[level-membership-for-cardiovascular-category]
CHAPTER 28 Congenital Cardiac Surgery
Pediatric cardiology as a specific discipline can track its beginnings to the first ligation of a patent ductus arteriosus by Gross in 1938.1 Much anatomic research had been done up to that time, but surgical treatment was now an option (Table 28-1). In 1945, Crafoord and Nylin2 reported the first surgical repair of coarctation of the aorta, and in the same year, surgical palliation of tetralogy of Fallot with an aortopulmonary shunt was described by Taussig and Blalock.
TABLE 28-1 Chronology of Selected Milestones in Pediatric Cardiology
| 1936 | Maude Abbott publishes landmark atlas with historical data on patients with congenital heart disease. |
| 1939 | Gross and Hubbard publish case reports of a 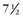 -year-old patient with successful ligation of a patent ductus. -year-old patient with successful ligation of a patent ductus. |
| 1945 | Crafoord and Nylin publish report of successful coarctation repair in two patients. |
| 1945 | Blalock and Taussig publish report of successful shunts in three tetralogy patients. |
| 1949 | Janeway invites Nadas to develop pediatric cardiology at Boston Children’s Hospital. |
| 1955 | Kirklin and associates report open heart surgery in eight patients with congenital heart disease. |
| 1964 | Mustard reports atrial repair of transposition of the great vessels in a 23-month-old. |
| 1966 | Raskind and Miller report balloon atrial septostomy in three infants. |
| 1966 | Ross and Somerville report homograft repair of pulmonary atresia in an 8-year-old. |
| 1968 | McGoon and associates report repair of truncus in an 8-year-old. |
| 1971 | Fontan and Baudet report successful repair of tricuspid atresia in two of three patients aged 12, 23, and 35 years. |
| 1975 | Jatene and associates report arterial switch for transposition of the great arteries. |
| 1975 | Norwood and associates report successful palliatives of hypoplastic left heart syndrome in two of three patients. Early 1970s M-mode echocardiography begins widespread use. |
| 1975 | Elliot and associates report ductal dilation with prostaglandin E in two patients. |
| 1976 | Bargeron and associates describe axial cineangiography; late 1970s, echocardiography is introduced. |
| 1982 | Kan and associates report percutaneous valvuloplasty for valvular pulmonary stenosis. |
| 1980s-1990s | Doppler studies, color flow, fetal studies, and transesophageal echocardiography become vital part of pediatric cardiology. |
| 1980s | Explosion of studies show possibility and success of percutaneous treatment of pulmonary artery stenosis, coarctation, and aortic stenosis. |
| 1990s | Interventional catheterization therapy for patent ductus arteriosus, pulmonary and aortic stenosis, pulmonary artery stenosis, and many atrial septal defects becomes standard part of management. |
From Graham TP Jr. Minimizing the morbidity of pediatric cardiovascular disease—historical perspective; pediatric cardiology. Prog Pediatr Cardiol 2005; 20:1-6.
For the repair of intracardiac defects, cardiopulmonary bypass was needed, and in 1955, Lillehei3 reported successful repair of ventricular septal defect, atrioventricular septal defect, and tetralogy of Fallot with use of this human cross-circulating technique. Kirklin4 demonstrated the successful use of mechanical cardiopulmonary bypass, reporting eight cases in 1955.
The development of prostaglandins has had an impact on pediatric cardiology and cardiac surgery most significantly. The introduction of prostaglandin E1 in routine clinical use in the mid-1970s5 has allowed proper diagnosis in a timely fashion of a child with congenital heart disease while permitting further clinical stabilization and refinement of the medical management and surgical intervention.
With imaging, cardiac catheterization was a necessary advance for the diagnosis and treatment of congenital cardiac defects, and by the 1950s,6 many centers were routinely studying children with heart defects and planning surgical interventions on the basis of these studies. However, the development of two-dimensional echocardiography and color flow Doppler imaging by the 1980s significantly changed the ability to diagnose infants and children with heart disease and refined the ability of surgeons to perform more complex procedures in infants and young children. Three- and four-dimensional multiplanar echocardiography is a developing imaging modality that is affecting how we visualize intracardiac anatomy and great vessel disease, and it is rapidly becoming an expectation of the surgeon as surgical intervention is planned.
