CHAPTER 18 Contrast Agents in Magnetic Resonance Imaging
INTRODUCTION
Intravenous contrast agents have become integral to the clinical practice of cardiovascular magnetic resonance (MR) imaging. Myocardial perfusion and myocardial delayed enhancement MR are important contrast-enhanced techniques for the clinical evaluation for myocardial disease. Contrast-enhanced magnetic resonance angiography (CE MRA) has become the preferred method for fast and accurate assessment of common arterial structures such as the thoracic aorta, abdominal aorta, carotid arteries, renal arteries, and peripheral arteries. These applications were facilitated by innovations in MRI instrumentation that improved acquisition speed and in optimization of cardiovascular imaging protocols.1 This chapter will focus solely on those MR contrast agents that have, or promise to have, value for cardiovascular applications.
The value of a contrast agent depends on its ability to generate image contrast for a given imaging modality. Radiographic contrast agents lead to an attenuation of the transmitted x-rays; MR agents rely on a completely different biophysical principle. MR contrast agents generate image contrast by locally changing the relaxivity of the recipient tissue. For example, all contrast agents for x-ray computer tomography (CT) use iodine as their central ion, whereas MR contrast agents depend on the magnetic moment of the central atom. For cardiovascular MR imaging, we classify today’s available agents into paramagnetic (mostly gadolinium-based) and super-paramagnetic (mostly iron oxide-based).2 We also experience a substantial variability in country specific availability of MR contrast agents for cardiovascular imaging as well as regulatory approvals, which is especially evident for cardiovascular imaging. Therefore, we will initially discuss the development and safety of MR contrast agents before we will review some of their type-specific characteristics for cardiovascular MR and MRA.
Regulatory Label of MR Contrast Agents
Using a medication or drug, which requires a physician order or prescription, outside its regulatory defined approved, labeled use is considered to be at least off-label; however it could also be against-label if such a use is defined as a contraindication or a defined warning applies. The vast majority of contrast-enhanced MRI examinations of the cardiovascular system performed globally to date has been done “off-label”.3 In 2010, none of the marketed MR contrast agents in the United States has an FDA-approved indication for use in cardiac MRI despite a huge number of clinical trials and clinical experience; and only one, gadofosveset trisodium (Ablavor, Lantheus Medical Imaging, North Billerica, MA) has an FDA-approved MRA indication and only “to evaluate aortoiliac occlusive disease (AIOD) in adults with known or suspected peripheral vascular disease.”
An off-label use has its foundation in the declaration of Helsinki and is basically founded in the physician’s prerogative to provide the most appropriate care to patients. Off-label use is a common and essential part of today’s practice of medicine, but it also requires appropriate diligence in its use. First of all, off-label use and clinical research are quite different. An investigational use (i.e., research use) requires a formal protocol approved by the Institutional Review Board (IRB) or an equivalent ethics board and typically informed consent; whereas off-label use requires only peer reviewed evidence. Why peer reviewed evidence? The regulatory agencies and legal interpretations expect objectivity without bias or conflict of interest. It is well recognized that regulatory agencies are appropriately very sensitive on the propagation of off-label use of drugs and therefore disallow drug vendors to advertise, market, or otherwise incentivize such off-label use as can be seen by a $2.3 billion4 fine recently enforced for off-label propagation of a therapeutic drug and numerous warning letters being posted by FDA.5 A key distinction of off-label use versus an investigational use is that off-label use pertains only to applications for patient care (i.e., clinical practice) and not for a research aim or objective.
