgery (44 ± 35 months after the initial surgery), in all 43 patients in whom an adenoma had been identified at the initial surgery, the tumor was found at the same site or contiguous to the same site,48 indicating that recurrence of CD is from growth of residual cells left in situ at the original surgery and that at repeat exploration the site occupied by the original tumor should be the focus of the exploration. Dural invasion by an ACTH-producing tumor was identified during repeated surgery in 42 (62%) of the 68 patients re-explored after prior surgery and recurrent or persistent CD. In addition, 39 (93%) of these 42 invasive adenomas were located laterally and involved the cavernous sinus. Adenoma invasion of the dura mater was found in 31 (54%) of 57 microadenomas and in all 11 macroadenomas at repeated surgery. The presence of tumor was not detected in 28 of the 59 patients studied with MRI and in none of these 59 patients was dural invasion evident on MRI. The results of this study thus established that recurrent and persistent CD consistently results from residual tumor. At repeated surgery the residual tumor can be found at, or immediately contiguous to, the site at which the tumor was originally found. Unappreciated dural invasion with growth of residual tumor within the cavernous sinus dura, which frequently occurs without residual tumor or dural invasion being evident on MR images or to the surgeon during surgery, is the basis of surgical failure in many patients with CD.
Radiation Therapy
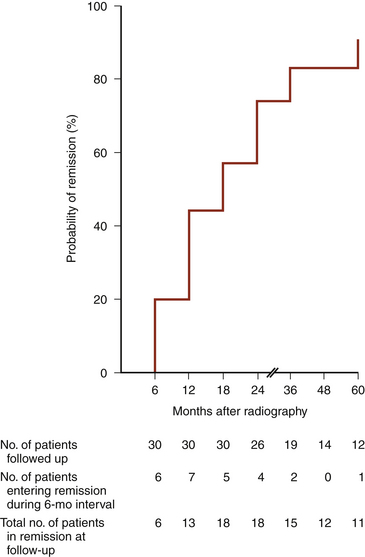
Fractionated radiation of the sella after failed trans-sphenoidal surgery achieves biochemical remission in most patients (80% at 4 years; Fig. 20-18).50 Because remission is delayed 6 months to several years after radiation therapy, medical therapy is used to block adrenal45 production of glucocorticoids permitting remission of the clinical syndrome of hypercortisolism until the effects of irradiation occur. Hypopituitarism is an expected side effect, but usually occurs 5 to 10 years after treatment. Other risks of sellar irradiation include, in decreasing likelihood, optic neuropathy, oculomotor neuropathy, and secondary neoplasia. Stereotactic radiosurgery may produce an earlier response than fractionated conventional radiation therapy, but whether it will reduce or increase the risk of radiation-induced complications has not been established. Radiosurgery of the sella, or of the focus of known residual tumor, is an option for treatment of CD after unsuccessful surgery, but, as in fractionated therapy remission of hypercortisolism, biochemical remission is delayed 1 to 3 years and the incidence of permanent hypopituitarism is expected to be similar to the incidence with fractionated irradiation therapy, and with new deficiencies being seen as long as 10 years after treatment.51–53 In patients who have failed microsurgery, gamma knife radiosurgery provides remission in 40% to 80% of patients.51–53
Medical Therapy
“Chemical adrenalectomy,” by medically blocking production of biologically active cortisol by the adrenal cortex, eliminates hypercortisolism and induces remission from the signs and symptoms of CD while awaiting trans-sphenoidal surgery or, more commonly, while awaiting the effects of pituitary irradiation. For this, ketoconazole, which blocks steroidogenesis, is used.54,55 Its use is limited primarily by hepatic toxicity. Other side effects include reduced androgen production and gynecomastia in men, and a disulfiram reaction. Recent experience using a combination therapy using a new somatostatin analogue, pasireotide, with a dopamine agonist, cabergoline, prior to ketoconozole, proved effective in normalizing urine-free cortisol in 15 of 17 patients with CD.56 Yet another option is metyrapone, which inhibits glucocorticoid and mineralocorticoid production, but its clinical usefulness is also limited by toxicity.
