[level-membership-for-pediatrics-category]Chapter 31
Congenital Brain Malformations
With advances in magnetic resonance imaging (MRI) and molecular biology and the availability of mutant mouse models of human cortical malformations, many malformations of brain development have been reclassified.1–3 Given the large number of congenital human brain malformations, the complexity of the molecular genetics, and the degree of anatomic variability, an in-depth discussion of some malformations is beyond the scope of this chapter. This chapter addresses malformations seen most often in clinical practice and those malformations that are less common but are distinctive and have a profound impact of early childhood development. The malformations presented here are grouped according to presumed dominant defect in embryologic or fetal development. Diffusion tensor imaging (DTI) is included in the diagnosis of malformations in which aberrant white matter tracts are a dominant feature.
A rudimentary description of relevant embryology is needed. Around 26 to 28 days after conception, the process of neurulation, in which the lateral edges of the neural plate elevate into neural folds, takes place.1 The folds then fuse medially to form the neural tube. At the cranial end of the neural tube, the primitive brain vesicles form and include the prosencephalon, mesencephalon, and rhombencephalon.1,2,4 The prosencephalon divides into the telencephalon, which gives rise to the cerebral hemispheres and the corpus striatum, and the diencephalon, which gives rise to the thalami and the hypothalamus. The cerebral peduncles and midbrain arise from the mesenchephalon. The rhombencephalon gives rise to the metencephalon, which forms the pons and cerebellum, and the myelencephalon, from which the medulla arises. Malformations of brain development may result from chromosomal aberrations, single gene mutations, teratogenic infections and agents, or ischemia.1,3 However, in 70% of malformations, the etiology is identifiable.
Defects of Neurulation
Complete failure of closure of the cranial end of the neuropore results in anencephaly, in which the forebrain, skull, and scalp are absent. Affected patients die soon after birth and postnatal imaging is not usually indicated. Less severe disorders of neural tube closure result in meningoceles and encephaloceles, which are protrusions of the meninges or brain, respectively, through a congenital defect of the skull and dura; the latter occurs in about 1 per 5000 live births.4 Encephaloceles tend to be midline. In the countries of the Western hemisphere, a predominance of posterior cephaloceles is seen, whereas in Asian children, encephaloceles tend to be anterior.1 Frontal encephaloceles include interorbital frontal, nasofrontal, nasoethmoidal, and naso-orbital lesions. Interorbital frontal encephaloceles protrude through a defect in frontal bone (Fig. 31-1). Nasofrontal encephaloceles involve the region of nasal bridge, the floor of the anterior cranial fossa, or both (Fig. 31-2). Nasofrontal encephaloceles are also referred to as nasal gliomas, although they are not neoplastic. These masses of neuroglial tissue are categorized as extranasal (60%), intranasal (30%), or mixed (10%).1,2,4 When telangiectasias occur on the skin overlaying an external nasal glioma, the lesion may be mistaken for a hemangioma. Often, hypertelorism is present with a broad nasal bridge. Intranasal glioma presents as an intranasal mass; biopsy should be avoided prior to imaging because of the risk of meningitis. With nasoethmoidal encephaloceles, frontal bone is intact. Neural tissue bulges into the ethmoid sinus through a defect in the floor of the anterior cranial fossa, and the nasal septum defines the posterior margin. A defect in the medial orbital wall results in a naso-orbital encephalocele, which protrudes into the orbit and produces unilateral exophthalmos. Other facial anomalies seen with anterior encephaloceles include a bifid nasal tip or complete midline splitting of the nose in an uncommon malformation known as frontonasal dysplasia (e-Fig. 31-3). Morning Glory syndrome includes midline facial defects, callosal agenesis, frontal encephaloceles, and characteristic eye anomalies.
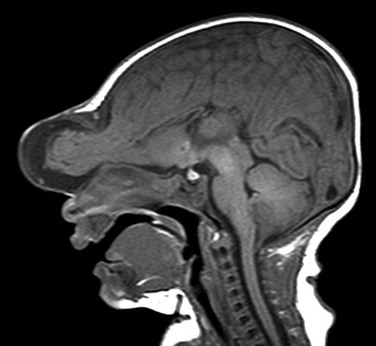
Figure 31-1 Frontal encephalocele.
A sagittal T1 image shows agenesis of the corpus callosum associated with encephalocele at the glabella.
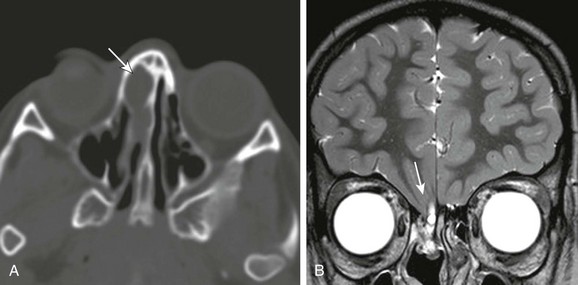
Figure 31-2 Incidental nasofrontal encephalocele found on head computed tomography done for trauma.
A, Axial computed tomography shows a focally expansile lesion within the right ethmoid sinus (arrow). B, A high-resolution coronal T2 image shows the clinically occult defect in the right cribriform plate through which has herniated the frontal cortex (arrow).
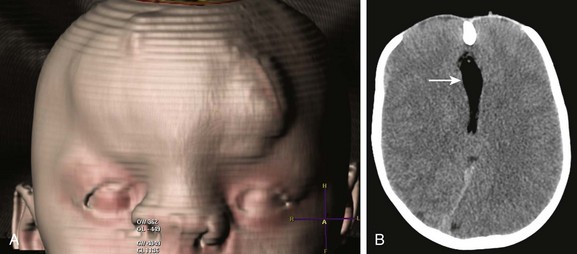
e-Figure 31-3 Median facial cleft.
A, Shaded surface rendering from computed tomography. The frontal encephalocele and median facial cleft are skin covered; marked hypertelorism and a broad nasal bridge are seen. B, Axial computed tomography shows the large defect in the frontal bone; an interhemispheric lipoma (arrow) associated with absence of the corpus callosum is seen.
Basal encephaloceles result from defects in the sphenoid bone (Fig. 31-4); the encephalocele herniates into the posterior nasopharynx anterior to the dorsum sella and may contain pituitary tissue, optic nerves, branches of the circle of Willis, or all (Fig. 31-5). Encephaloceles in the parietal bone range from large deforming “towering” lesions to small meningoceles (Fig. 31-6). Atretic encephaloceles or meningoceles present as small subcutaneous fibrofatty masses, which are often painful to palpation (Fig. 31-7). Occipital encephaloceles usually contain dysplastic cerebellar tissue alone or with the cerebral cortex (Fig. 31-8). Rarely, the brainstem may be within the encephalocele; this malformation is lethal.
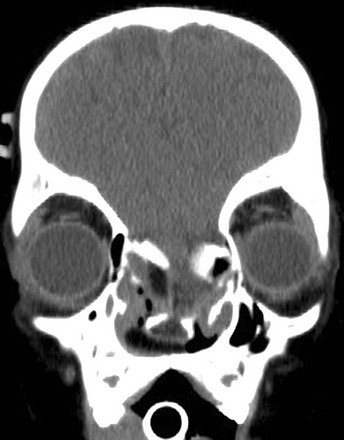
Figure 31-4 Basal encephalocele.
A coronal reformatted computed tomography image shows the floor of the anterior cranial fossa is deficient, the nasal turbinates are disorganized, and marked hypertelorism exists.
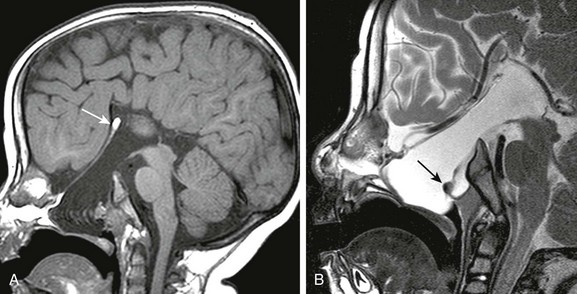
Figure 31-5 Basal encephalocele.
A, A sagittal T1-weighted image shows callosal agenesis with a tiny lipoma (arrow). A large defect in the basisphenoid is seen. Note the apparent absence of the pituitary, floor of the third ventricle, and optic pathways. B, A high-resolution sagittal T2-weighted image shows the pituitary-hypothalamic structures (arrow) and optic pathways are contained within the encephalocele.
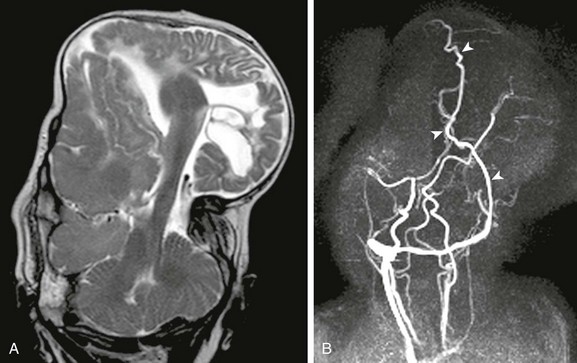
Figure 31-6 Parietal encephalocele.
A, A coronal T2-weighted image shows extremely dysmorphic hemisphere and diencephalic structures contained within the massive parietal encephalocele. B, Coronal maximum-intensity projection from two-dimensional magnetic resonance venogram shows dysplastic dural sinuses (arrowheads) contained within the encephalocele.
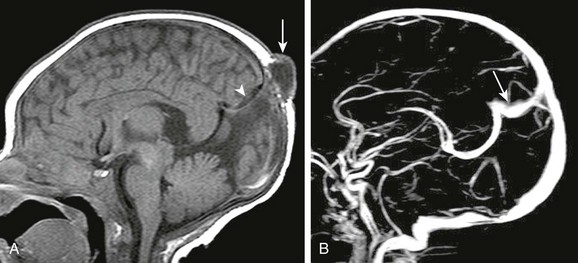
Figure 31-7 Atretic parietal meningocele.
A, A sagittal T1-weighted image shows the small meningocele (arrow) over the parietal vertex. Focal expansion of the posterior interhemispheric subarachnoid space is shown (arrowhead). B, A sagittal image from a gadolinium-enhanced magnetic resonance venogram shows a persistent falcine sinus (arrow) draining the deep medullary venous system; no straight sinus is present.