Use of a drug or contrast agent for a listed contraindication or excluded use cannot be considered to be off-label; they are against label however they might still be necessary and patient-specific appropriate if used with the proper diligence. A new situation arose when the FDA decided to put a black box warning label on all gadolinium-based contrast agents in 2007 as a response to the occurrence of cases with NSF (nephrogenic systemic fibrosis).6 Before we review the current status of this severe adverse reaction to gadolinium-containing MR contrast agents, let us finish with the labeling aspects. What is the intent of such a warning and what does it mean from a practical point of view? First, it is the strongest warning mechanism that the regulatory agencies have to ensure the user/health care professional is aware of a change in a label and product or class-associated warning, and second, gives regulatory guidance on the appropriate use. The FDA black box label means, for example, that any contrast dosage outside the contrast agent-specific label cannot be considered off-label anymore but will have to be considered against the label. In summary, the country-specific label of an MR contrast agent must be known and appropriately considered in the clinical practice of cardiovascular MR/MRA and will continue to change.
Safety of MR Contrast Agents
The nonimaging community was warned in a 2003 letter to the editor of the New England Journal of Medicine that severe pseudohypocalcemia was observed after gadolinium-enhanced MRA.7 The authors noted lower calcium values in blood samples obtained in patients immediately after they had an MRA performed with gadodiamide (Omniscan, GE Healthcare Medical Diagnostics) as MR contrast agent. The interaction of excess chelate in the gadodiamide with colorimetric calcium tests was recognized by experts but was neither included in the product label nor commonly known and caused multiple issues, especially in patients who had undergone CE MRA.8,9 A subsequent letter and editorial revealed that these drug laboratory test interactions are not specific to MRA, but to two contrast agent formulations, gadodiamide and gadoversetamide (Optimark, Mallinckrodt) that interact to lead to false lower calcium levels in colorimetric but not in ionic calcium tests.10 These observations and subsequent public discussion can be credited with increasing awareness about MR contrast agent safety, which was perceived as entirely safe with considerable complacency evolving.
One of the most essential safety aspects of a contrast agent is that it needs to be completely eliminated after injection into the patient. Although this sounds trivial, imaging agents did have some dark clouds in their history when thorium dioxide (Thorotrast, Heyden) was discovered and subsequently used as a capable x-ray contrast agent, however, its retention in the body and radioactivity (alpha-particle) was not readily recognized in the early part of the last century.11 Most MR imaging agents including gadolinium chelates are eliminated via renal clearance; iron oxides, with the liver and reticuloendothelial system (RES). It is important to understand the specific characteristics and elimination pathway of an agent as well as what happens if elimination is impaired. Therefore, it should not be a surprise that a drug that depends on renal elimination has the potential to change its biologic behavior if the pathway is impaired, consequently making agents with multiple or other elimination pathways highly desirable for patient populations with renal impairment. Contrast agents should always be given at the lowest effective dose to enable diagnostic-appropriate visualization of the target organ system, here the cardiovascular system; however, at this juncture, is also the pitfall. For a time some in our community suggested that “more is better” which frequently did improve the image quality obtainable by still evolving MR methodologies, however, the safety profile does change with changing populations and dosages. Similar to the speed rating of a tire, safety of medications can and does vary when we use it beyond recommended usage. From a safety perspective, the rapid elimination from the body, no or limited drug to drug interactions and no or limited toxicity are the key desirable safety aspects of a contrast agent.