Adrenalectomy
Although, adrenalectomy was the treatment for CD in the mid-20th century, the current use of bilateral adrenalectomy is limited to patients in whom other treatments have failed. Adrenalectomy may be used in lieu of irradiation to avoid hypopituitarism in young patients with CD refractory to surgery or it may be combined with irradiation to reduce the risk of development of Nelson’s syndrome, which occurs in 10% to 20% of patients after adrenalectomy for CD.57
Nelson’s Syndrome
As the adenomas of CD are under negative feedback, albeit incomplete negative feedback, by high circulating levels of glucocorticoids, plasma ACTH levels increase greatly after adrenalectomy for CD and some ACTH-secreting pituitary tumors aggressively enlarge after correction of hypercortisolism. Nelson’s syndrome, hyperpigmentation (Fig. 20-19) associated with unbridled production of pro-opiomelanocortin and very high levels of plasma ACTH and α-melatonin–stimulating hormone and the appearance of a rapidly growing pituitary macroadenoma after bilateral adrenalectomy for CD, was identified in the era before microneurosurgery, when bilateral adrenalectomy was the treatment of choice for CD. The risk of Nelson’s syndrome after adrenalectomy is 10% to 20% in most series, but is as high as 30% in others, and it may present 1 to 29 years after adrenalectomy.58 It is a serious complication that is characterized by uncontrolled tumor growth and parasellar invasion and compression syndromes that are otherwise atypical of CD. The success of surgical treatment Nelson’s syndrome is limited by the large size and invasiveness of these tumors; the incidence of remission from surgery is in the range of 20%,59 but is higher if the tumors are detected before parasellar spread. Surgery followed by irradiation is the preferred treatment,58 although many of these tumors are refractory to surgical and/or radiation therapy.

Bochicchio D., Losa M., Buchfelder M. Factors influencing the immediate and late outcome of Cushing’s disease treated by transsphenoidal surgery: a retrospective study by the European Cushing’s Disease Survey Group. J Clin Endocrinol Metab. 1995;80:3114-3120.
Chen J.C., et al. Transsphenoidal microsurgical treatment of Cushing disease: postoperative assessment of surgical efficacy by application of an overnight low-dose dexamethasone suppression test. J Neurosurg. 2003;98:967-973.
Dichek H.L., et al. A comparison of the standard high dose dexamethasone suppression test and the overnight 8-mg dexamethasone suppression test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1994;78:418-422.
Dickerman R.D., Oldfield E.H. Basis of persistent and recurrent Cushing disease: an analysis of findings at repeated pituitary surgery. J Neurosurg. 2002;97:1343-1349.
Estrada J., et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Engl J Med. 1997;336:172-177.
Hall W.A., Luciano M.G., Doppman J.L., et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994;120:817-820.
Jagannathan J., et al. Outcome of using the histological pseudocapsule as a surgical capsule in Cushing disease. J Neurosurg. 2009;111:531-539.
Krikorian A., Abdelmannan D., Selman W.R., Arafah B.M. Cushing disease: use of perioperative serum cortisol measurements in early determination of success following pituitary surgery. Neurosurg Focus. 2007;23:E6.
Liddle G.W. Pathogenesis of glucocorticoid disorders. Am J Med. 1972;53:638-648.
Liddle G.W. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. 1960;12:1539.
Losa M., Picozzi P., Redaelli M.G., et al. Pituitary radiotherapy for Cushing’s disease. Neuroendocrinology. 2010;92(Suppl 1):107-110.
Magiakou M.A., Mastorakos G., Chrousos G.P. Final stature in patients with endogenous Cushing’s syndrome. J Clin Endocrinol Metab. 1994;79:1082-1085.
Nieman L.K. Diagnostic tests for Cushing’s syndrome. Ann N Y Acad Sci. 2002;970:112-118.
Oldfield E.H., et al. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing’s syndrome. N Engl J Med. 1991;325:897-905.
Papanicolaou D.A., Mullen N., Kyrou I., Nieman L.K. Nighttime salivary cortisol: a useful test for the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. 2002;87:4515-4521.
Patronas N., et al. Spoiled gradient recalled acquisition in the steady state technique is superior to conventional postcontrast spin echo technique for magnetic resonance imaging detection of adrenocorticotropin-secreting pituitary tumors. J Clin Endocrinol Metab. 2003;88:1565-1569.
Ram Z., et al. Early repeat surgery for persistent Cushing’s disease. J Neurosurg. 1994;80:37-45.
Tyrrell J.B., Findling J.W., Aron D.C., et al. An overnight high-dose dexamethasone suppression test for rapid differential diagnosis of Cushing’s syndrome. Ann Intern Med. 1986;104:180-186.
Wilson C.B., Tyrrell J.B., Fitzgerald P.A., Pitts L.H. Cushing’s disease and Nelson’s syndrome. Clin Neurosurg. 1980;27:19-30.
1. Boscaro M., Barzon L., Fallo F., Sonino N. Cushing’s syndrome. Lancet. 2001;357:783-791.
2. Ayvazian L.F., Schneider B., Gewirtz G., Yalow R.S. Ectopic production of big ACTH in carcinoma of the lung. Its clinical usefulness as a biologic marker. Am Rev Respir Dis. 1975;111:279-287.
