CHAPTER 185 Craniofacial Surgery for Congenital and Acquired Deformities
Congenital craniofacial deformities commonly occur as isolated defects and less often as part of a syndrome. The Committee on Nomenclature and Classification of Craniofacial Anomalies of the American Cleft Palate Association has organized craniofacial malformations into the following five categories: (1) facial clefts/encephaloceles and dysostoses; (2) atrophy/hypoplasia; (3) neoplasia/hyperplasia; (4)craniosynostosis; and (5) unclassified. Clinical entities such as orbital hypertelorism often exist with a syndrome that clearly fits into one of the aforementioned classifications.1 Hypoplasia of the midface and micrognathia would be incorporated into the second category. In children, hematologic disorders and overly aggressive cerebrospinal fluid (CSF) shunting can result in secondary craniofacial disorders, which can be put into the third or fifth category. Acquired deformities of the craniofacial complex also include those inflicted by means of a traumatic event. Neoplasm (third category) and its treatment are classified as acquired deformities.
Epidemiology
Craniofacial anomalies have been reported to constitute approximately one third of all congenital defects. The incidence of assorted individual deformities and syndromes varies. However, the overall incidence is considered to be 0.2 to 0.5 per 1000 births.2 Interestingly, some craniofacial deformities occur at a uniform rate across racial and ethnic populations, whereas others occur at a frequency that varies as a function of race and ethnicity.
Several craniofacial malformations appear to have a genetic etiology. Several familial types have been well documented, and transmission can occur via autosomal dominant or recessive modes. With occasional exceptions males and females tend to have similar incidences for most anomalies. Craniofacial anomalies have been reported to occur as part of a genetic condition in about 20% of cases.2 However, with the discovery of new syndromes and the recognition that “isolated defects” often have a genetic etiology, the 20% figure is probably an underestimate. Furthermore, as discussed here, the pathogenesis of craniofacial anomalies is complex, and generally it is most accurate to describe their etiology as multifactorial.
Mechanisms of Postnatal Facial Growth
It was once thought that craniofacial growth (like all other growth) was regulated, to a large extent, by the central nervous system. It is now understood to be much more complex. On a cellular level, the signal-sensitive membranes of the many types of cells respond to several different cues. On a developmental level, the functional needs of a particular structure and its relationship to adjoining structures strongly influence patterns of growth. Craniofacial bony growth is secondary to neural tissue growth and expansion. The brain as an organ mass develops early in embryogenesis. The developing brain highly influences the morphology of the basicranium, which in turn functions as a template for subsequent facial growth. The face can be viewed as a series of stratified vertical levels or fields that are strongly affected by growth of the brain and basicranium.3
Physiologically, there are two primary forces at play during the growth of individual craniofacial bones and structures, displacement and remodeling (Fig. 185-1). Growth does not indicate uniform enlargement of a structure; rather, it refers to a combination of displacement and remodeling. Displacement occurs when the bone is pushed away from its articulation with other bones (by the combined effects of growth of the surrounding soft tissue and growth center/sutural activity). Remodeling occurs by resorption and deposition of new bone that result in a net vector of growth. It is in this way that the correct tissue mass is achieved and that enlargement occurs proportionally and in the appropriate direction.
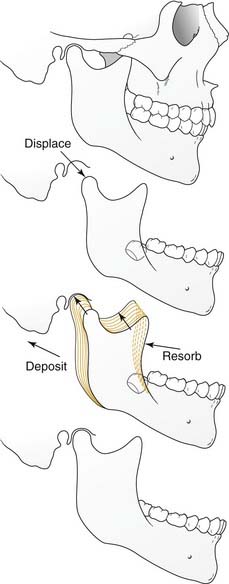
In the craniofacial complex the basicranium (along with the related soft tissues) influences the naso-orbito-maxillary complex and the ramus of the mandible, which conform through continual remodeling. Growth of the naso-orbito-maxillary complex displaces the maxillary dento-alveolus. This displacement together with growth of the mandibular ramus then affects growth of the corpus (body) of the mandible. The importance of soft tissues in growth is demonstrated in such clinical manifestations as the “adenoid face.” The entire system is plastic and in flux throughout the growing period (even after periods of growth, albeit to a far lesser extent). It is for this reason that rigid nonbiologic implants such as those of titanium have the potential to interfere with normal growth if used during active development; they lack the plasticity that is so vital to dynamic growth.4–6
Throughout the period of craniofacial growth an overriding homeostasis exists among the different fields of growth. Small aberrations in one field can be counterbalanced by compensatory growth in another, resulting in constant equilibrium. This is very important for the physician to bear in mind when planning surgical or orthodontic intervention. Treatment failure or relapse is likely if that equilibrium is significantly disrupted.7 Generally, interventions are more likely to succeed if the cause of the abnormality is addressed during periods of rapid growth. In contrast, the effect of the abnormality is more successfully treated during periods of slow growth.3
Craniofacial synostosis, or premature closure of a suture, is a good example of the morphologic consequence of defective skeletal growth. It also illustrates the importance of homeostasis in craniofacial development. The causes of craniosynostosis are likely multifactorial and the source of some controversy.8,9 It has been shown to be associated with abnormalities in sutural biology, various biomechanical forces,10 and defects in the primary growth centers of the involved bones. Sutural growth is also affected by surrounding tissue interactions. The contacting dura, for instance, influences the continued patency of sutures and helps regulate its closure.11–14
Fusion of the facial processes during embryogenesis is another fundamental aspect of facial growth that is critical to normal development. Orofacial clefting results from a defect in this process. For opposing facial processes to fuse in the midline, the contacting epithelia must be eliminated so that the underlying mesenchymal tissue can become continuous. In the palate this elimination has been shown to occur through apoptosis, epithelial to mesenchymal cell transdifferentiation, or cell migration.14–17 Underdevelopment of the mesenchyme of the involved subunits might preclude proper fusing, as can excessive apoptosis. Inadequate migration or proliferation of neural crest ectomesenchyme might have the same result. Facial clefting can also result from constricting anatomic defects. In the case of stunted mandibular growth, a Pierre Robin sequence can ensue whereby the palatal shelves are prevented from fusing in the midline.
Pathophysiology
Molecular Genetics
Several genetic defects have been identified that are directly associated with various craniofacial defects (Table 185-1). The products of these genes tend to fall into one of the following categories:
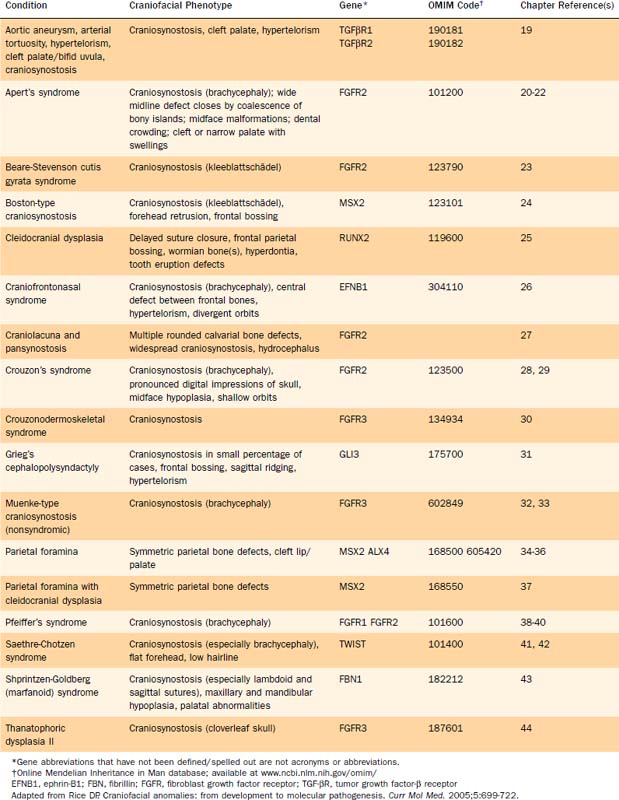
These genetic defects have been linked to syndromes as well as isolated defects. For instance, defective FGFRs have been identified as the etiologic basis of some types of craniosynostosis as well as various syndromes (see Table 185-1).18–45
FGFRs are transmembrane receptors for fibroblast growth factors (FGFs). FGFs are involved in regulating cell proliferation, differentiation, and migration through a number of pathways. The receptor is made up of an extracellular domain (receptor-ligand binding), a transmembrane domain, and an intracellular tyrosine kinase enzymatic domain (Fig. 185-2A). There are several variations of the FGFR protein—all with important cell growth functions. Signaling via FGFR1, for instance, has been shown to facilitate neural crest migration, and a defective receptor has been linked to midline facial clefting in mice.45 Defects in FGFR1 have been found to cause Kallmann’s syndrome 2, which can include cleft lip and palate (see Fig. 185-2B).46 Defects in FGFR1 and FGFR2 have been linked to syndromes such as Pfeiffer’s syndrome (in which both receptors are defective) and to conditions of which craniosynostosis is a feature.38,47 Defects in FGFR2 alone have been found in Apert’s syndrome,20,48 Jackson-Weiss syndrome,28 Crouzon’s syndrome,29 and Beare-Stevenson cutis gyrata syndrome.49 A defective FGFR3 protein is responsible for Crouzonodermoskeletal syndrome (Crouzon’s syndrome with acanthosis nigricans),30 for Muenke-type craniosynostosis,32 and also for the disease process seen in achondroplasia.50 In this disease the epiphyseal growth plates in long bones prematurely fuse. There are gross and histological similarities of long bone growth plates to the synchondroses of the calvaria. This work has led to the understanding that FGFR3 is involved in regulation of skeletal growth. In FGFR3 knockout mice (mice in which the gene that encodes this protein has been eliminated), there is severe retardation of skeletal maturation.48,51–53
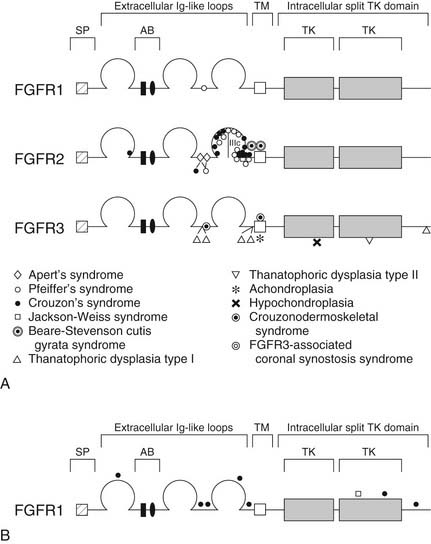
The link between known syndromes or anomalies and genetic defects, however, can be complex and difficult to characterize. Defects in each of the three domains (in different FGFRs) have been discovered in association with craniofacial abnormalities, yet defects in the same domain of one FGFR type can cause two separate syndromes. For instance, identical mutations have been discovered in patients with Crouzon’s, Pfeiffer’s, and Jackson-Weiss syndromes, suggesting the involvement of other factors in the ultimate phenotypic expression.38 Conversely, as is evident in Figure 185-2B, mutations in several distinct domains on one particular FGFR can result in the same phenotype.
