CHAPTER 268 Current Status and Future Direction of Management of Spinal Cord Injury
Spinal cord injury (SCI) costs society in excess of 7 billion dollars annually1,2 and bears the greater cost of human suffering related to impaired ambulation, sensation, and bowel, bladder, and sexual function. Advances in supportive care have significantly improved the survival of patients with SCI in recent years2; however, physicians currently have little else to offer these devastated patients. Fortunately, the last decade has seen tremendous scientific advancement in this field. Several emerging therapeutics are showing promise in early-phase clinical trials, and there is a real possibility that one or more efficacious therapies will soon enter clinical practice. It is thus increasingly important for neurosurgeons and purveyors of emergency and critical care to be familiar with these advances. Other advances such as cell replacement therapy are more distant on the horizon but also have strong potential for translation.
This chapter aims to review current treatments of acute SCI, as well as those that are on the horizon. Supportive and surgical care is reviewed in greater detail elsewhere in this text, as is the treatment of chronic SCI.3,4
Pathophysiology of Spinal Cord Injury and A Basis for Therapeutics
The pathophysiology of SCI can be conceptually divided into two critical phases: the initial primary injury and subsequent secondary injury.5–9 Primary injury refers to the initial traumatic insult to the spinal cord, which results in immediate severing of axons and death of spinal cord cells.10 Secondary injury invariably follows and leads to progressive tissue damage for weeks and even years after the initial insult.11–13 It results from a complex series of interrelated events seen on a number of physiologic levels. Hypotension and hypoxia are important preventable causes of secondary injury at a clinical level and must be recognized and either prevented or treated proactively. Numerous secondary injury mechanisms also occur on the cellular level, and although their delayed time course theoretically provides a window for therapeutic intervention,14 we are currently less capable of arresting these processes, which include vasospasm and localized ischemia,15,16 delayed cell damage and apoptosis,17–19 ion-mediated cell damage and excitotoxicity,13,20 and neuroinflammation,21 as well as mitochondrial dysfunction6 and oxidative cell damage (Fig. 268-1).22,23
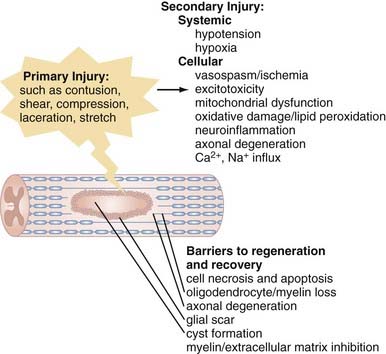
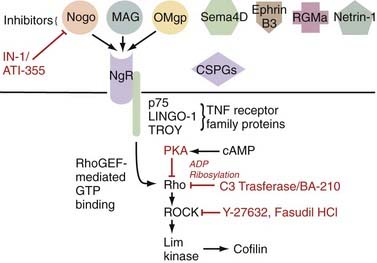
Additionally problematic is the fact that regeneration within the central nervous system (CNS) is poor after an insult.24 Trophic factor levels and the endogenous stem cell response are insufficient after injury.12 Moreover, the myelin normally essential for neuronal function becomes a major inhibitor of axonal growth after injury (Fig. 268-2).25,26 A number of myelin-associated inhibitors of axonal regrowth have been identified, the first being Nogo.27–29 Subsequently identified were myelin-associated glycoprotein and oligodendrocyte-myelin glycoprotein, as well as guidance molecules (semaphorin 4D, ephrin B3, repulsive guidance molecule A, netrin-1) and chondroitin sulfate proteoglycans of the extracellular matrix (NG2, versican, neurocan, brevican, phosphacan), which have the same inhibitory effects.30–33 Of therapeutic significance, all these inhibitors of axonal regeneration appear to signal via activation of the guanosine triphosphatase (GTPase) Rho.34 Glial scarring is also an obstacle in that it is a barrier to neural outgrowth and may promote neuropathic pain.35–37
Although these obstacles are formidable, neural preservation and repair are highly feasible. Only a fraction of spinal cord axons must be intact to facilitate ambulation,38 and residual axons cross the injury even in the face of severe SCI.38,39 Furthermore, many of these axons are viable but dysfunctional only because of oligodendrocyte loss and demyelination.40,41 Agents that improve cell survival, replace lost cells, or enhance function of the surviving neurological substrate even modestly may thus be of great functional significance. Additionally, in a landmark finding, Aguayo and David demonstrated that central neurons have the capacity to regenerate in environments free of the inhibitors typically found in the CNS.42 Therapies that neutralize myelin inhibitors and antagonize Rho are showing great promise in early-phase human clinical trials.43 With this in mind, optimism of both patients and physicians is thus justified.
Current Treatment of Spinal Cord Injury
Spine surgery is a rapidly changing field, and although operative nuances have seen dramatic change even in recent years, the reality is that decompression, realignment, and stabilization remain the primary offerings of the spine surgeon for the acutely spinal cord injured. SCI guidelines44–48 and pharmacotherapy have been more recent important additions to the clinical armamentarium.
Initial Management
The top priority in managing acute SCI is assessment and stabilization of vital signs, with strict adherence to the advanced trauma life support protocol.49,50 After stabilization, patients with SCI should be transferred expeditiously to the nearest trauma center that can provide definitive care.44 During transport, the cervical spine must be immobilized by stabilizing both the body and head44; merely stabilizing the head allows the body and thus the cervical spine to move and risks iatrogenic injury. After transfer it is also imperative to remove a patient from rigid spine boards immediately, preferably within 4 hours of being placed on such a board, to prevent pressure-induced ulceration.50 Initial management should be provided in a critical care unit experienced in intensive neurological monitoring and invasive blood pressure monitoring.
Breathing
Respiratory insufficiency is an important cause of early mortality after SCI. High cervical injuries (C3-5) paralyze the diaphragm and cause respiratory arrest. Lower cervical injuries affect the intercostal muscles, typified by paradoxical breathing. Paralysis of the intercostal musculature is associated with an approximate 70% decrease in forced vital capacity and maximal inspiratory force because inspiration causes chest wall collapse until the onset of spasticity.51 Patients who compensate initially can fatigue rapidly, and indeed, a third of patients with cervical injuries will require intubation in the first 24 hours. It is thus useful to monitor vital capacity in such patients; intubation should be considered in patients whose vital capacity is less than 1 L, particularly if there is evidence of fatigue.51 Hypoxia is an important cause of secondary SCI and must be avoided.
Circulation
Fluid administration is generally considered first-line therapy in this situation52; however, a restricted heart rate makes these patients susceptible to fluid overload.51 Early consideration of vasoactive agents is recommended, in particular an agent such as dopamine with both α- and β-adrenergic activity, to counter peripheral vasodilation, in addition to providing chronotropic support to the heart (Table 268-1).51 It is recommended that mean arterial pressure be maintained higher than 85 mm Hg for the first 7 days after SCI.44,53
| DRUG | DOSE RANGE | MECHANISM OF ACTION |
|---|---|---|
| Dopamine* | 1-10 µg/kg/min | α Agonist at low doses, β agonist at higher doses |
| Dobutamine | 5-15 µg/kg/min | Only β-adrenergic |
| Epinephrine | 1-8 µg/min | α Agonist and β agonist, may promote cardiac dysrhythmia |
| Norepinephrine | 1-20 µg/min | Mainly β-adrenergic, mildly α-adrenergic |
| Phenylephrine | 10-100 µg/min | Only α-adrenergic |
The ideal pressor for spinal cord injury counters peripheral dilation (α-agonists), as well as impaired cardiac chronotropy (β-agonists).
* Dopamine has both properties and is an excellent choice for many patients with spinal cord injury.
Adapted from Ball PA. Critical care of spinal cord injury. Spine. 2001;26:S27-S30.
Clinical Assessment
A number of valid clinical assessment tools have also been devised. The American Spinal Injury Association (ASIA) impairment scale, alternatively known as the International Standard for Neurological Classification of Spinal Cord Injury, was first published in 1982 and now represents the international standard for postinjury evaluation of neurological function.54 Its simplified form, the ASIA impairment scale, bears the same form as the earlier Frankel scale and maintains backward compatibility with it. The modified Benzel classification system, used in the GM1 ganglioside trial,84 may be used in future trials because it addresses some of the perceived deficits of the ASIA impairment scale: it subdivides ASIA D into three different grades to reduce the ceiling effect and additionally assesses walking and sphincter function.