3. Schulte H.M., et al. Clonal composition of pituitary adenomas in patients with Cushing’s disease: determination by X-chromosome inactivation analysis. J Clin Endocrinol Metab. 1991;73:1302-1308.
4. Bourdeau I., et al. Loss of brain volume in endogenous Cushing’s syndrome and its reversibility after correction of hypercortisolism. J Clin Endocrinol Metab. 2002;87:1949-1954.
5. Boscaro M., et al. Anticoagulant prophylaxis markedly reduces thromboembolic complications in Cushing’s syndrome. J Clin Endocrinol Metab. 2002;87:3662-3666.
6. Koch C.A., Doppman J.L., Watson J.C., et al. Spinal epidural lipomatosis in a patient with the ectopic corticotropin syndrome. N Engl J Med. 1999;341:1399-1400.
7. Magiakou M.A., Mastorakos G., Chrousos G.P. Final stature in patients with endogenous Cushing’s syndrome. J Clin Endocrinol Metab. 1994;79:1082-1085.
8. Plotz C.M., Knowlton A.I., Ragan C. The natural history of Cushing’s syndrome. Am J Med. 1952;13:597-614.
9. Gafni R.I., Papanicolaou D.A., Nieman L.K. Nighttime salivary cortisol measurement as a simple, noninvasive, outpatient screening test for Cushing’s syndrome in children and adolescents. J Pediatr. 2000;137:30-35.
10. Papanicolaou D.A., Mullen N., Kyrou I., Nieman L.K. Nighttime salivary cortisol: a useful test for the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. 2002;87:4515-4521.
11. Wood P.J., Barth J.H., Freedman D.B., et al. Evidence for the low dose dexamethasone suppression test to screen for Cushing’s syndrome—recommendations for a protocol for biochemistry laboratories. Ann Clin Biochem. 1997;34(Pt 3):222-229.
12. Liddle G.W. Pathogenesis of glucocorticoid disorders. Am J Med. 1972;53:638-648.
13. Liddle G.W. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. 1960;12:1539.
14. Tyrrell J.B., Findling J.W., Aron D.C., et al. An overnight high-dose dexamethasone suppression test for rapid differential diagnosis of Cushing’s syndrome. Ann Intern Med. 1986;104:180-186.
15. Dichek H.L., Nieman L.K., Oldfield E.H., et al. A comparison of the standard high dose dexamethasone suppression test and the overnight 8-mg dexamethasone suppression test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1994;78:418-422.
16. Chrousos G.P., Schulte H.M., Oldfield E.H., et al. The corticotropin-releasing factor stimulation test. An aid in the evaluation of patients with Cushing’s syndrome. N Engl J Med. 1984;310:622-626.
17. Nieman L.K. Diagnostic tests for Cushing’s syndrome. Ann N Y Acad Sci. 2002;970:112-118.
18. Nieman L.K., Oldfield E.H., Wesley R., et al. A simplified morning ovine corticotropin-releasing hormone stimulation test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1993;77:1308-1312.
19. Oldfield E.H., Doppman J.L., Nieman L.K., et al. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing’s syndrome. N Engl J Med. 1991;325:897-905.
20. Miller D.L., Doppman J.L., Peterman S.B., et al. Neurologic complications of petrosal sinus sampling. Radiology. 1992;185:143-147.
21. Doppman J.L., Chang R., Oldfield E.H., et al. The hypoplastic inferior petrosal sinus: a potential source of false- negative results in petrosal sampling for Cushing’s disease. J Clin Endocrinol Metab. 1999;84:533-540.
22. Oldfield E.H., Girton M.E., Doppman J.L. Absence of intercavernous venous mixing: evidence supporting lateralization of pituitary microadenomas by venous sampling. J Clin Endocrinol Metab. 1985;61:644-647.
23. Batista D., Gennari M., Riar J., et al. An assessment of petrosal sinus sampling for localization of pituitary microadenomas in children with Cushing disease. J Clin Endocrinol Metab. 2006;91:221-224.
24. Batista D., Courkoutsakis N.A., Oldfield E.H., et al. Detection of adrenocorticotropin-secreting pituitary adenomas by magnetic resonance imaging in children and adolescents with Cushing disease. J Clin Endocrinol Metab. 2005;90:5134-5140.
25. Patronas N., Bulakbasi N., Stratakis C.A., et al. Spoiled gradient recalled acquisition in the steady state technique is superior to conventional postcontrast spin echo technique for magnetic resonance imaging detection of adrenocorticotropin-secreting pituitary tumors. J Clin Endocrinol Metab. 2003;88:1565-1569.