As previously mentioned, defects in transcription factors and connective tissue proteins are also implicated in the pathogenesis of craniosynostosis. MSX2 is a homeobox gene that encodes a DNA binding transcription factor. It has been associated with nonsyndromic craniosynostosis (Boston type),24,54 and ALX4, another homeobox gene, has been linked to parietal foramina.34 Most cases of Saethre-Chotzen syndrome are caused by haplo-insufficiency of the TWIST gene, which appears to encode a transcription factor.41 The GLI-3 gene and the transcription factor that it encodes are defective in Grieg’s cephalopolysyndactyly (a rare syndrome that can include craniosynostosis).31,55
Genetic defects encoding several different extracellular matrix constituents can lead to anomalies of the craniofacial complex. Defective collagen, such as that seen in osteogenesis imperfecta, is clearly associated with bony abnormalities, although this disease only occasionally affects the craniofacial skeleton. Defective fibrillin (encoded by FBN1) results in Shprintzen-Goldberg syndrome with craniosynostosis and maxillary/mandibular hypoplasia.43 The defective genes responsible for Stickler’s syndrome are COL2A1 and COL11A2, which encode types II and XI collagen, respectively.56,57
Metabolic Disorders
Several metabolic derangements are known to interfere with craniofacial development. The best known is the group of disorders known as the mucopolysaccharidoses. This group of disorders is characterized by a deficiency of lysosomal enzymes that results in a buildup of mucopolysaccharides. Although there are several clinical entities, patients with some of the more severe variations of the disease have large and dolichocephalic (long and narrow) skulls with premature closure of the sagittal suture and poorly developed mastoids and paranasal sinuses. Mucolipidosis, hyperthyroidism, and rickets are also known causes of craniosynostosis.58,59
Intrauterine Causes
In utero environmental contributors to craniofacial maldevelopment fit into the following four general categories: teratogenic, infectious, nutritional, and mechanical. Fetal alcohol syndrome can involve a wide range of defects, including holoprosencephaly. Tobacco use during critical periods of embryogenesis has also been shown to adversely affect craniofacial development. Medications such as hydantoin, phenytoin, valproic acid, and isotretinoin (a vitamin A derivative used in the treatment of acne) can exert deleterious effects on embryogenesis, as can toluene, the environmental contaminant dioxin, and ionizing radiation.14,60,61
Viral infections during pregnancy have been shown to cause cleft lip/palate via mutations in PVRL1 and IRF6.62,63 Nutritional intake is also clearly linked to craniofacial development. Sonic hedgehog signaling, which is important in mediating parts of craniofacial development, is modulated by cholesterol. Therefore diets and medications that profoundly influence cholesterol levels can cause craniofacial malformations.64,65 The link between a diet deficient in folic acid and neural tube defects has been well documented and has led to the widespread supplemental addition of folic acid to many processed foods throughout the developed world.
Finally, intrauterine constraint can cause deformation of the craniofacial skeleton. Factors that have been associated with intrauterine constraint include breech presentation, persistent fetal lie, early pelvic engagement of the head, oligohydramnios, prima gravidity, multiple gestations, uterine malformations, amniotic bands, and defects in fetal neuromuscular development.66,67
Acquired Craniofacial Deformities
Several postnatal conditions predispose an infant to disrupted craniofacial growth. Hematologic diseases such as thalassemias, sickle cell anemia, congenital hemolytic icterus, and polycythemia vera are known to cause craniosynostosis. Hyperplasia of the bone marrow leads to bony overgrowth in the skull. This in turn can cause the calvarial sutures to fuse prematurely.68 Iatrogenic craniofacial anomalies can occur in patients who require ventricular shunts. If the shunt volumes in such patients are excessive, there is a lack of tension across the sutures, which produces an environment that mechanistically resembles the one seen in microcephaly. In these conditions there is an increased risk of secondary craniosynostosis.58 Etiologically, trauma and neoplasm can cause acquired craniofacial deformity, although they do so rarely.
Clinical Manifestations
Congenital Craniofacial Abnormalities
Craniofacial anomalies range from mild functionally asymptomatic defects (certain single-suture craniosynostoses) to severe anomalies that are not compatible with life (forms of holoprosencephaly). Typically, the pediatrician consults the craniofacial surgeon about a neonate who has an abnormally shaped head or unusual appearance. A fundamentally important point is to suspect the deformity to be part of a wider syndrome and to initiate a comprehensive examination in search of other abnormalities. However, the majority of cases of craniosynostosis are isolated with no associated anomalies.69 Descriptions of some of the more common craniofacial abnormalities follow.
Craniosynostosis
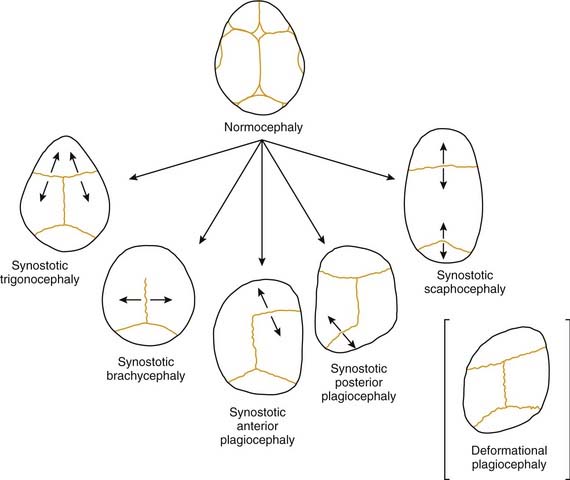
Craniosynostosis refers to premature closure of the involved bony suture. The consequence is that growth is inhibited in the direction perpendicular to the suture line (Fig. 185-3). Ensuing developmental growth occurs in a compensatory pattern that permits, to the extent possible, continued expansion of the intracranial content in unrestricted directions. It must be stressed that the compensatory growth that occurs is the normal biologic response to sutural constraint. The functional and aesthetic imbalance that occurs is, in part, a byproduct of normal physiology. The various types of craniosynostosis are as follows.
Scaphocephaly
Scaphocephaly is the most common of cranial synostotic conditions. The sagittal suture, or part of it, prematurely fuses (Fig. 185-4). This anomaly prevents calvarial growth in the transverse dimension. The compensatory growth occurs in the anteroposterior (AP) direction, leaving the patient with decreased biparietal and bitemporal dimensions. The resulting head shape is long and narrow. There are several variations (“saddle deformity,” “football deformity”), depending on the extent of premature sagittal fusion and also on whether the metopic suture is involved.
Trigonocephaly
Trigonocephaly refers to a triangular forehead (as viewed from above) and is associated with metopic suture synostosis. There is a decrease in bifrontal diameter. It is a mild form of craniosynostosis with rare functional consequences (Fig. 185-5). The suture area is thickened, resulting in an anterior midsagittal ridge. This appearance has been described as resembling the keel of a ship. There can be associated hypotelorism. In mild cases there may be only suture ridging without significant forehead deformity.
Plagiocephaly
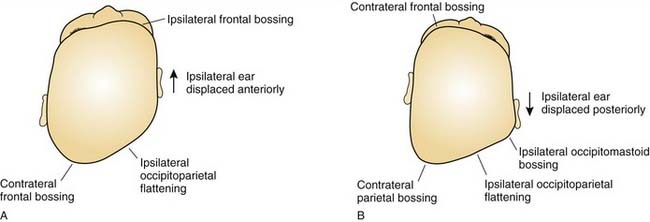
In plagiocephaly, the head is asymmetric, flattened in one or more regions. It can be either synostotic or deformational and can be anterior or posterior. Synostotic plagiocephaly refers to either unilateral coronal (Fig. 185-6) or lambdoid synostosis. In coronal synostosis the forehead of the affected side is flat whereas the supraorbital rim is elevated and displaced posteriorly. The rim lies at or posterior to the plane of the cornea although it should normally be about 1 cm anterior to the cornea. The nasal root can be deviated to the side of the defect. The contralateral forehead is bossed, and the supraorbital rim can be pushed down. The ipsilateral auricle is anteriorly displaced but the occiput is minimally affected.
