Chapter 39 Craniopharyngiomas and Suprasellar Tumors
• Craniopharyngiomas with different points of origin of tumor growth have substantially different topographical relationships with surrounding structures determining their surgical resectability and the outcome of treatment; originally infradiaphragmatic tumors are the most amenable for safe radical removal, which is the best treatment of craniopharyngiomas.
• The majority of craniopharyngiomas growing inside and outside the cavity of the third ventricle have started to grow as intrapial tumors, thus acquiring direct and intimate contact with hypothalamic structures.
• If the floor of the third ventricle cannot be seen directly on preoperative magnetic resonance imaging (MRI) scans, the topographical relationship of the tumor with hypothalamic structures may be predicted according to its relation with the chiasm and the presence or absence of hydrocephalus.
• The nature and intensity of the tumor adherence to surrounding structures vary in different cases within each topographical group. Decisions about the optimal extent of tumor removal may thus be made only at surgery.
• The natural history of craniopharyngiomas is unpredictable, the growth potential of the tumor or its residual tumor may differ significantly, and therefore radiotherapy should be indicated in case of tumor progression.
The region above the sella contains anatomical structures of the hypothalamus-pituitary system (hypothalamic structures and infundibulum with its pars compacta named also the pituitary stalk), the structures of the visual pathways (optic chiasm and adjacent portions of the optic nerves and the optic tracts), and important blood vessels. The tumors affecting the suprasellar region may belong to (a) tumors of dysembryogenetic origin (craniopharyngioma, epidermoid cyst, dermoid cyst, hamartoma, germ cell tumors, Rathke’s cleft cyst), (b) tumors originating from the tissues of suprasellar structures (gliomas of the visual pathways and hypothalamus, pituicytoma, granular cell tumor of the neurohypophysis, meningioma), (c) tumors extending into the suprasellar space secondarily, most often from the cavity of the sella (pituitary adenoma, Rathke’s cleft cyst, and other cysts), and (d) systemic tumors affecting the central nervous system (CNS metastasis, lymphoma, leukemia).1–3
The structures of optic pathways and the hypothalamus-pituitary system may be involved also by suprasellar meningiomas or large and giant pituitary adenomas. The latter may severely displace the floor of the third ventricle upward and may also lead to atrophy of the pia and invade the hypothalamus.4 Suprasellar meningiomas originating from the lower leaf of the diaphragm can mimic nonfunctioning pituitary adenomas.5 Extremely rare is a meningioma originating from the pituitary stalk having no connection to surrounding dura.6
Craniopharyngiomas
The intention to remove the craniopharyngioma totally whenever technically possible emerged in the past in conjunction with therapeutic and diagnostic improvements: steroid hormone replacement, microsurgery, magnetic resonance imaging.7–16 Another attitude to management of craniopharyngiomas, namely, intentional incomplete removal and radiotherapy, has been advocated in order to lower the surgical morbidity rate.17–23 However, radiotherapy cannot prevent recurrences in many instances24 and causes adverse effects.16 A compromise solution has been elaborated: radical removal was recommended only for the patients harboring the tumors not involving the hypothalamus.25 Nevertheless, the definition of “involvement of the hypothalamus” is not clear. Radiological signs of retrochiasmatic growth of the tumor in the direction of the hypothalamus and the impossibility of identifying the latter26 as a distinct form of the tumor do not indicate whether the hypothalamus is compressed or invaded.27
The management of craniopharyngiomas presented in this chapter is based on the results of our morphological studies,28 the results of the correlation of morphological data with neuroradiological and operative findings,27 and our clinical experience.
Incidence
Craniopharyngiomas account for 1% to 4.6% of all intracranial tumors and 13% of suprasellar tumors. Their incidence represents 0.5 to 2.5 new cases per 1 million population per year, being more frequent in Nigerian (18% of all CNS tumors) and Japanese children, with an annual incidence of 5.25 cases per 1 million per year. Papillary craniopharyngiomas occur virtually exclusively in adults, at a mean age of 40 to 55 years. A bimodal age distribution of adamantinomatous craniopharyngiomas is observed with peaks in children aged 5 to 15 years and in adults aged 40 to 55 years.29 In children, craniopharyngiomas account for 2.5% to 13% (average 7.5%) of all tumors and 56% of the sellar-chiasmatic tumors.30
Tumor Development
According to the widely accepted embryogenetic theory of Erdheim, craniopharyngiomas take their origin from the remnants of the craniopharyngeal duct or Rathke’s pouch. Epithelial cell rests have been reported to occur in the “nests” most frequently along the anterior part of the infundibulum near its origin from the brain and the anterosuperior surface of the adenohypophysis.29,31 When the cells of the vanishing hypophyseal duct are brought into immediate contact with the infundibular area before the primitive pia mater has developed, they will be intimately adherent to the neuroepithelium and subsequently developed pia mater will incorporate them into the subpial space.32 Alternatively, when the cell rests do not come into immediate contact with the neuroepithelium they will remain extrapial (Fig. 39.1). On the basis of histological findings, Grekhov33 distinguished four groups of craniopharyngiomas according the point of their original growth: infrasellar, intrasellar (intrasellar and suprasellar), pituitary stalk, and infundibular craniopharyngiomas. Intimate contact of the cells of tumor origin with neural elements probably accounts for the manner in which many of these tumors appear to blend with the tuber cinereum, so as to form an intrinsic portion of that structure, or even to replace it.34 These developmental concepts represent the clue to understanding the topographical variations of craniopharyngiomas.
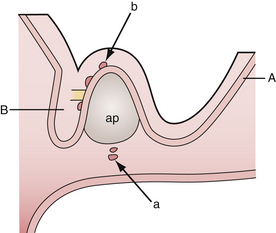
The metaplastic theory postulates that the nests of squamous epithelium are derived from mature cells of adenohypophysis which undergo metaplasia.35 However, these nests were not found in persons in their first decade, and rarely in those in the second; according to Kernohan36 these observations failed to support the origin of craniopharyngiomas from such cells. The theory explains the craniopharyngioma spectrum, attributing the adamantinomatous type (typical for childhood) to embryonic remnants, and the papillary type (common in adult age) to metaplasia.14
Pathology and Surgical Anatomy
Microscopic Anatomy
The brain parenchyma that surrounds both variants of craniopharyngioma is typically gliotic and often shows a profuse number of Rosenthal fibers.29,34 Tumor tissue, gliosis, and brain parenchyma may form one layer of tissue with a common vascular network. Finger-like protrusions of the tumor into the surrounding gliosis or even into the adjacent brain parenchyma are characteristic of adamantinomatous craniopharyngiomas.37 On histological sections, these outgrowths appear as isolated islands of epithelium in the zone of intense gliosis.34,36
Surgical Anatomy
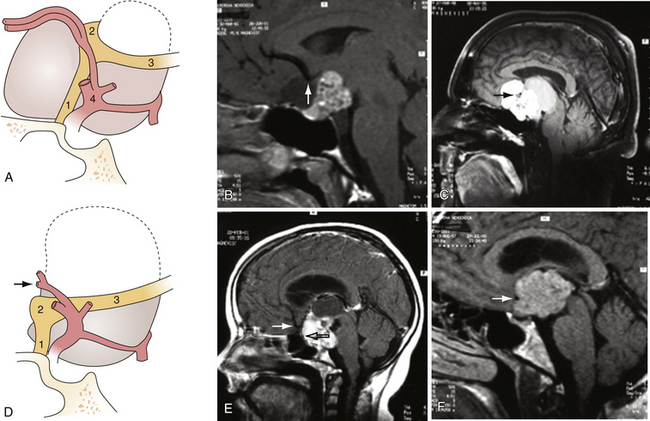
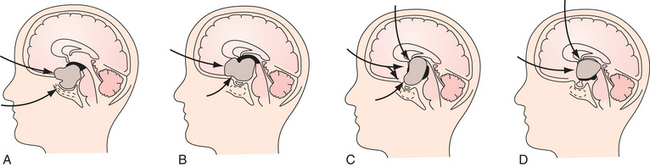
Craniopharyngiomas may start to grow below the sellar diaphragm (infradiaphragmatic tumors) or above it (supradiaphragmatic tumors). The point of origin of growth of the tumor influences its future relationship with surrounding structures.28 To a lesser degree the topography of the tumor depends on premorbid anatomy, first of all on the length of the optic nerves and hence the position of the chiasm.
Infradiaphragmatic Craniopharyngiomas
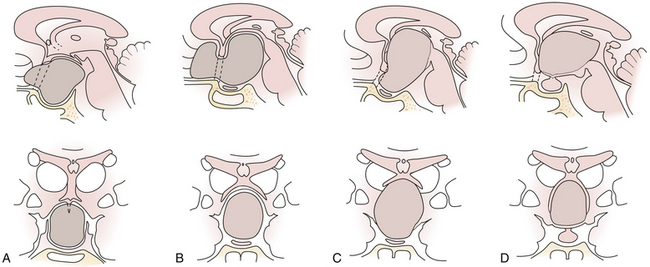
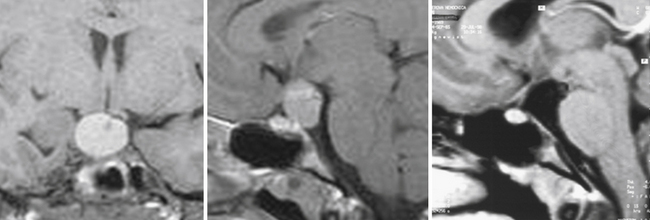
Enlargement of the infradiaphragmatic (i.e., intrasellar) craniopharyngiomas causes upward displacement of the sellar diaphragm and the arachnoid. Such an intrasellar and suprasellar craniopharyngioma grows below the chiasm and gradually compresses and displaces it upward as well as the floor of the third ventricle similar to pituitary adenomas (Fig. 39.2).
Supradiaphragmatic Craniopharyngiomas
The point of original growth of primarily supradiaphragmatic tumors may be in the pituitary stalk or inside the infundibulum (i.e., in the most basal part of the floor of the third ventricle). Tumors starting to grow from the pituitary stalk are located in the subarachnoid space and grow below the chiasm and the floor of the third ventricle (suprasellar extraventricular craniopharyngiomas, SECs). A part of such a tumor almost always extends anteriorly in front of the chiasm between the optic nerves, where it can be reached (see Fig. 39.2). Rare retrochiasmatic location of an entire SEC may be the consequence of premorbid anatomy of short optic nerves and pre-fixed chiasm.
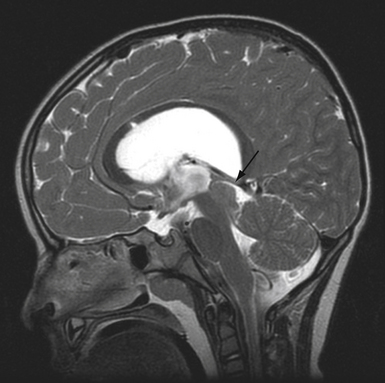
The intraventricular and extraventricular location of the craniopharyngiomas, according to a widespread idea, is a consequence of the disruption of a stretched and thinned-out floor of the third ventricle. This mechanism, which may also be occasionally observed in giant pituitary adenomas,4 is rather rare. More commonly, it is caused by the mechanism described previously. Our morphological27,28 and clinical observations showed that the structures of the third ventricular floor undergo atrophy of different degrees. The most affected structure is the tuber: its central part (postinfundibular eminence) is usually absent, and its remnants, the lateral eminences, are located on the lateral or lateral-basal surface of the tumor. The infundibulum (median eminence) is destroyed less frequently, whereas compressed mammillary bodies covering the posterior pole of the tumor can be found in all IEVCs.27
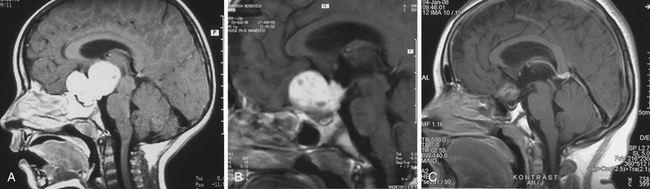
Some tumors classified in the literature as intraventricular craniopharyngiomas have been described as protruding into the interpeduncular cisterns.38 We classified such tumors as IEVCs, reserving the term intraventricular for rare craniopharyngiomas lying entirely within the ventricular cavity covered from below by the floor of the third ventricle.39–41 Such a tumor is attached to a partially atrophied floor of the third ventricle while it may only touch its lateral walls (Figs. 39.2 and 39.3). This observation28 has also been confirmed by others.42 We have seen only one such tumor in our clinical series.
Clinical Presentation
The most common signs and symptoms include endocrine deficiencies (over 90%) more frequently in children, and visual disturbances (up to 96%) more frequently in adults. Among hypothalamic disorders in children obesity is prevalent, and mental disorders occur in both age groups.43 Increased intracranial pressure is the most frequent presenting symptom in children.