26. Colombo N., Loli P., Vignati F., Scialfa G. MR of corticotropin-secreting pituitary microadenomas. AJNR Am J Neuroradiol. 1994;15:1591-1595.
27. Peck W.W., Dillon W.P., Norman D., et al. High-resolution MR imaging of pituitary microadenomas at 1.5 T: experience with Cushing disease. AJR Am J Roentgenol. 1989;152:145-151.
28. Doppman J.L., Frank J.A., Dwyer A.J., et al. Gadolinium DTPA enhanced MR imaging of ACTH-secreting microadenomas of the pituitary gland. J Comput Assist Tomogr. 1988;12:728-735.
29. Tabarin A., Laurent F., Catargi B., et al. Comparative evaluation of conventional and dynamic magnetic resonance imaging of the pituitary gland for the diagnosis of Cushing’s disease. Clin Endocrinol (Oxf). 1998;49:293-300.
30. Hall W.A., Luciano M.G., Doppman J.L., et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994;120:817-820.
31. Camaris C., Balleine R., Little D. Microadenomas of the human pituitary. Pathology. 1995;27:8-11.
32. Teramoto A., Hirakawa K., Sanno N., Osamura Y. Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology. 1994;193:161-164.
33. Watson J.C., Shawker T.H., Nieman L.K., et al. Localization of pituitary adenomas by using intraoperative ultrasound in patients with Cushing’s disease and no demonstrable pituitary tumor on magnetic resonance imaging. J Neurosurg. 1998;89:927-932.
34. Jagannathan J., Smith R., DeVroom H.L., et al. Outcome of using the histological pseudocapsule as a surgical capsule in Cushing disease. J Neurosurg. 2009;111:531-539.
35. Weil R.J., Vortmeyer A.O., Nieman L.K., et al. Surgical remission of pituitary adenomas confined to the neurohypophysis in Cushing’s disease. J Clin Endocrinol Metab. 2006;91:2656-2664.
36. Pluta R.M., Nieman L., Doppman J.L., et al. Extrapituitary parasellar microadenoma in Cushing’s disease. J Clin Endocrinol Metab. 1999;84:2912-2923.
37. Mason R.B., Nieman L.K., Doppman J.L., Oldfield E.H. Selective excision of adenomas originating in or extending into the pituitary stalk with preservation of pituitary function. J Neurosurg. 1997;87:343-351.
38. Oldfeld E.H. Cushing disease. J Neurosurg. 2003;98:948-951. discussion 951
39. Ram Z., Nieman L.K., Cutler G.B.Jr., et al. Early repeat surgery for persistent Cushing’s disease. J Neurosurg. 1994;80:37-45.
40. Arnott R.D., Pestell R.G., McKelvie P.A., et al. A critical evaluation of transsphenoidal pituitary surgery in the treatment of Cushing’s disease: prediction of outcome. Acta Endocrinol (Copenh). 1990;123:423-430.
41. Chen J.C., Amar A.P., Choi S., et al. Transsphenoidal microsurgical treatment of Cushing disease: postoperative assessment of surgical efficacy by application of an overnight low-dose dexamethasone suppression test. J Neurosurg. 2003;98:967-973.
42. Hout W.M., Arafah B.M., Salazar R., Selman W. Evaluation of the hypothalamic-pituitary-adrenal axis immediately after pituitary adenomectomy: is perioperative steroid therapy necessary? J Clin Endocrinol Metab. 1988;66:1208-1212.
43. Krikorian A., Abdelmannan D., Selman W.R., Arafah B.M. Cushing disease: use of perioperative serum cortisol measurements in early determination of success following pituitary surgery. Neurosurg Focus. 2007;23:E6.
44. Simmons N.E., Alden T.D., Thorner M.O., Laws E.R.Jr. Serum cortisol response to transsphenoidal surgery for Cushing disease. J Neurosurg. 2001;95:1-8.
45. Bochicchio D., Losa M., Buchfelder M. Factors influencing the immediate and late outcome of Cushing’s disease treated by transsphenoidal surgery: a retrospective study by the European Cushing’s Disease Survey Group. J Clin Endocrinol Metab. 1995;80:3114-3120.
46. Estrada J., Garcia-Uria J., Lamas C., et al. The complete normalization of the adrenocortical function as the criterion of cure after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab. 2001;86:5695-5699.
47. Friedman R.B., Oldfield E.H., Nieman L.K., et al. Repeat transsphenoidal surgery for Cushing’s disease. J Neurosurg. 1989;71:520-527.