Imaging
When SCI is present, imaging is used to confirm the injury and delineate the anatomy of the lesion. If possible, magnetic resonance imaging (MRI) is optimal and delineates soft tissues involved in the injury. Spinal cord compression is important to note and can be quantified with reproducible techniques.56 Moreover, several MRI parameters, including the degree of cord compression, the severity of spinal cord swelling, and the presence of intraparenchymal hemorrhage, have been shown to predict adverse neurological outcomes after SCI.57 As discussed later, a growing body of evidence suggests benefit from early decompression of the compressed spinal cord, which must then be considered. In the cervical cord, decompression may be performed by either closed or open means.
Spinal Cord Decompression
Closed Reduction
There are a number of contraindications to cervical traction, as outlined in Table 268-2, and great care must be taken in patients with abnormal spinal anatomy, such as those with ankylosing spondylitis, in whom the risk for iatrogenic injury is high. Most studies report a greater than 80% success rate, and about 80% of patients improve neurologically after traction.44 Reports of neurological worsening with traction have raised fear of exacerbating or inducing disk herniation and resulting SCI.58,59 The literature suggests that although the prevalence of disk herniation is high in patients with SCI, its clinical relevance is doubtful.60 Nonetheless, our group prefers to perform MRI before traction in neurologically intact patients who have “everything to lose” but prefer traction as a first step in patients with any SCI. Guidelines pertinent to closed decompression are presented in Table 268-3.
TABLE 268-3 Recommendations from the 2002 Acute Spinal Cord Injury Guidelines Pertinent to Closed Reduction; All Recommended as Therapeutic Options
MRI, magnetic resonance imaging.
From Hadley MN, Walters B, Grabb P, et al. Guidelines for the management of acute cervical spine and spinal cord injuries. Clin Neurosurg. 2002;49:407-498.
Timing of Decompression
A significant body of animal research has demonstrated neurological benefit from early decompression of the injured spinal cord; however, such benefit is less clear in humans, particularly in polytrauma patients, who are often medically unstable in the acute postinjury phase.61–63 Despite the fact that early spine surgery appears to be safe in polytrauma patients,61 the question of whether early decompression for acute human SCI is or is not beneficial for neurological recovery remains incompletely answered.61
Decompression of Central Cord Syndrome
Central cord syndrome is uniquely challenging with respect to determining the optimal timing of intervention given that most patients are initially seen without spinal instability and experience substantial spontaneous neurological improvement. In addition, a historic and influential publication from Schneider and associates in 1954, which first described central cord syndrome,64 reported several poor outcomes arising from early decompression. The result was a recommendation to consider central cord syndrome a unique clinical entity and to avoid early procedures because of perceived risk to the spinal cord. Despite the tenuous evidence supporting it, this recommendation has persisted in the literature, although recent evidence challenges this conclusion.65
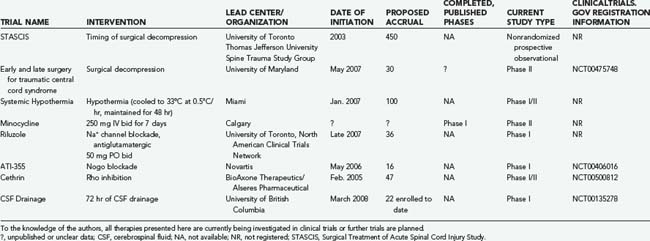
Even though attempts have been made to identify factors that influence neurological outcome after central cord syndrome65–68 and specifically address whether early surgery is beneficial,69–71 a prospective trial is needed. Fortunately, investigators at the University of Maryland have registered a phase II, single-center randomized clinical trial to examine the timing of decompression in patients with central cord syndrome. This trial seeks to randomize 30 patients to decompression within 5 days or after 6 weeks of injury. The study will compare ASIA, Functional Independence Measure, and Spinal Cord Independence Measure scores; the degree of canal compromise and spinal cord compression; and syrinx size. Patients will be monitored for 1 year.
Venous Thrombosis Prophylaxis
Patients with SCI are at very high risk for venous thromboembolism in light of their tissue injury and venous stasis resulting from paralysis. The acute SCI guidelines recommend a combination of low-molecular-weight heparin, rotating beds, adjusted-dose heparin, pneumatic compression stockings, or electrical stimulation as prophylaxis for deep venous thrombosis.44 Low-dose heparin therapy or oral anticoagulation alone is not recommended as prophylactic treatment, but vena cava filters are recommended for patients who fail anticoagulation or cannot undergo anticoagulation. A 3-month duration of prophylaxis for deep venous thrombosis and pulmonary embolism is recommended.
Guidelines
In recent years, the neurotrauma field has been significantly advanced by the establishment of guidelines. The acute SCI guidelines were published in 2002 and focus on pharmacotherapy, traction, intensive care management, and surgery.44 These guidelines will help standardize management of patients with SCI and will thus play an important role in future SCI clinical trials. Key recommendations from these guidelines have been discussed in the preceding sections.
A parallel effort in establishing guidelines for chronic SCI was published in 2005 by the Consortium for Spinal Cord Medicine, whose clinical practice guidelines deal with rehabilitative aspects of care. These guidelines cover outcome measures, autonomic dysreflexia, respiratory function, thromboembolism, pressure ulcers, bowel function, and depression.72
Pharmacotherapy and Completed Randomized Controlled Trials for Acute Spinal Cord Injury
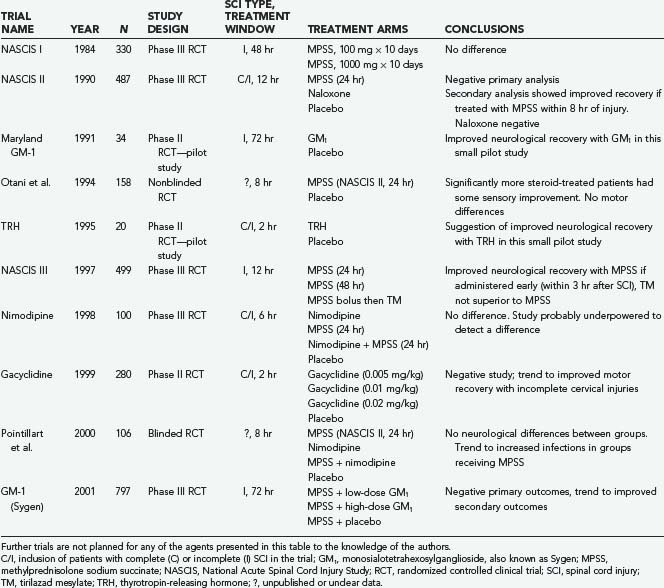
A number of pharmacotherapies have been developed with the aim of ameliorating secondary injury processes; however, only methylprednisolone sodium succinate (MPSS) has found clinical application thus far. Other agents investigated in large multicenter prospective randomized controlled trials include tirilazad mesylate, GM1 ganglioside, thyrotropin-releasing hormone (TRH), gacyclidine, naloxone, and nimodipine (Table 268-4).