Patients with craniopharyngioma generally present late, and visual symptoms are often preceded by a long history of systemic symptoms.44 Children rarely become aware of visual problems and often present after almost complete visual damage has taken place.45
Involvement of the optic pathway manifests itself by the defects of the visual fields and lowering of the visual acuity. Many different varieties of the visual field defects are found, among them bitemporal hemianopia, homonymous hemianopia, concentric contractions of fields, and central or paracentral scotomas.43 The commonest field defect is asymmetrical bitemporal hemianopia, as a result of the compression of the chiasm, whereas homonymous visual field defects are caused by the tumors located behind the chiasm. The structures of the optic pathway may be compressed not only by the tumor but also by the arteries of the anterior part of the circle of Willis. The tumors growing below the chiasm push the optic structures upward against the anterior cerebral arteries (A1) and the anterior communicating artery (AComA). A strangulation groove may be found on the upper surface of the chiasm or on the terminal portion of the optic nerves. Retrochiasmatic tumors push the chiasm forward and downward below the A1-AComA complex. Strangulation grooves may be found on the most anterior parts of the optic tracts (Figs. 39.4 and 39.5). In the most severe case of our series we observed practically complete amputation of the optic tract from the chiasm.46 Decompression of the optic pathway in such a case cannot be expected to improve the visual functions. Visual impairment may also be caused by compression of the lower chiasmatic arteries, branches of the internal carotid arteries (ICAs) between the upper surface of the tumor and the stretched chiasm. The decussating fibers in the central chiasm receive their arterial supply solely from this inferior group of vessels.47,48 Visual defects caused by ischemia may rapidly resolve after surgical decompression.
Endocrinopathies represented the most common manifestation at diagnosis (68%) in pediatric series of Di Rocco and associates.16 However, they are rarely the reason for referral. Growth deceleration and diabetes insipidus are often overlooked;49 gonadotropic deficiency cannot be observed in small children. Delayed puberty in appropriate age was observed in all children with craniopharyngiomas.49 Adults complain of decreased sexual drive and almost 90% of men complain of impotence, and most women complain of amenorrhea. Growth hormone deficiency in a large group of mostly adult patients was found in 82%.50 The frequency of presurgery diabetes insipidus varies from 8% to 35%.16,49,51,52 Compression of the pituitary stalk by a suprasellar mass causes hyperprolactinemia, which can be detected in more than 40% of patients with craniopharyngiomas.53 Obesity is present in up to 25% of the patients.51 Its evolution before and after surgery is related to the severity of the involvement of the hypothalamus.54,55 Growth hormone insufficiency can also lead to increased body fat.56
Signs of increased intracranial pressure from obstructive hydrocephalus, headache and vomiting, are more frequent in pediatric patients.51 This may be explained by the more common occurrence of the IEVCs causing hydrocephalus in children (73%) more frequently than in adults (49%). Extraventricular tumors, even large or giant, usually do not cause complete obstruction of the cerebrospinal fluid (CSF), as can be assumed according to the absence of enlargement of the lateral ventricles.
Cognitive impairment and personality changes occur more often in adults. In a series with the patients over 40 years of age, more than half suffered from impairment of memory.43 Memory disturbances are attributed to a lesion of the mammillary bodies or of their connections with the hippocampal system, the fornix, and the mammillary-thalamic tract.57,58
Radiographic Diagnosis
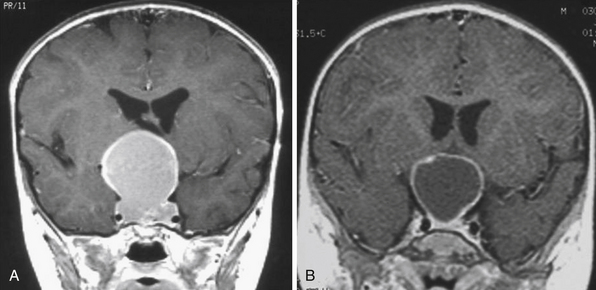
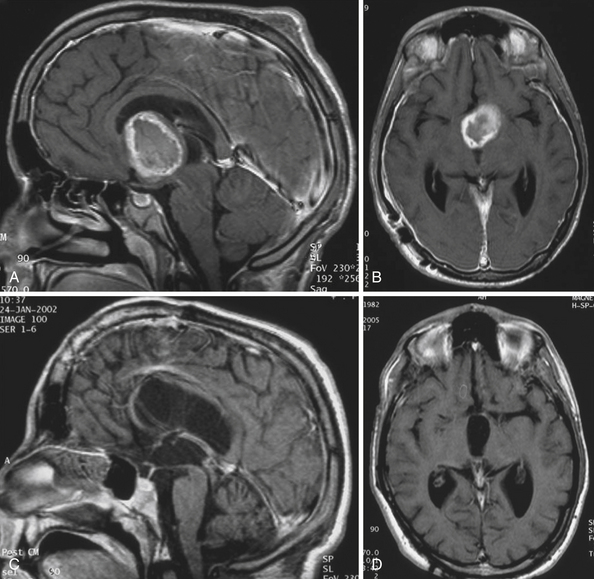
Infradiaphragmatic craniopharyngiomas (intrasellar, infrasellar, and suprasellar) enlarge the sella similar to pituitary adenomas. Supradiaphragmatic tumors displace the diaphragm and the pituitary downward and the sella is shallow with depressed tuberculum, acquiring a J or a pear shape on plain radiography. Calcifications are present in about 85% of childhood and 40% of adult craniopharyngiomas, and are more readily detected by computed tomography (CT). Calcium deposits are present in most adamantinomatous tumors. Eggshell-like calcification of the wall of a cystic tumor may occasionally delineate the entire lesion (Fig. 39.6). Cyst fluid is usually of low density; however, cystic portions may appear dense or solid if they contain a sufficient quantity of suspended calcium salts (Fig. 39.7). Solid tumor tissue and the cyst walls show contrast enhancement.
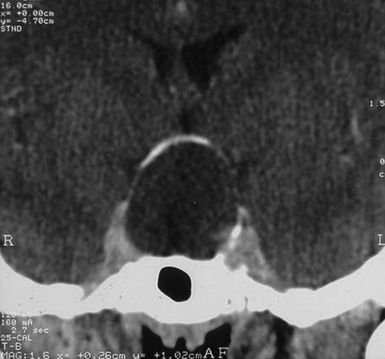
FIGURE 39.6 Eggshell-like calcification of the capsule of intrasellar ane suprasellar craniopharyngioma.
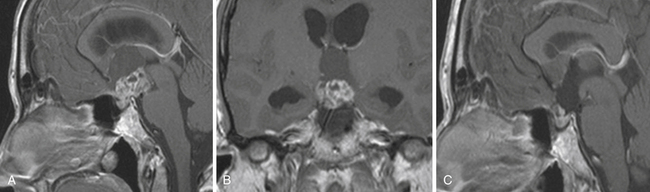
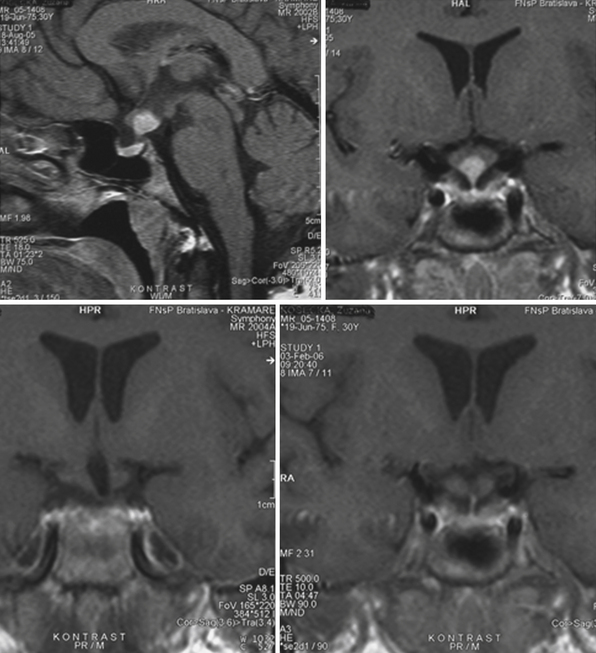
The heterogeneous nature of the craniopharyngiomas is best displayed by magnetic resonance imaging (MRI). Solid parts of the tumor are most often isointense on T1-weighted images and show contrast enhancement. Hyperintense ring enhancement of the cyst wall is common. The high signal of the cyst fluid seen on T1 images in some tumors is related to high protein content or blood breakdown products. MRI provides information on the topography essential for planning the surgical approach. A different relation to the sella and its diaphragm may be seen on coronal scans (Fig. 39.8). In a tumor of a large or a giant size, the chiasm and the structures of the floor of the third ventricle often cannot be identified. The position of the chiasm may be detected indirectly as the AComA is always located in its immediate vicinity, most often at its upper posterior surface (see Fig. 39.4). Furthermore, the relation of the tumor with the chiasm and presence or absence of hydrocephalus may indicate the relation of the tumor to the floor and the cavity of the third ventricle with a high degree of probability.27
Modalities and Options for Treatment
The argument for radical surgery is the recurrence rate, which is considerably lower after complete tumor removal compared to subtotal or partial tumor resection, reaching 0% to 43% and 40% to 100%, respectively, and a worse outcome of secondary surgery.9,10,12–16,59–63 The authors advocating conservative surgery and subsequent radiotherapy stress lower incidence of endocrine deficit, hypothalamic insufficiency, major disability, and even higher rate of local tumor control after more conservative management.17–22 There is almost general agreement on decreasing the number of recurrences and progression of residual tumors after radiotherapy. The recurrence rate in children after incomplete removal reaching 44% to 100% lowered to 16% to 37.5% after radiotherapy.60,64,65 The recurrence-free survival rate representing 38% after partial surgery increased to 77% after radiotherapy.66 Even a 100% progression-free survival rate after radiotherapy without previous surgery has been reported.17 Some other studies, however, found no relation between recurrence and adjuvant radiotherapy—neither globally nor in patients with incomplete resection.24
There is no agreement concerning the timing of radiation. Lin and colleagues63 could prevent the recurrence in all cases with radiotherapy applied initially; if radiotherapy was applied at relapse the tumor subsequently recurred in 22%. Some authors therefore recommend routine administration of postoperative radiation.18,22,67 On the other hand, residual tumor may remain stable for a very long period of time; therefore, others prefer the wait-and-see policy and do not recommend radiotherapy in stable residual tumor.16
Irradiation may be applied in fractions as conventional radiation therapy or by means of more advanced techniques: three-dimensional conformal radiation treatment, intensity modulated radiation therapy, or stereotactic radiotherapy. The tolerance of the optic chiasm is 54 Gy in 30 fractions.17 Gamma Knife stereotactic radiosurgery delivers to a tumor margin a dose from 9.5 to 35 Gy.68–70 Close tumor relationship to visual pathways, however, is a limiting factor. For targeted single high-dose irradiation the dose to the optic chiasm should not exceed 8 Gy.69
Shrinking of a cystic craniopharyngioma may be achieved by means of instillation of the solution of beta particle–emitting radioactive isotopes of yttrium-90, gold-198, or phosphorus-32. Long-term results after yttrium-90 colloid irradiation led to reduction of the initial cyst volume of 79%; in 47 patients in the reduction was more than 80%.71
All modalities of radiotherapy may cause damage to the pituitary, the hypothalamus, and the visual structures, and it may cause moyamoya syndrome, cavernous malformation, and secondary malignant astrocytoma.16,72–75 Most often affected is the function of the anterior pituitary.18 Deterioration of vision after instillation of beta wave emitters, attributed to irradiation and as the result of tumor progression, reached 22.5% to 52%.76,77 New neuro-ophthalmological deficits after yttrium-90 colloid irradiation occurred in 5.8%.71 The latter was comparable to 6.1% of visual deterioration after radiosurgery.68 Damage to the carotid was reported after yttrium-90 treatment.71 In our autopsy series of craniopharyngiomas, the occlusion of the supraclinoid ICA was noted in a 15-year-old boy 6 years after intracystic instillation of 5 mCi of yttrium-90 and 2 years after additional external radiotherapy.
Chemotherapy with bleomycin has been used in cystic craniopharyngiomas. It causes thickening of the tumor capsule, and the cyst contracts, pulling away from brain structures, which facilitates cyst removal.78 However, bleomycin is highly toxic for neural structures if it is allowed to leak from the cyst. In a review of 16 articles79 at least one severe toxic effect, including visual or hearing impairment, personality changes, seizures, somnolence, or even death, occurred in 19% of patients. A combination of intracystic bleomycin and phosphorus-32 used in nine patients led to an even higher rate of complications.80
Other surgical techniques are sometimes used in patients with primary or recurrent craniopharyngiomas. Ventriculoperitoneal shunt or even external ventricular drainage (EVD) may be inserted in cases with severe intracranial hypertension. We have had severe acute intracranial hypertension necessitating EVD in one patient. Some patients have had shunts implanted before referring to us for a tumor removal. Our policy is to restore the patency of the CSF pathways by removal of the tumor. Stereotactic technique is sometimes used for evacuation of a cyst or for instillation of radioisotopes or bleomycin into the tumor cyst.21,70,81 A repeatedly enlarging large cystic tumor may be treated by the implantation of an Ommaya reservoir, enabling simple evacuation of the cystic contents.
Cyst evacuation, different forms of irradiation, and chemotherapy are recommended mostly as an adjuvant treatment. We believe the best management of craniopharyngiomas is radical tumor removal whenever safely possible. A lower quality of life was observed after radical removal of craniopharyngiomas “involving the hypothalamus.”25,26 However, in cases with “hypothalamus no longer identifiable” on MRI scans the tumor may just compress the floor of the third ventricle and not invade it.27,28 It may be a good decision to remove the tumor radically in cases of extraventricular craniopharyngiomas and consider it contraindicated when the tumor is intraventricular.82 Nevertheless, the nature and intensity of the adherence of the tumor to diencephalic structures may differ within any of the topographical groups. Imaging studies do not show directly whether the tumor is simply compressing the hypothalamus or invading it, without a plane of cleavage. Therefore, the final decision about the extent of safe tumor removal cannot be made before the operation.