48. Dickerman R.D., Oldfield E.H. Basis of persistent and recurrent Cushing disease: an analysis of findings at repeated pituitary surgery. J Neurosurg. 2002;97:1343-1349.
49. Nakane T., Kuwayama A., Watanabe M., et al. Long term results of transsphenoidal adenomectomy in patients with Cushing’s disease. Neurosurgery. 1987;21:218-222.
50. Estrada J., Boronat M., Mielgo M., et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Engl J Med. 1997;336:172-177.
51. Hoybye C., Grenback E., Rahn T., et al. Adrenocorticotropic hormone-producing pituitary tumors: 12- to 22-year follow-up after treatment with stereotactic radiosurgery. Neurosurgery. 2001;49:284-291. discussion 291-292
52. Jagannathan J., Sheehan J.P., Pouratian N., et al. Gamma knife surgery for Cushing’s disease. J Neurosurg. 2007;106:980-987.
53. Losa M., Picozzi P., Redaelli M.G., et al. Pituitary radiotherapy for Cushing’s disease. Neuroendocrinology. 2010;92(Suppl 1):107-110.
54. Engelhardt D., Weber M.M. Therapy of Cushing’s syndrome with steroid biosynthesis inhibitors. J Steroid Biochem Mol Biol. 1994;49:261-267.
55. Sonino N., Boscaro M., Paoletta A., et al. Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients. Clin Endocrinol (Oxf). 1991;35:347-352.
56. Feelders R.A., de Bruin C., Pereira A.M., et al. Pasireotide alone or with cabergoline and ketoconazole in Cushing’s disease. N Engl J Med. 2010;362:1846-1848.
57. Nagesser S.K., van Seters A.P., Kievit J., et al. Long-term results of total adrenalectomy for Cushing’s disease. World J Surg. 2000;24:108-113.
58. Kemink S.A., Grotenhuis J.A., De Vries J., et al. Management of Nelson’s syndrome: observations in fifteen patients. Clin Endocrinol (Oxf). 2001;54:45-52.
59. Wilson C.B., Tyrrell J.B., Fitzgerald P.A., Pitts L.H. Cushing’s disease and Nelson’s syndrome. Clin Neurosurg. 1980;27:19-30.
60. Loriaux D.L., Cutler G.B.J. In Kohler PO, ed. Clinical Endocrinology. John Wiley and Sons, Inc; 1986. 167-238
61. Veldhuis J.D., Iranmanesh A., Johnson M.L., Lizarralde G. Amplitude, but not frequency, modulation of adrenocorticotropin secretory bursts gives rise to the nyctohemeral rhythm of the corticotropic axis in man. J Clin Endocrinol Metab. 1990;71:452-463.
62. Liu J.H., Kazer R.R., Rasmussen D.D. Characterization of the twenty-four hour secretion patterns of adrenocorticotropin and cortisol in normal women and patients with Cushing’s disease. J Clin Endocrinol Metab. 1987;64:1027-1035.
63. Melby J.C. Assessment of adrenocortical function. N Engl J Med. 1971;285:735-739.
64. Besser G.M., Edwards C.R.W. Cushing’s syndrome. Clinics Endocrinol Metab. 1972;1:451-490.
65. Oldfield E.H., Chrousos G.P., Schulte H.M., et al. Preoperative lateralization of ACTH-secreting pituitary microadenomas by bilateral and simultaneous inferior petrosal venous sinus sampling. N Engl J Med. 1985;312:100-103.
66. Oldfield E.H., Vortmeyer A.O. Development of a histological pseudocapsule and its use as a surgical capsule in the excision of pituitary tumors. J Neurosurg. 2006;104:7-19.
67. Avgerinos P.C., Chrousos G.P., Nieman L.K., et al. The corticotropin-releasing hormone test in the postoperative evaluation of patients with Cushing’s syndrome. J Clin Endocrinol Metab. 1987;65:906-913.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 20 Cushing’s Disease
Pathophysiology
Cushing’s syndrome, the syndrome produced by chronic exposure to excess glucocorticoids, has several etiologies (Table 20-1). The most common cause of Cushing’s syndrome is iatrogenic—that is, prescribed glucocorticoids for patients with chronic obstructive pulmonary disease, autoimmune disorders, and transplant patients requiring chronic immunosuppression, among others. Excluding iatrogenic causes, the most common cause of Cushing’s syndrome is an ACTH-secreting pituitary adenoma, defined as CD, which affects 60% to 80% of patients with spontaneous (noniatrogenic) Cushing’s syndrome (Table 20-1).1 The other common causes of Cushing’s syndrome, which must be distinguished from CD, are adrenal tumors and adrenal cortical hyperplasia, and ectopic ACTH secretion from a nonpituitary tumor, most commonly small-cell–lung cancer or bronchial carcinoid. Secretion of ACTH by lung carcinoma is not rare, but the ACTH is usually inactive, termed “big ACTH.”2 A rare cause of Cushing’s syndrome is ectopic secretion of corticotropin-releasing hormone causing corticotroph hyperplasia.
