[level-membership-for-neurology-category]
5 Cranial Nerves III, IV, and VI
Oculomotor, Trochlear, and Abducens Nerves: Ocular Mobility and Pupils
Cranial Nerve III: Oculomotor
Clinical Vignette
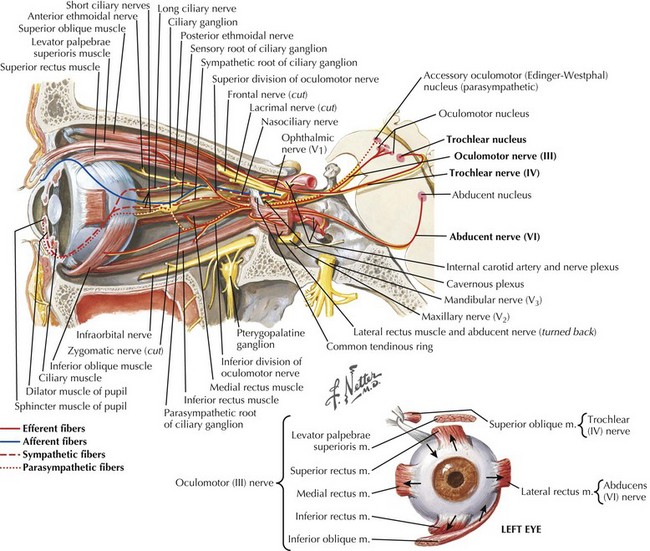
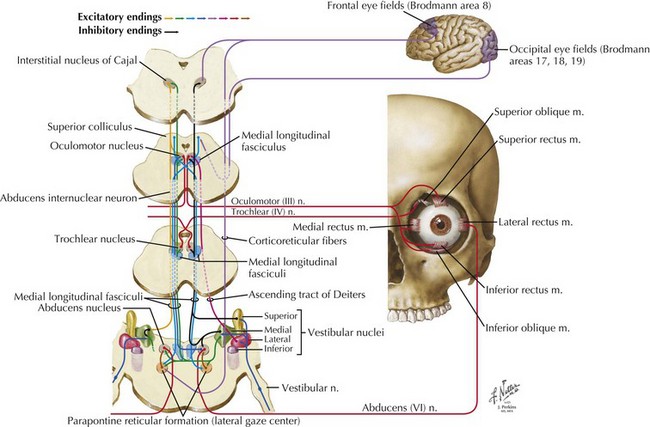
CN-III begins at its nucleus in the midline upper midbrain. The nucleus is a lepidopteroid collection of nine subnuclei located in the center of the rostral midbrain at the level of the superior colliculi (Fig. 5-1). The most ventral of these subnuclei is the central caudate nucleus, a midline structure that innervates both levator palpebrae muscles. Uniquely, axons from the medial subnuclei or columns decussate completely to innervate the contralateral superior rectus muscles. The other six subnuclei, three left-and-right pairs, innervate ipsilateral extraocular muscles. The ventral subnucleus, intermediate column, and dorsal subnucleus, respectively, control the medial rectus (eye adduction), inferior oblique (intorsion and some elevation), and inferior rectus (depression).
Etiology and Pathogenesis
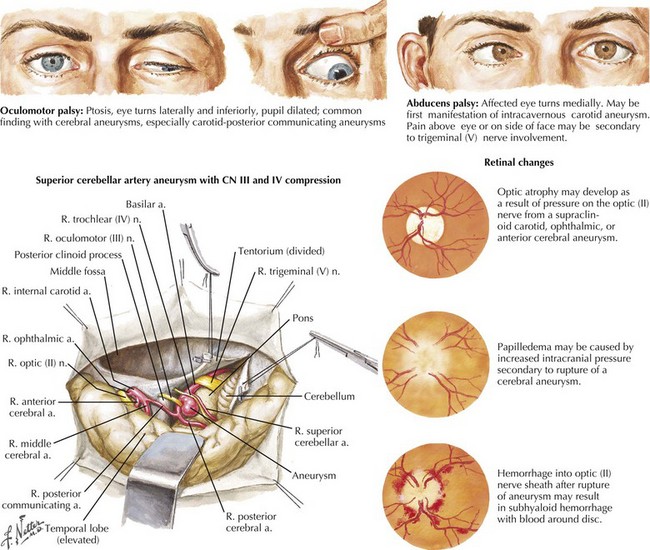
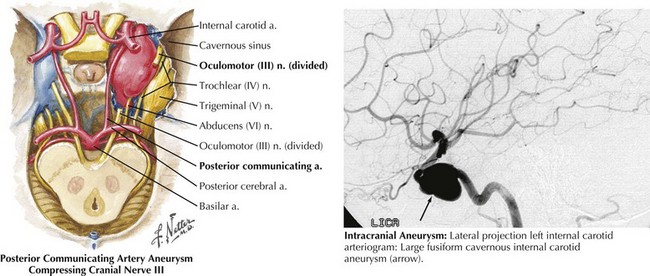
In the patient presenting with acute, severe headache and pupil-involved CN-III palsy, an expanding aneurysm, usually of the posterior communicating artery (p-com), is the most important cause (Fig. 5-2). The location of these aneurysms is the origin of p-com at the ICA (Fig. 5-3) and 90% of these aneurysms present with CN-III palsy. Aneurysm in other nearby arteries can likewise present as CN-III palsy, with up to 30% of acquired CN-III palsies being caused by aneurysms.