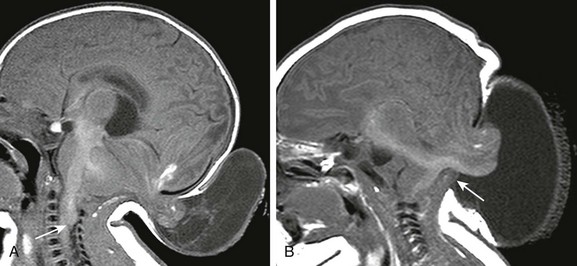
Figure 31-8 Occipital encephalocele.
A, A sagittal T1-weighted image shows an occipital encephalocele containing cerebrospinal fluid and dysplastic cerebellum. A beaked tectal plate and kinking of the cervicomedullary brainstem (arrow), similar to that seen in the Chiari II malformation, are present. B, A sagittal T1-weighted image in a different patient shows a large cephalocele containing both cerebellar tissue and brainstem (arrow); the patient died after closure of the defect.
Conditions and syndromes associated with encephaloceles include trisomy 13 and 18, amniotic band syndrome, Meckel-Gruber syndrome, dyssegmental dwarfism, Knobloch syndrome, Walker-Warburg (type II lissencephaly) syndrome, cryptophthalmos, and Voss syndrome. The prognosis depends on the severity of associated brain anomalies and the amount of dysplastic brain contained within the encephalocele.1,2,4
Imaging: High-resolution MRI is performed soon after birth to define contents of the encephalocele, the severity of the malformation, and the frequent associated anomalies. These include callosal agenesis, anomalies of cortical formation, and variable anomalies of the cerebellum, diencephalon, and brainstem. Although dysplastic nonfunctional neural tissue is usually resected during closure of the encephalocele, major dural venous sinuses are preserved. Therefore, magnetic resonance venography is essential for diagnosing large occipital and midline parietal encephaloceles that may contain dural venous sinuses. Basal encephaloceles containing optic chiasm or nerves or pituitary tissue cannot be closed without sacrificing these structures. Affected patients are at risk for meningitis and cerebrospinal fluid (CSF) leakage into the nasophayrnx. The typical intracranial manifestations of atretic parietal encephaloceles include posterior tenting of the tectal plate, a persistent falcine sinus with or without atresia of the straight sinus, and expansion of posterior interhemispheric CSF. Hydrocephalus is common after closure of large parietal and occipital encephaloceles.
Chiari II Malformations
Clinical Presentation: Chiari II malformations are the intracranial manifestations of posterior dysraphic defects such as myelomeningoceles. Neural tube defects have been reported with multiple chromosomal abnormalities, including trisomies 18, 13, and 9; triploidy; unbalanced translocations and deletions; and in Turner, DiGeorge, and velocardiofacial syndromes. The genes in the region of 22q11 have been implicated in the development of neural tube defects, although neural tube defects and the associated Chiari II malformation are probably caused by a combination of genetic polymorphisms and environmental factors, including dietary folate intake and maternal folate metabolism.5
The most commonly accepted unifying theory for the Chiari II malformation suggests that the failure of neural tube closure prevents the transient closure of the central canal, which is essential for the distension of the primitive ventricular system;6 this results in the premature fusion of the mesenchymal components that form the calvarium, whereas the hindbrain manifestations result from leakage of CSF through the neural tube defect. The Chiari II malformation is associated with a wide range of brain malformations;1,2,7,8 DTI shows disordered axonal migration, suggesting that the malformation is not explained by mechanical alterations caused by faulty CSF dynamics alone.8 The Chiari II malformation is characterized by mesodermal dysplasia, small and dysplastic lower cranial nerve ganglia, deficient tentorium cerebelli, hypoplastic and dysmorphic cerebellum, and thickened basal meninges.6 As in holoprosencephaly (HPE), these anomalies are attributable to a defective or deficient mesenchyme, which presumably deprives the skull base, hindbrain, and rhombencephalon of normal inductive effects.7 Clinical problems related specifically to the malformed hindbrain include apnea, aspiration, feeding difficulties, and recurrent respiratory infections.
Imaging: The calvarial manifestations of the Chiari II malformation include a bifid frontal bone and luckenschadel, or lacunar, skull. The latter is a manifestation of the mesodermal dysplasia of the membranous skull and is caused by nonossified fibrous bone in the inner and outer tables of the skull, resulting in the apparent cranial scalloping (Fig. 31-9); the affected cranium ossifies and appears normal by 6 months of age. Luckenschadel is not the result of increased intracranial pressure and is not synonymous with “the beaten copper skull.”
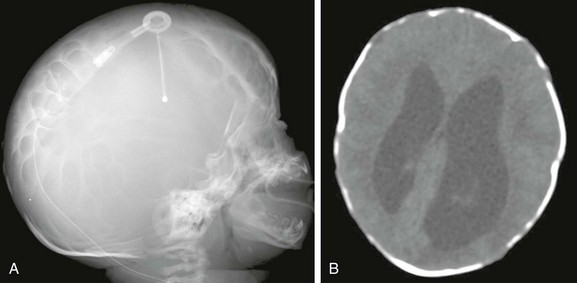
Figure 31-9 Luchenschadel.
A, A skull radiograph shows a lacelike appearance to the cranium. B, Axial computed tomography shows scalloping of the inner and outer tables of the calvarium.
The intracranial stigmata of the Chiari II malformation are complex and variable; the hallmarks of the malformation are infratentorial. In the most severe forms, the foramen magnum is enlarged and the posterior cranial fossa is constricted, with effacement of CSF spaces; the cerebellum wraps around the ventral aspect of the brainstem (see Fig. 31-9), and the clivus and petrous ridges are concave. MRI shows caudal displacement of a dysplastic brainstem and a hypoplastic cerebellum into the upper cervical canal with “kinking” of the cervicomedullary brainstem (Fig. 31-10). The fourth ventricle is effaced and caudally displaced. The torcula and transverse sinuses are low lying, which presents a potential surgical hazard during decompressive suboccipital craniectomy, which may be performed for relief of symptomatic hindbrain compression. A “beaked” tectal plate is present, and a variably thickened massa intermedia may be so thick that the thalami appear virtually fused with partial atresia of the third ventricle.1,7 Common supratentorial abnormities include callosal dysgenesis, neuronal migration anomalies, and hydrocephalus. After ventricular shunt placement, the cerebral hemispheres drop away from the inner table of the skull, allowing interdigitations of the cortex across the midline under the hypoplastic falx and projection of the superior cerebellar vermis superiorly across a widened tentorial incisura. These findings are nonspecific manifestations seen after CSF diversion of severe congenital obstructive hydrocephalus. DTI of Chiari II malformations associated with more severe degrees of cerebellar hypoplasia shows absence of the dorsal transverse fibers at the level of the pons with preservation of the corticospinal tracts and mediolateral lemniscus fibers (Fig. 31-11). The cingulum may be anomalous, crossing the midline above the corpus callosum (Fig. 31-12). These alterations in axonal migration evident by DTI are difficult to appreciate with conventional MRI.
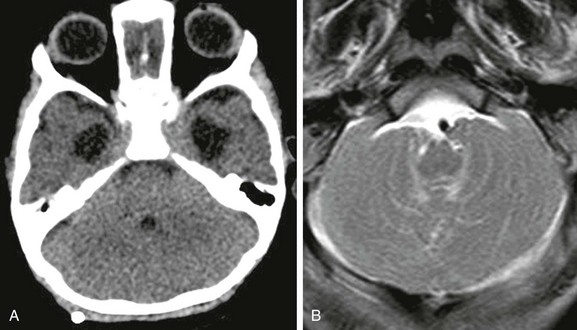
Figure 31-10 Chiari II malformation.
A, Axial computed tomography showing a constricted posterior fossa, the fourth ventricle is small and low lying, and a paucity of cerebrospinal fluid exists. B, An axial T2-weighted image through the posterior fossa shows cerebellar tissue wrapping around the brainstem.
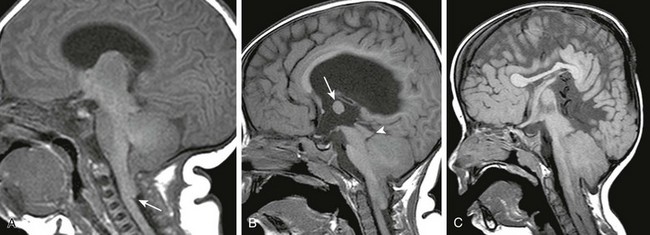
Figure 31-11 Chiari II malformation.
A, A sagittal T1-weighted image shows a small cerebellum, the fourth ventricle caudally displaced, and a kink at the cervicomedullary junction (arrow). The brainstem is well-formed. B, A sagittal T1-weighted image in another patient shows that the cerebellum and brainstem are more hypoplastic and the tectal plate is distorted (arrowhead). Note the large massa intermedia (arrow) and hydrocephalus. C, Sagittal T1-weighted image in another patient shows the brainstem is hypoplastic with a beaked tectal plate. The cerebellum is dysplastic and caudally herniated. The fourth ventricle is inapparent.
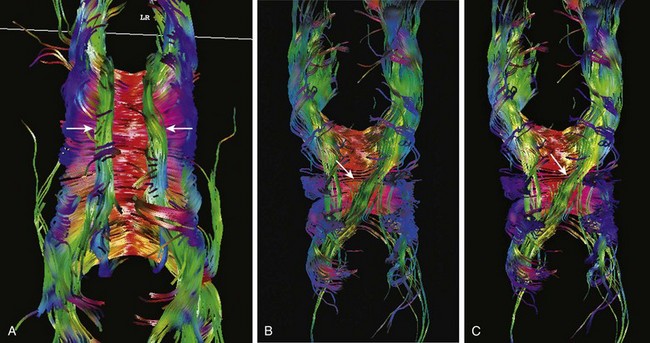
Figure 31-12 Diffusion tensor imaging in Chiari II malformation.
A, Diffusion tractography from a normal patient shows paired cingula (arrows), which are association fibers of the outer limbic system, above the corpus callosum. The green orientation indicates the fibers are oriented back-to-front. By definition, association fibers do not cross the midline. B, In a patient with Chiari II malformation, diffusion tractography shows the left cingulum (arrow) is medially displaced. C, In another patient with Chiari II malformation, the right cingulum is absent, and the left cingulum (arrow) crosses the midline.