Pharmacovigilance of MR Contrast Agents
Pharmacovigilance is the analysis of observed adverse events of an available drug, in this case MR contrast agent, and is the methodology employed to monitor the safety when a drug is broadly available. It is still a growing science as we continue to learn more about how to assess, manage, and predict the safety of drugs in large, diverse patient populations and with considerable changes in the way we practice medicine. Aside from post-marketing, phase IV studies, the information source is solely based on adverse event reporting. A healthcare provider is encouraged and sometimes mandated by country-specific laws to report any adverse event observed during the clinical use of medications/drugs—either directly to the vendor or to a regulatory body sponsored website such as MedWatch by the FDA.12 Although this spontaneous adverse event reporting has its shortcomings, it is the best and only broad-based mechanism currently available. Unfortunately, drug manufacturers and, as such, also the vendors of MR contrast agents do not commonly voluntarily release their adverse event reporting database which they are required to compile on a global basis. The manufacturer does know how many doses of a drug are sold and those sales data are then related to the adverse event reporting rates. In an adverse event report, the reporter documents the observations, some patient characteristics, severity of the adverse event, and assesses the relationship to the contrast agent. Depending on the severity and expectancy of the adverse event, the regulatory agency and/or manufacturer may further investigate such a report. As part of country-specific marketing approvals, a manufacturer may have to report the noted observations, however these are typically not publically available documents. The largest released reporting of pharmacovigilance data on an MR contrast agent is available on the use of Gd-DTPA (Magnevist, Bayer HealthCare Pharmaceutical) and has been voluntarily reported. These data indicate for specific event categories, such as cardiovascular reactions rates, of 4 to 8 events per 100,000 doses administered.13 Renal impairment was identified in adverse event reports from 0.1 to 0.8 events per 100,000 doses and was with angioedema, the only major category that showed an increasing trend in the recent years of adverse event reporting. Further analysis of those reports indicating renal impairment revealed that patients most commonly had preexisting renal conditions due to nephrotoxic medications and were receiving higher than labeled contrast agent doses. Unfortunately, these data are not publically available for the other commonly used MR contrast agents. The current annual global use of MR contrast agents is estimated to be around 12 million patient doses. Although no broad-based data are currently available on the cardiovascular MRA examinations being performed, estimates suggest an annual rate of about 2 to 3 million procedures. In order to further put adverse event reporting in perspective, it must be highlighted that those for the Gd-DTPA MR contrast agent are two to three times lower than those reported for nonionic monomeric iodinated contrast agents used in x-ray, and allergic reactions are reported about eight times more frequently for nonionic iodinated contrast media used in x-ray than for the Gd-DTPA, an MR contrast agent.14 Anaphylactoid reactions have been seen in Gd-DTPA at a reporting rate of 3 to 4 per million, whereas urticaria has been reported at a rate of 29 to 79 per million.
NEPHROGENIC SYSTEMIC FIBROSIS (NSF)
Nephrogenic systemic fibrosis (NSF) initially also referred to as nephrogenic fibrosing dermopathy (NFD) is a condition that, to date, has occurred only in people with kidney disease. NSF is a systemic disorder with its most prominent and visible effects in the skin, hence its original designation as a dermopathy.15 Our current knowledge recognizes that kidney disease seems to be a prerequisite for developing NSF and, therefore, it has been accepted as the terminology most reflective of the reality of the disorder. Although the pathophysiology of this disease mechanism is not yet fully understood and is still evolving, it is simultaneously subject to intense litigation, such as those consolidated by a judicial panel to the U.S. District court in Cleveland, Ohio.
CLASSIFICATION OF MR CONTRAST AGENTS FOR CARDIOVASCULAR IMAGING
MR contrast agents currently fall into two broad categories; those based on gadolinium, which are predominately paramagnetic in nature, and those based on iron oxide particles of different coating and size that are superparamagnetic (Fig. 18-1). The broadest use for cardiovascular imaging is based on gadolinium chelates which can be subclassified into agents revealing no interaction with proteins, those that have weak temporary interaction with proteins leading to increased relaxivity and/or having an additional extrarenal elimination pathway, and those that have strong protein binding. Table 18-1 summarizes the contrast agents that are currently available or have been in clinical trials at varying stages relevant for cardiovascular MR imaging.16