Management and Therapy
The management of symptomatic intracranial aneurysm is usually urgent, via endovascular or surgical intervention if the general state of the patient permits (see Chapter 57). Management of microvascular palsy usually centers on prevention of recurrent events via reduction of risk factors. Optimization of any causative disease, such as diabetes, is crucial, and daily aspirin is often recommended. Therapy for other underlying causes of CN-III palsy will vary, appropriate to etiology.
Cranial Nerve IV: Trochlear
Clinical Vignette
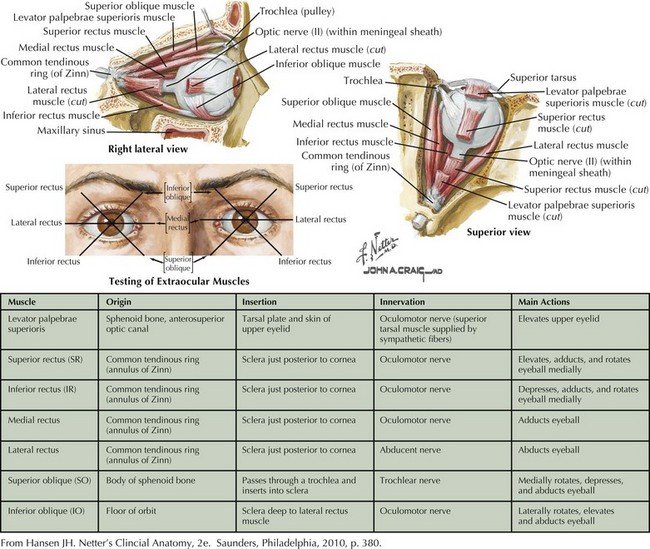
The superior oblique is chiefly a depressor of the globe and is most active when the eye is adducted and depressed. It has a secondary function of intorting the eye during ipsilateral head tilt and is a weak abductor of the eye in downgaze (Fig. 5-4). Therefore, CN-IV palsy will produce ipsilateral loss of depression (hyperopia) and excyclotorsion of the globe.
Etiology and Pathogenesis
In the subarachnoid space, CN-IV can be affected by carcinomatous meningitis, by aneurysm (especially of the superior cerebellar artery; Fig. 5-2), or by dolichoectasia of the basilar artery. The nerve itself may be the site of schwannomas. Once within the dural canal leading to the cavernous sinus, the nerve may be affected by tumor, especially meningioma. Compression of CN-IV can occur at the cavernous sinus itself, by dissections or aneurysm of the carotid artery, by extension of sellar and orbital tumors, and by metastases. Typically CN-IV, -III, -VI, and the ophthalmic branch of CN-V are involved in cavernous sinus lesions.
Cranial Nerve VI: Abducens
Clinical Vignette
The CN-VI nucleus, located just beneath the facial colliculi in the inferior pons is enveloped by the turning CN-VII fascicular fibers of the facial genu and contains two physiologically—but not topographically—distinct groups of neurons (Fig. 5-5). One group innervates the ipsilateral lateral rectus; the other sends axons across the midline to the contralateral medial longitudinal fasciculus (MLF). These latter axons ascend in the MLF to the ventral nucleus of the contralateral CN-III nuclear complex. These internuclear neurons connect the nuclei of CN-VI and -III, producing the almost simultaneous stimulation of the contralateral medial rectus during ipsilateral abducens nerve stimulation to produce lateral horizontal gaze.
The Pupils
Abnormalities of Pupil Function
Light Reflex Abnormalities, Afferent Limb
Light is detected at the retina, and the signal sent via the optic nerve to the brain. Although the majority of visual information is transmitted to the lateral geniculate nucleus, the pupillary afferent fibers reach the pretectal nucleus via an extrageniculate pathway. The input from each eye reaches both the left and right nuclei, so that stimulation of one retina results in both the ipsilateral and contralateral pupillary constriction, producing a direct and consensual pupillary response, respectively (Fig. 5-6). Because of this anatomic property, disease of the pupillary afferents, even if unilateral, does not produce pupils that are unequal in size (anisocoria).
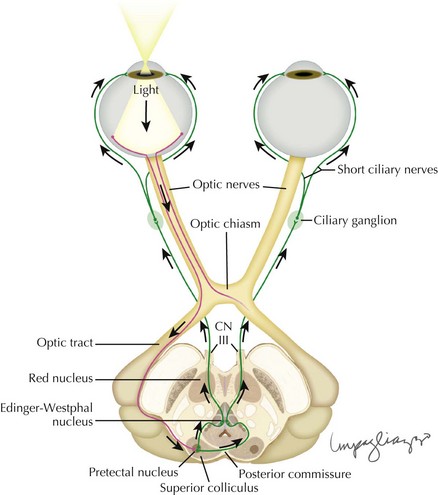
Efferent Limb Abnormalities: Parasympathetic
The pretectal nuclei receive input from both optic nerves, and in turn send efferents to both Edinger–Westphal nuclei. These nuclei send efferent pupilloconstrictive fibers via the oculomotor nerve (CN-III) to the ciliary ganglion, where they synapse with the short ciliary nerves carrying motor efferents to the pupillary sphincter (see Fig. 5-6).