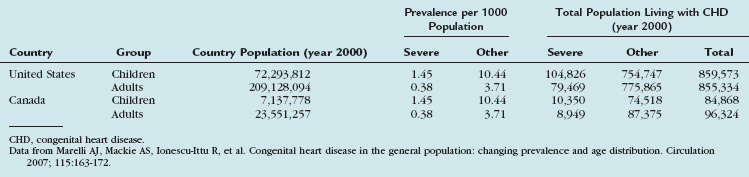
The more intriguing aspect of congenital heart disease is the fact that within the next few years, there will be more adults with congenital heart disease than children with congenital heart disease (Table 28-2).7 Survival to adulthood with a diagnosis of congenital heart disease is now an expectation.
TABLE 28-2 Extrapolated Prevalence of Adult Congenital Heart Disease in the General Population and Extrapolated Numbers to the United States and Canada
This chapter serves as a general overview of congenital heart disease, surgical considerations, and imaging strategies. More detailed aspects of these defects (Table 28-3) are addressed in subsequent chapters.
TABLE 28-3 Relative Frequency of Major Congenital Heart Lesions*
| Lesion | Percentage of all Lesions |
|---|---|
| Ventricular septal defect | 35-30 |
| Atrial septal defect (secundum) | 6-8 |
| Patent ductus arteriosus | 6-8 |
| Coarctation of aorta | 5-7 |
| Tetralogy of Fallot | 5-7 |
| Pulmonary valve stenosis | 5-7 |
| Aortic valve stenosis | 4-7 |
| D-Transposition of great arteries | 3-5 |
| Hypoplastic left ventricle | 1-3 |
| Hypoplastic right ventricle | 1-3 |
| Truncus arteriosus | 1-2 |
| Total anomalous pulmonary venous return | 1-2 |
| Tricuspid atresia | 1-2 |
| Single ventricle | 1-2 |
| Double-outlet right ventricle | 1-2 |
| Others | 5-10 |
* Excluding patent ductus arteriosus in preterm neonates, bicuspid aortic valve, physiologic peripheral pulmonic stenosis, and mitral valve prolapse.
SURGERY FOR ACYANOTIC CONGENITAL HEART LESIONS WITH A SHUNT
Description and Special Anatomic Considerations
Acyanotic congenital heart disease (Table 28-4) is characterized by a lack of cyanosis. In further defining these disorders, they are often classified on the basis of the presence or absence of left-to-right shunt.
TABLE 28-4 Acyanotic Congenital Heart Disease with a Left-to-Right Shunt
|
Atrial septal defect: causes diastolic overload of the right ventricle and increased pulmonary blood flow
|
Ventricular septal defect (VSD), the most common form of congenital heart disease, represents approximately one third of all major congenital cardiac defects. VSDs are generally classified into one of four groups, depending on their location in the interventricular septum (Fig. 28-1). These may be associated with other cardiac defects, such as atrioventricular valve defects, coarctation of the aorta, and other left-to-right shunts. The ventricular septum anatomy is complex, and many associated anatomic structures are key in the consideration of the repair, such as location of the conduction system of the heart.
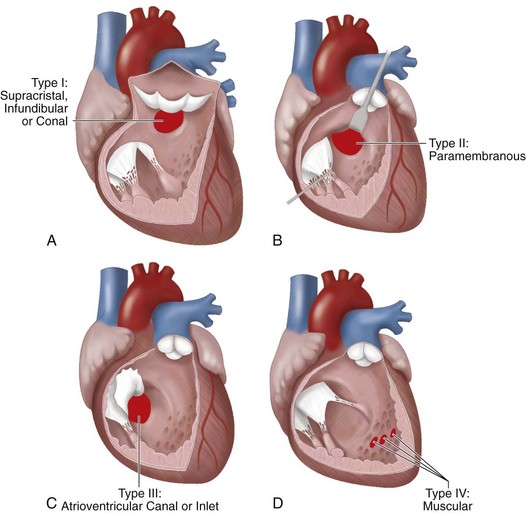
 FIGURE 28-1
FIGURE 28-1Indications
In this broad category of defects, the usual driving indication for early surgical intervention is congestive heart failure and how difficult it is to control with medical management. This is balanced against the surgical risks for the various procedures and the possible comorbidities that may exist as a part of a clinical syndrome or initial clinical presentation. An infant’s weight and gestational age also may play a role in the surgical timing for many of these defects. A typical scenario for surgical intervention based on prematurity and significant lung disease as a result of left-to-right shunt is a patent ductus arteriosus. The advent of indomethacin8 as medical management for closure of these defects has significantly reduced the need for surgical intervention.9 However, in extreme prematurity, renal disease, and severe diastolic “runoff” through a large patent ductus arteriosus, surgery may be a relative emergency.