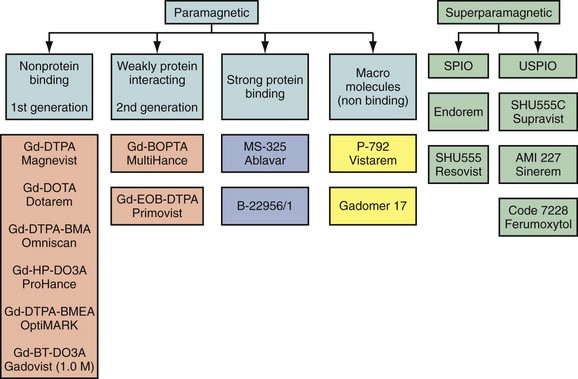
 FIGURE 18-1
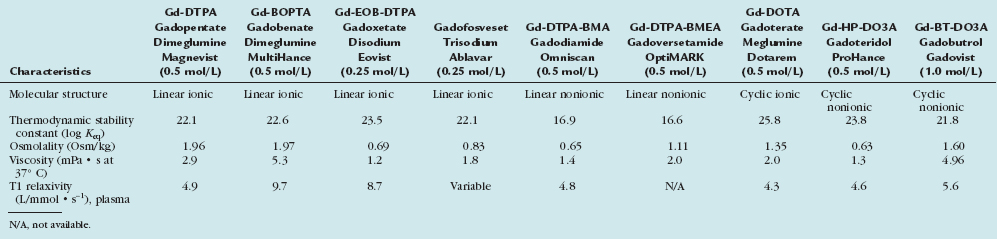
FIGURE 18-1TABLE 18-1 Physicochemical Characteristics of Clinically Developed Gadolinium-Based MR Contrast Agents
Nonprotein Interacting Standard Gadolinium Chelates
This group of “conventional” gadolinium chelate agents was introduced more than 20 years ago with nearly simultaneous approval of gadopentetate dimeglumine (Gd-DTPA, Magnevist, Bayer Healthcare) in all three key markets: European Union, United States, and Japan. Five of these agents are available as 0.5 molar formulations and one, gadobutrol (Gd-BT-DO3A, Gadovist, Bayer Healthcare) is being marketed at a 1.0 molar formulation. Although differences exist between these agents in terms of the molecular structure and chemical and physical properties (Tables 18-1 and 18-2), all agents are nonspecific and are eliminated unchanged via the renal pathway by glomerular filtration. The T1 relaxation rates of these agents are comparable and fall in the range between 4.3 and 5.6 L/mmol · s−1. These similarities lead to equivalent imaging characteristics at the same dose and injection rate.
Table 18-2 Physicochemical Characteristics of Small and Ultra-Small Particles of Iron Oxides Developed as MR Contrast Agents

From the molecular structure, the agents can be subclassified into ionic or nonionic, linear, or macrocyclic. The concept of the nonionic agents was that they would have an even better safety profile with fewer adverse events comparable to the impact of reducing ionicity in iodinated contrast agents. This idea could not be realized with the agents and the stability of the binding of the gadolinium central atom has become much more critical. From this perspective, the nonionic linear molecules are the least stable and the ionic macrocyclic agents are the most stable. Therefore, the binding strength of the gadolinium by its surrounding chelating complex has become a differentiating factor. The two agents, gadodiamide (Gd-DTPA-BMA, Omniscan, GE-Healthcare) and gadoversetamide (Gd-DTPA-BMEA) have substantially lower binding and, therefore, include excess chelate in the formulation to trap any dissociated gadolinium ion in the vial which has also been the causative factor for the interference with colorimetric calcium tests and the spurious hypocalcemia.10
Gadolinium Chelates with Weak Protein Interaction
This class represents a second generation of gadolinium chelates that possess a higher T1 relaxivity in blood such as for gadobenate (Gd-BOPTA, Multihance, Bracco) (9.7 L/mmol · s−1) due to the weak transient interaction between the agent and serum proteins, particularly albumin and a T1 relaxivity of 8.2 L/mmol · s−1 in human plasma for gadotexetate disodium (Eovist, Bayer Healthcare). Both agents are ionic, linear chelates and have a dual elimination pathway with partially hepatobiliary elimination, gadobenate is weaker than gadoxetate. The higher T1 relaxivity manifests as a significantly greater intravascular signal intensity enhancement compared to that achieved with conventional gadolinium chelates at equivalent doses with the benefits of a more pronounced effect in smaller vessels as well as in the margins of the tumors. To objectively assess if differences in intravascular image contrast exist between the first group of standard gadolinium chelates and the new group, an intraindividual cross-over study was performed that revealed that gadobenate dimeglumine presented a significantly more intense contrast enhancement with a higher, longer peak duration and larger area under the vascular contrast enhancement curve.17 This finding was confirmed in subsequent larger MRA studies for the run-off vasculature,18 pelvic and carotid vasculature. The practical impact is that for the same dose and administration approach, a more intense and longer duration intravascular signal intensity benefit was noted. The clinical advantages of the increased relaxivity also have been demonstrated for many vascular territories that range from the carotid vasculature16 to the distal run-off vessels.16 Like the conventional nonprotein interacting GBCAs, gadobenate dimeglumine has an excellent safety profile with a very low incidence of adverse events noted for the clinical development program as a whole,16 however the potential risk to cause NSF cannot be excluded and the same level of diligence also applies to this group. The fact that more signal/enhancement can be obtained for the same dosing more readily enables full diagnostic quality at lower doses, thereby reducing dose and accumulation-dependent potential effects.