Chari III malformations are high cervical or low occipital encephaloceles.9 The term Chiari IV malformation describes severe cerebellar hypoplasia in association with a neural tube defect. A better description of affected patients would be Chiari II malformation with cerebellar hypoplasia or aplasia (e-Fig. 31-13). Defects of neurulation have also been reported to coexist in patients with HPE; the latter malformation is considered a disorder of differentiation of the dorsal neural plate.10,11 The coexistence of these malformations, traditionally considered disparate in embryologic timing and insult, may be explained, in part, by mutations in genes implicated in both developmental pathways.12,13
Disorders of Differentiation of Dorsal Neural Plate
Clinical Presentation: HPE is the most common anomaly affecting the ventral forebrain, resulting from a primary defect in patterning and induction of the basal forebrain expressed around the fifth to sixth week of gestation, and occurs in 1 in 250 embryos and 1 in 8300 to 16,000 live births.1,13,14 HPE is thought to be caused by a deficient or defective prechordal mesoderm, with failure of induction or abnormal fusion of normally paired and separate neo-cortex, caudates, and claustrum. The consequences of the inductive failure of the median and paramedian structures are most pronounced in the ventral forebrain and decline in severity from rostral to caudal and from medial to lateral. HPE is caused by a combination of genetic polymorphisms and environmental factors; the incidence of HPE is increased 200-fold in maternal diabetes mellitus. HPE is notable for its genetic heterogeneity. Mutations in single genes result in syndromic forms of HPE, triploidies of entire chromosomes (e.g., 13 and 18), deletions or duplications of regions of a chromosome, and copy number variants. At least 12 HPE loci that play some role in the midline of the developing nervous system have been identified, including Shh, Otx2, Emx, Pax, Nkx-2.2, and some POU domain genes.13 Although 24% to 45% of affected live-born individuals have chromosomal abnormalities, no known correlation exists between the type or severity of the holoprosencephalic defect and the specific mutation. In addition to controlling prosencephalic separation and differentiation, the inductive effects of the prechordal mesoderm also influence the corpus striatum, thalami, eyes, face, and cerebral vasculature.13 The spectrum of associated facial anomalies range from flattening of the nasal bridge, hypotelorism without metopic synostosis, a single central maxillary incisor, cleft lip or palate to facial clefting, and cyclopia with a central proboscis.15 More severe facial anomalies are seen with more severe variants of HPE, but mild facial anomalies may occur in the absence of brain anomalies. Clinical problems associated with HPE include developmental delay, seizures, hypothalamic and brainstem dysfunction with swallowing and respiratory problems, thermal instability, pituitary dysfunction, and erratic sleeping patterns.13,15
Imaging: The hallmark of HPE is incomplete separation of the forebrain, absence of the anterior interhemispheric falx, and fusion of central gray nuclei. The septum pellucidum is absent. Considerable topographic variation exists in HPE, which is most often characterized as alobar, semilobar, or lobar.13–19 The malformation represents a continuum, ranging from overt hypotelorism, with severe facial anomalies, to mild hypotelorism, with subtle fusion of basal forebrain and central gray nuclei. Hydrocephalus may be present.
Alobar Holoprosencephaly
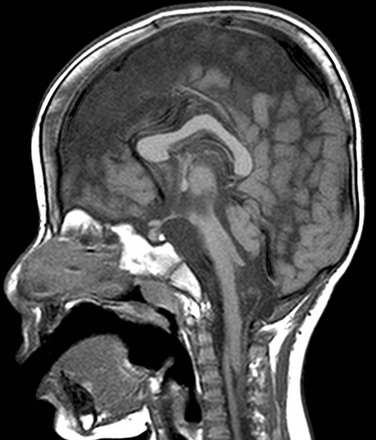
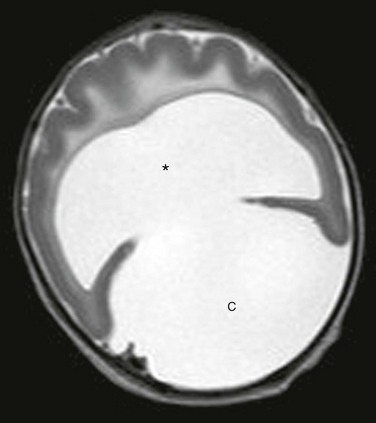
In this most severe form of HPE, separation between the cerebral hemispheres is completely lacking, with absence of the falx. The corpus callosum and septum pellucidum are absent, the central gray nuclei are fused, and the rudimentary single ventricle has a “U” configuration, which may communicate with a dorsal cyst (Fig. 31-14).1,14,17
Semilobar Holoprosencephaly
Relative preservation of the lateral and posterior cerebrum and the splenium exists in semilobar HPE. The posterior interhemispheric fissure and falx are present, whereas the hypoplastic frontal lobe is undivided. (Fig. 31-15).1,13,16 Lack of formation of the frontal lobes results in an anterior position of the Sylvian fissures, termed a “wide Sylvian angle” by Barkovich et al.17 The globus pallidi are absent or hypoplastic, and the caudate nuclei are fused, resulting in obliteration of or lack of formation of the septal region. The posterior limbs of the internal capsules are ventral to partially or totally fused thalami. The hippocampus is virtually always present, although usually incompletely or abnormally developed. A dorsal cyst may be present (e-Fig. 31-16).
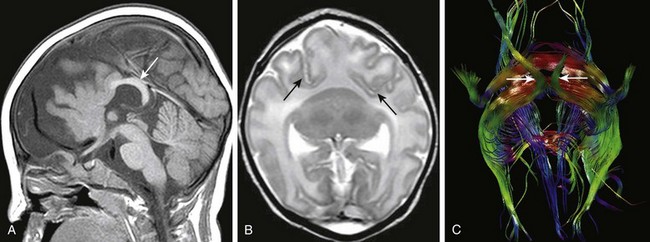
Figure 31-15 Semilobar holoprosencephaly.
A, A sagittal T1-weighted image shows underdeveloped frontal lobes with midface hypoplasia. The splenium (arrow) of the corpus callosum is formed. B, An axial T2-weighted image shows failure of separation of the frontal lobes; the caudate and thalami are fused. Arrows indicate the anomalous anterior position of the Sylvian fissures. C, Diffusion tractography. The brain is viewed from the inferior aspect; arrows indicate the optic pathways. The red fibers are midline extensions of anomalous association fibers.
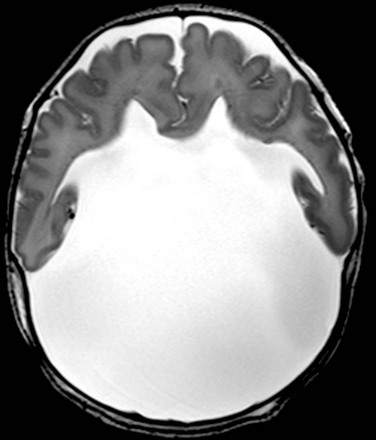
Lobar Holoprosencephaly
In this mildest form of HPE, hypoplasia of the frontal poles is present, along with agenesis of the septal pellucidum and incomplete separation of the basal forebrain, which is best depicted with high-resolution coronal images. The posterior frontal, parietal, and occipital lobes are more normally formed.1,14,17 The callosal body and splenium are preserved (Fig. 31-17).
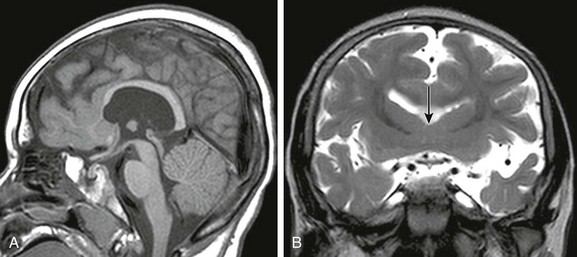
Syntelencephaly (Middle Frontal Variant)
The middle frontal variant is an unusual variant of lobar HPE characterized by separation of the frontal and occipital poles, fusion of the middle portions of the cerebral hemispheres (Fig. 31-18), preservation of portions of the commissural fibers of the corpus callosum, and neuronal migration anomalies; thalamic fusion is variable.18,19
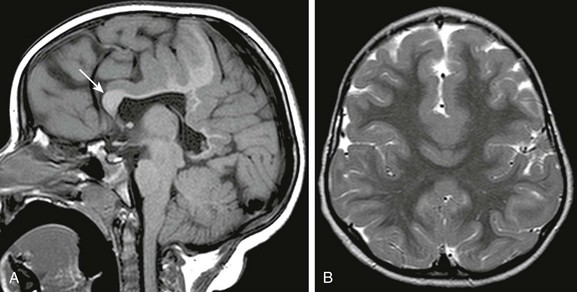
Figure 31-18 Middle frontal variant of holoprosencephaly.
A, A sagittal T1-weighted image shows that the callosal genu (arrow) is formed; the callosal body appears formed but is hypoplastic. Partial fusion of the frontal lobes is seen. B, An axial T2-weighted image shows fusion of cortex and white matter across the midline in the middle frontal region.
Septo-optic Dysplasia
Clinical Presentation: Septo-optic dysplasia (SOD) is considered along the continuum of disorders of ventral forebrain differentiation.20 Patients typically present in early childhood with nystagmus, optic nerve atrophy, short stature because of growth hormone deficiency, or panhypopituitarism without diabetes insipidus. Affected patients are at risk for Addisonion crisis from subclinical adrenal insufficiency, which may become clinically apparent only during illness or severe stress. Most cases of isolated SOD are sporadic.
Imaging: SOD is characterized by absence of the septum pellucidum and variable optic nerve hypoplasia (e-Fig. 31-19). The neurohypophysis is often ectopic or may be absent, in which case the pituitary stalk may be interrupted. However, agenesis of the septum pellucidum may be isolated with normal optic pathways and intact neuroendocrine function. Absence of the septal leaves results in downward displacement of the fornices into the third ventricle. SOD is associated with multiple malformations, such as schizencephaly, neuronal migration anomalies, and neural tube defects.