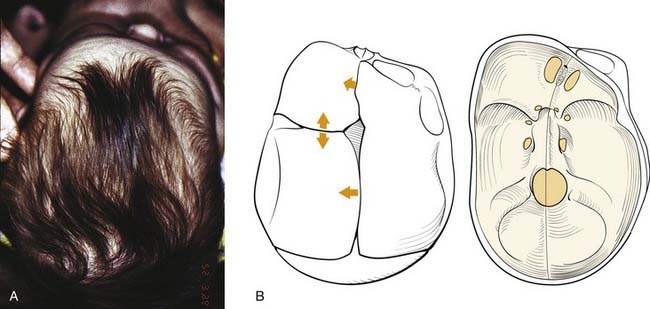
Deformational plagiocephaly is far more common and can be influenced by the previously noted intrauterine constraint factors, hypotonia, and prematurity. It is further compounded by sleeping in the supine positioning, with a predilection of the infant to sleep on one side owing to a prominent occiput and poor head control. This phenomenon has increased since the American Academy of Pediatrics, in an effort to reduce sudden infant death syndrome (SIDS), issued its recommendations to place sleeping infants in the supine position.58,69 Additionally, persistent fetal lie, vertebral anomalies, muscular disorders, ocular abnormalities as well as assorted other conditions, can cause torticollis, further aggravating sleep position problems. The sutures in deformational plagiocephaly remain open so there is no ridging. The differences in head shape between synostotic and deformational plagiocephaly are illustrated in Figure 185-7.
Brachycephaly
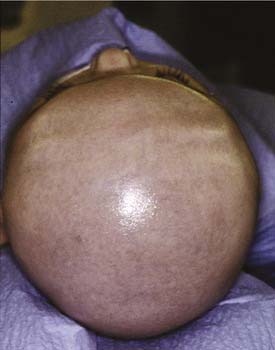
Brachycephaly is the term used for bilateral coronal synostosis (Fig. 185-8). It is often accompanied by turricephaly (tall head) or acrocephaly (pointed head). As a result of this sutural synostosis, the skull is unable to expand in the AP dimension. In order to accommodate the expanding intracranial content, the skull grows in the lateral and superior dimensions. The neonate’s cranium, therefore, has a smaller AP distance and greater lateral dimensions. The forehead comes right off the nose with a low hairline, and the glabellar depression is absent.
Acrocephaly
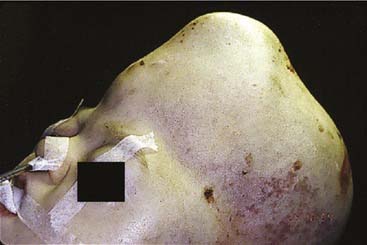
Acrocephaly (also known as oxycephaly and turricephaly) consists of a skull that is high and conical (Fig. 185-9). This abnormality can develop as a result of progressive postnatal involvement of both the sagittal and coronal sutures. Compensatory growth occurs through the anterior fontanel.
Kleeblattschädel
Kleeblattschädel is also known as cloverleaf skull (Fig. 185-10). It results from synostosis of all the sutures previously discussed except for the metopic suture. The bone overlying the sutures is very thick but the intervening bone is thin. The expanding intracranial content tends to cause a ballooning of the thin bone between the sutures. This compensatory growth, in addition to expansion through the remaining open sutures, gives the characteristic lobular appearance of a cloverleaf. The skull growth falls drastically short of the needs of intracranial expansion, giving rise to neurologic impairment, increased intracranial pressure (ICP), and, often, secondary hydrocephalus. These patients exhibit exorbitism and therefore are at risk for globe exposure, and their optic nerves can also be at risk for damage. This anomaly requires prompt surgical attention to release the binding skull.
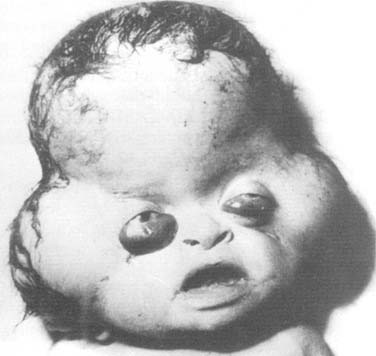
Encephalocele
An encephalocele is a protrusion of intracranial content through a defect in the craniofacial skeleton (Fig. 185-11). It is classified according to its position in the skull. If the protruding content contains meninges, it is referred to as a meningocele. If it contains meninges and brain, it is called a meningoencephalocele, and if the protrusion additionally contains ventricle, it is known as a meningoencephalocystocele.70
Syndromes
Apert’s Syndrome
Apert’s syndrome resembles Crouzon’s syndrome in several ways. Most cases of Apert’s syndrome arise sporadically through new mutations, although some familial cases with autosomal dominant transmission have been reported.71 The disorder is characterized by brachycephaly (with resultant turricephaly) and midfacial hypoplasia (with associated orbital and dental issues). Unlike patients affected with Crouzon’s syndrome, patients with Apert’s syndrome have symmetric syndactyly of the hands and feet as well as other axial skeletal abnormalities. The palate frequently has a high arch and may be cleft. The defects in Apert’s syndrome are present at birth, and intelligence is often decreased.
Velocardiofacial Syndrome
Velocardiofacial syndrome is autosomal dominant with variable expressivity and penetrance and may be the most common syndrome after Down syndrome. There are numerous associated anomalies aside from the well-documented craniofacial, cardiac, and vascular malformations. The craniofacial anomaly broadly consists of a more open angle of flexion in the basicranium. This has an impact on the appearance of the midface and lower face (mandible). Clefting of the secondary palate can be overt or can take the form of an occult cleft that is apparent only on nasopharyngoscopy.72 Velocardiofacial syndrome can easily be overlooked because affected patients are often not obviously abnormal in appearance.
Facio-Oculo-Auriculo-Vertebral Spectrum
Facio-oculo-auriculo-vertebral spectrum (FOAVS) refers to a group of anomalies that originate from unilateral defects in the first and second branchial arches. Most cases are sporadic, although familial cases have been reported. There is evidence that this syndrome results from a unilateral hemorrhagic event involving the stapedial artery during early craniofacial development.73 FOAVS (also known as hemifacial microsomia), which includes Goldenhar’s syndrome, can be quite diverse in terms of severity and scope. It is a rare disorder (1/5600 live births) that is present at birth. It involves the unilateral soft and skeletal tissues of the face and external and middle ear. It can manifest as temporal, zygomatic (malar), maxillary, and mandibular hypoplasia, hypoplastic facial musculature, and cleft lip and palate. It involves a wide variety of external and middle ear deformities related to structures that derive from the first and second branchial arch. Atresia of the external auditory canals and preauricular skin tags can all exist in FOAVS. The eye can be involved, with the occurrence of colobomas, epibulbar choristomas, blepharophimosis, and strabismus. Vertebral anomalies include fusion of vertebrae, spina bifida, and other more specific abnormalities. A variety of other, non-craniofacial abnormalities are associated with FOAVS.
Trauma
The incidence of facial fractures in the pediatric population has been estimated to range from 1% to 14.7%, and head injuries are frequently associated with them. The group least likely to suffer from facial trauma is children younger than 5 years. The preponderance of injuries in children has been reported to occur in boys, though some studies have found no gender predilection. According to some reports motor vehicle accidents are responsible for these injuries in up to 80% of cases. However, in the last 20 years, with the wider use of seatbelts and federally approved child-safe car seats, the incidence has significantly declined. Injuries that do result in pediatric maxillofacial trauma are usually quite severe and therefore are frequently accompanied by other systemic injuries.74,75
Neoplasm
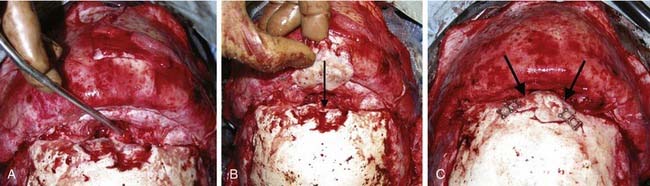
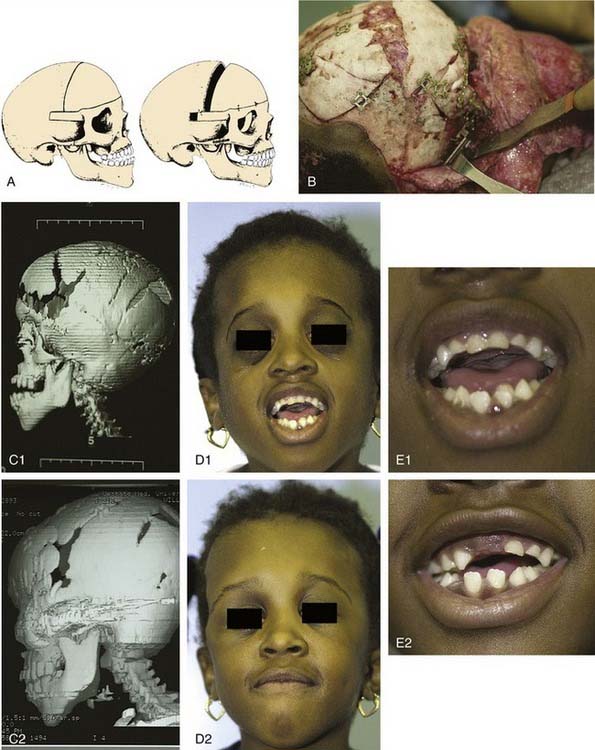
Craniofacial or skull base tumors are fortunately rare occurrences in the pediatric population. Head and neck tumors account for about 5% of all childhood cancers, and those involving the skull base are a small proportion of that figure. Of the solid nonhematologic lesions, brain tumors are the most common, followed by neuroblastoma and rhabdomyosarcoma, non-rhabdomyosarcoma sarcomas, salivary gland tumors, and teratomas. Benign tumors in the craniofacial region are also rare; they include fibrous dysplasia (Fig. 185-12), angiofibromas, granulomas, pituitary adenomas, and other unusual lesions.