Methylprednisolone Sodium Succinate (Solu-Medrol)
Corticosteroids have been used for neurotrauma for decades but have only recently been subject to intensive scientific scrutiny. Their neuroprotective effects include antioxidant properties, enhancement of spinal cord blood flow, reduced calcium influx, reduced axonal dieback, and attenuated lipid peroxidation.23,24 After the accumulation of preclinical data that were generally supportive of a neuroprotective role in animal models of acute SCI,73 MPSS was studied in five prospective human acute SCI trials,74 thus making it the most extensively studied drug for acute SCI.75
Three landmark National Acute Spinal Cord Injury Study (NASCIS) trials examined the use of MPSS for acute SCI. The first NASCIS trial, published in 1984,61 compared high-dose with low-dose MPSS; placebo was judged unethical because benefit from steroids was presumed.75 Neurological improvement was not significantly different in the two groups, although a statistically significant increase in wound infection was noted in the high-dose group, as well as increased rates of gastrointestinal hemorrhage, sepsis, pulmonary embolism, delayed wound healing, and death.62
Animal studies completed subsequent to NASCIS I suggested that higher doses may be required for neuroprotection.63 NASCIS II was thus designed to examine a higher dose of MPSS with comparison to placebo and the opioid antagonist naloxone given within 24 hours of injury.66 In the overall analysis there was no neurological benefit in the MPSS-treated group; however, a post hoc analysis74 (reportedly planned a priori65) found that patients receiving the drug within 8 hours of injury benefited neurologically, notably including those with complete injuries.79 As in NASCIS I, MPSS administration was associated with an increase in wound infection and pulmonary embolism.79
The NASCIS III trial was designed and powered to explore the beneficial effects of MPSS administration within 8 hours of injury reported in NASCIS II.68,69 This was the only NASCIS trial to assess functional outcome; it used the Functional Independence Measure and compared the 24-hour infusion used in NASCIS II with a 48-hour infusion of MPSS and a group receiving tirilazad mesylate, a 21-aminosteroid believed to be an antioxidant without glucocorticoid effects.70 Overall, this trial demonstrated no sustained benefit with MPSS administration. A post hoc analysis noted that patients receiving an MPSS bolus 3 to 8 hours after injury demonstrated improved neurological function at 6 weeks and 6 months but not at 1 year when administered MPSS for 48 rather than 24 hours. This led to the recommendation that within 3 hours of injury, a 24-hour infusion would suffice, but if initiated within 3 to 8 hours of injury, a 48-hour MPSS regimen was better than the 24-hour NASCIS II regimen. The 48-hour regimen represents the highest dose of MPSS prescribed for any clinical condition71 and was associated with a twofold higher rate of severe pneumonia, a fourfold higher rate of severe sepsis, and a sixfold higher incidence of death when compared with the 24-hour group.74
Two other prospective human SCI trials involving corticosteroids have been published. Although Otani and coauthors76 reported benefit from MPSS administration, Pointillart and colleagues77 reported none. Nonetheless, both studies were small and plagued by methodologic problems, which limits their interpretation as either positive or negative studies.
Scrutiny of the aforementioned trials has led to two predominant concerns regarding the use of MPSS for acute SCI.74,78–82 First, the benefits noted have been modest and have come from secondary analyses. Second, high rates of adverse events were consistently associated with MPSS administration. Our personal view is that MPSS administration remains justified for acute SCI (with 8 hours) in nondiabetic and nonimmunocompromised patients given the severity of SCI deficits and current lack of alternatives.83
GM1 Ganglioside (Sygen)
Gangliosides are complex glycolipids abundant in the membranes of nervous tissue. In 1991, exogenous administration of monosialotetrahexosylganglioside, also known as GM1 or Sygen, was examined in the prospective randomized Maryland GM-1 study of 37 patients. This trial demonstrated statistically significant improvement in ASIA motor scores when compared with placebo.84 Efficacy was noted as late as 48 hours after injury, and a predominant effect on lower extremity function suggested effect on axons traversing the injury.75 This led to the largest prospective randomized clinical trial in patients with acute SCI to date, the Sygen Multi-Center Acute Spinal Cord Injury Study, which enrolled more than 750 patients. This study failed to demonstrate a significant difference in its ambitious primary outcome measure—a two-point improvement on the modified Benzel walking scale. It is possible, however, that greater efficacy may have been seen with earlier administration; because the patients were obligated to receive MPSS first, GM1 was not administered, on average, until 55 hours after injury in this study. We are unaware of any plans for future trials with this agent.85
Thyrotropin-Releasing Hormone
In addition to its well-known hormonal functions, TRH has been shown to antagonize secondary injury mediators such as excitotoxic amino acids, peptidoleukotrienes, endogenous opioids, and platelet-activating factor.86 In 1995, the only clinical trial ever conducted with TRH for acute SCI was published.87 In this randomized double-blind, placebo-controlled trial, patients with incomplete but not complete SCI achieved statistically significant improvements on the NASCIS and Sunnybrook scales. This result must be interpreted with caution—it may represent a type I error because the trial was plagued by attrition and only 20 patients were ultimately analyzed. No further clinical SCI studies to investigate TRH have been initiated.
Gacyclidine (GK-11)
Glutamate is the main excitatory neurotransmitter in the CNS, but excess of this neurotransmitter after CNS injury leads to excitotoxicity. Previous trials of antiglutamatergic agents, even with competitive antagonists such as selfotel,88,89 have been unsuccessful because of significant cognitive side effects, including agitation, sedation, hallucinations, and memory deficits. Interestingly, a noncompetitive N-methyl-D-aspartate receptor antagonist, gacyclidine or GK-11 (Beaufour-Ipsen Pharma, Paris), has demonstrated substantially better tolerability.90 A double-blind phase II human trial randomized more than 200 patients to receive three escalating doses of gacyclidine.91 Early benefit was seen in the treatment groups, but it was not maintained at 1 year. Despite the fact that this negative result probably stems from insufficient statistical power, this agent is no longer being pursued for SCI.
Nimodipine
Intracellular calcium levels are tightly regulated because high intracellular concentrations can activate calpains and other destructive enzymes that lead to apoptosis. In addition, calcium influx contributes to excitotoxicity because release of glutamate is dependent on calcium. Nimodipine is an L-type calcium channel blocker that may antagonize these processes.92 A human trial in patients with SCI was completed in France in 1996.93 This trial, which included 100 patients, had four treatment arms: nimodipine, MPSS (NASCIS II protocol), both agents, and placebo. Benefit over placebo was not demonstrated in any treatment group, although it is quite likely that this study was also underpowered to reveal a therapeutic effect.
Opioid Antagonism
Dynorphin A, an endogenous opioid, is released after SCI and has neurotoxic effects; it also reduces spinal cord blood flow by nonopioid mechanisms.94 In the 1980s the opioid antagonist naloxone was examined in a phase I human SCI trial. The results suggested benefit,95 but imbalance between the experimental groups makes this result difficult to interpret. A more definitive examination of this agent was performed in the NASCIS II trial,96 which showed no benefit from its administration.
Toward the Next Generation of Trials
Much has been learned from completed studies on acute SCI. Intense scrutiny of their design and interpretation is playing a critical role in shaping the next generation of trials. Additionally, a number of authors have published recommendations for the scientific and ethical conduct of future trials, including Tator,97 Cesaro,98 and Sagen.99 A parallel effort has come from the International Campaign for Cures of Spinal Cord Injury Paralysis (ICCP), which published four documents in 2007 related to (1) statistical power needed for clinical trials in relation to injury severity and timing of administration of the experimental therapy, (2) appropriate outcome measures, (3) inclusion/exclusion criteria and ethics, and (4) trial design.45–48
Paradoxically, more experimental therapies for SCI than ever before are being pursued in an environment of questionable scientific rigor and ethics. Recent years have seen many patients travel appreciable distances at great personal cost and risk to seek cell or tissue transplantation therapies,100 which are at best unproven and at worst very dangerous.100,101 Additionally, information surrounding the conduct and results of many current SCI clinical trials is not in the public domain,97 in contravention of the current effort to promote registration of trials.102,103 In response, the ICCP has produced an additional document designed to educate patients considering enrollment in an SCI clinical trial and encourage them to participate in studies with high scientific and ethical quality.104
Ongoing Clinical Trials
More than 70 clinical trials on SCI are registered online at www.clinicaltrials.gov; however, the majority relate to interventions for chronic SCI and will not be reviewed. Here, we describe registered interventional trials involving acute and subacute SCI, as well as others that we have identified and of which we have some knowledge (Tables 268-5 and 268-6).
TABLE 268-5 Summary of Recent Published Experimental Trials for Acute and Subacute Spinal Cord Injury

TABLE 268-6 Summary of Ongoing, Well-Documented, Unpublished Experimental Trials for Acute and Subacute Spinal Cord Injury
Cerebrospinal Fluid Drainage
In thoracoabdominal aortic aneurysm surgery, drainage of cerebrospinal fluid (CSF) has been found to significantly reduce the incidence of ischemic paraplegia. This suggests that lowering intrathecal pressure improves spinal cord perfusion pressure and provides neuroprotection.105 Investigators at the University of British Columbia have initiated a safety and feasibility study to evaluate CSF drainage through a lumbar intrathecal line as a neuroprotective strategy after acute SCI. Twenty-two patients have been enrolled to date, and there have been no adverse events attributed to CSF drainage.