Surgery
Surgical strategy and tactics are based on the answers to two questions: Where to begin, and when to stop tumor removal? The answer to the first question is related to the choice of the optimal surgical approach. It should respect the topographical relationship of the tumor with the surrounding structures and at the same time it must provide sufficient tumor exposure. The answer to the second question is related to the fact that accessibility of a craniopharyngioma does not mean its resectability. Hypothalamic structures should be avoided during surgical approach and tumor resection (Fig. 39.9). There are two factors limiting preoperative assumption of the tumor–third ventricular floor relationship on rare occasions. First, it may be impossible to find out whether a retrochiasmatic supradiaphragmatic tumor not reaching the foramina of Monro and not causing hydrocephalus lies below the floor of the third ventricle or is embedded inside the infundibulum and the tuber cinereum. Distinction in such an MRI finding is possible only if the structures of the floor of the third ventricle are directly delineated. Second, besides the majority of tumors with expressed topographical features characteristic for one of the described groups, there are transitional types of craniopharyngiomas displaying the features of two topographical groups.
Intrasellar and Intrasellar and Suprasellar Craniopharyngiomas
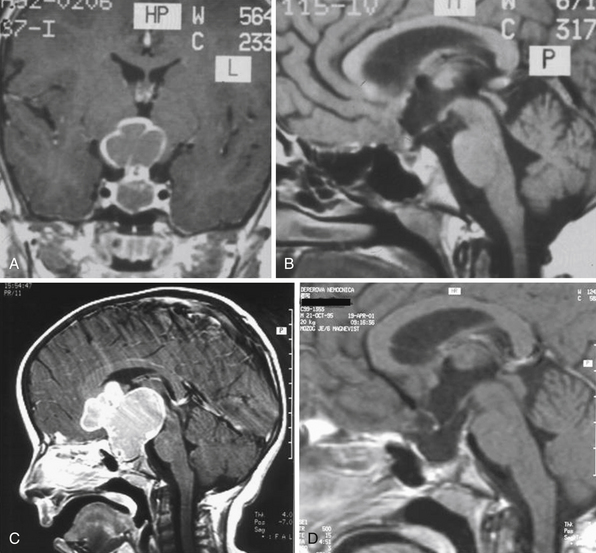
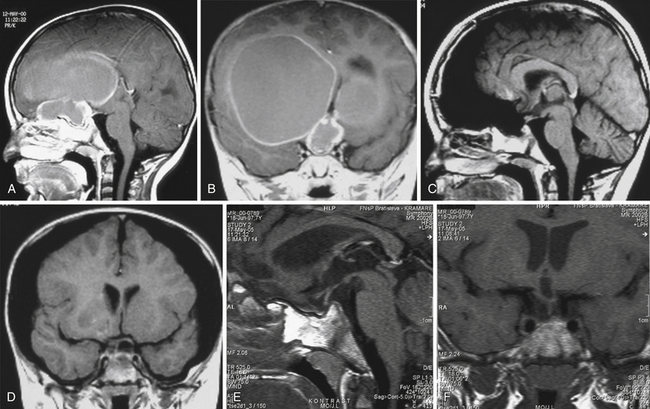
For intrasellar craniopharyngiomas the only appropriate approach is the transsphenoidal route, which exposes the entire tumor and allows identification of the remnants of the pituitary gland located anterior to the tumor or posterior to it.83 The transsphenoidal approach is suitable also for the intrasellar and suprasellar craniopharyngiomas, with the exception of giant tumors and the tumors of a dumbbell or multinodular shape, which are rather rare in intrasellar and suprasellar craniopharyngiomas (Fig. 39.10). These tumors can be resected through the extended transsphenoidal approach, which allows removing even supradiaphragmatic craniopharyngiomas.84,85 In our series of 43 infradiaphragmatic tumors, 4 were intrasellar and 39 extended above the sellar diaphragm. Some of them were large or even giant tumors.
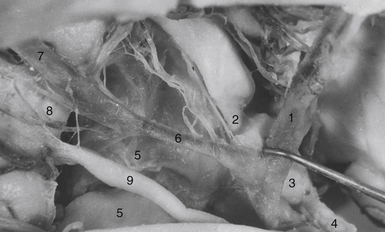
To expose the tumor via the transsphenoidal route we use the unilateral paraseptal, usually sublabial, approach performed under the operative microscope from the first incision. In order to get sufficient exposure of intrasellar contents a large opening of the sphenoid sinus and the sellar floor is mandatory.83 The anterior wall of the sphenoid sinus and the sellar floor is resected under neuronavigation guidance. In a majority of our cases in which a compressed pituitary gland was found (one third of the intrasellar and suprasellar tumors), it could be detected at the sellar floor immediately after dural incision. The gland, which was identified at the floor of the enlarged sella, was split in the midline and displaced to the side so the tumor capsule could be identified (Fig. 39.11). The capsule always adheres to surrounding dura covering the sellar cavity; nevertheless, the tumor does not invade the cavernous sinus as the pituitary adenomas do. The capsule is separated from the dura by pulling it into the sellar cavity and detaching it by blunt microdissection, although sometimes sharp dissection is necessary. If the capsule is thin and fragile, care must be taken not to lose its continuity as later on it may not be identified and may be unintentionally left in place. On the other hand, if there is a thick firmly adhering capsule, it may not be possible to dissect it free and a part of it may have to be left in place.
Suprasellar Extraventricular Craniopharyngiomas
Superior displacement of the chiasm and partial prechiasmatic extension of a great majority of the SECs allow tumor removal through the (1) prechiasmatic space between the optic nerves, anterior angle of the chiasm, and the sellar tubercle; (2) opticocarotid triangle between the lateral margin of the optic nerve and chiasm, supraclinoid carotid artery, and the A1 segment of the anterior cerebral artery; and (3) laterally to the carotid. All these extracerebral routes to the tumor are accessible by the unilateral subfrontal approach, which is our most commonly used craniotomy in craniopharyngiomas (Fig. 39.12). Removal of an extraventricular tumor through the lamina terminalis would jeopardize hypothalamic structures of the floor of the third ventricle covering the upper surface of the tumor. Therefore, with rare SECs growing exclusively behind the optic chiasm we approach below and behind the chiasm through pterional craniotomy (Figs. 39.13 and 39.14). Skull base approaches enabling an approach to the tumor more from below, such as the orbitozygomatic or transpetrosal approach, may be of great help in these cases.86 In a giant multicystic tumor a bifrontal craniotomy was necessary (Fig. 39.15).
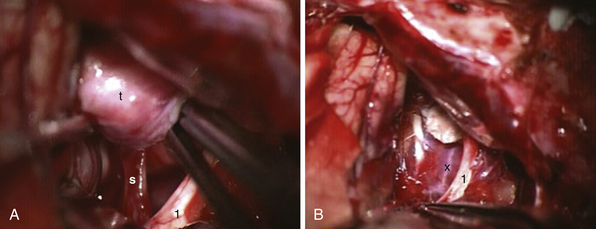
The tumor may adhere to large blood vessels. Intraoperative damage to the internal carotid artery has been reported. Removal of a calcified portion of the tumor from the wall of the carotid in children may be especially dangerous.8,87 We found such extensive calcifications more commonly in the IEVCs. The SECs could be dissected safely from the adventitia even if they completely envelop the large arteries at the base of the brain. Extreme care must be taken to look for the minute perforating vessels, the branches of the supraclinoid carotid, and the posterior communicating arteries supplying the visual pathways and the hypothalamus. It is important to dissect these tiny vessels from the capsule of the tumor because they often bifurcate, with one branch supplying the tumor and the other continuing to its original destination. The vessel may be occluded only after confirming that it does not supply adjacent neural structures. The branches of the A1 and the AComA come in such close contact only with the tumors growing in front of the chiasm. If the perforators adhere firmly and cannot be safely detached from the tumor surface, a part of the capsule is left attached to the vessels.
A more common cause of an incomplete removal of the SECs is the adherence of the upper pole of the tumor to the pia mater of the superiorly displaced floor of the third ventricle, or more rarely of the chiasm. If the capsule cannot be dissected without disruption of the pial vascular network, then part of it or even a small piece of a tumor tissue has to be left in place. On rare occasions the distended floor of the third ventricle falls down during removal of the upper pole of the tumor. In a majority of the cases we delineate the condition of the ventricular floor by angled endoscope. Unintentionally left tumor remnants may be removed, but those firmly adhering to the pia of the hypothalamus have to be left in place (Fig. 39.16).
Intraventricular and Extraventricular Craniopharyngiomas
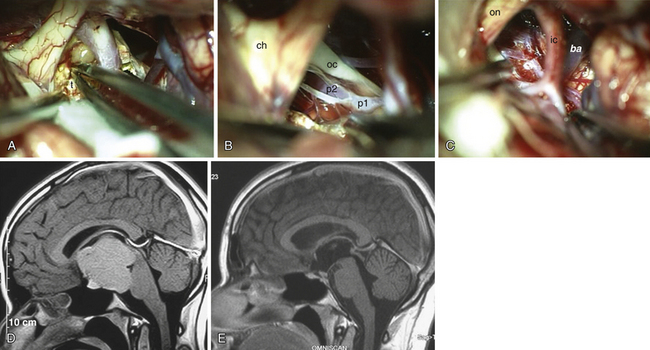
Atrophied hypothalamic structures within the remnants of the floor of the third ventricle in the IEVCs are displaced around the “equator” of the tumor. This topographical relationship in principle allows the use of both extracerebral and transventricular approaches to the tumor (Figs. 39.9 and 39.17). However, the retrochiasmatic location of the tumor, common low position of the chiasm, and a slit-like opticocarotid triangle most often preclude extracerebral tumor exposure. The central lower part of the lamina terminalis in the IEVCs is often composed of gliotic tissue and in fact represents the capsule of the anterior pole of the tumor. Its opening between the chiasm and the AComA usually allows good exposure of the anterior and the basal parts of the intraventricular mass and of the entire extraventricular portion of the tumor (Fig. 39.18).
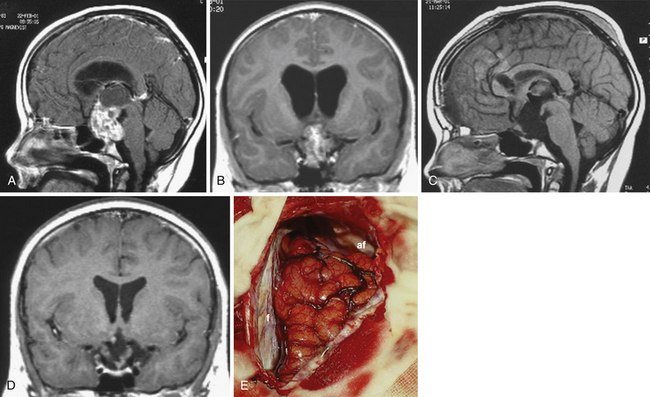
A disadvantage of this approach is an insufficient exposure of the superoposterior part of the third ventricle. The upper limit of the direct visual control of the ventricular cavity is represented by the line connecting the lower limit of the craniotomy and the AComA. The upper part of the tumor hidden above this borderline can thus be seen only after pulling it down to the lower part of the ventricle (Fig. 39.19) or by opening the lamina terminalis also above the AComA.88 For larger tumors we prefer the transcallosal approach. An advantage of this approach is the possibility of starting tumor removal at its upper pole where it is free from any neural structures at the enlarged foramina of Monro. After evacuation of the cyst following removal of the part of the tumor tissue through a larger foramen, the larger foramen provides a good access to a whole upper part of the tumor. This part of the tumor only touches and does not invade the fornix and the walls of the third ventricle. It helps to find the border and the plane of cleavage between the cerebral and the tumor tissue as one proceeds farther down to the more basal region where the tumor merges with the hypothalamus. The transcallosal exposure of the tumor eventually may allow removal of the whole tumor, which was the case in two of our patients (Fig. 39.20). If not, evacuation of the CSF from the lateral ventricles and removal of the most of the ventricular portion of the tumor provides a comfortable subsequent subfrontal approach.
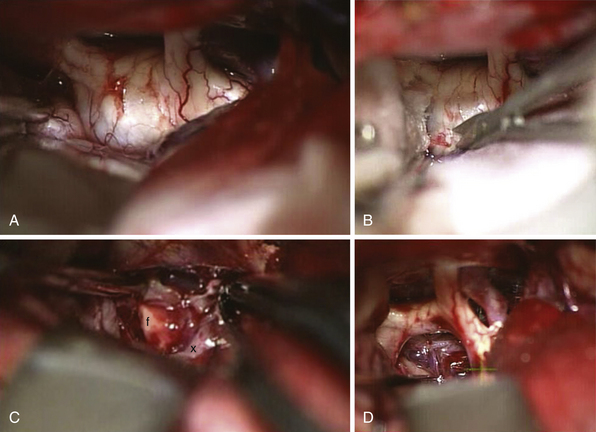
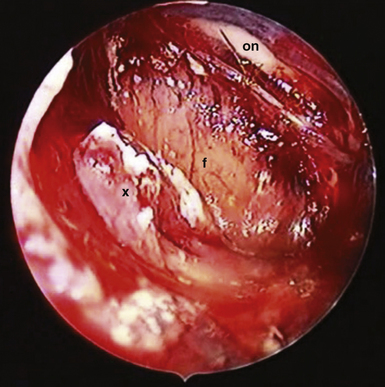
FIGURE 39.19 Intraoperative situation at the exposure of the lamina terminalis (A), its opening (B), removal of the last portion of the intraventricular and extraventricular craniopharyngioma (x) (C), and after tumor removal (D). f, Floor of the third ventricle. This is the same patient shown in Figure 39.18.