| ACTH-Dependent | 85% |
| Cushing’s disease | 80–85% |
| Ectopic ACTH-secreting tumor | 15–20% |
| Ectopic CRH-secreting tumor | Rare (<1%) |
| ACTH-Independent | 15% |
| Adrenal adenoma | 7% |
| Adrenocortical carcinoma | 7% |
| Bilateral micronodular adrenocortical hyperplasia | Rare (1%) |
| Bilateral macronodular adrenocortical hyperplasia | Rare (<1%) |
Pathology
The typical pituitary adenoma causing CD is a microadenoma (<1 cm greatest diameter) that stains for ACTH by immunohistochemistry. Larger adenomas are occasionally seen and may even present with apoplexy. These tumors are monoclonal.3 They disrupt the normal acinar pattern of the gland most clearly seen when stained for reticulin. They are typically basophilic by hematoxylin and eosin staining, but this terminology is no longer used, as immunohistochemistry for ACTH is more specific. Occasionally Cushing’s syndrome is attributed to corticotroph hyperplasia. When no definite tumor is identified at surgery and partial or total hypophysectomy is performed, the specimen must be meticulously and thoroughly examined with serial slices at closely spaced intervals, because tumors as small as 1 mm diameter, or less, may cause endocrinopathy from excess ACTH secretion. Corticotroph hyperplasia, which is usually a diagnosis of exclusion (i.e., only after no tumor can be found in the specimen from pituitary surgery), should be considered only after a careful search of the gland has ruled out a discrete adenoma. In our experience, hyperplasia is exceedingly rare (only two suspected, but unproven, cases from over 1200 operations for CD). A unique cytoplasmic staining pattern, known as Crooke’s hyaline change, occurs in corticotrophs of the normal pituitary—cells from which ACTH production has been shut down by chronic exposure to hypercortisolemia. The “hyaline” is comprised of intracytoplasmic microfilaments, which do not stain for ACTH. Crooke’s changes may also be found in the cells of the adenoma itself.
Mechanism of Hypercortisolemia in ACTH-Secreting Pituitary Adenomas
The basic endocrine disorder of the adenomas causing CD is that the sensitive negative feedback of cortisol on the production and release of ACTH is impaired (Fig. 20-1). However, ACTH-secreting pituitary adenomas are well-differentiated tumors derived from pituitary corticotrophs; thus, they typically retain negative feedback to glucocorticoids, it is just set at a higher threshold for suppression, as inhibition of ACTH release is retained in response to high doses of glucocorticoid. These well-differentiated tumor cells also retain their expression of CRH receptors and the cellular machinery necessary to respond to CRH. It is these features that underlie the typical diagnostic responses with provocative endocrine tests used for the differential diagnosis of Cushing’s syndrome (see below). Excessive production of ACTH leads to hyperplasia and overproduction of cortisol by the adrenal cortex, loss of normal diurnal plasma cortisol rhythm, and sustained hypercortisolemia. It is the excessive cortisol, rather than ACTH per se that causes the clinical manifestations of CD.
Clinical Manifestations
The typical patient with Cushing’s syndrome has truncal obesity with associated moon facies, enlarged dorsal fat pads (“buffalo hump”), and abdominal fat deposition with associated purple striae or “stretch marks” (Table 20-2). Hirsutism, especially noticeable on the face of women, is a common component, as are thin skin and easy bruisability, especially of the hands and forearms. Along with the outward appearance, mood or psychiatric disturbances are common, especially depression. A reversible form of brain atrophy is frequently displayed on imaging studies4 and may be a clue to the presence of Cushing’s syndrome in pediatric patients. Other signs include hypertension and hyperglycemia, often with frank diabetes mellitus. A hypercoagulable state has been described with Cushing’s syndrome, so prophylaxis for deep venous thrombosis in high-risk situations has been encouraged.5 Patients also may have complications related to immunosuppression, such as fungal or opportunistic infections. Spinal epidural lipomatosis may be symptomatic.6 Osteoporosis is common; related complications include vertebral compression fractures and susceptibility to traumatic long-bone fractures with minor trauma. Pediatric patients with CD stop growing linearly and gain weight, often producing morbid obesity.7 This “crossing of the weight and height curves” is so common in childhood Cushing’s syndrome that many consider it diagnostic of the condition. Affected children often appear cherubic. Symptoms caused by tumor growth and pressure on the optic nerves and chiasm are rare with CD, as the tumors are usually microadenomas.