Oscillating Field Stimulation
Neurites grow toward the negative pole (cathode) in an electrical field.106,107 Researchers at Indiana University Medical Center thus developed an implantable “oscillating field stimulator” capable of generating an electrical field along the rostrocaudal axis of the spinal cord. To promote axonal growth in both directions, the device “oscillates,” or changes polarity every 15 minutes. This device, initially tested in 10 patients with complete SCI, was implanted within 18 days and removed at 15 weeks after injury. Reported in 2005, this study noted that at 1 year mean improvement in light touch was 25.5 points (P = .02), mean improvement in pinprick sensation was 20.4 points (P = .02), and mean improvement in motor status was 6.3 points (P = .02).108 Based on comparison to historical controls from NASCIS III, efficacy is suggested but must be interpreted with caution. Few complications were associated with implantation of this device (one wound infection and one device failure). The Food and Drug Administration (FDA) has approved enrollment of additional patients in this study; Cyberkinetics Neurotechnology Systems, has purchased the intellectual property for this technology, and we look forward to additional data from the investigators.
A similar trial involving pulsed electrical stimulation has been conducted on 100 patients in Beijing by investigators Xu and Liu.109 The results of this work are uncertain, however.
Hypothermia
Hypothermia has long been explored for its putative neuroprotective effects despite risks that include coagulopathy, sepsis, and cardiac dysrhythmia. In addition to reducing the metabolic rate, hypothermia also appears to reduce extracellular glutamate, vasogenic edema, apoptosis, neutrophil and macrophage invasion and activation, and oxidative stress.110–118 Therapeutic hypothermia has become a treatment guideline for patients resuscitated from out-of-hospital cardiac arrest,33 but animal models of traumatic SCI have demonstrated mixed results.119,120 Researchers from the Miami Project to Cure Paralysis thus began a trial in 2007 to explore the role of systemic hypothermia in patients with SCI. This trial involves rapid cooling with chilled intravenous saline to drop core body temperature to around 34°C, with comparison to historical controls. We anxiously await the results of this trial.
Minocycline
Minocycline is a synthetic tetracycline derivative commonly used in dermatology. A number of independent laboratories have reported that minocycline attenuates secondary injury and enhances functional recovery in various animal models of SCI.121–124 Its mechanism of action appears to involve the inhibition of microglial activation,125–128 in addition to antiapoptotic properties.127,129
These promising preclinical results have led to two clinical trials, now ongoing. The first, led by researchers at the University of Calgary, is a prospective, randomized, placebo-controlled phase I/II human trial of intravenous minocycline in which patients are randomized within 12 hours of their injury. According to the principal investigators of this trial, enrollment will be halted in November 2008 and a decision made regarding whether a phase III study is warranted. Less is known about the second study, which is being conducted by investigators in Saudi Arabia to assess the effectiveness of minocycline when coadministered with the immunosuppressant tacrolimus (FK506), which inhibits the enzyme calcineurin.97
Riluzole
Riluzole (Rilutek; Sanofi-Aventis, Bridgewater, NJ) is a benzothiazole anticonvulsant that has been licensed for patients with amyotrophic lateral sclerosis for approximately 10 years130; it perhaps prolongs their lives by 2 to 3 months.131 Its neuroprotective effects appear to result from blockade of voltage-sensitive sodium channels,132 as well as antagonism of presynaptic calcium-dependent glutamate release.133 Of interest in SCI, it demonstrates synergy with MPSS,134 and in animal models of SCI it has provided neuroprotective effects when administered as late as 10 days after injury.132,135
In mid-2008, a human multicenter SCI trial began enrolling a target of 36 patients with ASIA A, B, or C injuries between the C4 and T10 neurological levels. Patients were enrolled within 12 hours of their injury and administered a 10-day course because glutamate- and sodium-mediated secondary injury is maximal during this period.20,132 One-year follow-up was planned, with assessments including the ASIA score, Spinal Cord Independence Measure, and Brief Pain Inventory.
Targeting Myelin-Associated Inhibitors of Regeneration
ATI-355
In the late 1980s, Pico Caroni and Martin Schwab demonstrated that oligodendrocytes and their myelin membranes are major inhibitors of axonal growth within the CNS.25,136 They then biochemically separated 35- and 250-kD inhibitory fractions within CNS myelin (NI-35 and NI-250) and developed a monoclonal antibody, IN-1, that could block their inhibitory properties in vitro.26 Subsequent in vivo application of IN-1 resulted in substantial axonal sprouting and some long-distance corticospinal axonal regeneration within the adult mammalian CNS,137 which led to improved function on a variety of tests.138,139 The IN-1 antibody was also used to characterize its target protein antigen,140 now known as Nogo.27–29
The humanized anti-Nogo antibody has been shown to promote axonal sprouting and functional recovery after SCI in numerous animal models, including primates.141,142 In May 2006, a human phase I clinical trial was initiated by Novartis in Europe and enrolled patients 4 to 14 days after complete SCI between C5 and T12. The agent is being administered by continuous intrathecal infusion in four increasing dose regimens, with the highest dose being delivered over a period of 28 days. The FDA has expressed concern with the external nature of the infusion pump, and hence clinical evaluation has been limited to Europe and Canada.
Cethrin
McKerracher’s group at the University of Montreal developed a therapy that exploits the fact that all known myelin and extracellular matrix inhibitors indentified thus far signal via activation of the GTPase Rho (see Fig. 268-2).143 Their work involves a toxin produced by Clostridium botulinum, C3 transferase, which is a specific inhibitor of Rho. Interruption of this “final common pathway” thus has the potential to be more potent than efforts to antagonize any single myelin inhibitor. Animal studies suggest that not only does this agent facilitate axonal growth and functional recovery100,143 but C3 transferase also has neuroprotective effects.144
Cellular Transplantation Therapies
Because limited substrate for neural repair is a significant obstacle after SCI, cell replacement strategies are thought to be among the most promising new strategies for treating SCI. Various cell types have been tried, with varying goals. Some aim to produce new neurons that will integrate into functional circuits, whereas groups such as our own have sought and achieved oligodendrocyte differentiation and remyelination.145 Despite diverse strategies, functional benefit has been seen consistently, although as yet the magnitude of this benefit has been uniformly modest.
Activated Autologous Macrophages (ProCord)
In the 1990s, pioneering work by Rapalino and colleagues demonstrated, in an animal model of SCI, that autologous macrophages activated ex vivo by peripheral myelin could promote functional recovery when injected into the injured spinal cord.146 Possible mechanisms include augmented clearance of myelin debris or enhanced synthesis of the beneficial trophic factors interleukin-1β and brain-derived neurotrophic factor and decreased synthesis of the harmful factor tumor necrosis factor-α.
This technology was then commercialized by Proneuron Biotechnologics, and a clinical trial for human SCI was launched in Israel, thus making activated autologous macrophages the first cellular substrate to be transplanted into human SCI patients in a carefully designed, rigorous clinical trial. In the initial trial eight patients with complete SCI were enrolled and underwent transplantation within 14 days of injury.147 The results were published in 2005, with three of the eight ASIA A patients recovering to ASIA C, and no major adverse events related to the cell transplants were encountered. This encouraging result led to the subsequent multicenter phase II ProCord trial in Israel and the United States. Unfortunately, this trial was suspended prematurely in spring 2006 for financial reasons, and there are currently no plans to continue this study. Fortunately, 1-year data on safety and neurological recovery in the 50 patients enrolled will be published in 2008 (personal communication, Dr. Dan Lammertse).
Schwann Cells
Schwann cells, the myelinating cells of the PNS, may represent an environment permissive of regeneration similar to the peripheral nerve grafts used by Aguayo’s group.148 Another potential benefit of these cells is the ability to harvest them from an autologous source, such as the sural nerve. The work of Dr. Bunge and colleagues at the Miami Project has explored this possibility since the early 1990s.42 A limitation of this technique, however, appears to be that regenerating CNS axons readily grow into the permissive environment that these cells provide but are not prone to growing out of it and back into the hostile CNS environment. Nonetheless, a formal clinical trial on Schwann cell transplantation for SCI is being planned at the Miami Project, which if initiated, would represent the realization of nearly a generation of pioneering scientific work in SCI for the investigators.