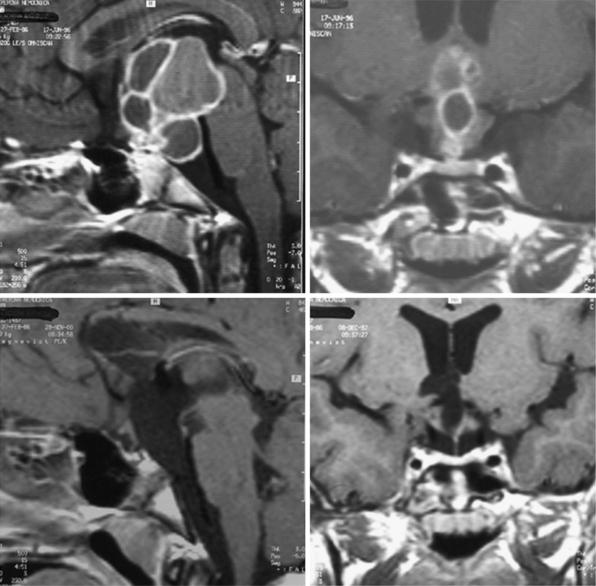
The transcallosal approach also has its limitations. The most inferior-anterior part of the tumor may not be exposed sufficiently and has to be approached through the opening of the lamina terminalis. A surgical approach that combines features of both basal and superior exposures is recommended for the suprasellar masses with simultaneous third ventricle involved.89 Removal of the tumor may be staged40,83 or performed at one sitting through two separate craniotomies12 or one larger60 craniotomy. In large or giant intraventricular and extraventricular craniopharyngiomas located entirely behind the chiasm/AComA reaching the roof of the third ventricle posterior to the foramina of Monro and causing hydrocephalus, we prefer a combined transcallosal and subfrontal approach through one unilateral craniotomy. We perform a large unilateral bone flap beginning at the lateral and the middle thirds of the orbital rim anteriorly, crossing the midline by 1.5 cm medially, and reaching or exceeding by 1 cm the coronal suture posteriorly. An opening of the dura approximately 7 cm long (in anteroposterior diameter) enables combined one-stage transcallosal and subfrontal tumor exposure (Fig. 39.21). One large craniotomy makes sequential or alternating transcallosal and subfrontal exposure easier, if necessary. The exposure of the tumor via a combined approach is as a rule sufficient, and no endoscopic assistance is necessary.
We have abandoned the transfrontal transcortical approach in order to avoid the seizure complications. Our results confirmed the observation of the others that a short incision up to 2 cm in the anterior part of the corpus callosum behind its genu causes no clinically apparent neuropsychological deficit.89–92 We have observed severe though transitional short memory disturbances in a single patient with craniopharyngioma in whom the tumor, an intraventricular and extraventricular craniopharyngioma, was removed through the lamina terminalis below and also above the AComA. The cause of memory disturbances lasting for almost 1 year could be a bilateral manipulation of the columnae of the fornix and the anterior commissure.93 We avoid the manipulation of both parts of the fornix whenever possible. During the transcallosal approach, we try to remove the tumor through one foramen of Monro using the other one for assessing the completeness of tumor removal.
Perioperative Management and Management of Complications
Severe acute hypothalamic failure may occur after removal of the tumor adherent to the hypothalamus. Life-threatening complication presents by mineral/water balance disturbances, which may be accompanied by hyperpyrexia, seizures, and decreased levels of consciousness. Careful monitoring and early correction of mineral and water balance disturbances presenting by diabetes insipidus, hypernatremia, and hypokalemia are absolutely mandatory. Such disturbances may be the consequence of a combination of disruption of pituitary stalk (lack of antidiuretic hormone) and damage of osmoreceptors in anterior hypothalamus (loss of thirst sensation). Disturbances of the fluid and electrolyte balance are treated in the intensive care unit. Detailed measurement of intake of fluid, output of urine, and blood and urine levels of sodium and potassium should start at the day of surgery and continue twice a day afterward. Measurement of osmolality of serum and urine should also be performed twice a day in cases with severe metabolic disturbances. Antidiuretic hormone (ADH) is administered if the diuresis exceeds 1 L in 6 hours and if there is tendency to hypernatremia and hyperosmolality. Hyponatremia and hypo-osmolality may follow after the initial phase of diabetes insipidus.94 It may be a part of different syndromes, such as syndrome of inappropriate ADH secretion (SIADH), cerebral salt-wasting syndrome (CSWS), and others. Management of the disturbance includes fluid restriction in transient or asymptomatic SIADH and aggressive replacement of urine water losses in CSWS. Sodium replacement is necessary in the advanced stage. These life-threatening metabolic disturbances should be managed by an anesthesiologist experienced in neurointensive care.
Long-Term Results
Surgical Mortality Rate
The outcome of surgery depends on the location of the tumor, its size, and the extent of surgery. The overall surgical mortality rate reported in most series is below 4%.15,16,83,87,95 In a large series with 45.7% radical removals the operative mortality rate was 0% after transsphenoidal surgery and 1.1% after primary transcranial surgery. The difference in outcomes of transsphenoidal and transcranial surgery is most probably due to the different topography and the size of the tumors operated by either approach. In another series with over 60% of large and giant tumors in which radical tumor removal was achieved in 90%, the surgical mortality and early postoperative mortality rates together reached 9%. On the other hand, the recurrence rate after such a radical approach was exceptionally low, around 7%.12 Analysis of our results showed the relation of the outcome to the location of the tumor. In our consecutive series of 105 patients operated since 1991 there were no deaths in 43 infradiaphragmatic tumors (24 transsphenoidal, 19 transcranial) including large and giant tumors with 81% radical removals (90% in primary surgery). Of 62 patients with supradiaphragmatic tumors (76% radical removals) 4 patients died after surgery (6.5%), with the mortality rate in a whole series reaching 3.8%.
Recurrences and Their Management
The extent of resection as seen in the first postoperative MRI is the most important factor concerning the recurrences in craniopharyngiomas.96 The recurrence rate after subtotal or partial tumor removal is higher than that after radical removal, representing 50% to 100% and 0% to 43%, respectively.14–16,25,26,59,62,63,65 In the series of Fahlbusch and co-workers83 the recurrence-free survival rate 10 years after total removal was 81.3%, but 5 years after subtotal and partial removal it reached only 48.8% and 41.5%, respectively. In the group of our patients who were followed for more than 1 year (12-201 months, mean 91 months) the tumor recurred in 19 of 94 patients after primary surgery (20.2%), in 16% after radical removal, and in 40% after incomplete removal.
Removal of recurrent craniopharyngioma is associated with a lower cure rate and a higher risk of complications than primary surgery because of the scarring from previous surgery and adherence of the tumor to surrounding structures. Radiation therapy therefore is recommended as the primary treatment for recurrences.65 However, some authors stress the possibility of safe resection of recurrent tumors and consider surgery to be the first therapeutic option for recurrent craniopharyngioma.9,13–15,62,97–100 Radiation therapy should also be considered but only as adjuvant therapy98 or second-line treatment for subsequent recurrence.97 Recurrence of craniopharyngioma can be safely managed by using meticulous contemporary microsurgical techniques even without additional radiotherapy.62 We performed 37 operations in 25 patients with 59.5% radical removals of recurrent tumor and the result comparable with primary surgery. One patient died after removal of a small residual tumor from pulmonary embolism. Permanent morbidity related to surgery occurred in two patients; both experienced visual deterioration in one eye. We decide to perform repeat surgery as soon as tumor recurrence or progression of the tumor remnant is proved by MRI. Some residual tumors remain stable for years without adjuvant therapy. The presence of a small residual calcification in children without enhancing tumor remnant does not have an impact on the risk of recurrence.101
Morbidity Rate
Pituitary hormone deficiency after craniopharyngioma removal occurs often. In a pediatric series some kind of hormonal replacement is necessary in 85% to 100%.16,51 A high number of multiple pituitary hormone deficiencies was found, regardless of total or partial surgery.51 After total tumor removal in both children and adults hormone deficits were found in 95%.102 Growth hormone deficiency is detected in up to 100%, hypogonadism in 65% to 80%, hypothyroidism in 38.5% to 95%, and hypocorticism in 55.2% to 78.1%.16,51–53,102,103 Diabetes insipidus at postsurgery follow-up is observed in 59.4% to 95% of patients.16,51,52,94,102,103 A new postsurgical diabetes insipidus in children occurred in 56%.16 Even if hormone levels seem to be adequate in the short term after treatment, deficiencies may develop over years and these patients need to be monitored closely.104 Diabetes insipidus may be reversible in some of the patients in whom the stalk was dissected as distally as possible from the tumor although ultimately sacrificed.105 Such recovery was not observed in another study103 and in our experience. New diabetes insipidus occurred in 20% of our patients. Permanent hormonal replacement was necessary in 90% of these patients.
Lower quality of life is observed most often after radical removal of retrochiasmatic craniopharyngiomas involving the hypothalamus. Besides the retrochiasmatic tumor location there are other different predictors of postsurgical morbidity: severe hydrocephalus, large size of the tumor, intraoperative adverse events, and young age.12,74,106–108 Recurrences and additional surgery were also associated with poorer quality of life.74
Obesity at follow-up after surgery is found in 58% to 62%.108–110 Weight gain of different degrees occurred after surgery in 20% of our pediatric patients; in one third of them it was evaluated as the consequence of increased appetite. A risk factor is also familial disposition for obesity.111 Significantly greater increase of the body mass index was observed in patients with the highest score of postsurgical hypothalamic damage as seen on follow-up MRI scans. Preoperative neuroimaging, however, demonstrated extensive hypothalamic “infiltration” by tumor in most of them.107 This is in concordance with our experience that the disruption of the third ventricular floor found on MRI scans after removal of IEVCs is most often due to the preoperative damage to the hypothalamus caused by the tumor itself. The postoperative weight gain appears to result from continued impact of preoperative hypothalamic damage.110 Another cause of obesity may be hypothalamic irradiation with the dose of 51 Gy or more.112
The data on mental disorders significantly differ in the literature. Significant school problems after radical tumor removal in children were observed in none15 to 50%.108 The neuropsychological outcome in a recent study was more benign than some previous studies have suggested.113 Satisfactory neuropsychological assessment is reported in almost all pediatric patients.114 Resolution of the preoperative impairment of cognitive functions in a few weeks in a majority of patients in a large series was reported. New cognitive disorders or personality changes were caused mostly by multiple recurrences treated with surgery and radiotherapy.95 According to Pierre-Kahn and associates, postoperative academic failure, when present, results more from a behavioral dysfunction than from an understanding incapability.82 Deleterious behavior (running away, theft, lies, irritability, aggressiveness, and bursts of rage) was observed in all 12 patients with the tumor extending into the third ventricle. Only 2 of their 14 children, both with extraventricular tumors, were attending normal schooling. We have observed mental disturbances much more rarely. The episodes of emotional lability and aggressiveness occurred in 1 child. Another 2 children who were less spontaneous and had less interest in fulfilling daily duties before surgery continued to be the same after the operation. Impaired cognitive functions present before surgery in 2 children improved. Long-lasting temporary memory disturbances occurred in 1 adult. We observed fatigue and worsened physical condition in some of the adult patients after otherwise successful surgical treatment of supradiaphragmatic craniopharyngiomas similar to those reported by others.115
Postsurgical behavioral problems may be attributed also to presumed implication of the frontal lobes arising from the surgical approach, which mediates aspects of impulsivity and cognitive flexibility to the hypothalamus which in turn plays an integral role in affect regulation.74
Visual functions after tumor removal are stabilized or improved in a majority of patients. Postsurgical visual impairment occurs in 5% to 66%.12,15,16,60,62,65,83,95,108 A risk factor is prechiasmatic extension of the tumor.16 In our patients vision improved after surgery in 13% and worsened in 7%. The patient operated on at the age of 11 months with “practical blindness,” paradoxical reaction to light, and still recordable visual evoked potentials can read without difficulties, although in one eye only light perception is present.
Cysts of the Suprasellar Region
In terminology and morphology of the cysts occurring in the suprasellar space a considerable overlap may be found. The term suprasellar cyst is sometimes used as a synonym for craniopharyngioma or for arachnoid cyst.116 Epidermoid cysts occurring in the suprasellar region are regarded as a variant of craniopharyngioma.2,117 Rathke’s cleft cysts and craniopharyngiomas share a common origin in embryological remnants of the fetal stomodeal cleft. Dumbbell lesions with both types of epithelia—the columnar in the intrasellar part and squamous in the suprasellar part—have been described.118
Epidermoid and Dermoid Cysts
Epidermoid cysts and the rarer dermoid cysts arise from the inclusion of ectodermally committed cells at time of closure of the neural groove.119 Epidermoid cysts account for 1% of all intracranial tumors. About one third of them are situated in some way within the cisterns of the suprasellar region.2
Clinical presentation is usually similar to that of craniopharyngiomas, with visual defects and pituitary or hypothalamic dysfunction. Less commonly, episodes of aseptic meningitis occur as a result of leakage of cyst contents into the CSF pathways. On the other hand, even after massive rupture of a suprasellar dermoid cyst excellent recovery could be achieved.120
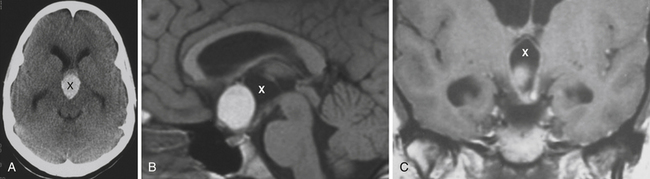
On both T1 and T2 MRI scans epidermoid cysts are slightly more hyperintense compared with CSF-containing arachnoid cysts (Fig. 39.22). With fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted pulse sequences, epidermoid cysts show higher signal intensity than does CSF. The signal intensity of dermoid cysts is usually like that of fat and may be similar to that of lipomas.