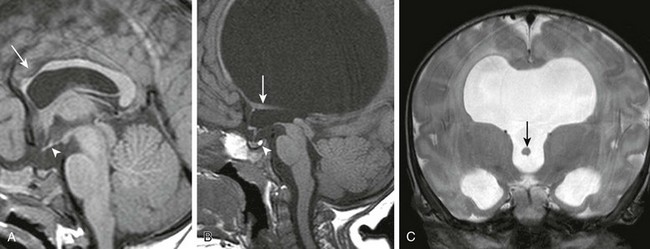
e-Figure 31-19 Variable appearance of septal agenesis.
A, A sagittal T1-weighted image shows that the genu and rostral portions of the corpus callosum are hypoplastic (arrow). An ectopic neurohypophysis (arrowhead) along the floor of the third ventricle is present. The optic pathways are hypoplastic. B, Septal agenesis with hydrocephalus has been caused by aqueductal stenosis. A normal hyperintense neurohypophysis (arrowhead) exists within the sella turcica. The optic pathways are normal. The fornices (arrow) are caudally displaced. C, A coronal T2-weighted image better shows the caudal displacement of the fornices (arrow) into the third ventricle.
Callosal Agenesis or Dysgenesis
Complete or partial malformation of the corpus callosum has an incidence of 1 in 4000.21 Normal corpus callosal development requires a precise sequence of cellular proliferation and migration, axonal growth and guidance, and midline glial development and patterning. Disruptions in this sequence may result from gene mutations, genetic polymorphisms, intrauterine infections, ischemia, and toxins.21
The telencephalic commissures normally develop in a predictable sequence.1,21 The anterior commissure (AC) forms around 55 days after conception, arising from the primitive hippocampal formations and ultimately connecting the anterior temporal lobes. Approximately 3 weeks later, the initial interhemispheric migration of pioneering callosal axons occurs, aided by the “glial sling,” which consists of primitive subependymal glial cells. The “glial wedge” is composed of radial glial cells that guide callosal axons across the midline and then repel the axons away from the midline into the contralateral hemisphere.1,21 After crossing the midline, callosal axons grow into the contralateral hemisphere; their ultimate destination mirror-images their region of origin and is within the same cortical layer from which the axon arose.21,22 The crossing axons follow a rostrocaudal gradient with the callosal rostrum forming before the splenium; formation of the splenium is complete by around 85 days after conception. Many of the crossing axons undergo programmed cell death after reaching their final destination; this apoptosis commences during the second trimester, with pruning of axonal axons continuing after birth.1,22
Imaging: The corpus callosum is the largest of the three telencephalic commissures, which include the anterior and hippocampal commissures. The AC is a variably-sized tract embedded in the cranial aspect of the lamina terminalis, which demarcates the anterior wall of the third ventricle and connects the anterior temporal lobes. The hippocampal commissure is a thin sheet of white matter connecting the two crura of the fornices and is not routinely visualized on MRI of the normal brain. Absence of the corpus callosum, AC, and hippocampal commissure is described as “complete commissural agenesis,” although some portion of the corpus callosum may be preserved (Fig. 31-20).1,2,22,23 Preservation of the AC in the absence of the corpus callosum and hippocampal commissure is “callosohippocampal agenesis” (e-Fig. 31-21), whereas the anterior and hippocampal commissures may be preserved, which results in “isolated callosal agenesis” (e-Fig. 31-22). Callosohippocampal agenesis is the most common malformation, although it might be argued these classifications are somewhat academic. The AC may be enlarged. On MRI, the sulci over the mesial surface of the frontal lobes have a radial configuration, and the lateral ventricles have a characteristic parallel orientation. Absence of the temporal segments of the cingulum results in dilatation of the temporal horns. Hypoplasia of association fibers connecting the occipital and temporal lobe allows dilatation of trigones of the lateral ventricles referred to as colpocephaly. When the axons fail to cross the midline and remain in their hemisphere of origin, they course along the medial aspect of the lateral ventricle, forming the longitudinal callosal bundles of Probst, which are thought to function as association fibers connecting regions of cortex within the same hemisphere (e-Fig. 31-23). Callosal agenesis may be associated with an interhemispheric diencephalic pseudocyst (e.g., high-riding third ventricle), or interhemispheric cysts that do not communicate with the ventricles (see e-Fig. 31-23). By DTI, the rudimentary cingulum and fornices are fused to the bundles of Probst (Fig. 31-24). In patients with preservation of a portion of the corpus callosum, diffusion tractography suggests the existence of aberrant connections between regions of cortex and subcortical structures in some patients, and heterotopic hemispheric connections that exist in such a manner that a frontal lobe may be connected with the contralateral parietal or occipital lobe (e-Fig. 31-25). These aberrant callosal fibers have been termed “asymmetric sigmoid bundles” and have also been reported in patients with a fully formed but abnormally shaped corpus callosum (e-Fig. 31-26).24,25
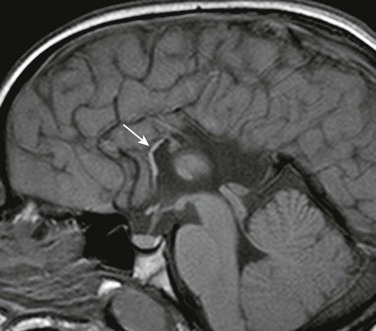
Figure 31-20 Near total callosal agenesis.
The callosal remnant contiguous with the superior aspect of the lamina terminalis (arrow), which demarcates the anterior wall of the third ventricle, may represent the ventral hippocampal commissure, which normally involutes.
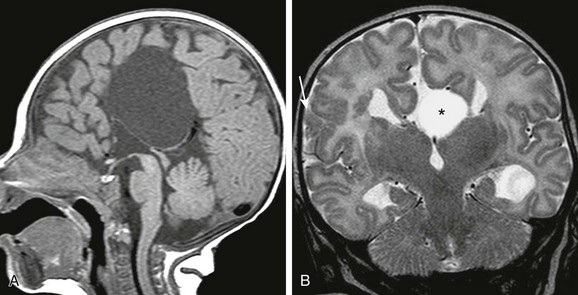
Figure 31-24 Corpus callosal agenesis with interhemispheric cyst.
A, A sagittal T1-weighted image shows a large interhemispheric cyst, which does not communicate with the ventricular system. B, A coronal T2-weighted image in another patient, the interhemispheric cyst (asterisk) represents a “high-riding” third ventricle. Note the cortical dysplasia (arrow) in the right cerebrum.
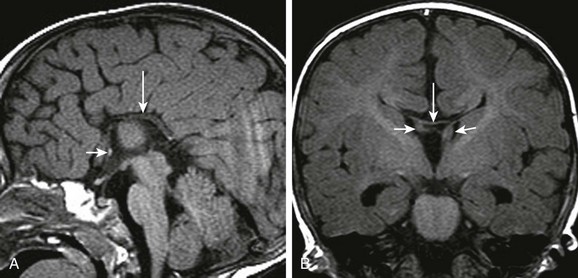
e-Figure 31-21 Callosal agenesis with preservation of the anterior and hippocampal commissures.
A, A thin midline commissural structure (arrows) represents the rostral portion of the hippocampal commissure, which has failed to involute, rather than a severely hypoplastic corpus callosum. B, The hippocampal commissure (arrows) connects the dysplastic fornices.
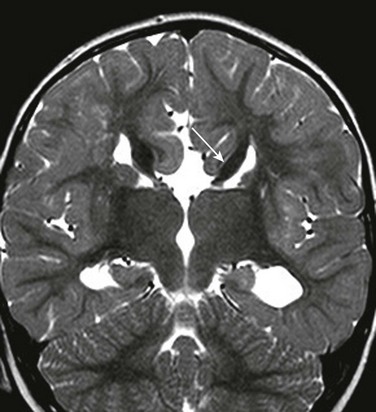
e-Figure 31-22 Longitudinal bundles of Probst (arrow) course along the medial walls of the lateral ventricles.
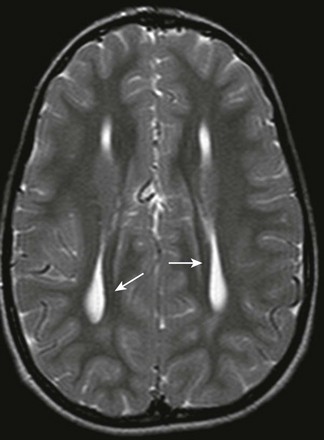
e-Figure 31-23 Characteristic appearance of the ventricles in callosal agenesis.
The lateral ventricles have a parallel orientation. The variable enlargement of the ventricles is caused by lack of regional white matter, rather than by hydrocephalus. Note Probst bundles (arrows) along the lateral ventricles.
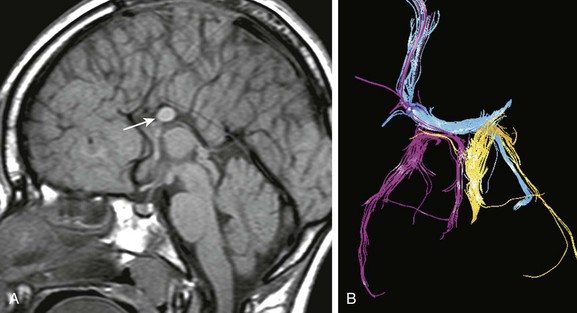
e-Figure 31-25 Callosal fragment containing with aberrant axons.
A, A sagittal T1-weighted image shows a callosal remnant (arrow). Note the radial configuration of the sulci over the medial surface of the hemisphere. B, Diffusion tractography of the callosal remnant shows fibers (blue) extending from the right frontal parasagittal cortex through the left fornix, fibers of the right fornix (pink), and fibers of the left fornix (yellow), indicating that axons from neocortical portions of the brain are admixed with fibers from archicortex. This indicates defective axonal migration.