Olfactory Ensheathing Cells
Olfactory ensheathing cells (OECs) are specialized glia of the olfactory system that may address the “PNS-to-CNS” barrier inherent with Schwann cells. OECs escort regenerating axons of olfactory receptor neurons from the PNS of the nasal epithelium to the “hostile” CNS of the olfactory bulb. A host of promising preclinical data culminated in a report of OEC transplantation promoting axonal regeneration and functional recovery in a model of complete spinal cord transection.150 Amid these encouraging findings, however, significant controversy exists about the myelinating potential of OECs; evidence suggests that remyelination may instead be mediated by Schwann cells that contaminate OEC cultures and also bear the p75 marker.149 Nonetheless, interest in this autologous transplantation strategy remains high, and a number of centers are currently implanting human SCI patients with OECs or tissue acquired from the olfactory region that consists of OECs and other cells.
In an uncontrolled Portuguese pilot study,151 seven patients with ASIA A injuries were treated with autologous olfactory mucosal implants at 6 months to 6.5 years after injury. Apparently, all patients had improvement in ASIA motor scores, with two progressing from ASIA A to ASIA C. Additionally, two patients reported return of sensation in their bladders, and one regained voluntary anal sphincter contraction. This work has not been subjected to independent analysis, and it is unclear what to conclude from this preliminary report.
An Australian center conducted a single-blind trial of “purified” autologous OECs implanted in three patients with complete thoracic SCI within 6 to 32 months of injury. Comparison was made to matched but untransplanted controls.34 One-year follow-up data reported no motor improvement, but an absence of surgical complications.152
Bone Marrow Stromal Cells
A Korean group has reported the results of autologous human BMSC transplantation combined with the administration of granulocyte-macrophage colony-stimulating factor in a phase I/II open-label, nonrandomized study.153,154 This trial involved 35 ASIA A patients who underwent transplantation within 14 days (n = 17), between 14 days and 8 weeks (n = 6), or at more than 8 weeks (n = 12) after injury, with comparison to 13 control patients treated by decompression and fusion only. BMSC transplantation led to improved neurological function in 30.4% of patients in the acute and subacute groups, but no significant improvement was observed in the chronic group.
Investigators in Prague have also pursued a human BMSC trial.97,145 Their trial included 20 patients with complete SCI who underwent transplantation 10 to 467 days after injury. Improvement in motor or sensory function (or both) was noted in 5 of 7 acute and 1 of 13 chronic patients, which led the authors to suggest a therapeutic window of 3 to 4 weeks after injury.
Human Embryonic Stem Cells
Researchers at the University of California at Irvine, led by Dr. Hans Keirstead, have reported promising results in a rat SCI model with the transplantation of human embryonic stem cells. These cells differentiate into oligodendrocyte progenitors and achieve remyelination of spared, demyelinated spinal cord axons.155,156 They have developed a technique to ensure high purity of their cell isolates, as well as techniques for culturing these cells without the need for feeder cells, which could theoretically lead to viral contamination or the presence of nonhuman polysaccharide epitopes on the surface of the transplanted cells.157 These steps have been critical in making these cells suitable for human transplantation.
Indeed, the biopharmaceutical corporation Geron is attempting to bring this cell type into human clinical trials—a phase I trial had been proposed as early as 2006.157 Geron had hoped to initiate this clinical trial in 2008; however, the FDA recently placed this trial on hold. The reasons for this hold were not known at the time of this writing but may involve concern regarding the potential for tumorigenesis. Formal release of this information will be of great importance to the field because the FDA’s response to this proposed trial will set precedents of monumental importance to future cell replacement therapies in humans.157
American College of Surgeons. ATLS Advanced Trauma Life Support Program for Doctors, 7th ed. Chicago: American College of Surgeons; 2004.
Ball PA. Critical care of spinal cord injury. Spine. 2001;26:S27-S30.
Baptiste DC, Fehlings M. Emerging drugs for spinal cord injury. Expert Opin Emerg Drugs. 2008;13:63-80.
Bracken MB, Shepard M, Collins W, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405-1411.
Caroni P, Schwab M. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85-96.
Eftekharpour E, Karimi-Abdolrezaee S, Wang J, et al. Myelination of congenitally dysmyelinated spinal cord axons by adult neural precursor cells results in formation of nodes of Ranvier and improved axonal conduction. J Neurosci. 2007;27:3416-3428.
Fawcett JW, Curt A, Steeves J, et al. Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: spontaneous recovery after spinal cord injury and statistical power needed for therapeutic clinical trials. Spinal Cord. 2007;45:190-205.
Fehlings M. Recommendations regarding the use of methylprednisolone in acute spinal cord injury: making sense out of the controversy [editorial]. Spine J. 2001;26:S56-S57.
Fehlings MG, Louw D. Initial stabilization and medical management of acute spinal cord injury. Am Fam Physician. 1996;54:155-162.
Fehlings MG, Perrin R. The role and timing of early decompression for cervical spinal cord injury: update with a review of recent clinical evidence. Injury. 2005;36:B13-B26.
Furlan JC, Fehlings M, Massicotte E, et al. A quantitative and reproducible method to assess cord compression and canal stenosis after cervical spine trauma: a study of interrater and intrarater reliability. Spine. 2007;32:2083-2091.
Hadley MN, Walters B, Grabb P, et al. Guidelines for the management of acute cervical spine and spinal cord injuries. Clin Neurosurg. 2002;49:407-498.
Hurlbert R. Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. J Neurosurg. 2000;93:1-7.
Jacobs WB, Fehlings M. The molecular basis of neural regeneration. Neurosurgery. 2003;53:943-948.
Karimi-Abdolrezaee S, Eftekharpour R, Wang J, et al. Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. J Neurosci. 2006;26:3377-3389.
Kwon BK, Mann C, Sohn H, et al. Hypothermia for spinal cord injury. Spine J. 2008;8:859-874.
Lammertse D, Tuszynski M, Steeves J, et al. Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: clinical trial design. Spinal Cord. 2007;45:232-242.
Richardson PM, McGuinness U, Aguayo A. Axons from CNS neurons regenerate into PNS grafts. Nature. 1980;284:264-265.
Sekhon LH, Fehlings M. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine. 2001;26:S2-12.
Steeves JD, Fawcett JW, Tuszynski MH, et al. Experimental Treatments for Spinal Cord Injury: What You Should Know If You Are Considering Participation in a Clinical Trial. Vancouver, Canada. International Campaign for Cures of Spinal Cord Injury Paralysis, 2007.
Steeves JD, Lammertse D, Curt A, et al. Guidelines for the conduct of clinical trials for spinal cord injury (SCI) as developed by the ICCP panel: clinical trial outcome measures. Spinal Cord. 2007;45:206-221.
Tator C. Review of treatment trials in human spinal cord injury: issues, difficulties and recommendations. Neurosurgery. 2006;59:957-987.
Tuszynski MH, Steeves J, Fawcett J, et al. Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: clinical trial inclusion/exclusion criteria and ethics. Spinal Cord. 2007;43:222-231.
Vale FL, Burns J, Jackson A, et al. Combined medical and surgical treatment after acute spinal cord injury: results of a prospective pilot study to assess the merits of aggressive medical resuscitation and blood pressure management. J Neurosurg. 1997;87:239-246.
Vogel G. Cell biology. Ready or not? Human ES cells head toward the clinic. Science. 2005;308:1534-1538.
1 DeVivo M. Causes and costs of spinal cord injury in the United States. Spinal Cord. 1997;35:809-813.
2 Sekhon LH, Fehlings M. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine. 2001;26:S2-12.
3 Houle JD, Tessler A. Repair of chronic spinal cord injury. Exp Neurol. 2003;182:247-260.
4 Barbeau H, Nadeau S, Garneau C. Physical determinants, emerging concepts, and training approaches in gait of individuals with spinal cord injury. J Neurotrauma. 2006;23:571-585.