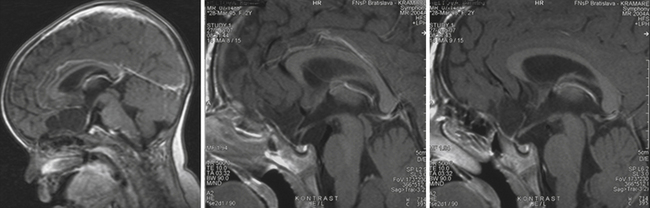
Rathke’s Cleft Cyst
Fragments of Rathke’s cleft persist in adults as microscopic cysts between the anterior and posterior lobes of the mature pituitary gland. It is presumed the remnants of the cleft persist also above the level of the sellar diaphragm in the pituitary stalk.117
The lining epithelium of the Rathke’s cleft cyst (RCC) consists of columnar or cuboidal cells with apical cilia, which may be stratified at places. In the suprasellar part of RCCs, the areas of stratified squamous epithelium may be found and occasionally may be very prominent. This has been interpreted as evidence of a close etiological relationship between craniopharyngiomas and RCCs.117 Keratinization is not seen. Lining epithelium may undergo extensive degeneration, leaving only a collagenous wall in some places.
MRI shows a single, uniloculated, round, sharply defined intra- or suprasellar mass that typically lies anterior to the infundibular stalk. The cystic contents may have variable signal intensity: either low signal intensity on T1 images and high signal intensity on T2 images resembling CSF, or high signal intensity on T1 images and variable signal intensity on T2 images owing to a high mucopolysaccharide content. Neither contrast enhancement nor calcifications are usually seen.3
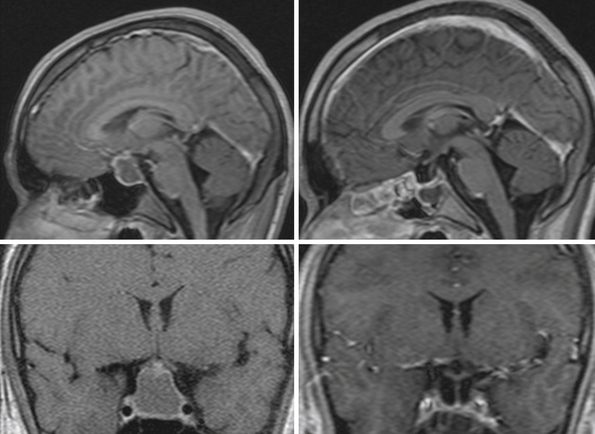
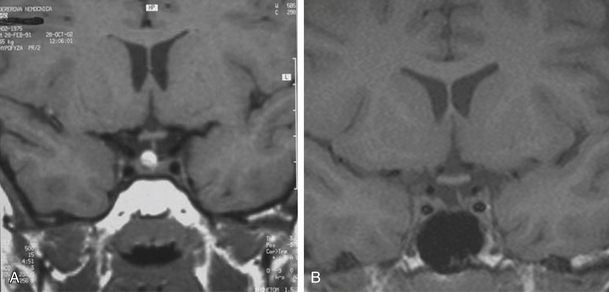
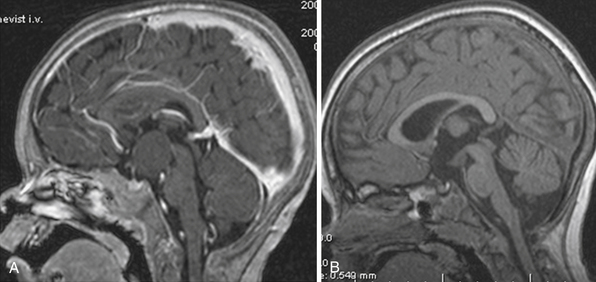
Intrasellar and suprasellar Rathke’s cleft cysts are approached transsphenoidally. Partial resection of the cyst wall and evacuation of the contents achieved microsurgically or by means of the endoscope is considered the best treatment.121–123 It allows adequate decompression of the pituitary tissue and the optic chiasm, with possible improvement of preoperative visual and endocrine deficit.121 Radical resection of the cyst wall may endanger the pituitary stalk. Incomplete removal of the cyst wall, however, leads to recurrences reported in all of the above-mentioned series. Suprasellar lesions are most often exposed through craniotomy.122,124 Preservation of the pituitary stalk in our patient did not result in improvement of preoperative endocrine deficiency (Fig. 39.23).
Gliomas of the Chiasm and Hypothalamus
Gliomas starting from the optic chiasm may involve the hypothalamus along direct optic fascicles from the lateral chiasm to the anterior hypothalamus.125 On the other hand, tumors arising within the walls of the third ventricle can infiltrate the chiasm. Usually it is impossible to determine whether the tumor started in the hypothalamus or in the optic chiasm, either clinically or radiologically.126 The gliomas of the chiasm and hypothalamus are therefore often referred to as a single disease entity named optic chiasmatic-hypothalamic gliomas,125,127,128 optic pathway/hypothalamic gliomas,126 chiasmal/hypothalamic gliomas,129 or hypothalamic/chiasmatic gliomas.130 Konovalov and colleagues129 distinguish nodular-type and diffuse-type tumors. The nodular type is classified into five groups according to the dominant directions of growth and possible place of origin along the visual pathway: (1) tumors with predominant anterior growth, (2) tumors growing anteriorly and penetrating the third ventricle, (3) tumors with the main part occupying the third ventricle but infiltrating the chiasm as well, (4) tumors of the optic tract, and (5) gliomas of the floor of the third ventricle. Kornienko and Pronin131 presented similar classification with the fifth group determined as the tumors of the third ventricle, growing into its lumen and causing occlusion of the CSF pathways.
Almost 90% of the chiasmatic/hypothalamic gliomas (CHGs) are found in the pediatric age group. The usual age of presentation is 2 to 4 years.132 CHGs in children represent 10% to 18.8% of the tumors of perisellar area.3,30 In children, the CHGs are typically pilocytic astrocytomas; in general about 40% are fibrillary and 60% are pilocytic in type. Anaplastic astrocytomas are rare (8%) and more often localized in the third ventricle (group 5).129
Our surgical experience showed that the gliomas occluding the greater part of or the entire third ventricular chamber may differ in relation to the structures of the optic pathway and the diencephalon. Besides the tumors infiltrating the chiasm or the hypothalamus or both, we found gliomas similar to group 5 of Konovalov and colleagues129 which infiltrated one lateral wall of the third ventricle and usually only touched the other one and the floor of the ventricle, or was just slightly attached to the latter. According to Yasargil90 astrocytic tumors of the third ventricle may arise from the ventricular wall as well as from the periventricular region. Fibrillary astrocytomas typically arise from the region of thalamus.133 We assume these tumors start to grow in the subependymal layer of the lateral wall of the third ventricle, break through it at an early stage of their development, and later grow inside the cavity of the third ventricle.
Differentiation between these two types of gliomas occupying the third ventricle is important from a surgical point of view. To a certain degree this can be assumed according to the relationship of the tumor mass with the AComA. A tumor growing inside the chiasm may expand anterior to the artery. All tumors that did not infiltrate the chiasm or the hypothalamus were located entirely behind the AComA. However, some tumors infiltrating the chiasm were located completely behind the vessel as well (Fig. 39.24). Consequently, in some instances it is not possible to differentiate between these topographic groups on the basis of preoperative MRI scans.
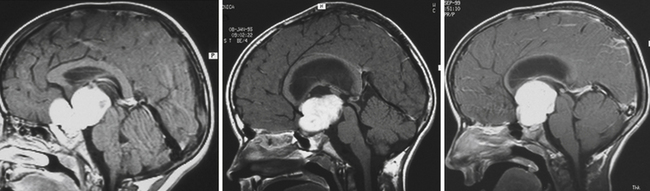
FIGURE 39.24 Variants of the relationship of chiasmatic/hypothalamic gliomas with the anterior communicating artery (see text).
CHGs are almost always hypointense with T1-weighted images and hyperintense with T2-weighted and FLAIR sequences. Large tumors are heterogeneous with cystic and solid components, with the latter enhancing markedly after contrast material injection.3
Clinical Presentation
Progressive loss of vision in tumors of the chiasm is accompanied by slowly developing optic atrophy. Visual fields show less typical hemianopic defects than in craniopharyngiomas.30 Defects in the temporal half of the visual field of the “better” eye combined with practical blindness in the other eye is typical for chiasmatic gliomas.129 Association of the visual disturbances with diabetes insipidus, obesity, and genital underdevelopment indicates hypothalamic involvement. Obesity is seen predominantly in adults. Sexual development is nearly normal, but a specific syndrome, precocious puberty, is seen in 8% of children.129 In CHGs “diencephalic syndrome” is observed in up to 25% of children aged less than 3 years. It is characterized by marked emaciation and loss of subcutaneous fat, which contrasts with normal height and normal (or near-normal) muscle mass. The appetite is normal. The child is alert, vigorous, hyperactive, and may be euphoric.132 The syndrome is associated with nystagmoid eye movements. Nystagmus is common also independently from the diencephalic syndrome.
Management of Chiasmatic/Hypothalamic Gliomas
The natural course of the CHGs is highly unpredictable. Some tumors remain static and quiescent for many years; others take an aggressive course, increasing rapidly in size and frequently leading to the patient’s death. Involution of a pilocytic astrocytoma134 and fibrillary astrocytoma135 of the chiasm after partial tumor removal or biopsy has been documented. Others have observed tumor regression only after “substantial” (more than 50%) tumor resection.126 The natural course is not always in accordance with the histological nature of the tumor. The patterns of malignancy were revealed in some pilocytic astrocytomas: frequent mitoses, pronounced endothelial proliferation, and necrotic foci.129 The expectation of more aggressive course of fibrillary astrocytomas has not proved to be entirely predictive.127 There is also disagreement about the significance of the association of the tumor with neurofibromatosis 1 (NF1).132 While some authors have reported better prognosis in these patients,126 according to others the association with NF1 is prognostically unfavorable134 and is a predictor of progressive disease.136
Prognostic uncertainty led to a divergence of opinion about the management of these tumors.127 Some authors recommend conservative surgery or biopsy followed by radiation and chemotherapy;130 others stress the important role of more radical resection.129,137
Surgery
The main aim of surgery was to obtain tissue for histological diagnosis and restoration of the patency of the CSF pathways. There is also a trend toward a more radical tumor removal which leads to a much lower recurrence rate and better outcome than partial resection.129,137,138 The indications for tumor resection are its nodular or exophytic growth without optic tract involvement136,138 or visual failure.127,128
Surgical Approach and the Extent of Tumor Removal
Surgical approaches to CHGs are those commonly used to access the suprasellar region: subfrontal, pterional, subtemporal, anterior interhemispheric, transcallosal, and combined transcallosal-subfrontal and transcallosal-pterional.127 Our preferred surgical approach to CHGs is subfrontal craniotomy as presented for craniopharyngiomas. The chiasm exposed below the frontal lobe is enlarged and infiltrated by the anterior part of the tumor in the majority of cases and it is not possible to discern the functioning parts of the visual pathway. From the enlarged and infiltrated chiasm we remove only a distinct tumor nodule found in a minority of patients. In cases in which no such nodule can be seen from the surface we enter the tumor through a small incision just below the AComA where the lamina terminalis is expected. From there we proceed posteriorly in the direction of the third ventricle. If an evident tumor is found inside the chiasm we remove only its central portion. In a majority of cases we leave the entire chiasmatic portion of the tumor (Fig. 39.25). This technique allowed us to preserve visual functions in all patients. The most common tumor in this location, the pilocytic astrocytomas, despite its benign nature infiltrates the optic pathways.139,140
The posterior part of a CHG often infiltrates the floor of the third ventricle. Gross total tumor removal was achieved in a patient in whom hypothalamic pilocytic astrocytoma did not infiltrate the chiasm and could be distinguished from the floor of the third ventricle (see video). In the great majority of the cases this part of the tumor is also left behind. We usually remove radically only the upper part of the tumor located inside the third ventricle where it is not intimately attached to the lateral walls. Although removal of the tumor from the third ventricular cavity is considered to be the most dangerous part of surgery,132 our experience showed that this part of the tumor behaves as “exophytic,” as also noted by others.136,138 This “ventricular component of the intraaxial tumor”89 may be safely removed. However, in a subgroup of the tumors adhering only to one lateral wall of the third ventricle radical resection was possible in more than half of the patients (see Fig. 39.26 and video).