For example, surgical timing for closure of a hemodynamically significant VSD may be indicated at a few weeks or as late as a few years. The later closure may be indicated for a late-identified supracristal VSD or a perimembranous VSD with a coexistent subaortic membrane or prolapse of the aortic valve leaflets. Both of these defects have been implicated in progressive aortic valve damage,10,11 even though they may be quite small and have no risks for long-term pulmonary vascular disease or congestive heart failure.
With the diagnosis of AVSD, there can be tremendous variability of the surgical timing based on the level of shunting (i.e., atrial vs. ventricular), the complexity of the associated common atrioventricular valve disease, and the associated clinical status (Fig. 28-2).
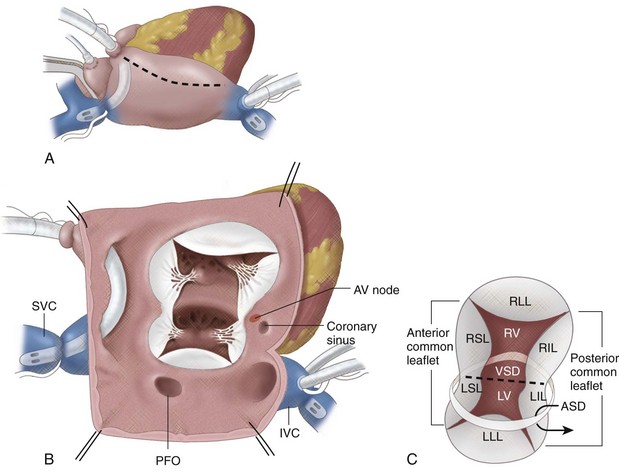
 FIGURE 28-2
FIGURE 28-2Imaging Findings
Preoperative Planning
The role of MRI is in diagnosis of anomalous pulmonary venous drainage in complex defects and particularly in the sinus venosus atrial septal defects, in which the incidence of partial anomalous pulmonary venous return may be as high as 85%.12 Their location within the mediastinum may make reliable echocardiographic diagnosis impossible, particularly in the operating room at the time of operative intervention.
SURGERY FOR ACYANOTIC CONGENITAL HEART LESIONS WITHOUT A SHUNT
Description and Special Anatomic Considerations
Acyanotic heart defects without a shunt generally have normal oxygen saturations at rest, unless they suffer from an A-a gradient secondary to pulmonary vascular congestion from their cardiac defect. These defects5 include left-sided and right-sided heart obstructive lesions and regurgitant valve disease (Table 28-5).
TABLE 28-5 Acyanotic Congenital Heart Disease without a Shunt
|
Left-Sided Heart Malformations |
Coarctation of the aorta may occur as an isolated defect or in association with various other lesions, most commonly bicuspid aortic valve and VSD.13 Surgical repair of coarctation of the aorta has been one of the oldest surgical procedures available. Early limitations revolved around the size or weight of the infant, but these considerations have virtually disappeared. The controversy that continues to smolder is the role of balloon dilation or stent for the native coarctation of the aorta. Additional intracardiac defects, such as a posterior malaligned VSD, may complicate the surgery and the surgical approach. Complete repair has become the generally accepted approach to these complex defects because the prior approach of repair of the coarctation of the aorta and pulmonary artery banding has resulted in complications such as double-outlet obstruction early postoperatively and a more complicated medical and surgical management approach long term.