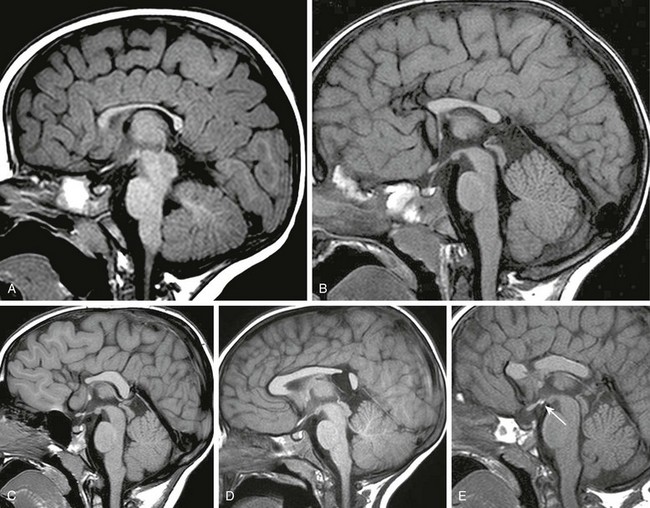
e-Figure 31-26 Variants of corpus callosal underdevelopment (all sagittal T1-weighted images).
A, Fully formed corpus callosum, which is diffusely hypoplastic or atrophic. B, Corpus callosum is truncated in the anteroposterior dimension. C, Preservation of the callosal splenium and dorsal body with hypoplasia of the genu and rostrum. D, Focal segmental defect in the isthmus. E, Focal defect in the rostral body associated with an ectopic neurohypophysis (arrow).
Callosal agenesis may be an isolated anomaly or associated with a myriad of other malformations, including aqueductal stenosis, Chiari II malformations, and malformations of cortical, brainstem, and cerebellar development.21–23 Numerous syndromes are associated with complete or partial agenesis of the corpus callosum. Aicardi syndrome is a rare sporadic X-linked dominant malformation seen in females, with an incidence of less than 1 in 200,000 births. Affected males have Klinefelter syndrome (XXY). Clinical findings include profound developmental delay, infantile spasms, micro-ophthalmia, coloboma, short philtrum, flat nose, and large ears. Imaging shows callosohippocampal agenesis, interhemispheric cysts, gray matter heterotopias, polymicrogyria (PMG), and cerebellar malformation. Ocular abnormalities include chorioretinic lacunae and coloboma. CRASH syndrome (corpus callosum agenesis, retardation, adducted thumbs, spastic paraplegia, and hydrocephalus) is caused by mutations in the L1 cell adhesion molecule gene that codes for a transmembrane cell adhesion protein involved in axonal migration.
Callosal Hypoplasia
The corpus callosum may be incompletely formed, diffusely hypoplastic, or segmentally deficient (e-Fig. 31-27).23 Partial callosal agenesis is usually characterized by absence of the splenium and dorsal body, with relative preservation of the genu and the rostral body. Diffuse callosal hypoplasia is characterized by diffuse thinning, whereas segmental callosal hypoplasia involves an intermediate portion of the corpus callosum. The segmental defects may result from a secondary callosal destruction or injury to regional white matter.
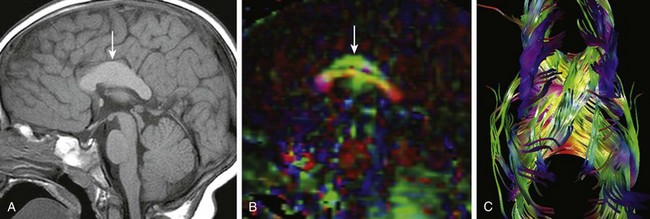
e-Figure 31-27 Corpus callosal hypertrophy.
A, The body of the corpus callosum (arrow) is markedly thickened. B, On the sagittal diffusion color map, the normal callosal fibers are red; the green fibers (arrow) represent aberrant association fibers. C, Diffusion tractography shows the aberrant supracallosal fibers accounting for the thickened corpus callosum on conventional magnetic resonance imaging.
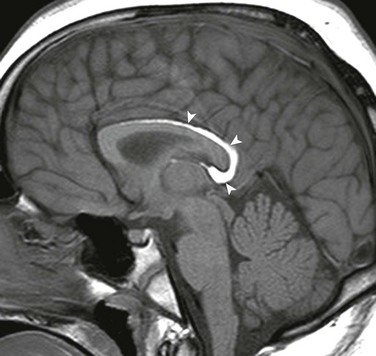
Callosal lipomas are rare and are thought to result from abnormal differentiation of pluripotential mesenchymal tissue. Most cases are associated with agenesis or incomplete formation of the corpus callosum. Anterior lipomas are more common than posterior. The lipoma itself may be an incidental finding and may be seen with a fully formed corpus callosum (Fig. 31-28).
Microcephaly Vera
Microcephaly is a nonspecific diagnosis defined by head circumference greater than 3 standard deviations below normal for age and gender and, as such, is a descriptor rather than a specific entity. Microcephaly may be caused by prenatal infections or intrauterine or perinatal insult or may be part of a syndrome. Microcephaly vera (“true” microcephaly) is an autosomal-recessive trait typically not associated with seizures.26 About 15% of patients with microcephaly vera have normal neurodevelopmental outcomes. MRI shows diffuse undersulcation of the brain, which may be subtle; brain structures are otherwise normal.
Encephaloclastic or Destructive Lesions
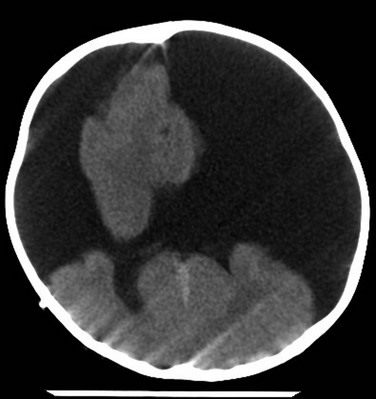
The term hydrancephaly describes near-complete to total absence of the cerebral hemispheres, with preservation of the brainstem, central gray nuclei, and the cerebellum and is thought to be caused by an intrauterine ischemic insult.1 Remnants of the inferior frontal, anterior temporal, or occipital lobes may exist (Fig. 31-29). Affected patients appear neurologically normal at birth because of the intact brainstem. The hydrocephalus in hydrancephaly results from aqueductal stenosis.
Neuronal Migration Anomalies or Anomalies of Cortical Development
These malformations include a wide spectrum of developmental malformations of the cortex caused by disruption of neuronal cell proliferation, migration, and organization.3,26,27 Neuronal proliferation occurs in the germinal matrix along the subependymal layer of the walls of the lateral ventricles during week 7 of gestation. Around week 8, the neurons begin to migrate from the germinal zone to the surface of the brain along bipolar radial glial fibers, which provide scaffolding for neuronal migration. A smaller population of neurons also migrates orthogonal to the radial glial cells. Neurons in cortical layer 1 are the first to reach the cortex, followed by those destined for layers 5, 4, 3, and 2 in an “inside out” sequence. At the completion of migration, the cortex becomes organized with synaptic contacts, which develop between neurons throughout the six-layered cortex. Disruptions of neuronal proliferation, migration, and organization may result from infections such as cytomegalovirus (CMV), toxoplasmosis, intrauterine ischemic insults, toxins, radiation exposure, or genetic anomalies, or from no known cause. Affected patients have seizures, developmental delay, and variable focal neurologic deficits. The resulting malformation depends on which process is disrupted:
• Disorders of proliferation include microlissencephaly, in which neuronal proliferation is diffusely decreased; hemimegancephaly, in which proliferation is increased; and focal cortical dysplasia (FCD), in which proliferation is abnormal.
• Disorders of migration include type I or classic lissencephaly (LIS1), in which neuronal migration is diffusely decreased; type II cobblestone lissencephaly, in which diffuse overmigration of neurons occurs; and gray matter heterotopias, in which migration is focally ectopic.
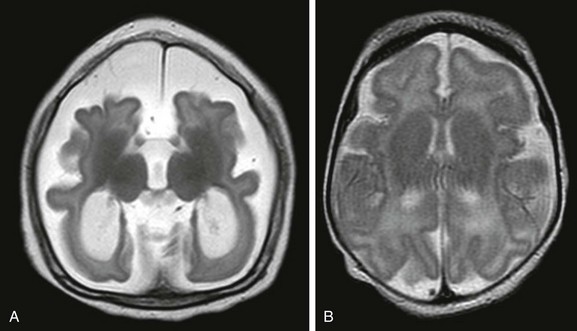
Microlissencephaly
Clinical Presentation: Patients with microlissencephaly have severe microcephaly at birth, with dysmorphic craniofacial features, abnormal genitalia, and arthrogryposis.26 These patients suffer from seizures and have developmental delay that is pervasive and profound. The mode of inheritance is thought to be autosomal recessive, related in some patients to mutations of the RELN gene. Congenital infections are known to cause microlissencephaly, especially CMV.
Imaging: The diagnosis of is made on the basis of a diffusely thinned cerebral mantle, with abnormal sulcation consisting of agyria-pachygyria (Fig. 31-30), variable callosal agenesis, and variable hypoplasia of the brainstem and cerebellum. Type A (Norman-Roberts syndrome) is microlissencephaly without infratentorial anomalies, and type B (Barth syndrome) is microlissencephaly with severe cerebellar and brainstem hypoplasia. Milder forms of microlissencephaly are referred to as microcephaly with a simplified gyral pattern; the cortex is usually of normal thickness.
Hemimegalencephaly
Clinical Presentation: Hemimegalencephaly is hamartomatous overgrowth of a cerebral hemisphere or lobe of the brain.1,2,28 It occurs as an isolated anomaly or with neurocutaneous syndromes such as epidermal nevus syndrome, Proteus syndrome, unilateral hypomelanosis of Ito, neurofibromatosis type I, Klippel-Trenaunay syndrome, and tuberous sclerosis. The malformation is thought to arise from faulty neuronal proliferation prior to radial neuroblast migration. Hemispherectomy is required for relief of the severe intractable epilepsy.
Imaging: MRI shows diffusely enlarged hemisphere or lobe (Fig. 31-31), with abnormal sulcation pattern that may include PMG, lissencephaly, and gray matter heterotopias. The regional white matter is thickened and gliotic. A hallmark is enlargement of the ipsilateral ventricle that becomes more pronounced with age with expansion of the overlaying hemicranium.