During the translamina terminalis approach the most superoposterior part of a large tumor reaching the roof of the third ventricle may not be exposed.141 It is hidden behind and above the AComA. The vessel may react even to gentle stretching by spasm. These tumors may be exposed via the transcallosal approach. The interhemispheric approach to the corpus callosum in some patients without previous shunt insertion may require evacuation of the CSF by ventricular puncture. The transcallosal route, however, in some cases does not enable appropriate exposure of the inferoanterior part of the third ventricle. Therefore, a combined transcallosal and translamina terminalis approach may be necessary for the exposure of the entire tumor.129
Adjuvant Therapy
Unlike in the benign gliomas of other locations radiotherapy and chemotherapy are often recommended in CHGs. In general, radiation therapy arrests the tumor growth, although there are reports of irradiated tumors shrinking or completely disappearing.139 The most utilized dosages range between 45 and 56.6 Gy (median 54 Gy) for chiasmatic gliomas and 55 and 60 Gy for hypothalamic tumors. The efficiency of radiation therapy is reported to be as low as 40% of cases.132,139 The course of radiation therapy in our series was recommended by medical/radiation oncologists in eight patients with pilocytic and in six others with fibrillary astrocytomas. In eight patients the tumor regressed (five pilocytic, three fibrillary), in five it remained stable (two pilocytic, three fibrillary), and in one (pilocytic) it progressed and had to be reoperated.
Radiation therapy carries a risk of serious adverse effects.2,16,137 We have observed postradiation complications in two children. MRI performed 11 months after the dose of 50 Gy in a 6-year-old boy showed complete regression of the residual pilocytic astrocytoma and a pathological signal in the right thalamus and the mesencephalon. Afterward the patient lost the sight of both eyes, became obese, and now, 12 years after radiation, he has outbursts of rage. Another patient with residual fibrillary astrocytoma developed diabetes insipidus 2 months after completion of radiation treatment.
Despite their benign nature, most of the CHGs can respond surprisingly well to chemotherapy, allowing for tumor shrinkage and stabilization, or even disappearance in some instances.130,132,137 Thus, even if radiotherapy is necessary, it can be postponed beyond the age of 5 years to limit its deleterious side effects on intellectual, visual, and endocrine functions.127,132
Complications
Another serious complication is bleeding into the tumor bed, which should be evacuated. Removal of CHGs often results in brain collapse, subdural CSF and blood collection, or tension pneumocephalus, which require additional surgical intervention in 5% of cases. The risk factors are ventriculomegaly and tumor size more than 5 cm.129 There were no surgical deaths in our series of 34 patients.
Outcome
Radical tumor removal of CHGs may lead to a much better outcome than generally expected. Radicality of surgery dramatically influences the long-term outcome with 5- and 10-year progression-free survival probabilities for the radical, subtotal, and partial surgery of 100%, 74%, and 51%, respectively.129 In our series including gliomas not infiltrating the chiasm and hypothalamus, after radical tumor removal 14 of 15 (93.3%) patients survived for 46 to 203 (mean 111.6) months; after incomplete removal 13 of 19 (68.4%) patients survived for 42 to 170 (mean 115.6) months. Good clinical condition at the end of follow-up after radical and incomplete removal was achieved in 66.7% and 42.1%, respectively. The cause of death during follow-up was progression of the disease in six patients; one died from pneumonia, and another one died from a nonmedical accident, both after being in good clinical condition.
In the series of Konovalov and colleagues129 visual functions in the early postoperative period improved in 14% of patients and worsened in 23%. During the follow-up period further improvement was observed; altogether, vision improved in 28%. Deterioration of vision seen at the end of follow-up in 22% was due to recurrence of tumor. Growth retardation and diabetes insipidus (12%) were rare, unlike in craniopharyngiomas, but most patients developed obesity and some precocious puberty (18%).
Germinomas
Pure germinomas are the most common primary CNS germ cell tumors (GCTs), which account for up to 0.5% of all CNS tumors in general and 3% of CNS tumors in children. Germinoma tissue may also be found in mixed GCTs.140 The large majority of CNS GCTs occurs about a midline intracranial axis that traverses the third ventricle. Occasionally there is a synchronous pineal and suprasellar region tumor.
It is important to differentiate pure germinomas from those containing an admixture of other GCTs or syncytiotrophoblastic giant cells (STGCs) secreting human chorionic gonadotropin (hCG).142 Pure germinomas are typically nonsecreting tumors, whereas in nongerminatous GCTs elevation of either α-fetoprotein or human β-hCG in the serum or the CSF is found. “Marker negative” germinomas are prognostically most favorable.
Suprasellar germinomas are infiltrative lesions that tend to involve a variety of structures, including the floor of the third ventricle, pituitary stalk, pituitary gland, and the optic nerves, chiasm, and tracts.2 Clinical manifestations include visual disturbances, diabetes insipidus, obesity, and pituitary insufficiency. At the time of diagnosis the diabetes insipidus is more frequent and obesity occurs less often than in children with craniopharyngiomas.143 Precocious puberty almost exclusively occurring in boys may be the consequence of involvement of the hypothalamus30 and the effect of hCG.144 Tumors blocking the third ventricle produce the symptoms of increased intracranial pressure.
Most suprasellar germinomas are clearly detected on MRI as homogeneous isointensive or mildly hypointensive on T1-weighted images, and isointensive or hyperintensive on T2-weighted images. On rare occasions they contain small cysts.3,131 Usually they intensively accumulate contrast medium. Neuroimaging studies may demonstrate CSF-borne metastases along ventricular surfaces or in subarachnoid space.
Germinomas are extremely radiosensitive; therefore, until recently radiotherapy was the cornerstone of treatment. In the past a single fractionated dose has been delivered to the tumor site to assess its radiosensitivity (“biological biopsy”). If a radiographic response was demonstrated, the tumor was considered to have a germinal origin, and high-dose focal radiotherapy was initiated. Because of associated long-term sequelae and morbidity of craniospinal radiotherapy in young children, however, recent strategies have been developed to avoid or reduce the role of radiotherapy in the treatment of germinomas.140 In our patient, an 18-year-old girl, residual germinoma resolved completely after focal and whole-brain radiation therapy. Seven years after treatment multiple cavernous malformations were found on MRI.
It has become more prevalent to initiate chemotherapy as the primary treatment for patients with CNS GCTs. Pure germinomas demonstrate an 80% complete radiographic response with chemotherapeutic regimens, regardless of the extent of tumor extension. Patients with pure germinomas, included in a good-prognosis group by Matsutani,145 are treated by three courses of carboplatin-etoposide chemotherapy followed by local radiation dose (24 Gy) delivered to a generous local field. Germinomas with STGC receive 30 Gy to a generous local field and 20 Gy to the tumor site and two additional courses of chemotherapy.
Hypothalamic Hamartomas
Hamartomas are non-neoplastic nonprogressive congenital malformations that are composed of disordered neurons, glial cells, and myelinated tracts. Hamartomas occurring in the region of the hypothalamus (HH) occur in two general locations: those below the tuber cinereum are usually pedunculated, and those within the third ventricle are sessile.146,147
Pedunculated lesions are more likely to be small (less than 2 cm) and to cause precocious puberty but no other neurological symptoms. Etiologically important is a presence of neurons immunolabeling for gonadotropin-releasing factors within the hamartoma. Sessile lesions are more often large (2-5 cm) and associated with seizures, particularly gelastic seizures. Subsequently the seizures are of longer duration and develop secondary, generalized epileptic manifestations. Cognitive deterioration ensues and severe behavioral problems commonly develop as well. Approximately two thirds of children with sessile HH have developmental delays, and half also have precocious puberty.146,147
Hamartoma is isointense or mildly hypointense on T1-weighted images and isointense to hyperintense on T2-weighted images, with no contrast enhancement, which is helpful in differential diagnosis from other lesions.3
Pedunculated HH may be approached via a pterional or subtemporal route. After total removal of the lesion, half of the patients were cured. Subtotal resection rarely leads to clinical and hormonal success.146 Sessile hamartomas within the third ventricle can be approached through a transcallosal route.148 After complete or near complete resection via interfornicial route three children were seizure-free, and the seizures in the other two were only occasional, brief, and mild. The children also exhibited marked improvements in behavior, school performance, and quality of life.148 The main difficulty of surgery seems to be the differentiation between the tissue of hamartoma and normal brain tissue.146,149 We had the same experience with the sessile HH approached through opening of the lamina terminalis. A small part of the lesion had to be left at the region of the mammillary bodies (Fig. 39.27). Endoscopic removal or disconnection of the hamartoma from the hypothalamus may also lead to clinical improvement.150
Pituicytomas (Tumors of the Neurohypophysis)
The tumors of neurohypophysis originate in the pituicytes, modified glial cells of the posterior pituitary gland and infundibulum. Therefore, they may start to grow within the sella or above it. The term pituicytoma was historically also used for other tumors of neurohypophysis (granular cell tumors, pilocytic astrocytomas);151 the terms choristoma and infundibuloma were also used.3 Pituicytoma, a spindle cell neoplasm, is now distinguished from the granular cell tumor of the neurohypophysis composed of nests of large cells with granular, eosinophilic cytoplasm.152 Both tumors are benign (WHO grade I). Pituicytomas are reported to be extremely rare, occurring only in adults, and granular cell tumors may exceptionally occur also in children.
The clinical presentation in both tumor types resembles that of other slowly expanding lesions of the sellar/suprasellar region. The most common presenting symptom is visual field deficit; other symptoms include various types of hypopituitarism, galactorrhea (“stalk effect”), headache, and diabetes insipidus, which is rather uncommon.152
The tumors are very rare, and surgical experience is therefore limited. The firm and vascular nature of the granular cell tumors, sometimes combined with absence of an obvious dissection plane from adjacent brain, may hamper gross total resection.152 In another case report of a suprasellar granular cell tumor there is no mention about intraoperative bleeding; the gummy, yellowish, slightly glassy tumor could be removed completely.153 Significant bleeding occurred also in reported cases of pituicytomas; one of them necessitated embolization prior to repeated surgery.154,155
The pituicytoma seen in our patient, a 30-year-old woman, presented with diabetes insipidus and hypopituitarism. MRI showed isointense retrochiasmatic infundibular lesion on both T1- and T2-weighted images with significant contrast enhancement. The tumor was removed via the translamina terminalis approach without significant bleeding. Although the pituitary stalk could be preserved (Fig. 39.28), no substantial improvement in endocrinological status was achieved.
Fahlbusch R., Honegger J., Paulus W., Buchfelder M. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg. 1999;90:237-250.
Konovalov A.N. Operative Management of Craniopharyngiomas. Advances and Technical Standards in Neurosurgery. Vol. 8. New York: Springer-Verlag; 1981. 281–318
Šteňo J., Maláček M., Bízik I. Tumor-third ventricular relationships in supradiaphragmatic craniopharyngiomas: correlation of morphological, magnetic resonance imaging, and operative findings. Neurosurgery. 2004;54:1051-1060.
Šteňo J. Microsurgical topography of craniopharyngiomas. Acta Neurochir Suppl (Wien). 1985;35:94-100.
Yasargil M.G., Curcic M., Kis M., et al. Total removal of craniopharyngiomas. J Neurosurg. 1990;73:3-11.
Please go to expertconsult.com to view the complete list of references.
1. Burger P.C., Scheithauer B.W., Vogel F.S. Surgical Pathology of the Nervous System and Its Coverings. Region of the Sella Turcica. Philadelphia: Churchill Livingstone; 2002. 437–498
2. Lopes M.B.S., Thapar K., Horvath E., Kovacs K. Neoplasms of the sellar region. In: McLendon R., Rosenblum M.K., Bigner D.D., editors. Russel and Rubinstein’s Pathology of Tumors of the Nervous System. 2nd ed. London: Hodder Arnold; 2006:663-764.
3. Saleem S.N., Said A.-H.M., Lee D.H. Lesions of the hypothalamus: MR imaging diagnostic features. Radiographics. 2007;27:1087-1108.
4. Šteňo J., Maláček M., Majerčík M. Surgical experience with giant pituitary adenomas. In: Samii M., editor. Skull Base Surgery. Anatomy, Diagnosis and Treatment. Basel: Karger; 1994:402-407.
5. Kinjo T., Al-Mefty O., Ciric I. Diaphragma sellae meningiomas. Neurosurgery. 1995;36:1082-1092.
6. Beems T., Grotenhuis J.A., Wesseling P. Meningioma of the pituitary stalk without dural attachment: case report and review of the literature. Neurosurgery. 1999;45:1474-1477.
7. Matson D.D., Crigler J.F.Jr. Management of craniopharyngioma in childhood. J Neurosurg. 1969;30:377-390.
8. Hoffman H.J., Hendrick E.B., Humphreys R.P., et al. Management of craniopharyngioma in children. J Neurosurg. 1977;47:218-227.
9. Konovalov A.N. Operative Management of Craniopharyngiomas. Advances and Technical Standards in Neurosurgery. Vol. 8. New York: Springer-Verlag; 1981. 281–318
10. Konovalov A.N. Technique and strategies of direct surgical management of craniopharyngiomas. In: Apuzzo M.L.J., editor. Surgery of the Third Ventricle. 2nd ed. Baltimore: Williams & Wilkins; 1998:1133-1142.
11. Symon L., Sprich W. Radical excision of craniopharyngioma. Results in 20 patients. J Neurosurg. 1985;62:174-181.
12. Yasargil M.G., Curcic M., Kis M., et al. Total removal of craniopharyngiomas. J Neurosurg. 1990;73:3-11.
13. Samii M., Samii A. Surgical management of craniopharyngiomas. Schmidek H.H., Sweet W.H., editors, Operative Neurosurgical Techniques: Indications, Methods, and Results. 3rd ed. – Vol. 1. Philadelphia: WB Saunders; 1995:357-370.