The extreme forms of obstructive left-sided heart lesions include hypoplastic left heart syndrome and its many variants.14 These patients can have varying degrees of hypoplasia or atresia of the left ventricle, aorta, and mitral or aortic valves, usually in some combination thereof. They all have coarctation of the aorta. Because these are duct-dependent lesions, they tend to be manifested in the first few days of life, although they may on occasion present late at several weeks of age. It is a lethal condition. Norwood first presented his approach to palliation of this entity in 1979. It essentially created a hemodynamically stable single ventricle, which could proceed down the single ventricle pathway of palliation with a bidirectional caval anastomosis and a subsequent Fontan procedure.
Anomalies of the tricuspid valve, such as Ebstein anomaly,15 tend to be manifested with cyanosis in the newborn period, and the infants can be quite ill. If acyanotic, they are typically diagnosed after an echocardiogram is obtained as part of evaluation of a murmur. The diagnosis of Ebstein anomaly may be a coincident finding made as part of a work-up for Wolff-Parkinson-White syndrome–mediated supraventricular tachycardia. The mean age at diagnosis of the acyanotic forms of this disease is the middle teenage years. These anomalies are almost uniformly associated with atrial septal defects.
Indications and Contraindications
Catheter-based intervention for severe aortic valve stenosis in the newborn16 is typically the preferred option. However, often less than perfect reduction in the stenosis gradient is tolerated such that one minimizes the risks for significant aortic insufficiency.
Aortic insufficiency is a much more difficult disease to treat in infants and children. A variety of clinical parameters have been used to prompt surgical intervention.17 Decreasing clinical activity level, decreasing left ventricular performance (as measured by echocardiography or MRI), and progressive left ventricular dilation and secondary mitral insufficiency or left atrial hypertension have been considered useful in formulating surgical intervention plans.
The Ross procedure has been an effective surgical procedure to provide good relief of aortic valve disease in children while avoiding many of the long-term medical management concerns of a mechanical aortic valve replacement. However, this has some significant controversy associated with it.18 More recent surgical approaches have aimed at “reconstruction” of the valve leaflets, although this is controversial as well and appears to be dependent on the surgical center’s abilities.
Pulmonary valve stenosis has long been a disease that lends itself to interventional catheterization procedures.19 More recent use of radiofrequency catheters to perforate pulmonary atresia, as a method to pass a wire and then a balloon catheter across such an obstruction to achieve some degree of palliation, is an exciting advancement in the hands of the skilled interventionalist. Surgical open pulmonary valvotomy is still an option, but it is often reserved for unique circumstances at this time.
SURGERY FOR CYANOTIC CONGENITAL HEART LESIONS WITH INCREASED PULMONARY BLOOD FLOW
Description and Special Anatomic Considerations
These congenital cardiac lesions are characterized by cyanosis or bluish coloration of the skin due to arterial oxygen desaturation resulting from the shunting of systemic venous blood to the arterial circulation. To help understand the basic physiology involved in these lesions, they are typically classified by degree of pulmonary blood flow (Table 28-6).
TABLE 28-6 Cyanotic Congenital Heart Disease with Increased Pulmonary Blood Flow
Complete transposition of the great arteries is the most common cyanotic congenital heart lesion that presents in neonates.20 This entity was first described more than 200 years ago. However, until the development of the surgical atrial septectomy in the 1950s and the balloon atrial septostomy in the 1960s, early death was expected. These palliative interventions allowed the development of physiologic palliative procedures, such as the atrial switch operation (Mustard and Senning),21 and later the anatomic correction with the arterial switch procedure. Although survival rates for the arterial switch procedure approach 95%, significant anatomic variations influence the outcome. These include associated cardiac anomalies, relationship of the great arteries to each other, and coronary artery anatomy variants.22 Echocardiography is the mainstay of this diagnosis, which is one that often may be made in utero by fetal echocardiography.
Single-ventricle defects are rare defects and can present with either increased or decreased pulmonary blood flow. To strictly fit this diagnosis, the single ventricle is missing the nontrabeculated inflow region of either ventricle. This differentiates these defects from tricuspid atresia patients, who typically have a smooth inlet of the remaining ventricle. These cardiac defects invariably have other associated cardiac defects, such as complex outflow tract obstruction or interrupted aortic arch. They also may have significant noncardiac congenital defects.23 Two-dimensional echocardiography is diagnostic for single ventricle and usually provides most detail of the associated cardiac defects.