Gadolinium Chelates with Strong Protein Interaction
The contrast agents in this category exhibit strong affinity for serum proteins which increase the relaxivity and also have extended intravascular half-life making them by design cardiovascular imaging agents. Gadofosveset trisodium, developed under the identifier MS-325 (then under the proposed product name of Vasovist and now under the new product name of Ablavar) has gone through full clinical development and is approved in several countries, including the United States, for specific MRA indications. This agent is available in a 0.25 mol/L concentration, has been reported to be 88% to 96% noncovalently bound to albumin in human plasma and to exhibit a relaxivity at 0.5T that is 6 to 10 times that of gadopentetate dimeglumine. The agent has a recommended dosing of 0.03 mmol/kg body weight19 and achieves its desired intravascular contrast at a substantially lower dose because of its higher relaxivity. The elimination pathway is primarily renal but it also has some hepatobiliary elimination. This agent can be used for first pass contrast-enhanced MRA and for steady-state imaging in a number of vascular territories. Although this agent has been investigated in trials in many vascular territories, its 2008 FDA approval and label states the indication as “MRA to evaluate aortoiliac occlusive disease (AIOD) in adults with known or suspected peripheral vascular disease”. The European Medicines Agency (EMA) had already approved the agent in 2005 with the labeled indication “for contrast-enhanced magnetic resonance angiography for visualization of abdominal or limb vessels in patients with suspected or known vascular disease”, which is a much broader indication. The agent also exhibits an extravasation in the case of blood brain-barrier breakdown and is currently the only approved agent that will allow first pass and steady-state imaging.
The second agent with strong affinity for serum proteins and increased relaxivity is gadocoletic acid (B22956, Bracco). This agent has undergone phase II trials for enhanced coronary MRA and has been shown to have even stronger affinity for serum albumin than gadofosveset (approximately 94% bound noncovalently) with a similarly long intravascular residence time.20
Gadolinium Contrast Agents with Macromolecular Structures
Examples of gadolinium-based blood pool agents with macromolecular structures are P792 (Vistarem, Guebert) and Gadomer-17 (Bayer Healthcare).16 These agents differ from the currently available low molecular weight gadolinium agents in possessing large molecular structures that prevent extravasation of the molecules from the intravascular space following injection, but do have slow, reduced leakage in case of blood-brain barrier breakdown. The molecular weights of P792 and gadomer-17 are 6.5 kDa and 35 kDa, respectively, which compare with weights of between approximately 0.56 kDa and 1.0 kDa for the purely first pass gadolinium agents. Whereas the structure of P792 is based on that of gadoterate substituted with four large hydrophilic spacer arms, gadomer-17 is a much larger polymer of 24 gadolinium cascades. In addition to differences in molecular weight and structure, these two agents appear to differ in terms of their rates of vascular clearance, with P792 considered a rapid clearance blood pool agent. Despite these differences, both agents have cardiovascular imaging capabilities and have been evaluated for these indications in clinical trials. Currently, it is not clear if and when any of these agents will receive regulatory approval or would be marketed.