14. Samii M., Tatagiba M. Craniopharyngioma. In: Kaye A.H., Laws E.R.Jr., editors. Brain Tumors. An Encyclopedic Approach. New York: Churchill Livingstone; 1995:873-894.
15. Zuccaro G. Radical resection of craniopharyngioma. Childs Nerv Syst. 2005;21:679-690.
16. Di Rocco C., Caldarelli M., Tamburrini G., Massini L. Surgical management of craniopharyngiomas—experience with a pediatric series. J Pediatr Endocrinol Metab. 2006;19(Suppl 1):355-366.
17. Kalapurakal J.A. Radiation therapy in the management of pediatric craniopharyngiomas—a review. Childs Nerv Syst. 2005;21:808-816.
18. Scarzello G., Buzzaccarini M.S., Perilongo G., et al. Acute and late morbidity after limited resection and focal radiation therapy in craniopharyngiomas. J Pediatr Endocrinol Metab. 2006;19(Suppl 1):399-405.
19. Muller H.L. Childhood craniopharyngioma. Recent advances in diagnosis, treatment and follow-up. Horm Res. 2008;69:193-202.
20. Muller H.L., Bruhnken G., Emser A., et al. Longitudinal study on quality of life in 102 survivors of childhood craniopharyngioma. Chils Nerv Syst. 2005;21:975-980.
21. Schubert T., Trippel M., Tacke U., et al. Neurosurgical treatment strategies in childhood craniopharyngiomas: is less more? Childs Nerv Syst. 2009;25:1419-1427.
22. Mark R.J., Lutge W.R., Shimizu K.T., et al. Craniopharyngioma: treatment in the CT and MR era. Radiology. 1995;197:195-198.
23. Alexander T.D., Weir B.K.A. What is the best treatment for craniopharyngiomas involving the third ventricle? In: Apuzzo M.L.J., editor. Surgery of the Third Ventricle. 2nd ed. Baltimore: Williams & Wilkins; 1998:1183-1191.
24. Tena-Suck M.L., Salinas-Lara C., Arce-Arellano R.I., et al. Clinico-pathological and immunohistological characteristics associated to recurrence/regrowth of craniopharyngiomas. Clin Neurol Neurosurg. 2006;22:661-669.
25. Sainte-Rose C., Puget S., Wray A., et al. Craniopharyngioma: the pendulum of surgical management. Childs Nerv Syst. 2005;21:691-695.
26. Puget S., Garnett M., Wray A., et al. Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg Pediatr. 2007;106(Suppl 1):3-12.
27. Šteňo J., Maláček M., Bízik I. Tumor-third ventricular relationships in supradiaphragmatic craniopharyngiomas: correlation of morphological, magnetic resonance imaging, and operative findings. Neurosurgery. 2004;54:1051-1060.
28. Šteňo J. Microsurgical topography of craniopharyngiomas. Acta Neurochir Suppl (Wien). 1985;35:94-100.
29. Rushing E.J., Giancaspero F., Paulus W., Burger P.C. Craniopharyngioma. In: Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K., editors. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon: IARC Press; 2007:238-240.
30. Koos W.T., Miller M.H. Intracranial tumors of infants and children. Stuttgart: Thieme; 1971. 415
31. Prabhu V.C., Brown H.G. The pathogenesis of craniopharyngiomas. Childs Nerv Syst. 2005;21:622-627.
32. Ciric I.S., Cozzens J.W. Craniopharyngiomas: transsphenoidal method of approach—for the virtuoso only? Clin Neurosurg. 1980;27:169-187.
33. Grekhov V.V. Topography of craniopharyngiomas [in Russian]. Vopr Neirokhir. 1959;6:12-17.
34. Northfield D.W.C. Rathke-pouch tumors. Brain. 1957;80:293-312.
35. Luse S.A., Kernohan J.W. Squamous cell nests of the pituitary gland. Cancer. 1955;8:623-628.
36. Kernohan J.W. Craniopharyngiomas. Minckler J., editor, Pathology of the Nervous System. – Vol. 2. New York: McGraw-Hill; 1971:1927-1937.
37. Janzer R.C., Burger P.C., Giancaspero F., Paulus W. Craniopharyngioma. In: Kleihues P., Cavenee W.K., editors. Pathology and Genetics of Tumours of the Nervous System. Lyon: IARC Press; 2000:244-246.
38. Maira G., Anile C., Colosimo C., Cabezas D. Craniopharyngiomas of the third ventricle. Trans-lamina terminalis approach. Neurosurgery. 2000;47:857-865.
39. Cashion E.L., Young J.M. Intraventricular craniopharyngioma: report of two cases. J Neurosurg. 1971;34:84-87.
40. Fukushima T., Hirakawa K., Kimura M., Tomonaga M. Intraventricular craniopharyngioma: its characteristic in magnetic resonance imaging and successful total removal. Surg Neurol. 1990;33:22-27.
41. Migliore A., Calzolari F., Marzola A. Intrinsic III ventricle craniopharyngioma. Childs Nerv Syst. 1992;8:56-58.
42. Pascual J.M., Gonzales-Llanos F., Barrios L., Roda J.M. Intraventricular craniopharyngiomas: topographical classification and surgical approach selection based on an extensive overview. Acta Neurochir (Wien). 2004;146:785-802.
43. Ross Russell R.W., Pennybacker J.B. Craniopharyngioma in the elderly. J Neurol Neurosurg Psychiatry. 1961;24:1-13.
44. Chen C., Okera S., Davies P.E., et al. Craniopharyngioma: a review of long-term visual outcome. Clin Experiment Ophthalmol. 2003;31:220-228.
45. Defoort-Dhellemmes S., Mority F., Bouacha I., Vinchon M. Craniopharyngioma: ophthalmological aspects at diagnosis. J Pediat Endocrinol Metab. 2006;19:321-324.
46. Šteňo J. Compression of the structures of the optic pathway by arteries of the circle of Willis in suprasellar tumors [article in Slovak]. Cesk Slov Neurol Neurochir. 1989;52/84:143-147.
47. Dawson B.H. The blood vessels of the human optic chiasma and their relation to those of hypophysis and hypothalamus. Brain. 1958;81:207-217.
48. Bergland R., Ray B.S. The arterial supply of the human optic chiasm. J Neurosurg. 1969;31:327-334.
49. de Vries L., Lazar L., Phillip M. Craniopharyngioma: presentation and endocrine sequelae in 36 children. J Pediatr Endocrinol Metab. 2003;16:703-710.
50. Paja M., Lucas T., Garcia-Uria J., et al. Hypothalamic-pituitary dysfunction in patients with craniopharyngioma. Clin Endocrinol. 1995;42:467-473.
51. Di Battista E., Naselli A., Queirolo S., et al. Endocrine and growth features in childhood craniopharyngioma: a mono-institutional study. J Pediatr Endocrinol Metab. 2006;19:431-437.
52. Gonc E.N., Yordam N., Ozon A., et al. Endocrinological outcome of different treatment options in children with craniopharyngioma: a retrospective analysis of 66 cases. Pediatr Neurosurg. 2004;40:112-119.
53. Honneger J., Buchfelder M., Fahlbusch R. Surgical treatment of craniopharyngiomas: endocrinological results. J Neurosurg. 1999;90:251-257.
54. Muller H.L., Emser A., Faldum A., et al. Longitudinal study on growth and body mass index before and after diagnosis of childhood craniopharyngioma. J Clin Endocrinol Metab. 2004;89:3298-3305.
55. Meuric S., Brauner R., Trivin C., et al. Influence of tumor location on the presentation and evolution of craniopharyngiomas. J Neurosurg. 2005;103(Suppl 5):421-426.
56. Bray G.A. Etiology and pathogenesis of obesity. Clin Cornerstone. 1999;2:1-15.
57. Saeki N., Sunami K., Kubota M., et al. Heavily T2-weighted MR imaging of white matter tracts in the hypothalamus: normal and pathologic demonstrations. AJNR Am J Neuroradiol. 2001;22:1468-1475.
58. Tanaka Y., Miyazawa Y., Akaoka F., Yamada T. Amnesia following damage to the mammillary bodies. Neurology. 1997;48:160-165.
59. Dhellemnes P., Vinchon M. Radical resection for craniopharyngiomas in children: surgical technique and clinical results. J Pediatr Endocrinol Metab. 2006;19(Suppl 1):329-335.
60. Choux M., Lena G., Genitori L. Craniopharyngioma in children. Neurochirurgie. 1991;37:1-174.
61. Shirane R., Su C.C., Kusaka Y., et al. Surgical outcome in 31 patients with craniopharyngiomas extending outside the suprasellar cistern: an evaluation of the frontobasal interhemispheric approach. J Neurosurg. 2002;96:704-712.
62. Minamida Y., Mikami T., Hashi K., Houkin K. Surgical management of the recurrence and regrowth of craniocraniopharyngiomas. J Neurosurg. 2005;103:224-232.
63. Lin L.L., El Naqa I., Leonard J.R., et al. Long-term outcome in children treated for craniopharyngioma with and without radiotherapy. J Neurosurg Pediatr. 2008;1:126-130.
64. Regine W.F., Mohiuddin M., Kramer S. Long-term results of pediatric and adult craniopharyngiomas treated with combined surgery and radiation. Radiother Oncol. 1993;27:13-21.
65. Tomita T., Bowman R.M. Craniopharyngiomas in children: surgical experience at Children’s Memorial Hospital. Childs Nerv Syst. 2005;21:729-746.
66. Karavitaki N., Brufani C., Warner J.T., et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol. 2005;62:397-409.
67. Khafaga Y., Jenkin D., Kanaan I., et al. Craniopharyngioma in children. Int J Radiation Oncology Biol Phys. 1998;42:601-606.
68. Kobayashi T., Kida Y., Mori Y., Hasegawa T. Long-term results of Gamma Knife surgery for the treatment of craniopharyngioma in 98 consecutive cases. J Neurosurg. 2005;103:482-488.
69. Chung W.-Y., Pan D.H.-C., Shiau C.-I., et al. Gamma Knife radiosurgery for craniopharyngiomas. J Neurosurg. 2000;93(Suppl 3):47-56.
70. Chytka T., Liščák R., Vladyka V., et al. Radiosurgery of craniopharyngiomas in combination with stereotactic methods. Cesk Slov Neurol Neurosurg. 2008;71(104):565-575.
71. Julow J., Backlund E.O., Lányi F., et al. Long-term results and late complications after intracavitary yttrium-90 colloid irradiation of recurrent cystic craniopharyngiomas. Neurosurgery. 2007;61:288-295.
72. Rittinger O., Kranzinger M., Jones R., Jones N. Malignant astrocytoma arising 10 years after combined treatment of craniopharyngioma. J Pediatr Endocrinol Metab. 2003;16:97-101.
73. Kranzinger M., Jones N., Rittinger O., et al. Malignant glioma as a secondary malignant neoplasm after radiation therapy for craniopharyngioma: report of a case and review of reported cases. Onkologie. 2001;24:66-72.
74. Sands S., Milner J.S., Goldberg J., et al. Quality of life and behavioral follow-up study of pediatric survivors of craniopharyngioma. J Neurosurg Pediatr. 2005;103(4):302-311.
75. Sasagawa Y., Akai T., Itou S., Iizuka H. Gamma Knife radiosurgery-induced cavernous hemangioma: case report. Neurosurgery. 2009;64:1006-1007.
76. Van den Berge J.H., Blaauw G., Breeman W.A., et al. Intracavitary brachytherapy of cystic craniopharyngiomas. J Neurosurg. 1992;77:545-550.
77. Hasegawa T., Kondziolka D., Hadjipanayis C.G., Lunsford L.D. Management of cystic craniopharyngiomas with phosphorus-32 intracavitary irradiation. Neurosurgery. 2004;54:813-820.
78. Broggi G., Giorgi C., Franzini A., et al. Preliminary results of intracavitary treatment of craniopharyngioma with bleomycin. J Neurosurg Sci. 1989;33:145-148.
79. Hargrave D.R. Does chemotherapy have a role in the management of craniopharyngioma? J Pediatr Endocrinol Metab. 2006;19(Suppl 1):407-412.
80. Jiang R., Liu Z., Zhu C. Preliminary exploration of the clinical effect of bleomycin on craniopharyngiomas. Stereotact Funct Neurosurg. 2002;78:84-94.
81. Pollock B.E., Natt N., Scomberg P.J. Stereotactic management of craniopharyngiomas. Stereotact Funct Neurosurg. 2002;79:25-32.
82. Pierre-Kahn A., Recassens C., Pinto G., et al. Social and psycho-intellectual outcome following radical removal of craniopharyngiomas in childhood. A prospective series. Childs Nerv Syst. 2005;21:817-824.
83. Fahlbusch R., Honegger J., Paulus W., Buchfelder M. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg. 1999;90:237-250.
84. Couldwell W.T., Weiss M.H., Rabb C., et al. Variations of the standard transsphenoidal approach to the sellar region, with emphasis on the extended approaches and parasellar approaches: surgical experience in 105 cases. Neurosurgery. 2004;55:539-547.
85. de Divitiis E., Capabianca P., Cavallo L.M., et al. Extended endoscopic transsphenoidal approach for extrasellar craniopharyngiomas. Neurosurgery. 2007;61(5 Suppl 2):219-227.
86. Al-Mefty O., Ayoubi S., Kadri P.A. The petrosal approach for the total removal of giant retrochiasmatic craniopharyngiomas in children. J Neurosurg. 2007;106:87-92.
87. Choux M., Lena G. Craniopharyngioma. In: Apuzzo M.L.J., editor. Surgery of the Third Ventricle. 2nd ed. Baltimore: Williams & Wilkins; 1998:1143-1181.