Indications
Complete repair of the patient with transposition of the great arteries with the arterial switch operation is the goal of the experienced cardiothoracic surgeon. The age at which to attempt complete repair is generally within the first 1 to 2 weeks of life because the left ventricular function and mass change as pulmonary vascular resistance changes with age. However, many variables have an impact on these decisions.24
The single-ventricle pathway,25 as it has become known, requires careful medical and surgical management of pulmonary vascular resistance and single-ventricle systolic function, with an attempt to preserve single-ventricle diastolic function. The staged approach to ultimate palliation with a Fontan variant repair of these defects is now common. These palliative procedures may include pulmonary artery banding, pulmonary artery transection and creation of systemic-to-pulmonary artery shunt, and the Damus-Kaye-Stansel approach for selected defects. Caval anastomoses, hemi-Fontan, and the Fontan variant are the typical considerations for subsequent surgery for these patients. However, when this palliation fails or the poor hemodynamics of the patient preclude consideration of a Fontan intervention, cardiac transplantation remains a viable option.
Outcomes and Complications
The single-ventricle patients have more issues involving close follow-up of the passive cavopulmonary circuits, which are necessary to provide adequate cardiac output and oxygenation of blood.26 The role of MRI and, for selected patients, CT has become common as a regular method to serially observe them longitudinally and to plan for any necessary interventions.
SURGERY FOR CYANOTIC CONGENITAL HEART LESIONS WITH DECREASED PULMONARY BLOOD FLOW
Description and Special Anatomic Considerations
The typical congenital cardiac lesions considered in this category of defects include tetralogy of Fallot, tricuspid atresia, and Ebstein anomaly (Table 28-7). Acyanotic patients can also present these defects, as noted before, and their manifestations are influenced by the degree of pulmonary blood flow.
TABLE 28-7 Cyanotic Congenital Heart Disease with Decreased Pulmonary Blood Flow
Indications
Definitive surgical repair for the patients with tetralogy of Fallot has become the goal of the pediatric cardiologist and cardiothoracic surgeon.27 However, palliation is still needed for a variety of complicating cardiac and extracardiac issues.
Tricuspid atresia patients are almost always confined to a single-ventricle palliation pathway. This may involve a Blalock-Taussig shunt to augment pulmonary blood flow but limit damage to the pulmonary vascular bed for chronic pressure or volume overload of the pulmonary arteries.28
Outcomes and Complications
Tricuspid atresia patients tend to do quite well with the single-ventricle pathway for palliation. Because these patients have a normally functioning left ventricle, they tend to be the ideal patients for excellent long-term survival after the Fontan procedure, with a good quality of life. Three patients with tricuspid atresia were the first to undergo the Fontan operation, which he published in 1971. It is not uncommon for these patients who have done well to be able to successfully carry a pregnancy to term with little adverse hemodynamic effect. However, this does require close pre-conception planning and cardiac surveillance during the pregnancy.29





Keane JF, Fyler DC, Lock JE. Nadas’ Pediatric Cardiology, 2nd ed. Philadelphia: Saunders; 2006.
Lai W, Mertens L, Cohen M, et al, editors. Echocardiography in Pediatric and Congenital Heart Disease: From Fetus to Adult. Hoboken, NJ: Wiley-Blackwell, 2009.
Nichols DG, Cameron DE, editors. Critical Heart Disease in Infants and Children, 2nd ed, St Louis: Mosby, 2006.
1 Gross RE, Hubbard JP. Surgical ligation of a patent ductus arteriosus. Report of first successful case. JAMA. 1939;112:729-731.
2 Crafoord C, Nylin G. Congenital coarctation of the aorta and its surgical treatment. J Thorac Surg. 1945;14:347-361.
3 Lillehei CW, Cohen M, Warn HE, Varco RL. The direct vision intracardiac correction of congenital anomalies by controlled cross circulation. Surgery. 1955;38:11-29.
4 Kirklin JW, DuShane JW, Patrick RT, et al. Intracardiac surgery with the aid of a mechanical pump oxygenator system (Gibbon type): report of 8 cases. Proc Staff Meet Mayo Clin. 1955;30:201-206.