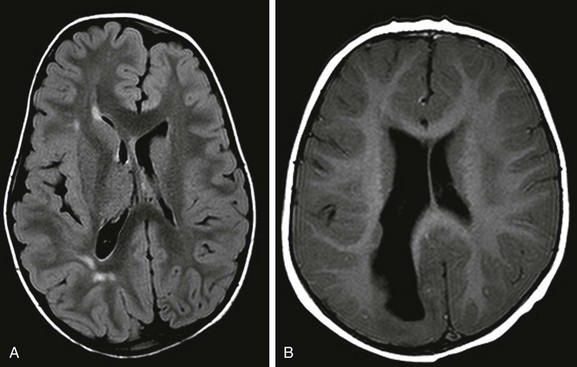
Figure 31-31 Hemimegancephaly.
A, The hemimegancephaly is lobar, with sparing of the right frontal lobe. The right occipital horn is dilated, and expansion of the right hemicranium and pachygyria, as well as periventricular grey matter heterotopias, are seen. B, In this patient, the entire right cerebrum is affected. The white matter is thickened and focally gliotic.
Focal Cortical Dysplasia
The most common clinical presentation of FCD is medication-resistant focal epilepsy.29,30 FCD is probably caused by mutations in multiple genes, but unlike lissencephaly and periventricular nodular heterotopias, FCD is not associated with a known single genetic mutation.29,30 The histologic hallmark of FCD is lack of normal cortical lamination. FCD is classified into types I, II, and, more recently, type III, based on cortical laminar structure, cytoarchitectural disruption, cell composition, and the presence of associated destructive brain lesions.30 Type I FCD has alterations in cortical layering, with distortion of the normal radial cortical lamination and lack of the normal six-layered neocortex; by definition, type I FCD has no balloon cells. Type II FCD is characterized by cortical dyslamination and dysmorphic neurons with or without balloon cells. FCD Type III is found in association with encephaloclastic lesions such as traumatic brain injury, perinatal ischemia, Rasmussen encephalitis, or low-grade tumors.30
Imaging: Identification of FCD requires high-resolution MRI, using T1-weighted, T2-weighted, and FLAIR (fluid-attenuated inversion recovery) sequences (Fig. 31-32); the diagnostic accuracy of MRI is increased with scrutiny of thin sections of the region of brain suspected of harboring the epileptogenic focus. Findings of type I FCD include focal cortical thickening, often within the depth of a sulcus, and blurring of the gray–white junction. FCD may be clinically occult and found on MRI performed for reasons other than seizures or may be associated with seizures and variable cognitive impairment. The most common histopathologic finding in surgical specimens removed from patients with cryptogenic epilepsy (e.g., epilepsy in which no lesion is identifiable by imaging) is type I FCD.29,30
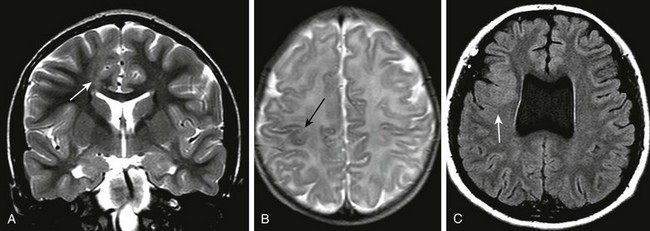
Figure 31-32 The variable appearance of focal cortical dysplasia.
A, On the coronal T2-weighted image and axial fluid-attenuated inversion recovery images, focal thickening of left parasagittal cortex (arrow) is seen. B, The small area of dysplastic cortex (arrow) was seen only on the T2 image in this neonate with seizures. C, Right frontal cortical dysplasia (arrow) with absence of the septum pellucidum. Focal expansion of the overlying subarachnoid space has occurred, and anomalous superficial cortical veins may simulate a vascular malformation.
Type II FCD is more readily apparent with MRI, especially in the presence of balloon cells. MRI shows increased T2 signal within the subcortical white matter underlying abnormal gyri and is often best seen on T2-weighted and FLAIR images. The radial extension of balloon cells and ectopic neurons into the deep white matter constitutes the ”transmantle sign” of FCD and may be the sole visible evidence of FCD.29 The differential diagnosis of type II FCD is low-grade tumor. Although type II FCD may be suggested on the basis of MRI findings, overlap exists in imaging findings between type I and II, and the ultimate diagnosis is based on histopathology.30
Gray Matter Heterotopias
Gray matter heterotopias are ectopic neurons resulting from mutations in the genes that code for proteins involved in neuronal migration (Fig. 31-33). Periventricular nodular heterotopias are gray matter heteropias along the walls of the lateral ventricles. These are nodular masses of normal neurons and glial cells devoid of laminar organization and are intrinsically epileptogenic. The seizures result from hyperexcitable circuitries between the ectopic neurons and the overlaying cortex.31,32 The clinical impact depends on the amount and location of the heterotopias and on the degree of extension to the overlying cortex. Bilateral heterotopias are associated with more severe and generalized seizure activity and cognitive delay compared with unilateral heterotopias.33
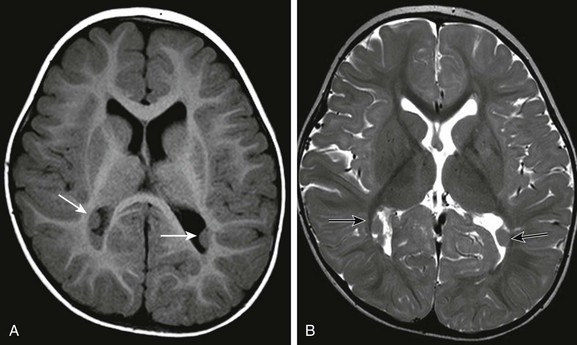
Disorders of Neuronal Migration
Type I (Classic) Lissencephaly
LIS1 is caused by disruption of the migration of neuroblasts from the ventricular surface to the pial surface, which normally occurs between 10 and 14 weeks’ gestational age.34–38 LIS1 includes lissencephalies with defined genetic abnormalities, isolated lissencephalies without any identifiable genetic defect, and lissencephalies associated with syndromes of multiple malformations.33–37 Mutations resulting in isolated LIS and Miller-Dieker syndrome (MDS) map to chromosome 17p13.3, which, along with mutations in the double cortin (DCX) gene, accounts for about 70% of LIS1. The DCX and Aristaless-related homeobox (ARX) genes are X-linked. The less common Reelin (RELN) gene maps to chromosome 7 and the α-tubulin 1a (TUBA1A) gene to chromosome 12. LIS1 occurs in 1 in 500,000 live births, with gender predominance only in the less common X-linked forms. Histopathology of LIS1 shows inversion of cortical laminar architecture; the normal six-layered cortex is replaced with a thick four-layered cortex. In the lissencephalic brain, layer I is a superficial molecular layer. Deep under layer I is layer II, which is a layer of pyramidal cells resembling the fifth and sixth layers of the normal neocortex, followed by a sparse cellular layer (layer III) and a thick band of disorganized neurons of variable size (layer IV), which extends into the regional white matter.
Clinical Presentation: The hallmarks of LIS1 are profound mental retardation, with a normal-small head size, intractable seizures, hypertonia, and hyperreflexia. Patients with isolated lissencephaly have normal facial structures, whereas patients with MDS have LIS1 with abnormal facies: wrinkled skin over the glabella and frontal suture, prominent occiput, narrow forehead, downward slanting palpebral fissures, and small nose and chin. Males with DCX mutation have classic LIS1 as seen on MRI, whereas females with DCX mutation have variable cognitive delay without seizures and subcortical band heterotopias (SBH) as delineated by MRI. The presence of an unexplained seizure disorder or cognitive problems in the mother of a male child with lissencephaly should trigger a screening MRI of the mother for possible SBH. Females with ARX mutation have ambiguous genitalia without lissencepahy; males have lissencephaly with ambiguous genitalia (XLAG) syndrome. Lissencephaly caused by RELN mutation is associated with congenital lymphedema.33–37
Imaging: The phenotypic expression of lissencephaly caused by LIS1 is similar in males and females and consists of a markedly thickened cerebral cortex, which may be diffuse (Fig. 31-34) or most pronounced posteriorly (Fig. 31-35); and Sylvian fissures fail to occur.1,2,36–38 The thickened cortex shows a highly organized pattern, as shown by DTI, because of the persistence of the radial pattern of the neurons in arrested migration.39 The lissencephalic pattern in males with DCX mutation is similar to L1S1 and more severe anteriorly. Females with DCX mutation have SBH or “double cortex.”37,38 MRI shows bands of heterotopic neurons within the cerebral white matter between a normal-appearing cortex and the ventricles (Fig. 31-36).40 MDS and the ARX mutation are associated with more severe lissencephaly posteriorly. With LIS1 mutations of MDS and DCX, the pons may be normal or mildly hypoplastic; severe cerebellar and pontine hypoplasia suggest a RELN mutation. MRI abnormalities described in the rare mutations of the TUBA1A gene include classic lissencephaly, with brainstem dysplasia and cerebellar hypoplasia.
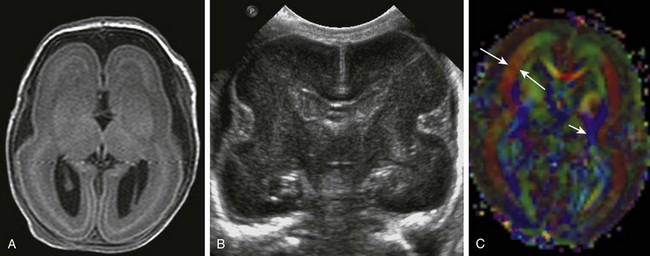
Figure 31-34 Classic lissencephaly caused by undermigration of neurons.
A, A T2-weighted fluid-attenuated inversion recovery image from a term neonate shows abnormally thickened laminated cerebrum with no sulcation. B, A cranial sonogram shows that the Sylvian fissures are unapposed, which is abnormal for a term infant. The undersulcated appearance suggests the brain of an extremely preterm infant. C, An axial color map from diffusion tensor imaging shows thick bands of brushlike fibers (long arrows) caused by the neurons arrested during radial migration. The blue fibers (short arrow) are projection fibers that are vertically oriented.