88. Dehdashti A.R., de Tribolet N. Frontobasal interhemispheric trans-lamina terminalis approach for suprasellar lesions. Neurosurgery. 2005;56(Suppl 2):418-423.
89. Apuzzo M.L.J., Levy M.L., Tung H. Surgical strategies and technical methodologies in optimal management of craniopharyngioma and masses affecting the third ventricular chamber. Acta Neurochir Suppl (Wien). 1991;53:77-88.
90. Yasargil M.G. Microneurosurgery. Vol. IVA. Stuttgart: Thieme; 1994.
91. Oepen G., Schulz-Weiling R., Zimmermann P., et al. Long-term effects of partial callosal lesions. Preliminary report. Acta Neurochir (Wien). 1985;77:22-28.
92. Kasowski H., Piepmeier J.M. Transcallosal approach for tumors of the lateral and third ventricles. Neurosurg Focus. 2001;10:E3.
93. Wojciechowsky C., Vogel S., Meyer B.U., Lehmann R. Neuropsychological consequences of partial callosotomy. J Neurosurg Sci. 1997;41:75-80.
94. Ghirardello S., Hopper N., Albanese A., Maghnie M. Diabetes insipidus in craniopharyngioma: postoperative management of water and electrolyte disorders. J Pediatr Endocrin Metab. 2006;19(Suppl 1):413-421.
95. Van Effenterre R., Boch A.L. Craniopharyngiomas in adults and children: a study of 122 surgical cases. J Neurosurg. 2002;97:3-11.
96. Lena G., Paz Paredes A., Scavarda D., Giusiano B. Craniopharyngioma in children: Marseille experience. Childs Nerv Syst. 2005;21:778-784.
97. Wisoff J.H. Surgical management of recurrent craniopharyngiomas. Pediatr Neurosurg. 1994;21(Suppl 1):108-113.
98. Caldarelli M., Massimi L., Tamburini G., et al. Long-term results of the surgical treatment of craniopharyngioma: the experience at the Policlinico Gemelli, Catholic University, Rome. Childs Nerv Syst. 2005;21:747-757.
99. Caldarelli M., di Rocco C., Papacci F., Colosimo C.Jr. Management of recurrent craniopharyngioma. Acta Neurochir (Wien). 1998;140:447-454.
100. Elliott R.E., Hsieh K., Hochm T., et al. Efficacy and safety of radical resection of primary and recurrent craniopharyngiomas in 86 children. J Neurosurg Pediatr. 2010;5:30-48.
101. Elliott R.E., Moshel Y.A., Wisoff J.H. Minimal residual calcification and recurrence after gross-total resection of craniopharyngioma in children. J Neurosurg Pediatr. 2009;3:276-283.
102. Zhou Z., Shi X. Changes of hypothalamus-pituitary hormones in patients after total removal of craniopharyngiomas. Chinese Med J. 2004;117:357-360.
103. Honneger J., Barocka A., Sadri B., Fahlbusch R. Neuropsychological results of craniopharyngioma surgery in adults: a prospective study. Surg Neurol. 1998;50:19-29.
104. Halac I., Zimmerman D. Endocrine manifestations of craniopharyngioma. Childs Nerv Syst. 2005;21:640-648.
105. Nishizawa S., Ohta S., Oki Y. Spontaneous resolution of diabetes insipidus after pituitary stalk sectioning during surgery for large craniopharyngioma. Neurol Med Chir (Tokyo). 2006;46:126-135.
106. De Vile C.J., Grant D.B., Kendall B.E., et al. Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J Neurosurg. 1996;85:73-81.
107. De Vile C.J., Grant D.B., Hayward R.D., et al. Obesity in childhood craniopharyngioma: relation to post-operative hypothalamic damage shown by magnetic resonance imaging. J Clin Endocrinol Metab. 1996;81:2734-2737.
108. Poretti A., Grotzer M.A., Ribi K., et al. Outcome of craniopharyngioma in children: long-term complications and quality of life. Dev Med Child Neurol. 2004;46:220-229.
109. Ahmet A., Blaser S., Stephens D., et al. Weight gain in craniopharyngioma—a model for hypothalamic obesity. J Pediatr Endocrinol Metab. 2006;19:121-127.
110. Vinchon M., Weill J., Delestret I., Dhellemmes P. Craniopharyngioma and hypothalamic obesity in children. Childs Nerv Syst. 2009;25:347-352.
111. Muller H.L., Bueb K., Bartels U., et al. Obesity after childhood craniopharyngioma—German multicenter study on pre-operative risk factors and quality of life. Klin Pediatr. 2001;213:244-249.
112. Lustig R.H., Post S.R., Srivannaboon K., et al. Risk factors for the development of obesity in children surviving brain tumors. J Clin Endocrinol Metab. 2003;88:611-616.
113. Bawden H.N., Salisbury S., Eskes G., Morehouse R. Neuropsychological functioning following craniopharyngioma removal. J Clin Exp Neuropsychol. 2009;31:140-144.
114. Pancucci G., Massimi L., Caldarelli M., et al. Pediatric craniopharyngioma: long-term results in 61 cases [article in Italian]. Minerva Pediatr. 2007;59:219-231.
115. Dekkers O.M., Biermasz N.R., Smit J.W., et al. Quality of life in treated adult craniopharyngioma patients. Eur J Endocrinol. 2006;154:483-489.
116. Hellwig D., Schulte M., Trikotai W. Surgical management of arachnoid, suprasellar, and Rathke’s cleft cysts. In: Schmidek H.H., Roberts D.W., editors. Schmidek and Sweet Operative Neurosurgical Techniques. Indications, Methods and Results. 5th ed. Philadelphia: WB Saunders; 2006:455-476.
117. Ironside J.W., Moss T.H., Louis D.N., et al. Diagnostic Pathology of Nervous System Tumours. London: Churchill Livingstone; 2002. 509–562
118. Russell D.S., Rubinstein L.J. Pathology of Tumours of the Nervous System, 3rd ed., London: Edward Arnold; 1971:19-25.
119. Fuller G.N., Ribalta T. Dermoid cyst, epidermoid cyst, and dermal sinus. In: McLendon R., Rosenblum M.K., Bigner D.D., editors. Russel and Rubinstein’s Pathology of Tumors of the Nervous System. 2nd ed. London: Hodder Arnold; 2006:583-590.
120. Cohen J.E., Abdallah J.A., Garrote M. Massive rupture of suprasellar dermoid cyst into ventricles. Case illustration. J Neurosurg. 1997;87:963.
121. El-Mahdy W., Powel M. Transsphenoidal management of 28 symptomatic Rathke’s cleft cysts, with special reference to visual and hormonal recovery. Neurosurgery. 1998;42:7-16.
122. Zada G., Ditty B., McNatt S.A., et al. Surgical treatment of Rathke cleft cyst in children. Neurosurgery. 2009;64:1132-1137.
123. Frank G., Sciaretta V., Mazzatenta D., et al. Transsphenoidal endoscopic approach in the treatment of Rathke’s cleft cyst. Neurosurgery. 2005;56:124-128.
124. Itoh J., Usui K. An entirely suprasellar symptomatic Rathke’s cleft cyst: case report. Neurosurgery. 1992;30:581-584.
125. Yasargil M.G. Optic Gliomas in Microneurosurgery. Vol. IVB. Stuttgart: Thieme; 1996. 224–233
126. Hoffman H.J., Humphreys R.P., Drake J.M., et al. Optic pathway/hypothalamic gliomas: a dilemma in management. Pediatr Neurosurg. 1993;19:186-195.
127. Alshail E., Rutka J.T., Becker L.E., Hoffman H.J. Optic chiasmatic-hypothalamic glioma. Brain Pathol. 1997;7:799-806.
128. Rutka J.T., Becker L.E., Hoffman H.J. Optic chiasmatic-hypothalamic gliomas. Brain Pathol. 2008;7:799-806.
129. Konovalov A.N., Gorelyshev S.K., Khuhlaeva E.A. Surgical management of brainstem, thalamic, and hypothalamic tumors. Chiasmal/hypothalamic gliomas. In: Schmidek H.H., Roberts D.W., editors. Schmidek and Sweet Operative Neurosurgical Techniques. Indications, Methods and Results. 5th ed. Philadelphia: WB Saunders; 2006:821-835.
130. Sutton L.N., Molloy P.T., Sernyak H., et al. Long-term outcome of hypothalamic/chiasmatic astrocytomas in children treated with conservative surgery. J Neurosurg. 1995;83:583-589.
131. Kornienko V.N., Pronin I.N. Diagnostic Neuroradiology. Berlin: Springer-Verlag; 2009.
132. Caldarelli M., Pezzotta S. Optic pathway and hypothalamic tumors. In: Choux M., Di Rocco C., Hockley A., Walker M., editors. Pediatric Neurosurgery. Philadelphia: Churchill Livingstone; 1999:509-529.
133. Amar A.P., Ghosh S., Apuzzo M.L.J. Ventricular tumors. Winn H.R., editor, Youmans Neurological Surgery. 5th ed. – Vol. 1. Philadelphia: WB Saunders; 2004:1237-1263.
134. Borit A., Richardson E.P.Jr. The biological and clinical behaviour of pilocytic astrocytomas of the optic pathways. Brain. 1982;105:161-187.
135. Venes J.L., Latack J., Kandt R.S. Postoperative regression of opticochiasmatic astrocytoma: a case for expectant therapy. Neurosurgery. 1984;15:421-423.
136. Valdueza J.M., Lohmann F., Dammann O., et al. Analysis of 20 primarily surgically treated chiasmatic hypothalamic pilocytic astrocytomas. Acta Neurochir (Wien). 1994;126:44-50.
137. Hoffman H.J., Soloniuk D.S., Humphreys R.P., et al. Management and outcome of low-grade astocytomas of the midline in children: a retrospective review. Neurosurgery. 1993;33:964-971.
138. Wisoff J.H., Abbott R., Epstein F. Surgical managenent of exophytic chiasmatic hypothalamic tumors of childhood. J Neurosurg. 1990;73:661-667.
139. Smith E.R., Ebb D.H., Tarbell N.J., Barker F.G.II. Pilocytic astrocytoma. In: Berger M.S., Prados M.D., editors. Textbook of Neuro-oncology. Philadelphia: WB Saunders; 2005:149-155.
140. Suh D.Y., Mapstone T. Pediatric supratentorial intraventricular tumors. Neurosurg Focus. 2001;10:E4.
141. Konovalov A.N., Gorelyshev S.K. Surgical treatment of anterior third ventricle tumours. Acta Neurochir (Wien). 1992;118:33-39.
142. Rosenblum M.K. Germ cell tumors. In: McLendon R., Rosenblum M.K., Bigner D.D., editors. Russel and Rubinstein’s Pathology of Tumors of the Nervous System. 2nd ed. London: Hodder Arnold; 2006:551-577.
143. Ono N., Kohga H., Zama A., et al. A comparison of children with suprasellar germ cell tumors and craniopharyngiomas: final height, weight, endocrine, and visual sequelae after treatment. Surg Neurol. 1996;46:370-377.
144. Kubo O., Yamasaki N., Kamijo Y., et al. Human chorionic gonadotropin produced by ectopic pinealoma in a girl with precocious puberty. Case report. J Neurosurg. 1977;47:101-105.
145. Matsutani M. Germ cell tumors. In: Berger M.S., Prados M.D., editors. Textbook of Neuro-oncology. Philadelphia: WB Saunders; 2005:310-320.
146. Albright A.L. Hypothalamic hamartomas. In: Berger M.S., Prados M.D., editors. Textbook of Neuro-oncology. Philadelphia: WB Saunders; 2005:652-655.
147. Rosenblum M.K. Hypothalamic neuronal hamartoma. In: McLendon R., Rosenblum M.K., Bigner D.D., editors. Russel and Rubinstein’s Pathology of Tumors of the Nervous System. 2nd ed. London: Hodder Arnold; 2006:391-399.
148. Rosenfeld J.V., Harvey A.S., Wrennal J., et al. Transcallosal resection of hypothalamic hamartoma, with control of seizures, in children with gelastic epilepsy. Neurosurgery. 2001;48:108-118.
149. Rutka J.T., Comment on:Rosenfeld J.V., Harvey A.S., Wrennal J., et al. Transcallosal resection of hypothalamic hamartoma, with control of seizures, in children with gelastic epilepsy. Neurosurgery. 2001;48:118.
150. Lekovic G.P., Gonzales L.F., Feiz-Erfan I., Rekate H.L. Endoscopic resection of hypothalamic hamartoma using a novel variable aspiration tissue resector. Neurosurgery. 2006;58(1):ONS165-ONS168.
151. Wesseling P., Brat D.J., Fuller G.N. Pituicytoma. In: Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K., editors. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon: IARC Press; 2007:243-244.
152. Fuller G.N., Wesseling P. Granular cell tumour of the neurohypophysis. In: Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K., editors. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon: IARC Press; 2007:241-242.
153. Boecher-Schwarz H., Fries G., Bornemann A., et al. Suprasellar granular cell tumor. Neurosurgery. 1992;31:751-754.
154. Ulm A.J., Yachnis A.T., Brat D.J., Rhoton A.L.Jr. Pituicytoma: report of two cases and clues regarding histogenesis. Neurosurgery. 2004;54:753-758.
155. Wolfe S.Q., Bruce J., Morcos J.J. Pituicytoma: case report. Neurosurgery. 2008;63:173-174.