5 Olley PM, Coceani F, Bodach E. E-type prostaglandin—a new emergency therapy for certain congenital cardiac malformations. Circulation. 1976;53:728-731.
6 Bargeron LM, Elliott LP, Soto B, et al. Axial cineangiography in congenital heart disease. Circulation. 1977;56:1075-1083.
7 Marelli AJ, Mackie AS, Ionescu-Ittu R, et al. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation. 2007;115:163-172.
8 Douidar SM, Richardson J, Snodgrass WR. Role of indomethacin in ductus closure: an update evaluation. Dev Pharmacol Ther. 1988;11:196-212.
9 Malviya M, Ohlsson A, Shah S. Surgical versus medical treatment with cyclooxygenase inhibitors for symptomatic patent ductus arteriosus in preterm infants. Cochrane Database Syst Rev 2008; 1:CD003951.
10 Mori K, Matsuoka S, Tatara K, et al. Echocardiographic evaluation of the development of aortic valve prolapse in supracristal ventricular septal defect. Eur J Pediatr. 1995;154:176-181.
11 Tweddell JS, Pelech AN, Frommelt PC. Ventricular septal defect and aortic valve regurgitation: pathophysiology and indications for surgery. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2006:147-152.
12 Dellegrottaglie S, Pedrotti P, Pedretti S, et al. Atrial septal defect combined with partial anomalous pulmonary venous return: complete anatomic and functional characterization by cardiac magnetic resonance. J Cardiovasc Med (Hagerstown). 2008;9:1184-1186.
13 Syamasundar P, Rao MD Coarctation of the aorta Available at http://emedicine.medscape.com/article/895502-overview
14 Wernovsky G, Ghanayem N, Ohye RG, et al. Hypoplastic left heart syndrome: consensus and controversies in 2007. Cardiol Young. 2007;17(Suppl 2):75-86.
15 Paranon S, Acar P. Ebstein’s anomaly of the tricuspid valve: from fetus to adult: congenital heart disease. Heart. 2008;94:237-243.
16 Zain Z, Zadinello M, Menahem S, Brizard C. Neonatal isolated critical aortic valve stenosis: balloon valvuloplasty or surgical valvotomy. Heart Lung Circ. 2006;15:18-23. Epub 2005 Jul 25
17 Backer CL. Techniques for repairing the aortic and truncal valves. Cardiol Young. 2005;15(Suppl 1):125-131.
18 Luciani GB, Mazzucco A. Aortic root disease after the Ross procedure. Curr Opin Cardiol. 2006;21:555-560.
19 Kutty S, Zahn EM. Interventional therapy for neonates with critical congenital heart disease. Catheter Cardiovasc Interv. 2008;72:663-674.
20 Skinner J, Hornung T, Rumball E. Transposition of the great arteries: from fetus to adult. Heart. 2008;94:1227-1235.
21 Mustard WT. Successful two-stage correction of transposition of the great vessels. Surgery. 1964;55:469-472.
22 Angelini P, de la Cruz MV, Valencia AM, et al. Coronary arteries in transposition of the great arteries. Am J Cardiol. 1994;74:1037-1041.
23 Cohen MS, Anderson RH, Cohen MI, et al. Controversies, genetics, diagnostic assessment, and outcomes relating to the heterotaxy syndrome. Cardiol Young. 2007;17(Suppl 2):29-43.
24 Warnes CA. Transposition of the great arteries (review). Circulation. 2006;114:2699-2709.
25 Rodefeld MD, Ruzmetov M, Schamberger MS, et al. Staged surgical repair of functional single ventricle in infants with unobstructed pulmonary blood flow. Eur J Cardiothorac Surg. 2005;27:949-955.
26 Driscoll DJ. Long-term results of the Fontan operation. Pediatr Cardiol. 2007;28:438-442.
27 van Doorn C. The unnatural history of tetralogy of Fallot: surgical repair is not as definitive as previously thought. Heart. 2002;88:447-448.