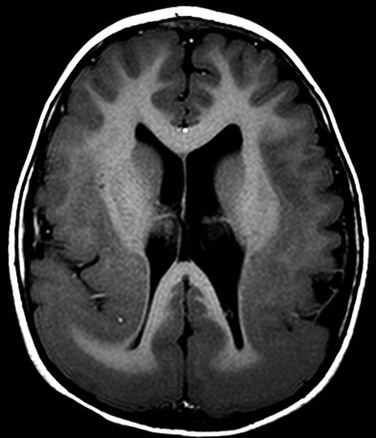
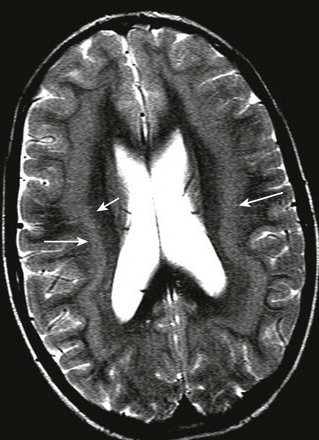
Type II Cobblestone Lissencephaly (Congenital Muscular Dystrophy)
The cobblestone type II lissencephalies (dystroglycanopathies) result from excessive neuronal migration, in contrast to LIS1, in which neuronal migration is deficient.41 Dystroglycan is a glycoprotein in the extracellular matrix receptor of muscle and the central nervous system (CNS), which mechanically stabilizes the muscle sarcolemma against contraction-stretch stress and which also plays a role in signal transduction during cell migration. Dystroglycan requires the addition of glycosol groups to function; the CNS manifestations of the defective glycosylation are overmigration of neurons through the defective glial–pial limiting membrane into the subarachnoid space, resulting in a cobblestone appearance to the surface of the brain. The malformation includes variable pachygyria and PMG, with a thickened cortex that lacks the normal six-layered architecture, fibroglial proliferation of the leptomeninges, and focal interhemispheric fusion.
Clinical Presentation: The clinical presentation of the cobblestone lissencephalies is congenital muscular dystrophy (CMD), which comprises a heterogeneous group of inherited disorders presenting with diffuse symmetric hypotonia at birth or in infancy.1,41 CMD is categorized as (1) CMD without CNS abnormalities, and (2) CMD with CNS abnormalities; the latter includes Fukuyama (FCMD), muscle-eye-brain disease (MEBD), and Walker-Warburg syndrome (WWS).41 Depending on the specific syndrome, affected patients also have psychomotor retardation, seizures, micro-ophthalmia, optic nerve hypoplasia, and colobomas. Scoliosis and contractures develop during early childhood. Death often results from respiratory failure, caused by chest wall rigidity and weakness of the diaphragm or aspiration, or from cardiomyopathy. The specific diagnosis is made on the basis of clinical findings, serum creatine kinase, neuroimaging, muscle and skin biopsy, and molecular genetic testing.41
FCMD is an autosomal-recessive disorder most common in Japan, where the incidence approaches 3 in 100,000 individuals.41 The syndrome results from mutations in the FKTN gene on chromosome 9, which codes for the protein fukutin, which interacts with α-dystroglycan in the extracellular matrix. MEB disease is an autosomal-recessive disorder, which is rare outside of Finland and is caused by mutations in the POMT1, POMT2, POMGnT1, fukutin, and FKRP genes; phenotypes may overlap with FCMD. WWS is the most severe form of CMD with CNS manifestations and is also associated with mutations in the POMT1, POMT2, fukutin, and fukutin-related protein genes.41
Imaging: MRI abnormalities are less severe in FCMD than in WWS and MEBD.41 In mild cases of FCMD, the cerebrum may be relatively normal or have a simplified gryal pattern. More severe malformations have variable pachygyria and PMG; the white matter is gliotic with cysts. Neurons may be seen lining the ependymal surface of the ventricle, contrasted on T2-weighted images by hyperintense signal of the CSF and the abnormal white matter (e-Fig. 31-37). The lateral ventricles are enlarged. The cerebellum consistently shows small cysts with variable PMG. The most severe cerebellar hypoplasia is seen with WWS, which is also associated with occipital encephalocele (Fig. 31-38). The brainstem is usually normal in FCMD, hypoplastic in MEBD, and small and kinked posteriorly in WWS. Hydrocephalus is rare in FCMD, common in MEBD, and almost invariable in WWS; the hydrocephalus may mask the malformation.41
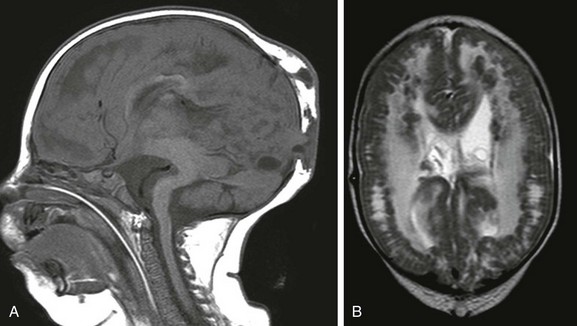
Figure 31-38 Walker-Warburg lissencephaly is the most severe form of congenital muscular dystrophy with central nervous system involvement.
A, A sagittal image shows the typical cerebellar hypoplasia and dorsal beaking of the brainstem. Small parieto-occipital meningoceles are present. B, An axial T2-weighted image showing the cobblestone appearance at the grey-white junction. The patient required shunt placement at birth for associated hydrocephalus.
e-Figure 31-37 Type II (cobblestone) lissencephaly is caused by overmigration of neurons.
A, The corpus callosum is thickened. Brainstem hypoplastia and a dysplastic cerebellum exist in this case of muscle-eye-brain disease. B, An axial T2-weighted image shows tiny cysts within the cerebellar folia and a small dysplastic pons. C, The frontal cortex is more severely affected; the white matter is thickened and gliotic with cysts.
Deranged Neuronal Organization (Polymicrogyria, Schizencephaly)
Clinical Presentation: PMG results from disruption of normal terminal neuronal migration and organization and is defined by excessive convolutions of the cerebral cortex.42–45 PMG may be unilateral and focal or bilateral and diffuse; bilateral patterns are most often frontoparietal and peri-Sylvian. The severity of seizures and developmental impairment correlates with the location and severity of the malformation. Focal PMG may be clinically occult or associated with seizures that are often medically uncontrollable. Bilateral peri-Sylvian PMG is associated with epilepsy, delayed development, strabismus, dysphagia and speech problems, and paresis. PMG may be isolated or associated with known disorders, including callosal agenesis, Aicardi syndrome, Joubert syndrome, FCMD, and Zellweger spectrum. Bilateral peri-Sylvian PMG is X-linked, whereas frontoparietal PMG maps to chromosome 16. At least 11 mutations in the G protein–coupled receptor 56 (GPR56) gene have been identified in bilateral PMG. PMG may also occur from CMV infection and intrauterine ischemic injury.42,43
Imaging: MRI shows increased cortical thickness and irregularity at the cortex–white matter junction (Fig. 31-39). The pattern of the PMG appears to differ with location.44 The appearance of the PMG may change with brain maturation, and microgyri obvious in unmyelinated brain may be less conspicuous as myelination progresses.
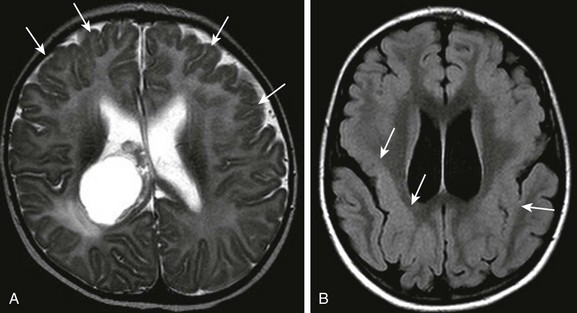
Figure 31-39 Variants of polymicrogyria.
A, Bifrontal polymicrogyria (arrows) with a choroid plexus cyst in the right lateral ventricle. B, Peri-Sylvian polymicrogyria (arrows) on T2-weighted fluid-attenuated inversion recovery image.
Schizencephaly results from congenital clefts in the cerebral hemisphere, which extend from the pial surface to the ventricles.46 Affected patients may present with seizures, motor deficits, or both or may be asymptomatic. Larger or bilateral defects are associated with poorer neurodevelopmental outcome.1,2
Imaging: The lips of the defects may be in apposition (closed lip) or separated (open lip). The clefts are lined with dysplastic gray matter, which extends the full length of the cleft (Fig. 31-40). Discontinuity of the ventricular ependyma and the subpial membrane in open-lip schizencephaly results in communication between the lateral ventricles and the subarachnoid space. The pulsations of the CSF may cause remodeling and outward expansion of the calvarium overlaying the open cleft. The septum pellucidum is absent in most cases of schizencephaly involving the frontoparietal regions. Unilateral schizencephaly is often associated with contralateral peri-Sylvian cortical dysplasia.46
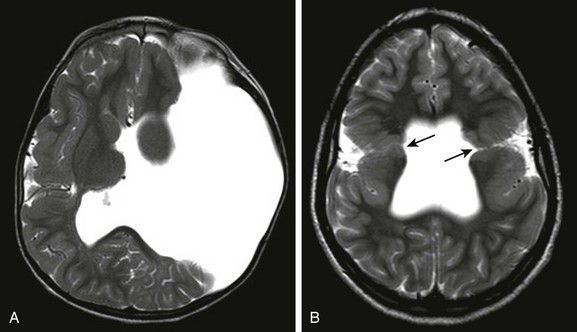
Figure 31-40 Schizencephaly.
A, Bilateral closed lip schizencephalic defects (arrows) are associated with agenesis of the septum pellucidum. B, Large open lip defect is also associated with septal agenesis; the expansion of the left hemicranium is caused by the pulsations of the underlying cerebrospinal fluid.
Hindbrain and Cerebellar Malformations
Brainstem malformations may be classified into four groups based on embryology and genetics.47
1. Group I comprises malformations resulting from defects in early differentiation of the neural tube (e.g., Chiari II).
2. Group II comprises generalized malformations affecting the brainstem, the cerebellum, the cerebrum, or all. These include type I lissencephalies with cerebellar hypoplasia, CMDs with CNS involvement (cobblestone lissencephaly), CMV, and so on.
3. Group III includes localized malformations affecting the brainstem and the cerebellum. The hallmark of these disorders is deficient or defective formation of cranial nerve nuclei, resulting in problems with ocular movement and lid control, facial palsy, neurosensory hearing loss, or all.