5 Kwon BK, Tetzlaff W, Grauer J, et al. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004;4:451-464.
6 Sullivan PG, Rabchevsky A, Waldemeier P, et al. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79:231-239.
7 Dumont RJ, Okonkwo D, Verma S, et al. Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol. 2001;24:254-264.
8 Dubendorf P. Spinal cord injury pathophysiology. Crit Care Nurs Q. 1999;22:31-35.
9 Tator CH. Update on the pathophysiology and pathology of acute spinal cord injury. Brain Pathol. 1995;5:407-413.
10 Balentine J. Pathology of experimental spinal cord trauma. I. The necrotic lesion as a function of vascular injury. Lab Invest. 1978;39:236-253.
11 Amar AP, Levy M. Pathogenesis and pharmacological strategies for mitigating secondary damage in acute spinal cord injury. Neurosurgery. 1999;44:1027-1039.
12 Hagg T, Oudega M. Degenerative and spontaneous regenerative processes after spinal cord injury. J Neurotrauma. 2006;23:264-280.
13 Schwartz G, Fehlings M. Secondary injury mechanisms of spinal cord trauma: a novel therapeutic approach for the management of secondary pathophysiology with the sodium channel blocker riluzole. Prog Brain Res. 2002;137:177-190.
14 Dumont AS, Dumont R. Will improved understanding of the pathophysiological mechanisms involved in acute spinal cord injury improve the potential for therapeutic intervention? Curr Opin Neurol. 2002;15:713-720.
15 Mautes AE, Weinzierl M, Donovan F, et al. Vascular events after spinal cord injury: contribution to secondary pathogenesis. Phys Ther. 2000;80:673-687.
16 Tator CH, Fehlings M. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg. 1991;75:15-26.
17 Buss A, Schwab M. Sequential loss of myelin proteins during wallerian degeneration in the rat spinal cord. Glia. 2003;42:424-432.
18 Casha S, Yu W, Fehlings M. Oligodendroglial apoptosis occurs along degenerating axons and is asociated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience. 2001;103:203-218.
19 Ackery AR, Fehlings M. Inhibition of Fas-mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury. J Neurotrauma. 2006;23:604-616.
20 Park E, Velumian A, Fehlings M. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma. 2004;21:754-774.
21 Popovich PG. Immunological regulation of neuronal degeneration and regeneration in the injured spinal cord. Prog Brain Res. 2000;128:43-58.
22 McCord JM, Edeas M. SOD, oxidative stress and human pathologies: a brief history and a future vision. Biomed Pharmacother. 2005;59:139-142.
23 Pehar M, Vargas M, Robinson K, et al. Peroxynitrite transforms nerve growth factor into an apoptotic factor for motor neurons. Free Radic Biol Med. 2006;41:1632-1644.
24 Jacobs WB, Fehlings M. The molecular basis of neural regeneration. Neurosurgery. 2003;53:943-948.
25 Caroni P, Schwab M. Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol. 1988;106:121-128.
26 Caroni P, Schwab M. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85-96.
27 GrandPré T, Nakamura F, Vartanian T, et al. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439-444.
28 Chen MS, Huber AB, van der Haar ME, et al. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434-439.
29 Prinjha R, Moore SE, Vinson M, et al. Inhibitor of neurite outgrowth in humans. Nature. 2000;403:383-384.
30 Jones LL, Margolis R, Tuszynski M. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol. 2003;182:399-411.
31 Tan AM, Colletti M, Rorai A, et al. Antibodies against the NG2 proteoglycan can promote the regeneration of sensory axons within the dorsal columns of the spinal cord. J Neurosci. 2006;26:4729-4739.
32 Löw K, Culbertson M, Bradke F, et al. Netrin-1 is a novel myelin-associated inhibitor to axon growth. J Neurosci. 2008;28:1099-1108.
33 Xie R, Zheng B. White matter inhibitors in CNS axon regeneration failure. Exp Neurol. 2008;209:302-312.
34 Féron F, Perry C, Cochrane J, et al. Autologous olfactory ensheathing cell transplantation in human spinal cord injury. Brain. 2005;128:2951-2960.
35 Davies SJ, Fitch M, Memberg S, et al. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680-683.
36 Hofstetter CP, Holmström N, Lilja J, et al. Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nat Neurosci. 2005;8:346-353.
37 Macias MY, Syring M, Pizzi M, et al. Pain with no gain: allodynia following neural stem cell transplantation in spinal cord injury. Exp Neurol. 2006;201:335-348.
38 Fehlings MG, Tator C. The relationships among the severity of spinal cord injury, residual neurological function, axons counts, and counts of retrogradely labeled neurons after experimental spinal cord injury. Exp Neurol. 1995;132:220-228.
39 Nashmi R, Fehlings M. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience. 2001;104:235-251.
40 Karimi-Abdolrezaee S, Eftekharpour R, Wang J, et al. Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. J Neurosci. 2006;26:3377-3389.
41 Eftekharpour E, Karimi-Abdolrezaee S, Wang J, et al. Myelination of congenitally dysmyelinated spinal cord axons by adult neural precursor cells results in formation of nodes of Ranvier and improved axonal conduction. J Neurosci. 2007;27:3416-3428.
42 Benfey M, Aguayo AJ. Extensive enlongation of axons from rat brain into peripheral nerve grafts. Nature. 1982;296:150-152.
43 Baptiste DC, Fehlings M. Emerging drugs for spinal cord injury. Expert Opin Emerg Drugs. 2008;13:63-80.
44 Hadley MN, Walters B, Grabb P, et al. Guidelines for the management of acute cervical spine and spinal cord injuries. Clin Neurosurg. 2002;49:407-498.
45 Steeves JD, Lammertse D, Curt A, et al. Guidelines for the conduct of clinical trials for spinal cord injury (SCI) as developed by the ICCP panel: clinical trial outcome measures. Spinal Cord. 2007;45:206-221.
46 Lammertse D, Tuszynski M, Steeves J, et al. Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: clinical trial design. Spinal Cord. 2007;45:232-242.
47 Tuszynski MH, Steeves J, Fawcett J, et al. Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: clinical trial inclusion/exclusion criteria and ethics. Spinal Cord. 2007;43:222-231.
48 Fawcett JW, Curt A, Steeves J, et al. Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: spontaneous recovery after spinal cord injury and statistical power needed to therapeutic clinical trials. Spinal Cord. 2007;45:190-205.
49 Fehlings MG, Louw D. Initial stabilization and medical management of acute spinal cord injury. Am Fam Physician. 1996;54:155-162.
50 American College of Surgeons. ATLS Advanced Trauma Life Support Program for Doctors, 7th ed. Chicago: American College of Surgeons; 2004.
51 Ball PA. Critical care of spinal cord injury. Spine. 2001;26:S27-S30.
52 Yeomans L. Yeomans Neurological Surgery, 5th ed. Philadelphia: WB Saunders; 2003.
53 Vale FL, Burns J, Jackson A, et al. Combined medical and surgical treatment after acute spinal cord injury: results of a prospective pilot study to assess the merits of aggressive medical resuscitation and blood pressure management. J Neurosurg. 1997;87:239-246.
54 Baptiste DC, Fehlings M. Update on the treatment of spinal cord injury. Prog Brain Res. 2007;161:217-233.
55 Heiss WD, Theil A, Grond M, et al. Which targets are relevant for therapy of acute ischemic stroke? Stroke. 1999;30:1486-1489.
56 Furlan JC, Fehlings M, Massicotte E, et al. A quantitative and reproducible method to assess cord compression and canal stenosis after cervical spine trauma: a study of interrater and intrarater reliability. Spine. 2007;32:2083-2091.
57 Miyanji F, Furlan J, Aarabi B, et al. Acute cervical traumatic spinal cord injury: MR imaging findings correlated with neurologic outcome—prospective study with 100 consecutive patients. Radiology. 2007;243:820-827.
58 Doran SE, Papadopoulos S, Ducker T, et al. Magnetic resonance imaging documentation of coexistent traumatic locked facets of the cervical spine and disc herniation. J Neurosurg. 1993;79:341-345.
59 Maiman DJ, Barolat G, Larson S. Management of bilateral locked facets of the cervical spine. Neurosurgery. 1986;18:542-547.
60 Vaccaro AR, Falatyn S, Flanders A, et al. Magnetic resonance evaluation of the intervertebral disc, spinal ligaments, and spinal cord before and after closed traction reduction of cervical spine dislocations. Spine. 1999;24:1210-1217.