| Fat Distribution | Skin Manifestations |
| Centripetal obesity | Purple striae |
| Moon facies | Plethora |
| “Buffalo hump” | Hirsutism |
| Supraclavicular fat pads | Acne |
| Epidural lipomatosis | Bruising |
| Musculoskeletal | Metabolic/Circulatory |
| Osteoporosis; fractures | Hypertension |
| Proximal muscle weakness Pituitary Dysfunction Amenorrhea Decreased libido, impotence Hypothyroidism Dwarfism (children) |
Glucose intolerance Hypokalemic alkalosis Mental Changes Irritability Psychosis |
Life expectancy is greatly foreshortened by untreated CD. If left untreated, most patients succumb early to complications of the disease (diabetes, hypertension, myocardial infarction, stroke, or complications associated with immunosuppression).8
Diagnosis
Establishing Hypercortisolism
Urine-free cortisol (UFC) measurements are assayed using a variety of techniques. The normal upper levels vary with the technique used and with the laboratory performing the assay (Fig. 20-2). Hypercortisolism is associated with loss of normal diurnal variation in cortisol secretion, which is demonstrated by obtaining morning (8 to 9 a.m.) and evening (11 to 12 p.m.) plasma cortisol levels (Fig. 20-3). Salivary cortisol levels are also reliably used for this and are well suited for outpatient screening of adult and pediatric patients for hypercortisolism.9 A study of more than 140 patients demonstrated a sensitivity of 93% and a specificity of 100% using this test to determine the presence of Cushing’s syndrome (Fig. 20-4).10 Because of its simplicity, the overnight low-dose (1 mg) dexamethasone suppression test is commonly used to screen patients for hypercortisolism (Fig. 20-5). In persons with a normal hypothalamic-pituitary-adrenal axis, AM cortisol levels are suppressed by the overnight low-dose dexamethasone suppression test (1.0 mg given the night before a morning [7 to 8 a.m.] cortisol measurement). A morning plasma cortisol level greater than 1.8 μg/dl after the bedtime (11 p.m.) administration of 1 mg of dexamethasone detects most patients with Cushing’s syndrome and justifies further diagnostic evaluation.11
(Modified from Loriaux DL, Cutler GB Jr. Diseases of the adrenal glands. In: Kohler PO, ed. Clinical Endocrinology. New York: Wiley; 1986:167-238, with permission.)60
Differential Diagnosis of Hypercortisolism
Once excess cortisol production has been established, plasma ACTH levels are measured to distinguish between an ACTH-dependent and ACTH-independent etiology (Fig. 20-6; see also Fig. 20-1). With CD or ectopic ACTH secretion, the ACTH levels will be normal or elevated relative to the degree of glucocorticoid secretion. For this reason, these two entities are categorized as “ACTH-dependent” Cushing’s syndrome (see Table 20-1), in contrast to adrenal disease, in which plasma ACTH is low (<5 pg/ml) or undetectable (“ACTH-independent” Cushing’s syndrome, as the adrenal cortical cortisol secretion is autonomous).