Patient Evaluation
Imaging
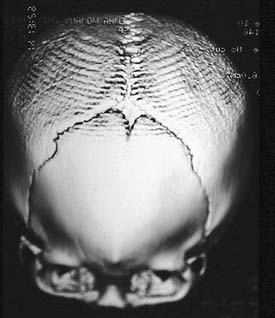
Plain radiographs can be useful in certain circumstances. The absence of normal sutural lucency may indicate synostosis. In plagiocephaly and brachycephaly, premature fusion of the frontoethmoid and frontosphenoid sutures causes elevation of the lesser wing of the sphenoid, producing the harlequin mask appearance (either unilateral or bilateral) on a plain AP film of the skull (Fig. 185-13). The hammered-copper or thumb-printing appearance of the inner table of the skull on cranial radiographs is associated with increased ICP and craniosynostosis; although its presence is easily detected, its clinical significance has been questioned.76 Lateral and AP cephalometric radiographs continue to be useful, especially for evaluation of the progress of craniofacial growth.
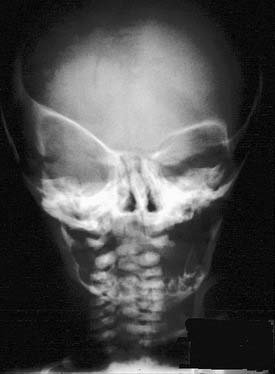
Figure 185-13. Skull film demonstrating the “harlequin mask” appearance seen in patients with unilateral coronal suture synostosis.
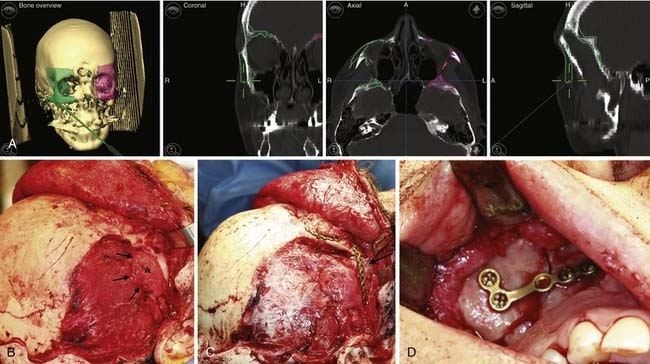
However, for most defects computed tomography is the ideal modality for evaluation. Given its ubiquity and declining costs it has largely replaced plain radiography. Additionally, the software for three-dimensional reconstruction is widely accessible and provides detail of the bony anatomy that is unsurpassed (Fig. 185-14). Various cephalometric and volumetric analyses can be easily achieved. If necessary, a stereolithographic model based on three-dimensional studies can be built that provides an accurate template of the patient’s craniofacial skeleton. Sophisticated software is also available to help in planning reconstruction (Fig. 185-15). Planning software is particularly helpful when one is preparing to reconstruct unilateral or some asymmetric defects. Although valuable information about the central nervous system can be obtained with computed tomography, magnetic resonance imaging is usually the modality of choice for evaluating the brain.
Indications for and Timing of Intervention
The complexity arises because the consequences of not intervening or of the intervention itself are not always fully understood. Significant fronto-orbital retrusion is at the more severe end of the spectrum, as it may portend serious functional, neurologic, or ophthalmic problems. The presence of more than one synostosis is often associated with neurocognitive compromise. Anomalies of the maxilla and mandible not only affect appearance but also can impact the airway, dentition, and speech and therefore frequently require therapeutic intervention. Hypertelorism and other orbital dystopias can manifest as significant visual issues, in which case surgical reconstruction is necessary. Nonsyndromic single-suture craniosynostosis (SSC) is particularly difficult to classify because the association between the anomalies and functional sequelae is controversial. The incidence of increased ICP and neurocognitive deficiencies in nonsyndromic single-suture craniosynostosis is still debated, and further investigation of this issue is warranted.77–79
When the neonate has the mildest of craniosynostoses, such as some types of single-suture craniosynostosis or partial synostosis, positional plagiocephaly, mild FOAVS, and other mild craniofacial anomalies, the problem can be classified as aesthetic in nature. In such cases the craniofacial team and the involved family members must weigh the severity of aesthetic deformity against the risks of surgical intervention. The temptation to develop a strict algorithm based on the defect alone has to be resisted, as demonstrated by metopic suture synostosis. This deformity manifests with a wide range of severity in fronto-orbital malposition and hypotelorism, and the metopic suture closes earlier than previously thought.80 The implication that prior standards used to guide the surgeon in deciding when to intervene surgically are not quite as relevant. In the case of positional plagiocephaly, the treatment does tend to be more clear-cut. Changing sleep position, treating causes of torticollis, and cranial molding orthotics can all be used quite effectively when appropriately applied.
Surgical Techniques
Congenital Craniofacial Abnormalities
Coronal Incision
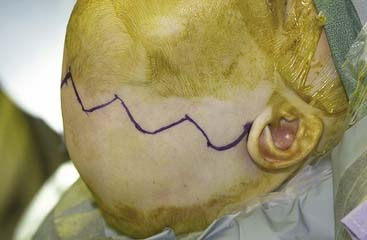
The coronal incision can extend pre- or postauricularly. When extended into the postauricular area, the incision is hidden, and adequate exposure of the cranium and the upper and part of the middle thirds of the face can still be obtained. The incision is designed as a wavy line that extends across the vertex of the head (Fig. 185-16).13,81 The overall incision can be rotated more anteriorly to allow more anterior/inferior retraction of the flap. Some surgeons sharply zigzag the incision so as to make multiple V-Y closures possible if the advancement is so significant as to preclude normal closure of the coronal flap. The flap can then be elevated in a subperiosteal or subgaleal plane over the calvarium. The dissection is continued to the deep temporalis fascia elevating the temporal-parietal fascia. Dissection in this plane enables the frontal branch of the facial nerve to be elevated in the flap. If access to the zygomatic arch is required, the superficial layer of the deep temporalis fascia is elevated starting at the temporal line of fusion (see Fig. 185-15). This maneuver has the added advantage of adding an extra layer of tissue in between the dissection and the facial nerve branch. In some types of cranial vault remodeling, it is necessary to elevate the temporalis muscle as well, but this step is performed independent of the scalp elevation.
Sagittal Synostosis
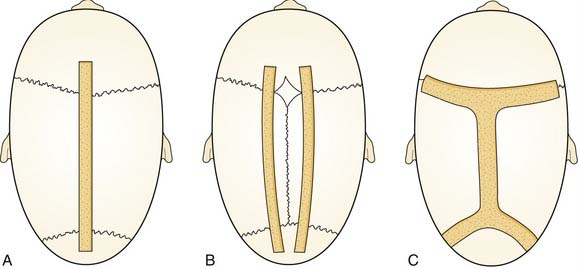
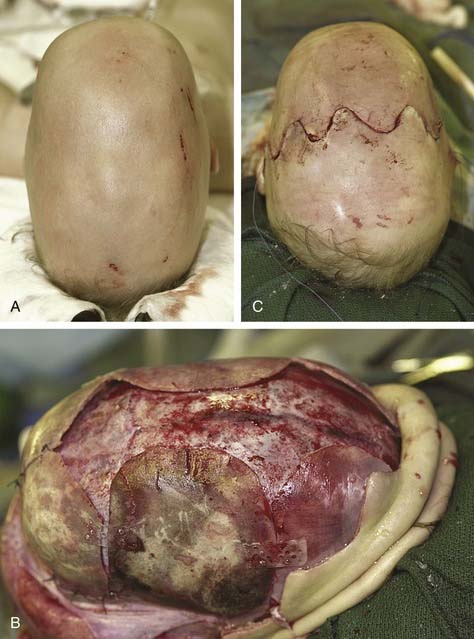
Sagittal synostosis requires that the skull be allowed to expand in the transverse dimension and reduced in the AP dimension. There is a spectrum of procedures for scaphocephalic correction. Mild cases treated before 3 months of age may be managed with strip craniectomy alone (Fig. 185-17). A strip craniectomy is performed by removing sagittal or parasagittal sections of bone. Severe cases and patients treated after 9 to 12 months may require total cranial vault remodeling. In these cases coronal and lambdoid osteotomies can be made, and the parietal bones out-fractured. In severe cases the frontal bone can be significantly bossed and often has to be reshaped or transposed to improve the contour of the forehead. This requires a coronal incision (Fig. 185-18).