28 Warnes CA. Tricuspid atresia and univentricular heart after the Fontan procedure. Cardiol Clin. 1993;11:665-673.
29 Drenthen W, Pieper PG, Roos-Hesselink JW, et al. Pregnancy and delivery in women after Fontan palliation. Heart. 2006;92:1290-1294. Epub 2006 Jan 31
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
CHAPTER 28 Congenital Cardiac Surgery
Pediatric cardiology as a specific discipline can track its beginnings to the first ligation of a patent ductus arteriosus by Gross in 1938.1 Much anatomic research had been done up to that time, but surgical treatment was now an option (Table 28-1). In 1945, Crafoord and Nylin2 reported the first surgical repair of coarctation of the aorta, and in the same year, surgical palliation of tetralogy of Fallot with an aortopulmonary shunt was described by Taussig and Blalock.
TABLE 28-1 Chronology of Selected Milestones in Pediatric Cardiology
| 1936 | Maude Abbott publishes landmark atlas with historical data on patients with congenital heart disease. |
| 1939 | Gross and Hubbard publish case reports of a -year-old patient with successful ligation of a patent ductus. |
| 1945 | Crafoord and Nylin publish report of successful coarctation repair in two patients. |
| 1945 | Blalock and Taussig publish report of successful shunts in three tetralogy patients. |
| 1949 | Janeway invites Nadas to develop pediatric cardiology at Boston Children’s Hospital. |
| 1955 | Kirklin and associates report open heart surgery in eight patients with congenital heart disease. |
| 1964 | Mustard reports atrial repair of transposition of the great vessels in a 23-month-old. |
| 1966 | Raskind and Miller report balloon atrial septostomy in three infants. |
| 1966 | Ross and Somerville report homograft repair of pulmonary atresia in an 8-year-old. |
| 1968 | McGoon and associates report repair of truncus in an 8-year-old. |
| 1971 | Fontan and Baudet report successful repair of tricuspid atresia in two of three patients aged 12, 23, and 35 years. |
| 1975 | Jatene and associates report arterial switch for transposition of the great arteries. |
| 1975 | Norwood and associates report successful palliatives of hypoplastic left heart syndrome in two of three patients. Early 1970s M-mode echocardiography begins widespread use. |
| 1975 | Elliot and associates report ductal dilation with prostaglandin E in two patients. |
| 1976 | Bargeron and associates describe axial cineangiography; late 1970s, echocardiography is introduced. |
| 1982 | Kan and associates report percutaneous valvuloplasty for valvular pulmonary stenosis. |
| 1980s-1990s | Doppler studies, color flow, fetal studies, and transesophageal echocardiography become vital part of pediatric cardiology. |
| 1980s | Explosion of studies show possibility and success of percutaneous treatment of pulmonary artery stenosis, coarctation, and aortic stenosis. |
| 1990s | Interventional catheterization therapy for patent ductus arteriosus, pulmonary and aortic stenosis, pulmonary artery stenosis, and many atrial septal defects becomes standard part of management. |
From Graham TP Jr. Minimizing the morbidity of pediatric cardiovascular disease—historical perspective; pediatric cardiology. Prog Pediatr Cardiol 2005; 20:1-6.
For the repair of intracardiac defects, cardiopulmonary bypass was needed, and in 1955, Lillehei3 reported successful repair of ventricular septal defect, atrioventricular septal defect, and tetralogy of Fallot with use of this human cross-circulating technique. Kirklin4 demonstrated the successful use of mechanical cardiopulmonary bypass, reporting eight cases in 1955.
The development of prostaglandins has had an impact on pediatric cardiology and cardiac surgery most significantly. The introduction of prostaglandin E1 in routine clinical use in the mid-1970s5 has allowed proper diagnosis in a timely fashion of a child with congenital heart disease while permitting further clinical stabilization and refinement of the medical management and surgical intervention.
With imaging, cardiac catheterization was a necessary advance for the diagnosis and treatment of congenital cardiac defects, and by the 1950s,6 many centers were routinely studying children with heart defects and planning surgical interventions on the basis of these studies. However, the development of two-dimensional echocardiography and color flow Doppler imaging by the 1980s significantly changed the ability to diagnose infants and children with heart disease and refined the ability of surgeons to perform more complex procedures in infants and young children. Three- and four-dimensional multiplanar echocardiography is a developing imaging modality that is affecting how we visualize intracardiac anatomy and great vessel disease, and it is rapidly becoming an expectation of the surgeon as surgical intervention is planned.