(From Besser GM, Edwards CRW. Cushing’s syndrome. Clin Endocrinol Metab. 1972;1:451-490, with permission.)64
At one time, the presence of Cushing’s syndrome and then differentiating CD from adrenal disease or ectopic ACTH secretion were examined with the 6-day dexamethasone suppression test, as described by Liddle.12,13 The first 2 days of the test were used for measurement of basal cortisol secretion. In most patients with CD, cortisol secretion, an indirect measure of ACTH secretion by the pituitary gland or the tumor, is not suppressed during the 2 days of low-dose dexamethasone (0.5 mg every 6 hours for 48 hours), but 24-hour urinary cortisol secretion is suppressed to less than 10% of baseline values by 2 days of high-dose dexamethasone (2 mg every 6 hours for 48 hours). In contrast, high-dose dexamethasone fails to suppress cortisol secretion in most cases of ectopic ACTH secretion. Because of the difficulty in successfully completing this test today, it is now rarely used, but has been replaced with the high-dose overnight dexamethasone suppression test. For this test, 8 mg of dexamethasone is administered orally at 11 p.m. and a morning (7 to 8 a.m.) plasma cortisol measurement is obtained;14 for greatest diagnostic accuracy, suppression of morning serum cortisol of greater than 68% is required to assign a diagnosis of CD (Fig. 20-7).14,15
Another test available today to distinguish ectopic ACTH secretion from an ACTH-secreting pituitary tumor is the CRH stimulation test.16–18 Since they are well-differentiated tumors derived from pituitary corticotrophs, most ACTH-secreting pituitary adenomas retain receptors for, and response to, CRH, whereas most ectopic tumors, tumors that are not derived from pituitary tissue, do not express receptors for CRH and do not respond to it (Fig. 20-8). The sensitivity and specificity of the test are optimal using ≥35% for the maximum ACTH response from the 15- or 30-minute samples to indicate CD (see Fig. 20-8B).18
The test with the greatest diagnostic accuracy for the differential diagnosis of CD versus ectopic ACTH syndrome is bilateral simultaneous inferior petrosal sinus sampling performed with and without intravenous CRH administration.19 The test is performed by placement of catheters with their tips in the inferior petrosal sinuses and in a peripheral vein (Fig. 20-9A), and then obtaining serial, simultaneous samples for central and peripheral plasma ACTH concentrations at 2 and 0 minutes before and at 3, 5, and 10 minutes after intravenous CRH administration (1 μg/kg body weight). IPSS is only used in patients with confirmed hypercortisolism, as the test cannot discriminate between normal subjects and patients with CD. Further, since this is an invasive procedure with rare but serious associated risks,20 it is generally used in patients in whom the results of provocative endocrine testing to distinguish ectopic ACTH secretion from CD are conflicting or equivocal and in instances in which the sella MRI is negative. In this test the levels of ACTH in the primary venous drainage of the pituitary, the inferior petrosal sinuses, are compared to simultaneous ACTH measurements in the peripheral blood. A peak ratio of 2:1 petrosal:peripheral during baseline (before CRH) or 3:1 before or after CRH indicates a pituitary source of the excess ACTH, that is, CD.19 The sensitivity of the test is increased by sampling after administration of CRH, which, because it stimulates a pituitary adenoma to secrete ACTH, enhances the ACTH concentration differential between the central (inferior petrosal sinus) and the peripheral blood (Fig. 20-9B).19 It originally seemed that the procedure had a diagnostic accuracy 100%. However, reports of diagnostic errors, almost all false negative, have appeared, although in our experience the diagnostic accuracy approaches 100%. The accuracy of the test also relies upon successful placement of the catheter tips in the petrosal sinus bilaterally, since a false negative result may arise if the blood from both inferior petrosal sinuses cannot be catheterized successfully. Thus, venography is performed to ensure correct catheter placement and to evaluate the venous anatomy. A hypoplastic or anomalous inferior petrosal sinus in 0.8% of 501 patients was associated with false-negative results in patients with proven CD.21 It was briefly thought that comparison of petrosal sinus ACTH from the right to left sides would accurately indicate the side of the pituitary in which a small pituitary tumor was located, permitting a more focused search for it at surgery, or permitting removal of the half of the pituitary containing an adenoma that was too small to identify despite a thorough search of the gland during surgery.22 However, more experience with the technique for lateralization indicated that the lateralization accuracy is only about 70%, and even less so in pediatric patients,23 compromising its usefulness as a localizing measure during surgery.19
Imaging
Sella magnetic resonance imaging (MRI) is the imaging procedure of choice for detecting and localizing the pituitary adenoma in patients with CD. MRI should be performed with and without contrast, as the adenomas typically have decreased enhancement compared to the normal gland (Fig. 20-10). The resolution of a 1.5-T magnet may reveal tumors as small as 3 mm in diameter. MRI provides other important anatomical information for the surgeon: aeration of the sphenoid, parasellar anatomy, location of the carotid arteries or coexisting aneurysms, extent of supra- or para-sellar extension of an adenoma, and ectopic parasellar tumors. Recently the spoiled gradient recalled acquisition (SPGR) technique, used with 1-mm nonoverlapping slices was shown to be more sensitive than the conventional spin echo approach (sensitivity 80% vs. 49%), a finding also true in pediatric CD.24,25 However, the incidence of false positives was also higher (4% vs. 2%). Since the sensitivity of the conventional MRI techniques (spin echo) for CD is only 50% to 75%,26–29 many patients have a negative MRI. Furthermore, MRI is not always available or possible, as in patients with an MRI incompatible cardiac pacemaker or a morbidly obese patient who cannot be accommodated by the scanner. In these patients, a sella computerized tomography image (CT) with and without contrast may demonstrate a tumor, but CT is less sensitive than MRI. Furthermore, since the specificity of MRI is not 100% (MRI abnormalities consistent with adenomas occur in the pituitary gland in 10% of normal volunteers30
[/not-level-membership-for-neurosurgery-category]