Fronto-Orbital Advancement
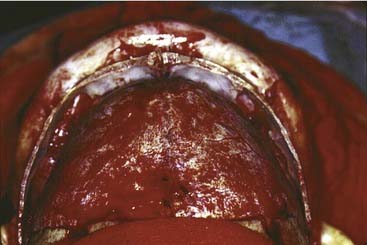
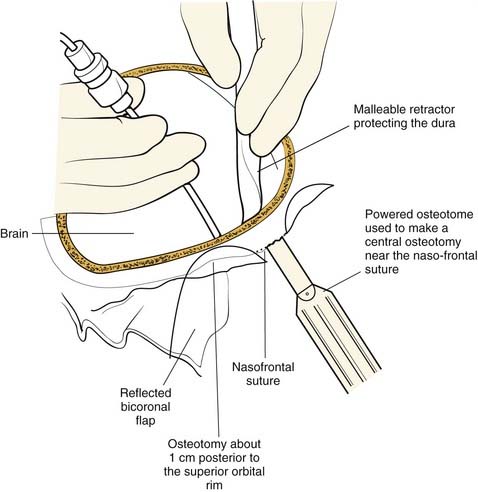
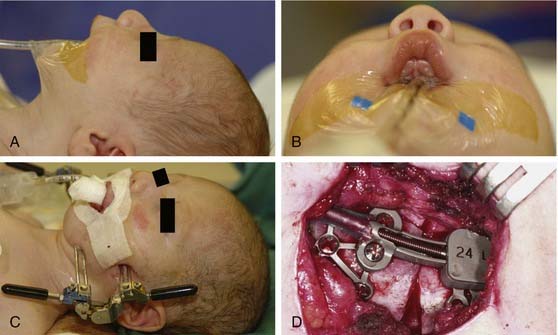
Once the dissection is complete, intended osteotomy sites for both the supraorbital bar and the bifrontal craniotomy are marked. A side-cutting power craniotome is typically used once strategic bur holes have been drilled. The bifrontal craniotomy (Fig. 185-19) is done initially, with care taken to leave a transverse bar of bone above the supraorbital rims that is between 10 and 15 mm in width. The dura is freed from the anterior aspect of the anterior cranial fossa but left adherent over the cribriform plate. The supraorbital bar or bandeau is then marked for osteotomy. The superior osteotomy for the supraorbital bar was created by the bifrontal craniotomy. If a strip of bone was left over, the sagittal sinus it is cut from the bandeau. Laterally, the superior bandeau osteotomy is extended into the squamous portion of the temporal bone. Inferiorly, the central transverse osteotomy is made at or just above the frontonasal suture (Fig. 185-20). This cut extends directly to the anterior cranial fossa at a point anterior to the crista galli. This location preserves olfaction as well as the barrier between the brain and the nasal cavity. The osteotomy is then carried into the medial orbit just above the lacrimal fossa. The osteotomy turns superiorly 5 to 10 mm inside the orbital rim, traverses the orbital roof around 10 mm posterior to the superior orbital rim, and extends down the lateral orbital wall to the level of the zygomaticofrontal suture. The lateral orbital rim is cut at the suture, and that cut extends posteriorly into the greater wing of the sphenoid and the squamous portion of the temporal bone parallel to and 10 to 15 mm inferior to the previous cut into this region. This site corresponds intracranially with the junction of the anterior and middle cranial fossae near the pterion. Enough length of the temporal portion of the bandeau will allow a tongue-and-groove action at this lateral point in order to increase the bony contact once the advancement has been completed. This maneuver facilitates bony stability. Removal of the bandeau is frequently hindered by incomplete osteotomy at the trifurcation (of the anterior extent) of the middle cranial fossa, the lateral orbital wall, and the greater wing of the sphenoid. The anterior tip of the temporal lobe must be protected while this cut is completed.
Once the bony segments are removed, they are reshaped on a separate table. The supraorbital bar can be reshaped to the extent required by the deformity (Fig. 185-21). Bone is removed as needed in thickened areas to facilitate bone bending. In unilateral synostosis (plagiocephaly) the central angle of the forehead is mildly affected, and only small alterations are necessary to increase the central angle to 150 to 170 degrees. Although only one side is affected in plagiocephaly, minor changes are required on the unaffected side because of compensatory frontal bossing. More drastic measures might be required for trigonocephaly, in which the central angle of the forehead can be as acute as 90 degrees. It is sometimes necessary to split the bandeau in the midline and add a bone graft in order to widen the central angle (Fig. 185-22). The components are then fixed to one another with absorbable plates, sutures, or both. Just above the lateral orbital rims, the angle between the supraorbital rim and the temporal fossa is adjusted to between 100 and 110 degrees. The orbital bar is then replaced in an advanced position.
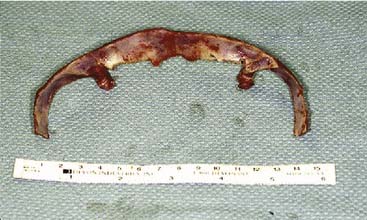
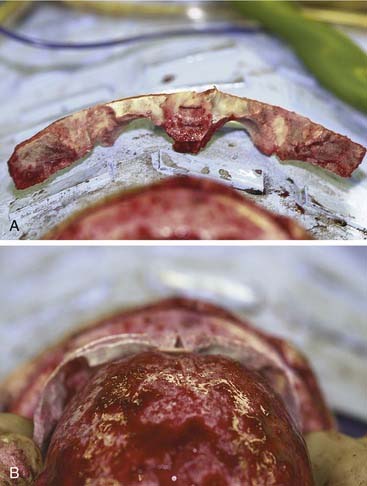
Figure 185-22. A, The bandeau can be split and a graft used to stabilize the new supraorbital construct. B, The bandeau is in place.
The extent of advancement obviously depends on the nature of the anomaly. In trigonocephaly and plagiocephaly, the orbital advancement is predominantly lateral, with the medial portion advancing little if any. In brachycephaly the entire bandeau is usually advanced, but the movement is still greater laterally than medially (Fig. 185-23). Maintaining contact between the supraorbital bar and the root of the nose is also important from the perspective of frontal sinus development, which later enhances central forehead projection.82 It is important to establish symmetry with the advancement. Both rims should be about 1 cm anterior to the cornea. In severe cases the bandeau can be advanced as much as 2 cm, but bone grafting may be necessary. The supraorbital bar is fixed to the skull with absorbable plates, sutures, or both at the nasion and laterally (Fig. 185-24).
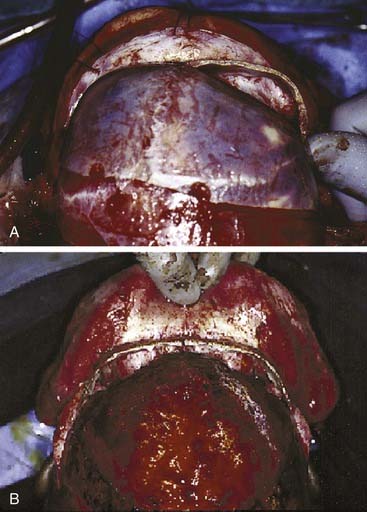
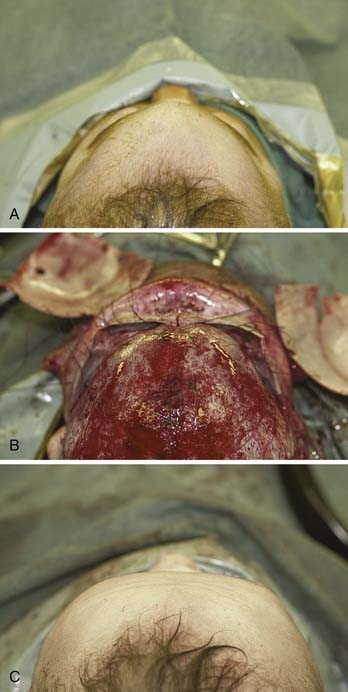
Figure 185-24. Trigonocephalic repair. A, Preoperative view. B. Intraoperative view. C, Postoperative view.
Resorbable plate technology represents a breakthrough in this treatment.83 It offers dependable rigid fixation without the use of metallic plates.84 Resorbable plates are less likely to interfere with craniofacial growth and development the way that titanium plates can, and they also rarely require removal.85 The problem of intracranial plate migration with growth is avoided as well—typically the plates lose a large proportion of their strength within the first 6 months and are resorbed in less than 18 months. The calvarial bone flaps are then reshaped to provide normal contour to the head. Sometimes it is necessary to further osteotomize the flaps. The various components can be replaced in different positions to give the best contour. The bones are then fixed to the surrounding skull with absorbable plates or sutures. Maintaining large pieces of bone that are bent to the desired conformation, rather than creating small segments of bone with many osteotomies, provides for a more stable construct with less fixation than would be required if the bones were cut into smaller pieces.
Because of the advancement, there will be defects in the bone at various places. In patients younger than 1 year, bony defects are generally not of major concern because of bone regeneration. However, in older children and adults, large defects must be addressed. The orbital roofs represent a clear example. If there is a persistent communication between the orbit and the brain, pulsatile ophthalmopathy can develop. Therefore, these defects must be repaired with bone grafts (Fig. 185-25). Calvarial defects in the child or adult can be repaired with bone grafts or other bone replacement materials.