Efferent Limb Abnormalities: Sympathetic
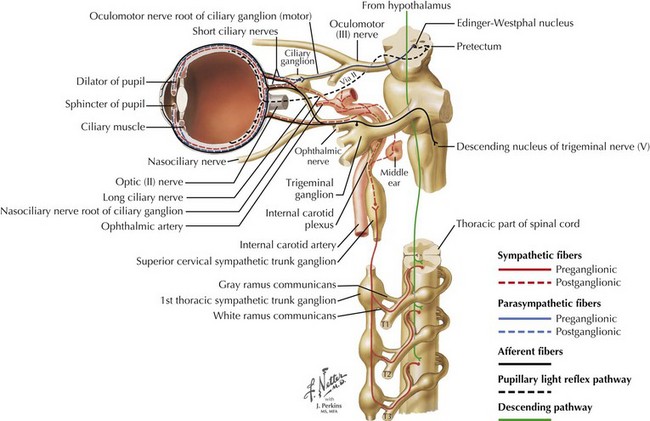
The sympathetic innervation of the pupillary dilator muscle involves a three-chain pathway. The primary neuron runs from the hypothalamus to the ciliospinal center of Budge and Waller at the junction of the thoracic and cervical spinal cord; the secondary neuron starts there, crosses over the lung apex and synapses in the superior cervical ganglion; and the tertiary neuron follows the ICA to the cavernous sinus, eventually reaching the eye after passing through the ciliary ganglion without synapsing (Fig. 5-7).
Girkin CA, Perry JD, Miller NR. A relative afferent pupillary defect without any visual sensory deficit. Arch Ophthalmol. 1998 Nov;116(11):1544-1545. Clinical description of a lesion of the extrageniculate pupillary afferent pathway
Miller SD, Thompson HS. Pupil cycle time in optic neuritis. Am J Ophthalmol. 1978;85:635-642. Description of this clinical test
Morales J, Brown SM, Abdul-Rahim AS, et al. Ocular effects of apraclonidine in Horner syndrome. Arch Ophthalmol. 2000 Jul;118(7):951-954. First description of the usefulness of this agent in the diagnosis of Horner pupil
Thompson HS, Kardon RH. The Argyll Robertson pupil. J Neuroophthalmol. 2006 Jun;26(2):134-138. Modern neuro-anatomic review of a classic pupillary syndrome
Thompson S, Pilley SF. Unequal pupils (A flow chart for sorting out the anisocorias). Surv Ophthalmol. 1976;21:45-48. The diagnostic paradigm that has become a classic
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
5 Cranial Nerves III, IV, and VI
Oculomotor, Trochlear, and Abducens Nerves: Ocular Mobility and Pupils
Cranial Nerve III: Oculomotor
Clinical Vignette
CN-III begins at its nucleus in the midline upper midbrain. The nucleus is a lepidopteroid collection of nine subnuclei located in the center of the rostral midbrain at the level of the superior colliculi (Fig. 5-1). The most ventral of these subnuclei is the central caudate nucleus, a midline structure that innervates both levator palpebrae muscles. Uniquely, axons from the medial subnuclei or columns decussate completely to innervate the contralateral superior rectus muscles. The other six subnuclei, three left-and-right pairs, innervate ipsilateral extraocular muscles. The ventral subnucleus, intermediate column, and dorsal subnucleus, respectively, control the medial rectus (eye adduction), inferior oblique (intorsion and some elevation), and inferior rectus (depression).
Etiology and Pathogenesis
In the patient presenting with acute, severe headache and pupil-involved CN-III palsy, an expanding aneurysm, usually of the posterior communicating artery (p-com), is the most important cause (Fig. 5-2). The location of these aneurysms is the origin of p-com at the ICA (Fig. 5-3) and 90% of these aneurysms present with CN-III palsy. Aneurysm in other nearby arteries can likewise present as CN-III palsy, with up to 30% of acquired CN-III palsies being caused by aneurysms.
Management and Therapy
The management of symptomatic intracranial aneurysm is usually urgent, via endovascular or surgical intervention if the general state of the patient permits (see Chapter 57). Management of microvascular palsy usually centers on prevention of recurrent events via reduction of risk factors. Optimization of any causative disease, such as diabetes, is crucial, and daily aspirin is often recommended. Therapy for other underlying causes of CN-III palsy will vary, appropriate to etiology.
Cranial Nerve IV: Trochlear
Clinical Vignette
The superior oblique is chiefly a depressor of the globe and is most active when the eye is adducted and depressed. It has a secondary function of intorting the eye during ipsilateral head tilt and is a weak abductor of the eye in downgaze (Fig. 5-4
[/not-level-membership-for-neurology-category]