61 Bracken MB, Shepard MJ, Hellenbrand KG, et al. Methylprednisolone and neurological function 1 year after spinal cord injury: results of the National Acute Spinal Cord Injury Study. J Neurosurg. 1985;63:704-713.
62 Dimar JR2nd, Glassman S, Raque G, et al. The influence of spinal canal narrowing and timing of decompression on neurologic recovery after spinal cord contusion in a rat model. Spine J. 1999;24:1623-1633.
63 Braughler JM, Hall ED. Effects of multi-dose methylprednisolone sodium succinate administration on injured cat spinal cord neurofilament degradation and energy metabolism. J Neurosurg. 1984;61:290-295.
64 Schneider RC, Cherry G, Pantek H. The syndrome of acute central cervical spinal cord injury; with special reference to the mechanisms involved in hyperextension injuries of cervical spine. J Neurosurg. 1954;11:546-577.
65 Bracken MB. Methylprednisolone and acute spinal cord injury: an update of the randomized evidence. Spine. 2001;26:S47-S54.
66 Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinalcord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405-1411.
67 Aito S, D’Andrea M, Werhagen L, et al. Neurological and functional outcome in traumatic central cord syndrome. Spinal Cord. 2007;45:292-297.
68 Bracken MB, Shepard MJ, Holford TR, et al. Administration of methylprednisolone for 24 or 48 hours or tirilizad mesylate for 48 hours in the treatment of acute spinal cord injury. JAMA. 1997;277:1597-1604.
69 Bracken MB, Shepard MJ, Holford TR, et al. Methylprednisolone or tirilizad mesylate administration after acute spinal cord injury: 1-year follow-up. J Neurosurg. 1998;89:699-706.
70 Hall ED. Neuroprotective actions of glucocorticoid and nonglucocorticoid steroids in acute neuronal injury. Cell Mol Neurobiol. 1993;13:415-432.
71 Qian T, Campagnolo D, Kirshblum S. High-dose methylprednisolone may do more harm for spinal cord injury. Med Hypotheses. 2000;55:452-453.
72 Paralyzed Veterans of America Consortium for Spinal Cord Medicine. Preservation of upper limb function following spinal cord injury: a clinical practice guideline for health-care professionals. J Spinal Cord Med. 2005;28:434-470.
73 Hall E. The neuroprotective pharmacology of methylprednisolone. J Neurosurg. 1992;76:13-22.
74 Hurlbert R. The role of steroids in acute spinal cord injury: an evidence-based analysis. Spine. 2001;26:S39-S46.
75 National Institute of Neurological Disorders and Stroke. NINDS Workshop Translating Promising strategies for Spinal Cord Injury Therapy. Feb 3-4, 2003, Bethesda, MD.
76 Otani K, Abe H, Kadoya S, et al. Beneficial effects of methylprednisolone sodium succinate in the treatment of acute spinal cord injury. Sekitsy Sekizui. 1994;7:633-647.
77 Pointillart V, Petitjean M, Wiart L, et al. Pharmacological therapy of spinal cord injury during the acute phase. Spinal Cord. 2000;38:71-76.
78 Coleman WP, Benzel D, Cahill D, et al. A critical appraisal of the reporting of the National Acute Spinal Cord Injury Studies (II and III) of methylprednisolone in acute spinal cord injury. J Spinal Disord. 2000;13:185-199.
79 Hurlbert R. Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. Neurosurg. 2000;93:1-7.
80 Hurlbert RJ, Moulton R. Why do you prescribe methylprednisolone for acute spinal cord injury? A Canadian perspective and a position statement. Can J Neurol Sci. 2002;29:236-239.
81 Nesathurai S. Steroids and spinal cord injury: revisiting the NASCIS 2 and NASCIS 3 trials. J Trauma. 1998;45:1088-1093.
82 Short DJ, El Masry W, Jones P. High dose methylprednisolone in the management of acute spinal cord injury—a systematic review from a clinical perspective. Spinal Cord. 2000;38:273-286.
83 Fehlings M. Recommendations regarding the use of methylprednisolone in acute spinal cord injury: making sense out of the controversy [editorial]. Spine J. 2001;26:S56-S57.
84 Geisler FH, Coleman W, Grieco G, et al. The Sygen multicenter acute spinal cord injury study. Spine. 2001;26:S87-S98.
85 Baptiste DC, Fehlings M. Pharmacological approaches to repair the injured spinal cord. J Neurotrauma. 2006;23:318-334.
86 Dumont RJ, Verma S, Okonkwo D, et al. Acute spinal cord injury, part II: contemporary pharmacotherapy. Clin Neuropharmacol. 2001;24:265-279.
87 Pitts LH, Ross A, Chase G, et al. Treatment with thyrotropin-releasing hormone (TRH) in patients with traumatic spinal cord injuries. J Neurotrauma. 1995;12:235-243.
88 Davis SM, Albers G, Diener H, et al. Termination of acute stroke studies involving selfotel treatment. Lancet Neurol. 1997;349:32.
89 Morris GF, Bullock R, Marshall S, et al. Failure of the competitive N-methyl-D-aspartate antagonist selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. J Neurosurg. 1999;91:737-743.
90 Hirbec H, Gaviria M, Vignon J. Gacyclidine: a new neuroprotective agent acting at the N-methyl-D-aspartate receptor. CNS Drug Rev. 2001;7:172-198.
91 Tadie M, D’Arbigny P, Mathé J, et al. Acute spinal cord injury: early care and treatment in a multicenter study with gacyclidine. Soc Neurosci. 1999;25:1090.
92 Lyden PD, Jackson-Friedman C, Shin C, et al. Synergistic combinatorial stroke therapy. A quantal bioassay of a GABA agonist and a glutamate antagonist. Exp Neurol. 2000;163:477-489.
93 Petitjean ME, Pointillart V, Dixmerias F, et al. Medical treatment of spinal cord injury in the acute stage. Ann Fr Anesth Reanim. 1998;17:114-122.
94 Long JB, Kinney R, Malcolm D, et al. Intrathecal dynorphin A (1-13) and (3-13) reduce spinal cord blood flow by non-opioid mechanisms. NIDA Res Monogr. 1986;75:524-526.
95 Flamm ES, Young W, Collins W, et al. A phase I trial of naloxone treatment in acute spinal cord injury. J Neurosurg. 1985;63:390-397.
96 Bracken MB, Shepard M, Collins W, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405-1411.
97 Tator C. Review of treatment trials in human spinal cord injury: issues, difficulties and recommendations. Neurosurgery. 2006;59:957-987.
98 Cesaro P. The design of clinical trials for cell transplantation into the central nervous system. NeuroRX. 2004;1:492-499.
99 Sagen J. Cellular therapies for spinal cord injury: what will the FDA need to approve moving from the laboratory to the human? J Rehabil Res Dev. 2003;40:71-79.
100 Dobkin BH, Curt A, Guest J. Cellular transplants in China: observational study from the largest human experiment in chronic spinal cord injury. Neurorehabil Neural Repair. 2006;20:5-13.
101 Anstett P. We have to overcome this hope barrier. Detroit Free Press; 2005.
102 Jiang S, Khan M, Middlemiss P, et al. AIT-082 and methylprednisolone singly, but not in combination, enhance functional and histological improvement after acute spinal cord injury in rats. Int J Immunopathol Pharmacol. 2004;17:353-366.
103 Grundman M, Capparelli E, Kim H, et al. A multicenter, randomized, placebo controlled, multi-dose, safety and pharmacokinetic study of AIT-082 (Neotrofin) in mild Alzheimer’s disease patients. Life Sci. 2003;73:539-553.
104 Steeves JD, Fawcett JW, Tuszynski M, et al. Experimental Treatments for Spinal Cord Injury: What You Should Know If You Are Considering Participation in a Clinical Trial. Vancouver, Canada. International Campaign for Cures of Spinal Cord Injury Paralysis, 2007.
105 Coselli JS, Lemaire S, Koksoy C, et al. Cerebrospinal fluid drainage reduces paraplegia after thoracoabdominal aortic aneurysm repair: results of a randomized clinical trial. J Vasc Surg. 2002;35:631-639.
106 Hinkle L, McCaig C, Robinson K. The direction of growth of differentiating neurones and myoblasts from frog embryos in an applied electric field. J Physiol. 1981;314:121-135.