The more intriguing aspect of congenital heart disease is the fact that within the next few years, there will be more adults with congenital heart disease than children with congenital heart disease (Table 28-2).7 Survival to adulthood with a diagnosis of congenital heart disease is now an expectation.
TABLE 28-2 Extrapolated Prevalence of Adult Congenital Heart Disease in the General Population and Extrapolated Numbers to the United States and Canada
This chapter serves as a general overview of congenital heart disease, surgical considerations, and imaging strategies. More detailed aspects of these defects (Table 28-3) are addressed in subsequent chapters.
TABLE 28-3 Relative Frequency of Major Congenital Heart Lesions*
| Lesion | Percentage of all Lesions |
|---|---|
| Ventricular septal defect | 35-30 |
| Atrial septal defect (secundum) | 6-8 |
| Patent ductus arteriosus | 6-8 |
| Coarctation of aorta | 5-7 |
| Tetralogy of Fallot | 5-7 |
| Pulmonary valve stenosis | 5-7 |
| Aortic valve stenosis | 4-7 |
| D-Transposition of great arteries | 3-5 |
| Hypoplastic left ventricle | 1-3 |
| Hypoplastic right ventricle | 1-3 |
| Truncus arteriosus | 1-2 |
| Total anomalous pulmonary venous return | 1-2 |
| Tricuspid atresia | 1-2 |
| Single ventricle | 1-2 |
| Double-outlet right ventricle | 1-2 |
| Others | 5-10 |
* Excluding patent ductus arteriosus in preterm neonates, bicuspid aortic valve, physiologic peripheral pulmonic stenosis, and mitral valve prolapse.
SURGERY FOR ACYANOTIC CONGENITAL HEART LESIONS WITH A SHUNT
Description and Special Anatomic Considerations
Acyanotic congenital heart disease (Table 28-4) is characterized by a lack of cyanosis. In further defining these disorders, they are often classified on the basis of the presence or absence of left-to-right shunt.
TABLE 28-4 Acyanotic Congenital Heart Disease with a Left-to-Right Shunt
|
Atrial septal defect: causes diastolic overload of the right ventricle and increased pulmonary blood flow
|
Ventricular septal defect (VSD), the most common form of congenital heart disease, represents approximately one third of all major congenital cardiac defects. VSDs are generally classified into one of four groups, depending on their location in the interventricular septum (Fig. 28-1). These may be associated with other cardiac defects, such as atrioventricular valve defects, coarctation of the aorta, and other left-to-right shunts. The ventricular septum anatomy is complex, and many associated anatomic structures are key in the consideration of the repair, such as location of the conduction system of the heart.
Indications
In this broad category of defects, the usual driving indication for early surgical intervention is congestive heart failure and how difficult it is to control with medical management. This is balanced against the surgical risks for the various procedures and the possible comorbidities that may exist as a part of a clinical syndrome or initial clinical presentation. An infant’s weight and gestational age also may play a role in the surgical timing for many of these defects. A typical scenario for surgical intervention based on prematurity and significant lung disease as a result of left-to-right shunt is a patent ductus arteriosus. The advent of indomethacin8 as medical management for closure of these defects has significantly reduced the need for surgical intervention.9 However, in extreme prematurity, renal disease, and severe diastolic “runoff” through a large patent ductus arteriosus, surgery may be a relative emergency.
For example, surgical timing for closure of a hemodynamically significant VSD may be indicated at a few weeks or as late as a few years. The later closure may be indicated for a late-identified supracristal VSD or a perimembranous VSD with a coexistent subaortic membrane or prolapse of the aortic valve leaflets. Both of these defects have been implicated in progressive aortic valve damage,10,11 even though they may be quite small and have no risks for long-term pulmonary vascular disease or congestive heart failure.
With the diagnosis of AVSD, there can be tremendous variability of the surgical timing based on the level of shunting (i.e., atrial vs. ventricular), the complexity of the associated common atrioventricular valve disease, and the associated clinical status (Fig. 28-2).