Superparamagnetic Iron Oxide Agents
The first approved and marketed iron oxide-based contrast agent was AMI 25, also known as ferumoxide and marketed as Endorem (Guebert) or Feridex (Bayer Healthcare), with an indication for T2*-weighted liver imaging. This SPIO has also been used for cell-tracking and has a demonstrated potential for molecular-based cardiovascular imaging applications.21 Although there were no regulatory issues, the sole manufacturer of this agent, AMAG Pharmaceuticals, decided in November 2008 to cease its manufacture.
All iron oxides have been used as carrier molecules for targeted imaging and it remains a highly exciting research area with great potential for molecular targeted cardiovascular imaging. AMI 227 (Ferumoxtran), also known as Combidex or Sinerem, is another USPIO that has been specifically evaluated for lymphatic MR imaging22,23 but has not yet received final regulatory approval. The fifth iron oxide agent that has been evaluated for MRA imaging is ferumoxytol, formerly known as Code 7228 and now as Feraheme (AMAG). Although its initial development goal envisioned it to be an imaging agent, it was subsequently developed as an iron replacement therapeutic drug indicated for the treatment of iron deficiency anemia in adult patients with chronic kidney disease (CKD), the very same population at higher risk for NSF from Gd chelate imaging agents. This agent received its therapeutic FDA approval in 2009 and is being marketed with its labeled therapeutic indications. The potential for cardiovascular applications of this agent are high because it has a first pass and steady-state imaging ability and a well established safety profile at even higher doses than needed for imaging in a high-risk population for Gd chelates. Overall, it can be speculated that iron oxides, especially the USPIOs, will have an important place in cardiovascular MRA in the future, not only for intravascular contrast but also as a molecular targeted MR contrast agent. The contrast agent field will continue to evolve and the efforts over the last decade are leading to exciting new, safe, and robust imaging approaches, further increasing the clinical importance of safe, effective, and noninvasive MR cardiovascular imaging.
KEY POINTS
 Most commonly, gadolinium-chelate contrast agents are used off-label for cardiac MRI and contrast-enhanced MRA in clinical practice. Off-label use requires peer-review evidence. New contrast agents and cardiovascular indications are evolving and vigilance by practitioners is recommended to ensure that they have the most current information.
Most commonly, gadolinium-chelate contrast agents are used off-label for cardiac MRI and contrast-enhanced MRA in clinical practice. Off-label use requires peer-review evidence. New contrast agents and cardiovascular indications are evolving and vigilance by practitioners is recommended to ensure that they have the most current information.

Bleicher AG, Kanal E. Assessment of adverse reaction rates to a newly approved MRI contrast agent: review of 23,553 administrations of gadobenate dimeglumine. AJR Am J Roentgenol. 2008:191-W307.
Knoff MV, Balzer T, Esser M, Kashanian FK, et al. Assessment of utilization and pharmacovigilance based on spontaneous adverse event reporting of gadopentetate dimeglumine as a magnetic resonance contrast agent after 45 million administrations and 15 years of clinical use. Invest Radiol. 2006;41:491-499.
Neuwelt EA, Hamilton BE, Varallyay CG, et al. Ultrasmall superparamagnetic iron oxides (USPIOs): a future alternative magnetic resonance (MR) contrast agent for patients at risk for nephrogenic systemic fibrosis (NSF)? Kidney Int. 2009;75:465-474.
Prompona M, Cyran C, Nikolaou K, Bauner K, et al. Contrast-enhanced whole-heart MR coronary angiography at 3.0 T using the intravascular contrast agent gadofosveset. Invest Radiol. 2009;44:369-374.
1 Runge VM, Kirsch JE, Lee C. Contrast-enhanced MR angiography. J Magn Reson Imaging. 1993;3(1):233-239.
2 Knopp MV, von Tengg-Kobligk H, Floemer F, Schoenberg SO. Contrast agents for MRA: future directions. J Magn Reson Imaging. 1999;10(3):314-316.