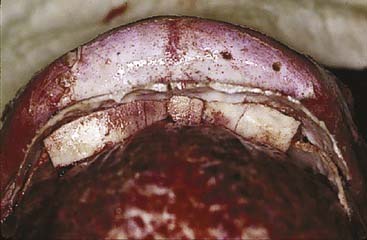
Monobloc Advancement
Older children (>2-4 years) who present with fronto-orbital retrusion in conjunction with midface retrusion are potential candidates for a one-stage procedure. The (frontofacial) monobloc advancement originally described by Ortiz-Monasterio and colleagues86 combines a fronto-orbital advancement with a Le Fort III level facial advancement (Fig. 185-26). The procedure is performed through the coronal incision. The craniotomy is similar to the one performed in a fronto-orbital advancement. The zygomatic arches are cut. The frontal bar is included with the Le Fort III segment. Circumferential orbital bony cuts are made with use of both extracranial and intracranial approaches. The lateral and inferior orbital walls are osteotomized starting in the inferior orbital fissure. The medial wall is cut just posterior to the posterior lacrimal crest. The orbital roof osteotomy is made intracranially. The nasal septum is cut via the frontoethmoid osteotomy. Finally, through the infratemporal fossa or the mouth, the maxilla is separated from the pterygomaxillary junction. The monobloc segment is then advanced and rigidly fixed with use of absorbable or titanium plates.
In some craniofacial deformities, such as those commonly seen in patients with Apert’s syndrome, hypertelorism is present in association with maxillary deformities. Facial bipartition is a technique used to simultaneously correct hypertelorism and maxillary arch abnormalities (Fig. 185-27). Bone grafts are used to help maintain the advancement. Pericranial flaps and tissue glues are also used to create a seal between the anterior cranial fossa and the nasal cavities.
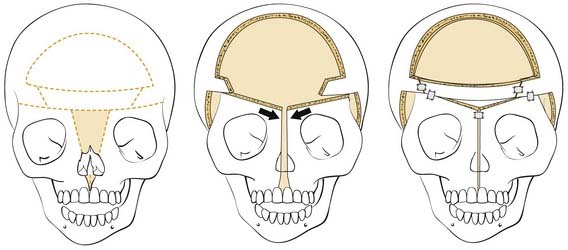
Although combining the two corrections into one procedure, has an obvious advantage, it is also associated with a higher rate of infection, presumably because of an incomplete seal of the central nervous system from the nasal cavities. It is for this reason that patients must be carefully selected for such a procedure. In situations in which the monobloc advancement is clearly the procedure of choice, distraction (in place of craniofacial advancement during the operation) may offer a safer way to achieve the desired results (see Fig. 185-26). The approach and the osteotomies are no different from the en bloc advancement just described.
Distraction Osteogenesis
Distraction osteogenesis has been employed in the long bones for just under a century now. It was first applied to the craniofacial skeleton more than 80 years ago87 but has come into widespread clinical use only in the last 25 years.88 Distraction osteogenesis refers to creation of a controlled fracture in a bony structure followed by separation of the bony segments in a controlled and gradual manner. This process induces the formation of new bone in between the distracted segments. The advantages of using distraction osteogenesis in the craniofacial skeleton are numerous. It allows for skeletal lengthening and advancement in three dimensions. The process is a gradual one that allows the skin–soft tissue envelope to adapt to and accommodate skeletal movement.89 It is also safer than many of the traditional skeletal techniques. Distraction osteogenesis is less involved and requires less operative time (generating less blood loss) than the techniques it has replaced. As a result it is an attractive treatment alternative for children and infants with some craniofacial anomalies.
Distraction osteogenesis can be carried out with internal or external devices (Fig. 185-28). Internal devices have the advantage of not requiring big and bulky hardware protruding from the face and can, at times, be applied intraorally. The extent of control over the vectors of distraction is limited with internal devices. The external devices allow for three-dimensional distraction and better control of the vectors of distraction (see Fig. 185-28). The disadvantage lies in their bulkiness. Pin migration scars can be significant as well. However, this is a rapidly evolving technique, and new devices are constantly being introduced. As the internal devices become more sophisticated, their roles will continue to expand.
Trauma
The goal of treatment is the restoration of function and three-dimensional architecture. For severe and displaced fractures, internal fixation is often necessary.90 The window of time in which fractures can be repaired is shorter in children as a result of the rapidity of their healing time. Many surgeons advocate using resorbable plates and screws for internal bony fixation, although others prefer using small titanium plates, arguing that complications from permanent (mini)plates are minimal.75,85,91–93
Neoplasm
Approaches to the skull base for tumor extirpation also require an intimate understanding of craniofacial anatomy because normal anatomy is often effaced. Several craniofacial approaches have been described in the pediatric population that can be used to gain access to tumors of the anterior and middle cranial fossae.94 Historically, tumors of the anterior skull base were approached with a frontal craniotomy combined with a facial incision such as a lateral rhinotomy with or without a Weber-Ferguson facial incision. However, tumors of the anterior cranial fossa can be adequately exposed with a subcranial (subfrontal) approach without facial incisions (see Fig. 185-12). As previously mentioned, this latter exposure involves removal of the frontonasal complex. The extent of frontal and orbital rim bone removal is determined by the size, location, and nature of the lesion. In an extended subcranial approach, large sections of the bony orbit can be removed.95
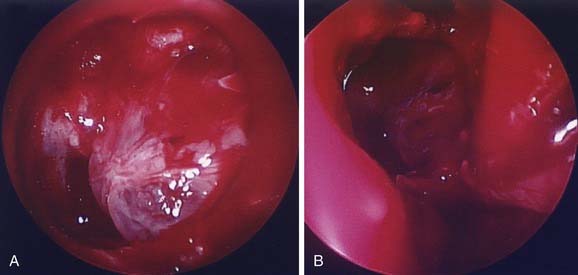
The middle cranial fossa can be approached through a variety of techniques. The endoscopic transnasal-transsphenoidal approach can be effective in resection of midline tumors associated with the sella turcica, sphenoid cavity, and posterior nasal cavity. Figure 185-11 demonstrates repair of a sphenoid sinus encephalocele via this endoscopic approach. Even malignant or locally destructive lesions are occasionally accessible via this approach. Le Fort I, transpalatal, and midfacial degloving techniques can be used to gain limited access to lesions of the skull base.96 More aggressive lesions that are off the midline (such as petroclival lesions) can be approached via the infratemporal fossa (or a modified version) or other transbasal approaches.97 The cranio-orbito-zygomatic and transcondylar approaches provide excellent exposure of the infratemporal fossa. A frontotemporal craniotomy extends the exposure to the middle cranial fossa and the petrous internal carotid artery as well as other petroclival structures. When a lesion involves the middle cranial fossa, the infratemporal fossa, and the nasopharynx, a wider exposure is frequently necessary. The facial translocation procedure provides very wide exposure to lesions in this area and is well suited for malignancies, for which a margin of healthy tissue must be removed as well. It involves extended facial incisions as well as transection of facial nerve branches that are later reanastomosed. A maxillary-zygomatic bony segment is removed in order to gain access to the necessary extracranial lesion. A subtemporal craniotomy then provides intracranial exposure. In reconstruction of the bony framework, the same principles must be followed as for any other craniofacial procedure.
Biomaterials in Skeletal or Cartilaginous Defect Repair
The third factor is the material itself. On a general level a material that is nontoxic, noncarcinogenic, and nonimmunogenic to biologic systems is said to be biocompatible. Inflammation, which in previous discussions of the subject was considered a negative quality, can actually be advantageous as long as it is resolvable. The concept of biocompatibility, however, is more complex than even this definition implies. As previously discussed, a material that is biocompatible in one area may not be so in another. The use of expanded polytetrafluorethylene (ePTFE) is an excellent example. ePTFE has been successfully used for many years in vascular surgery,98 but the use of Proplast, a closely related material, in reconstruction of the temporo-mandibular joint produced disastrous complications.99 In some applications and locations, a material that causes a local but temporary reaction is desirable, whereas in others, a totally inert material is required. On the basis of this analysis, it is more precise to include application and local factors in the definition of biocompatibility.
Furthermore, it is most useful to think of biocompatibility as a broad spectrum with autogenous grafts on one end and alloplastic (synthetic) materials on the other. Allografts (homografts) and xenografts fall in the middle of that spectrum. Autogenous materials represent the “gold standard,” but this category is not homogeneous. Cancellous bone grafts are quite different from cortical grafts. Cortical grafts can be further subclassified as endochondral or intramembraneous bone, with functional differences between the two.100–102 Although there is a vast and successful experience with autogenous grafting,103 this method still has its limitations. Donor site morbidity should not be taken lightly. In a prospective study evaluating iliac crest donor site pain, Sasso and colleagues104 reported that a significant proportion of patients still had donor site pain 2 years after surgery. In addition, incomplete resorption and remodeling of cortical bone occurs in autogenous grafts, and unpredictable remodeling can be an issue as well.105,106
Polymers include a diverse set of materials some of which are nonresorbable and some resorbable. They have a successful track record for use in facial augmentation.107 Methacrylates are nonresorbable and have been widely used for calvarial reconstruction since World War II. High-density porous polyethylene is a straight-chain aliphatic hydrocarbon that is inert and also nonresorbable. It has been used successfully in some craniofacial applications. The primary problem with these materials is associated with their risk for infection or extrusion during the life of the recipient. Implants fabricated from polylactic acid and polyglycolic acid are resorbable and have sufficient strength for some load-sharing applications. This material has been promising as a means of bony fixation without the use of a permanent material.
Ceramics include calcium phosphates such as hydroxyapatite and bioglass. Hydroxyapatite, which has the advantage of being closely related to the carbonated apatite found in bone, has been shown to be reasonably effective in animal models and has proven clinical efficacy in some applications.108,109 However, in the sintered and cement forms, it is not entirely accessible to the remodeling activities of osteoclasts. Its osteoconductivity and osteoinductivity are often less than ideal.106,110,111 Bioglasses are very osteoconductive, but some of their properties, such as stiffness and elasticity, render them inappropriate for many craniofacial applications.