107 Jaffe LF, Poo M. Neurites grow faster towards the cathode than the anode in a steady field. J Exp Zool. 1979;209:377-386.
108 Shapiro S, Borgens R, Pascuzzi R, et al. Oscillating field stimulation for complete spinal cord injury in humans; a phase 1 trial. J Neurosurg Spine. 2005;2:3-10.
109 Tator CH. Review of treatment trials in human spinal cord injury: issues, difficulties, and recommendations. Neurosurgery. 2006;59(5):957-982.
110 Erecinska M, Thorensen M, Silver I. Effects of hypothermia on energy metabolism in Mammalian central nervous system. J Cereb Blood Flow Metab. 2003;23:513-530.
111 Liu L, Yenari M. Therapeutic hypothermia: neuroprotective mechanisms. Front Biosci. 2007;12:816-825.
112 Hachimi-Idrissi S, Van H, Michotte A, et al. Postischemic mild hypothermia reduces neurotransmitter release and astroglial cell proliferation during reperfusion after asphyxial cardiac arrest in rats. Brain Res Mol Brain Res. 2004;1019:217-225.
113 Zornow M. Inhibition of glutamate release: a possible mechanism of hypothermic neuroprotection. J Neurosurg Anesthesiol. 1995;7:148-151.
114 Huang ZG, Xue D, Preston E, et al. Biphasic opening of the blood-brain barrier following transient focal ischemia: effects of hypothermia. Can J Neurol Sci. 1999;26:298-304.
115 Ohmura A, Nakajima W, Ishida A, et al. Prolonged hypothermia protects neonatal rat brain against hypoxic-ischemia by reducing both apoptosis and necrosis. Brain Dev. 2005;27:517-526.
116 Inamasu J, Suga S, Sato S, et al. Post-ischemic hypothermia delayed neutrophil accumulation and microglial activation following transient focal ischemia in rats. J Neuroimmunol. 2000;109:66-74.
117 Globus MY, Alonso O, Dietrich W, et al. Glutamate release and free radical production following brain injury: effects of posttraumatic hypothermia. J Neurochem. 1995;65:1704-1711.
118 Lei B, Tan X, Cai H, et al. Effect of moderate hypothermia on lipid peroxidation in canine brain tissue after cardiac arrest and resuscitation. Stroke. 1994;25:147-152.
119 Inamasu J, Nakamura Y, Ichikizaki K. Induced hypothermia in experimental traumatic spinal cord injury: an update. J Neurol Sci. 2003;209:55-60.
120 Kwon BK, Mann C, Sohn H, et al. Hypothermia for spinal cord injury. Spine J. 2008;8:859-874.
121 Wells JE, Hurlbert R, Fehlings M, et al. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126:1628-1637.
122 Stirling DP, Khodarahmi K, Liu J, et al. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci. 2004;24:2181-2190.
123 Teng YD, Choi H, Onario R, et al. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A. 2004;101:3071-3076.
124 Lee SM, Yune T, Kim S, et al. Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma. 2003;20:1017-1027.
125 Yrjänheikki J, Keinänen R, Pellikka M, et al. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95:15769-15774.
126 He Y, Appel S, Le W. Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res. 2001;909:187-193.
127 Festoff BW, Ameenuddin S, Arnold P, et al. Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem. 2006;97:1314-1326.
128 Tikka TM, Koistinaho J. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527-7533.
129 Yune TY, Lee J, Jung G, et al. Minocycline alleviates death of oligodendrocytes by inhibiting pro–nerve growth factor production in microglia after spinal cord injury. J Neurosci. 2007;27:7751-7761.
130 Bhatt JM, Gordon P. Current clinical trials in amyotrophic lateral sclerosis. Expert Opin Investi Drugs. 2007;16:1197-1207.
131 Miller RG, Mitchell J, Lyon M, et al. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2007;1:CD0001447.
132 Schwartz G, Fehlings M. Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: improved behavioral and neuroanatomical recovery with riluzole. J Neurosurg. 2001;94:245-256.
133 Wang SJ, Wang K, Wang W. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neuroscience. 2004;125:191-201.
134 Mu X, Azbill R, Springer J. Riluzole and methylprednisolone combined treatment improves functional recovery in traumatic spinal cord injury. J Neurotrauma. 2000;17:773-780.
135 Nógrádi A, Szabó A, Pintér S, et al. Delayed riluzole treatment is able to rescue injured rat spinal motoneurons. Neuroscience. 2007;144:431-438.
136 Caroni P, Savio T, Schwab M. Central nervous system regeneration: oligodendrocytes and myelin as non-permissive substrates for neurite growth. Prog Brain Res. 1988;78:363-370.
137 Schnell L, Schwab M. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269-272.
138 Bregman BS, Kunkel-Bagden E, Schnell L, et al. Recovery from spinal cord injury mediated by antibodies to neurite growth inhibitors. Nature. 1995;378:498-501.
139 Fouad K, Dietz V, Schwab M. Improving axonal growth and functional recovery after experimental spinal cord injury by neutralizing myelin associated inhibitors. Brain Res Brain Res Rev. 2001;36:204-212.
140 Spillmann AA, Bandtlow CE, Lottspeich F, et al. Identification and characterization of a bovine neurite growth inhibitor (bNI-220). J Biol Chem. 1998;273:19283-19293.
141 Freund P, Schmidlin E, Wannier T, et al. Nogo-A–specific antibody treatment enhances sprouting and functional recovery after cervical lesion in adult primates. Nat Med. 2006;12:790-792.
142 Freund P, Wannier T, Schmidlin E, et al. Anti–Nogo-A antibody treatment enhances sprouting of corticospinal axons rostral to a unilateral cervical spinal cord lesion in adult macaque monkey. J Comp Neurol. 2007;502:644-659.
143 Dergham P, Ellezam B, Essagian C, et al. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci Res. 2002;22:6570-6577.
144 Dubreuil CI, Winton M, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233-243.
145 Syková E, Homola A, Mazanec R, et al. Autologous bone marrow transplantation in patients with subacute and chronic spinal cord injury. Cell Transplant. 2006;15:675-687.
146 Rapalino O, Lazarov-Spiegler O, Agranov E, et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med. 1998;4:814-821.
147 Knoller N, Auerbach G, Fulga V, et al. Clinical experience using incubated autologous macrophages as a treatment for complete spinal cord injury: phase I study results. J Neurosurg Spine. 2005;3:173-181.
148 Richardson PM, McGuinness U, Aguayo A. Axons from CNS neurons regenerate into PNS grafts. Nature. 1980;284:264-265.
149 Boyd JG, Doucette R, Kawaja M. Defining the role of olfactory ensheathing cells in facilitating axon remyelination following damage to the spinal cord. FASEB J. 2005;19:694-703.
150 Ramón-Cueto A, Cordero M, Santos-Benito F, et al. Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron. 2000;25:425-435.
151 Lima C, Pratas-Vital J, Escada P, et al. Olfactory mucosa autografts in human spinal cord injury: a pilot clinical study. J Spinal Cord Med. 2006;29:191-203.
152 Rabinovich SS, Seledtsov V, Poveschenko O, et al. Transplantation treatment of spinal cord injury patients. Biomed Pharmacother. 2003;57:428-433.
153 Park HC, Shim Y, Ha Y, et al. Treatment of complete spinal cord injury patients by autologous bone marrow cell transplantation and administration of granulocyte-macrophage colony stimulating factor. Tissue Eng. 2006;11:913-922.
154 Yoon SH, Shim Y, Park Y, et al. Complete spinal cord injury treatment using autologous bone marrow cell transplantation and bone marrow stimulation with granulocyte macrophage-colony stimulating factor: Phase I/II clinical trial. Stem Cells. 2007;25:2066-2073.
155 Keirstead HS, Nistor G, Bernal G, et al. Human embryonic stem cell–derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25:4694-4705.
156 Jane S, Lebkowski J, Gold J, et al. Human embryonic stem cells: culture, differentiation, and genetic modification for regenerative medicine applications. Cancer J. 2004;7:83-98.
157 Vogel G. Cell biology. Ready or not? Human ES cells head toward the clinic. Science. 2005;308:1534-1538.