3 Knopp MV, Runge VM. Off-label use and reimbursement of contrast media in MR. J Magn Reson Imaging. 1999;10(3):48-95.
4 http://www.businessweek.com/bwdaily/dnflash/content/sep2009/db2009092_913433.htm.
5 www.fda.gov/downloads/ICECI/EnforcementActions/WarningLetters/2000/UCM068176.pdf.
6 www.fda.gov/bbs/topics/NEWS/2007/NEW01638.html.
7 Doorenbos CJ, Ozyilmaz A, van Wijnen M. Severe pseudohypocalcemia after gadolinium-enhanced magnetic resonance angiography. N Engl J Med. 2004;350(1):87-88.
8 Choyke P, Knopp M. Pseudohypocalcemia with MR imaging contrast agents: a cautionary tale. Radiology. 2003;227:627-628.
9 Prince M, Erel H, Lent R, Blumenfeld J, et al. Gadodiamide administration causes spurious hypocalcemia. Radiology. 2003;227:639-646.
10 Prince MR, Choyke PL, Knopp MV. More on pseudohypocalcemia and gadolinium-enhanced MRI. N Engl J Med. 2004;350(1):87-88.
11 Becker N, Liebermann D, Wesch H, Van Kaick G. Mortality among Thorotrast-exposed patients and an unexposed comparison group in the German Thorotrast study. Eur J Cancer. 2008;44(9):1259-1268.
12 www.fda.gov/safety/MedWatch/default.htm.
13 Knopp M, Balzer T, Esser M, et al. Assessment of utilization and pharmacovigilance based on spontaneous adverse event reporting of gadopentetate dimeglumine as a magnetic resonance contrast agent after 45 million administrations and 15 years of clinical use. Invest Radiol. 2006;41:491-499.
14 Niendorf HP, Haustein J, Corenlius I, et al. Safety of gadolinium-DTPA: extended clinical experience. Magn Reson Med. 1991;22(2):222-228.
15 Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17:2359-2362.
16 Knopp M, Kirchin M. Contrast agents for magnetic resonance angiography: current status and future perspectives. In Arlart I, Bongartz G, Marchal G, editors: Magnetic Resonance Angiography, 2nd rev ed., Berlin: Springer, 2005.
17 Knopp MV, Schoenberg SO, Rehm C, et al. Assessment of gadobenate dimeglumine for magnetic resonance angiography: phase I studies. Invest Radiol. 2002;37(12):706-715.
18 Knopp MV, Giesel FL, von Tengg-Kobligk H, et al. Contrast-enhanced MR angiography of the run-off vasculature: intraindividual comparison of gadobenate dimeglumine with gadopentetate dimeglumine. J Magn Reson Imaging. 2003;17(6):694-702.
19 Hartman M, Wiethoff A, Hentrich H, Rohrer M. Initial imaging recommendations for vasovist angiography. Eur Radiol [Suppl 2]. 2006:B15-BB23.
20 de Haën C, Anelli PL, Lorusso V, et al. Gadocoletic acid trisodium salt (b22956/1): a new blood pool magnetic resonance contrast agent with application in coronary angiography. Invest Radiol. 2006;41(3):279-291.
21 Anderson S, Glod J, Arbab A. Noninvasive MR imaging of magnetically labeled stem cells to identify neovasculature in a glioma model. Blood. 2005;105:420-425.
22 Bellin M, Roy C, Kinkel K, Thoumas D, et al. Lymph node metastases: safety and effectiveness of mr imaging with ultrasmall superparamagnetic iron oxide particles—initial clinical experience. Radiology. 1998;207:799-808.
23 Harisinghani M, Barentsz J, Hahn P, et al. Noninvasive detection of clinically occult lymph-node metastases in prostate cancer. N Engl J Med. 2003;348:2491-2499.