Bauer TW, Muschler GF. Bone graft materials: an overview of the basic science. Clin Orthop Relat Res. 2000:10-27.
Cohen MMJr. Etiopathogenesis of craniosynostosis. Neurosurg Clin North Am. 1991;2:507-513.
Cohen M. Sutural pathology. In: Cohen M, MacLean R, editors. Craniosynostosis: Diagnosis, Evaluation, and Management. New York: Oxford University Press, 2000.
Hunt JA, Hobar PC. Common craniofacial anomalies: facial clefts and encephaloceles. Plast Reconstr Surg. 2003;112:606-615.
Imola MJ, Tatum SA. Craniofacial distraction osteogenesis. Facial Plast Surg Clin North Am. 2002;10:287-301.
McCarthy JG, Schreiber J, Karp N, et al. Lengthening the human mandible by gradual distraction. Plast Reconstr Surg. 1992;89(1):1-8.
Ortiz-Monasterio F, del Campo AF, Carrillo A. Advancement of the orbits and the midface in one piece, combined with frontal repositioning, for the correction of Crouzon’s deformities. Plast Reconstr Surg. 1978;61:507-516.
Obwegeser JA. Maxillary and midface deformities: characteristics and treatment strategies. Clin Plast Surg. 2007;34:519-533.
Posnick J. Craniofacial and Maxillofacial Surgery in Children and Young Adults. Philadelphia: W.B. Saunders; 2000.
Slater BJ, et al. Cranial sutures: a brief review. Plast Reconstr Surg. 2008;121:170e-178e.
Tessier P, Kawamoto H, Matthews D, et al. Autogenous bone grafts and bone substitutes—tools and techniques: I: a 20,000-case experience in maxillofacial and craniofacial surgery. Plast Reconstr Surg. 2005;116(Suppl):6S-24S.
1. Whitaker LA, Pashayan H, Reichman J. A proposed new classification of craniofacial anomalies. Cleft Palate J. 1981;18:161-176.
2. Larsen WJ, Sherman LS, Potter SS, et al. Human Embryology. New York: Churchill Livingston; 1993.
3. Enlow DH. The facial growth process. In: Enlow DH, editor. Facial Growth. Philadelphia: WB Saunders, 1990.
4. Marschall MA, Chidyllo SA, Figueroa AA, et al. Long-term effects of rigid fixation on the growing craniomaxillofacial skeleton. J Craniofac Surg. 1991;2:63-68.
5. Posnick JC, Yaremchuk MJ. The effects of nonresorbable internal fixation devices placed on and within a child’s cranial vault: brain function, morbidity, and growth restriction. Plast Reconstr Surg. 1995;96:966-968.
6. Yaremchuk MJ, Fiala TG, Barker F, et al. The effects of rigid fixation on craniofacial growth of rhesus monkeys. Plast Reconstr Surg. 1994;93:1-10.
7. Whitaker LA. Biological boundaries: a concept in facial skeletal restructuring. Clin Plast Surg. 1989;16:1-10.
8. Moss ML. The pathogenesis of premature cranial synostosis in man. Acta Anat (Basel). 1959;37:351-370.
9. Virchow R. Uber den cretinismus, namentlich in Franken, und uber pathologische schadelformen. Verh Phys Med Gesellsch Wurzburg. 1981:2.
10. Ozaki W, Buchman SR, Muraszko KM, et al. Investigation of the influences of biomechanical force on the ultrastructure of human sagittal craniosynostosis. Plast Reconstr Surg. 1998;102:1385-1394.
11. Opperman LA, Passarelli RW, Morgan EP, et al. Cranial sutures require tissue interactions with dura mater to resist osseous obliteration in vitro. J Bone Miner Res. 1995;10(12):1978-1987.
12. Kim HJ, Rice DP, Kettunen PJ, et al. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development. 1998;125(7):1241-1251.
13. Bradley JP, Han VK, Roth DA, et al. Increased IGF-I and IGF-II mRNA and IGF-I peptide in fusing rat cranial sutures suggest evidence for a paracrine role of insulin-like growth factors in suture fusion. Plast Reconstr Surg. 1999;104:129-138.
14. Rice DP. Craniofacial anomalies: from development to molecular pathogenesis. Curr Mol Med. 2005;5:699-722.
15. Cuervo R, Valencia C, Chandraratna RA, et al. Programmed cell death is required for palate shelf fusion and is regulated by retinoic acid. Dev Biol. 2002;245:145-156.
16. Martinez-Alvarez C, Tudela C, Pérez-Miguelsanz J, et al. Medial edge epithelial cell fate during palatal fusion. Dev Biol. 2000;220:343-357.
17. Shuler CF, Guo Y, Majumder A, et al. Molecular and morphologic changes during the epithelial-mesenchymal transformation of palatal shelf medial edge epithelium in vitro. Int J Dev Biol. 1991;35:463-472.
18. Britto JA. Advances in the molecular pathogenesis of craniofacial conditions. Oral Maxillofac Surg Clin North Am. 2004;16:567-586.
19. Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275-281.
20. Wilkie AO, Slaney SF, Oldridge M, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165-172.
21. Park WJ, Theda C, Maestri NE, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995;57:321-328.
22. Moloney DM, Slaney SF, Oldridge M, et al. Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet. 1996;13:48-53.
23. Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nat Genet. 1996;13:492-494.
24. Jabs EW, Müller U, Li X, et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell. 1993;75:443-450.
25. Mundlos S, Otto F, Mundlos C, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773-779.
26. Twigg SR, Kan R, Babbs C, et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci U S A. 2004;101:8652-8657.
27. Steinberger D, Müller U, Jünger TH, et al. Mutation of FGFR2 (cys278phe) in craniolacunia and pansynostosis. J Med Genet. 1999;36:499-500.
28. Jabs EW, Li X, Scott AF, et al. Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat Genet. 1994;8:275-279.
29. Reardon W, Winter RM, Rutland P, et al. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 1994;8:98-103.
30. Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet. 1995;11:462-464.
31. Vortkamp A, Gessler M, Grzeschik KH. GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature. 1991;352:539-540.
32. Muenke M, Gripp KW, McDonald-McGinn DM, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet. 1997;60:555-564.
33. Reardon W, Wilkes D, Rutland P, et al. Craniosynostosis associated with FGFR3 pro250arg mutation results in a range of clinical presentations including unisutural sporadic craniosynostosis. J Med Genet. 1997;34:632-636.
34. Wuyts W, Cleiren E, Homfray T, et al. The ALX4 homeobox gene is mutated in patients with ossification defects of the skull (foramina parietalia permagna, OMIM 168500). J Med Genet. 2000;37:916-920.
35. Wilkie AO, Tang Z, Elanko N, et al. Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat Genet. 2000;24:387-390.
36. Mavrogiannis LA, Antonopoulou I, Baxová A, et al. Haploinsufficiency of the human homeobox gene ALX4 causes skull ossification defects. Nat Genet. 2001;27:17-18.
37. Garcia-Miñaur S, Mavrogiannis LA, Rannan-Eliya SV, et al. Parietal foramina with cleidocranial dysplasia is caused by mutation in MSX2. Eur J Hum Genet. 2003;11:892-895.
38. Rutland P, Pulleyn LJ, Reardon W, et al. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nat Genet. 1995;9:173-176.
39. Muenke M, Schell U, Hehr A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet. 1994;8:269-274.
40. Lajeunie E, Ma HW, Bonaventure J, et al. FGFR2 mutations in Pfeiffer syndrome. Nat Genet. 1995;9:108.
41. Howard TD, Paznekas WA, Green ED, et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36-41.
42. el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, et al. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15:42-46.
43. Sood S, Eldadah ZA, Krause WL, et al. Mutation in fibrillin-1 and the Marfanoid-craniosynostosis (Shprintzen-Goldberg) syndrome. Nat Genet. 1996;12:209-211.
44. Tavormina PL, Shiang R, Thompson LM, et al. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet. 1995;9:321-328.
45. Trokovic N, Trokovic R, Mai P, et al. FGFR1 regulates patterning of the pharyngeal region. Genes Dev. 2003;17:141-153.
46. Dode C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463-465.
47. Robin NH, Feldman GJ, Mitchell HF, et al. Linkage of Pfeiffer syndrome to chromosome 8 centromere and evidence for genetic heterogeneity. Hum Mol Genet. 1994;3:2153-2158.
48. Wilkie AO. Craniosynostosis: genes and mechanisms. Hum Mol Genet. 1997;6:1647-1656.
49. Bratanic B, Praprotnik M, Novosel-Sever M. Congenital craniofacial dysostosis and cutis gyratum: the Beare-Stevenson syndrome. Eur J Pediatr. 1994;153:184-186.
50. Shiang R, Thompson LM, Zhu YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78(2):335-342.
51. Deng C, Wynshaw-Boris A, Zhou F, et al. LederFibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911-921.
52. Colvin JS, Bohne BA, Harding GW, et al. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390-397.
53. Robin NH. Molecular genetic advances in understanding craniosynostosis. Plast Reconstr Surg. 1999;103:1060-1070.
54. Warman ML, Mulliken JB, Hayward PG, et al. Newly recognized autosomal dominant disorder with craniosynostosis. Am J Med Genet. 1993;46:444-449.
55. Brueton L, Huson SM, Winter RM, et al. Chromosomal localisation of a developmental gene in man: direct DNA analysis demonstrates that Greig cephalopolysyndactyly maps to 7p13. Am J Med Genet. 1988;31:799-804.
56. Mulliken JB, Warman ML. Molecular genetics and craniofacial surgery. Plast Reconstr Surg. 1996;97:666-675.
57. Vikkula M, Mariman EC, Lui VC, et al. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell. 1995;80:431-437.
58. Cohen M. Sutural Pathology. In: Cohen M, MacLean R, editors. Craniosynostosis: Diagnosis, Evaluation, and Management. New York: Oxford University Press; 2000:51-68.
59. McCarthy JG, Reid CA. Craniofacial synostosis in association with vitamin D–resistant rickets. Ann Plast Surg. 1980;4:149-153.
60. Mimura J, Yamashita K, Nakamura K, et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells. 1997;2:645-654.
61. Abbott BD, Perdew GH, Birnbaum LS. Ah receptor in embryonic mouse palate and effects of TCDD on receptor expression. Toxicol Appl Pharmacol. 1994;126:16-25.
62. Hu D, Helms JA. The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development. 1999;126:4873-4884.
63. Rice R, Spencer-Dene B, Connor EC, et al. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113:1692-1700.
64. McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1-114.
65. Edison RJ, Muenke M. Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N Engl J Med. 2004;350:1579-1582.
66. Cohen MMJr. Etiopathogenesis of craniosynostosis. Neurosurg Clin North Am. 1991;2:507-513.
67. Higginbottom MC, Jones KL, James HE. Intrauterine constraint and craniosynostosis. Neurosurgery. 1980;6:39-44.
68. Reimann F, Gokmen M, Inceman S. On the behavior of the cranial sutures in thalassemias. IV. Further studies on changes in the skeletal system in severe blood diseases [in German]. Blut. 1968;17:214-226.
69. Cohen MMJr. Lambdoid synostosis is an overdiagnosed condition. Am J Med Genet. 1996;61:98-99.
70. Hunt JA, Hobar PC. Common craniofacial anomalies: facial clefts and encephaloceles. Plast Reconstr Surg. 2003;112:606-615.
71. Katzen JT, McCarthy JG. Syndromes involving craniosynostosis and midface hypoplasia. Otolaryngol Clin North Am. 2000;33:1257-1284. vi
72. Shprintzen RJ. Velocardiofacial syndrome. Otolaryngol Clin North Am. 2000;33:1217-1240. vi
73. Poswillo D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg Oral Med Oral Pathol. 1973;35:302-328.
74. Gussack GS, Luterman A, Powell RW, et al. Pediatric maxillofacial trauma: unique features in diagnosis and treatment. Laryngoscope. 1987;97:925-930.
75. Haug RH, Foss J. Maxillofacial injuries in the pediatric patient. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:126-134.
76. Tuite GF, Evanson J, Chong WK, et al. The beaten copper cranium: a correlation between intracranial pressure, cranial radiographs, and computed tomographic scans in children with craniosynostosis. Neurosurgery. 1996;39:691-699.
77. David LR, Genecov DG, Camastra AA, et al. Positron emission tomography studies confirm the need for early surgical intervention in patients with single-suture craniosynostosis. J Craniofac Surg. 1999;10:38-42.
78. Pittman T. Single suture synostosis and intracranial hypertension. J Ky Med Assoc. 2003;101:63-70.
79. Kapp-Simon KA, Speltz ML, Cunningham ML, et al. Neurodevelopment of children with single suture craniosynostosis: a review. Childs Nerv Syst. 2007;23:269-281.
80. Vu HL, Panchal J, Parker EE, et al. The timing of physiologic closure of the metopic suture: a review of 159 patients using reconstructed 3D CT scans of the craniofacial region. J Craniofac Surg. 2001;12:527-532.
81. Fox AJ, Tatum SA. The coronal incision: sinusoidal, sawtooth, and postauricular techniques. Arch Facial Plast Surg. 2003;5:259-262.
82. Marchac D, Renier D, Arnaud E. Evaluation of the effect of early mobilization of the supraorbital bar on the frontal sinus and frontal growth. Plast Reconstr Surg. 1995;95:802-811.
83. Salyer KE, Bardach J, Squier CA, et al. A comparative study of the effects of biodegradable and titanium plating systems on cranial growth and structure: experimental study in beagles. Plast Reconstr Surg. 1994;93:705-713.
84. Tatum SA, Kellman RM, Freije JE. Maxillofacial fixation with absorbable miniplates: computed tomographic follow-up. J Craniofac Surg. 1997;8:135-140.
85. Imola MJ, et al. Resorbable plate fixation in pediatric craniofacial surgery: long-term outcome. Arch Facial Plast Surg. 2001;3(2):79-90.
86. Ortiz-Monasterio F, del Campo AF, Carrillo A. Advancement of the orbits and the midface in one piece, combined with frontal repositioning, for the correction of Crouzon’s deformities. Plast Reconstr Surg. 1978;61:507-516.
87. Honig JF, Grohmann UA, Merten HA. Facial bone distraction osteogenesis for correction of malocclusion: a more than 70-year-old concept in craniofacial surgery. Plast Reconstr Surg. 2002;109:41-44.
88. McCarthy JG, Schreiber J, Karp N, et al. Lengthening the human mandible by gradual distraction. Plast Reconstr Surg. 1992;89:1-8.
89. Swennen G, Schliephake H, Dempf R, et al. Craniofacial distraction osteogenesis: a review of the literature: part 1: clinical studies. Int J Oral Maxillofac Surg. 2001;30:89-103.
90. Winzenburg SM, Imola MJ. Internal fixation in pediatric maxillofacial fractures. Facial Plast Surg. 1998;14:45-58.
91. Berryhill WE, Rimell FL, Ness J, et al. Fate of rigid fixation in pediatric craniofacial surgery. Otolaryngol Head Neck Surg. 1999;121:269-273.
92. Lakhani RS, Tatum SA. Update on craniofacial surgery. Curr Opin Otolaryngol Head Neck Surg. 2003;11:291-294.
93. Thaller SR, Huang V, Tesluk H. Use of biodegradable plates and screws in a rabbit model. J Craniofac Surg. 1992;2:168-173.
94. Teo C, Dornhoffer J, Hanna E, et al. Application of skull base techniques to pediatric neurosurgery. Childs Nerv Syst. 1999;15:103-109.
95. Kellman RM, Marentette L. The transglabellar/subcranial approach to the anterior skull base: a review of 72 cases. Arch Otolaryngol Head Neck Surg. 2001;127:687-690.
96. Lewark TM, Allen GC, Chowdhury K, et al. Le Fort I osteotomy and skull base tumors: a pediatric experience. Arch Otolaryngol Head Neck Surg. 2000;126:1004-1008.
97. Kai Y, Nagahiro S, Yoshioka S, et al. Application of the skull base technique to the repair of transsphenoidal meningoencephaloceles. Pediatr Neurosurg. 1996;25:54-56.
98. Palmaz JC. Review of polymeric graft materials for endovascular applications. J Vasc Interv Radiol. 1998;9:7-13.
99. Trumpy IG, Lyberg T. In vivo deterioration of Proplast-Teflon temporomandibular joint interpositional implants: a scanning electron microscopic and energy-dispersive X-ray analysis. J Oral Maxillofac Surg. 1993;51:624-629.
100. Wong RW, Rabie AB. A quantitative assessment of the healing of intramembranous and endochondral autogenous bone grafts. Eur J Orthod. 1999;21:119-126.
101. Zins JE, Whitaker LA. Membranous versus endochondral bone: implications for craniofacial reconstruction. Plast Reconstr Surg. 1983;72:778-785.
102. Kusiak JF, Zins JE, Whitaker LA. The early revascularization of membranous bone. Plast Reconstr Surg. 1985;76:510-516.
103. Tessier P, Kawamoto H, Matthews D, et al. Autogenous bone grafts and bone substitutes—tools and techniques: I: A 20,000-case experience in maxillofacial and craniofacial surgery. Plast Reconstr Surg. 2005;116(Suppl):6S-24S.
104. Sasso RC, LeHuec JC, Shaffrey C. Iliac crest bone graft donor site pain after anterior lumbar interbody fusion: a prospective patient satisfaction outcome assessment. J Spinal Disord Tech. 2005;18(Suppl):S77-S81.
105. Enneking WF, Mindell ER. Observations on massive retrieved human allografts. J Bone Joint Surg Am. 1991;73:1123-1142.
106. Bauer TW, Muschler GF. Bone graft materials: an overview of the basic science. Clin Orthop Relat Res. 2000:10-27.
107. Romo T3rd, Baskin JZ, Sclafani AP. Augmentation of the cheeks, chin and pre-jowl sulcus, and nasolabial folds. Facial Plast Surg. 2001;17:67-78.
108. Hobar PC, Hunt JA, Antrobus S. Assessment of the effects on growth of porous hydroxyapatite granule cranioplasty in the immature guinea pig craniofacial skeleton. Plast Reconstr Surg. 2003;111:1667-1675.
109. Reddi SP, Stevens MR, Kline SN, et al. Hydroxyapatite cement in craniofacial trauma surgery: indications and early experience. J Craniomaxillofac Trauma. 1999;5:7-12.
110. Holmes RE, Hagler HK. Porous hydroxyapatite as a bone graft substitute in cranial reconstruction: a histometric study. Plast Reconstr Surg. 1988;81:662-671.
111. Lu J, et al. The biodegradation mechanism of calcium phosphate biomaterials in bone. J Biomed Mater Res. 2002;63:408-412.