4. Group IV includes combined brainstem hypoplasia and atrophy in degenerative disorders of prenatal onset.
Joubert Syndrome and Related Disorders
Clinical Presentation: Joubert syndrome is a rare malformation that is usually autosomal recessive; it involves one of multiple genes involved in ciliary function integral to chemical signaling during neuronal and axonal migration. The clinical syndrome is characterized by hypotonia, episodic hypoventilation, and hyperventilation, which tend to improve with age; truncal ataxia, which develops during early childhood; and oculomotor apraxia, nystagmus, pigmentary retinopathy, endocrinopathies, and abnormal facial features, including ptosis, hypertelorism, and low-set ears. Joubert syndrome and related disorders (JSRD) describes the constellation of clinical problems associated with Joubert syndrome, which are diverse. The marked clinical variability probably results in underdiagnosis.
Imaging: The diagnosis of Joubert syndrome is made on the basis of characteristic MRI findings, which include hypoplasia of the superior vermis, with enlargement of the superior aspect of the fourth ventricle and the superior cerebellar peduncles.48–50 The molar tooth sign (Fig. 31-41), seen on an axial image through the pontomesencephalic junction, is also seen in a number of cerebello-oculo-renal disorders. DTI of Joubert syndrome shows variable absence of transverse pontine fibers; the decussating fibers of the superior cerebellar peduncles are absent.50 In addition to the clinical variability of JSRD, considerable variability also exists in the appearance of the brainstem.
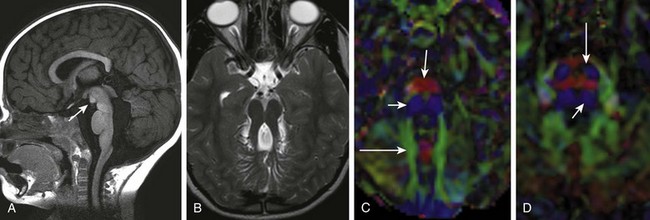
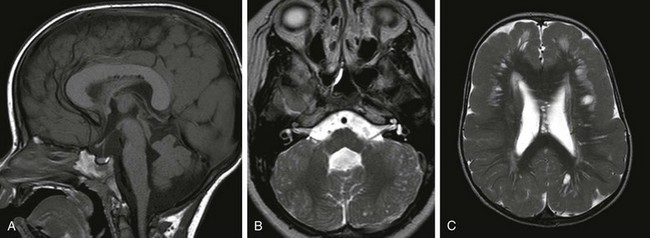
Figure 31-41 Joubert syndrome.
A, A sagittal image shows an elongated mesencephalon with a ventral bulge (arrow). The roof of the fourth ventricle is dilated, and the superior cerebellar vermis is underdeveloped and dysplastic. B, An axial T2-weighted image shows the classic “molar tooth deformity.” C and D, An axial color map from diffusion tensor imaging shows a single ventrally positioned transverse fiber bundle. The vertically oriented (blue) tracts consist of the corticospinal tracts (long arrows) and mediolateral lemniscus bundles (short arrows), which in a normal brainstem (D) are separated by a second transverse pontine fiber bundle.
Horizontal Gaze Palsy with Progressive Scoliosis
Clinical Presentation: Horizontal gaze palsy with progressive scoliosis (HGPPS) is the expression of mutation of the ROBO3 gene that codes for proteins involved in axonal guidance and neuronal migration.51,52 Expression of the defective gene appears to be limited to decussating fibers in the brainstem. Patients with HGPPS tend to be cognitively normal but have congenital nystagmus and are unable to perform lateral eye movements because of lack of decussating fibers in the brainstem. Progressive scoliosis develops during early childhood.
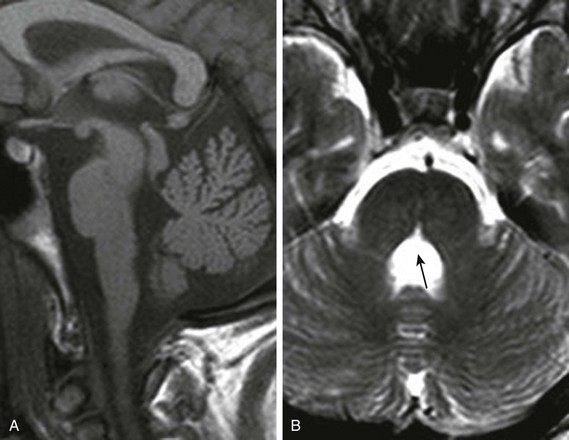
Imaging: MRI shows hypoplasia of the pons, which is partially divided by a midsagittal cleft (e-Fig. 31-42). Facial colliculi are absent, the inferior olivary nuclei are prominent, and the medulla lacks the normal dorsal convexities. DTI of HGPPS shows hypoplasia of the ventral transverse pontine fibers and absence of the dorsal transverse pontine fibers, small middle and superior cerebellar peduncles, and decussating fibers of the superior cerebellar peduncle within the midbrain.51 The appearance of the brainstem by diffusion imaging in HGPPS is similar to that of other less well-characterized brainstem malformations.
Pontine Tegmental Cap Dysplasia
Pontine tegmental cap dysplasia is a rare brainstem malformation associated with variable developmental delay, ataxia, restricted horizontal gaze, failure initiating fast eye movements, facial weakness, deafness, and swallowing problems, which reflect deficient lower cranial nerves.53
Imaging: MRI shows pontine hypoplasia; the ventral surface is flattened, and ectopic tissue protrudes dorsally into the fourth ventricle, which is referred to as the tegmental “cap.” The superior cerebellar vermis is hypoplastic, which, along with the elongated and laterally misplaced superior cerebellar peduncle, results in a molar tooth–like deformity. As shown by DTI, the tegemental cap is composed of transversely oriented ectopic pontine fibers. DTI also shows the absence of the normal decussations of the superior and middle cerebellar peduncles.54 The pattern of white matter tract abnormalities, as shown by DTI, is similar to those reported in some cases of JSRD.
Chiari I
The Chiari I malformation is not associated with defective neural tube closure, and Chari I “malformations” may be acquired or spontaneously resolve. Chiari I malformation is defined as downward displacement of cerebellar tonsils below the foramen magnum.55–57 Although many patients with Chiari I malformations are asymptomatic, some patients experience suboccipital headaches, neck pain, ataxia, dysmetria, nystagmus, and dysequilibrium because of brainstem or cerebellar compression, upper extremity weakness and pain caused by cervical cord myelopathy, or both. Treatment of a symptomatic Chiari I malformation with or without syrinx is a suboccipital decompressive craniectomy, with or without duraplasty. In many centers, cerebellar tonsils are also resected.
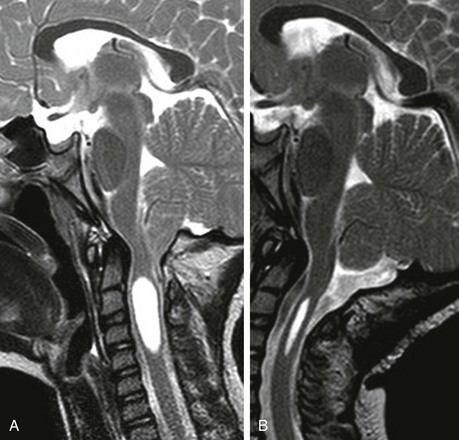
Imaging: The Chiari I malformation is defined by cerebellar tonsils greater than 6 mm below the posterior lip of the foramen magnum. In symptomatic patients, the low-lying tonsils disrupt CSF flow across the foramen magnum, which predisposes to syringomyelia in the thoracic or cervical spinal cord (Fig. 31-43). Flow of CSF across the foramen magnum is further impaired when the odontoid is elongated and dorsally angulated or with any significant basilar invagination. The dynamic significance of the low-lying tonsils is best depicted using single-slice, multiple-phase, peripheral, or cardiac-gated two-dimensional phase contrast in the sagittal and axial planes with a velocity encoding of 5 centimeters per second (cm/s). Obstruction of CSF flow results in abnormal bidirectional movement of the hindbrain across the foramen magnum, which is seen as changes in signal intensity on the phase contrast sequence. In patients with Chari I malformations, increased intracranial pressure, caused by a tumor or hydrocephalus resulting in secondary tonsillar herniation, should be excluded with screening images through the brain (Table 31-1).
Table 31-1
Comparison of Chiari I and II Malformations
| Chiari I | Chiari II |
| Headache and neck pain increased by cough or Valsalva maneuver; dysarthria, dysphagia, downbeat nystagmus, upper extremity weakness or numbness | Brainstem dysfunction; swallowing or feeding difficulties, stridor, apnea, weak cry, nystagmus |
| Downward herniation of cerebellar tonsils resulting in compression of cervicomedullary brainstem; impeded cerebrospinal fluid flow | Downward herniation of dysplastic lower brainstem and cerebellar vermis |
| Brain usually normal Syringomyelia in 30%-70% |
Callosal dysgenesis, neuronal migration anomalies, hydrocephalus |
Cerebellar Malformations
This section addresses malformations of the cerebellum in which the cerebellum is hypoplastic or dysplastic but will not address cerebellar atrophy, a classification proposed by Patel and Barkovich.58 Cerebellar hypoplasia is defined by a small cerebellum with fissures of normal size compared with the folia. Cerebellar dysplasia describes disorganized development and includes an abnormal folial pattern, cerebellar gray matter heterotopias, which may be generalized involving cerebellar hemispheres and vermis, or focal and limited to a hemisphere or vermis. The latter would include the isolated inferior vermian hypoplasia often suggested by fetal MRI. Cerebellar hypoplasia includes the Dandy-Walker continuum and isolated focal cerebellar hypoplasia.
Dandy-Walker Continuum
The Dandy-Walker continuum includes the Dandy-Walker malformation and the Dandy-Walker variant.59 The Dandy-Walker malformation is defined by complete or partial agenesis of the vermis, cystic dilatation of the fourth ventricle, and an enlarged posterior fossa with cranial displacement of the torcula (Fig. 31-44) and is often associated with supratentorial hydrocephalus. Associated anomalies occurring in 70% of patients include an abnormal or absent corpus callosum, callosal lipoma, and neuronal migration anomalies. The Dandy-Walker variant is defined as inferior vermian hypoplasia with nonobstructive cystic dilatation of the fourth ventricle; the posterior fossa is normal or somewhat enlarged (Fig. 31-45). In contrast to the Dandy-Walker continuum, the posterior fossa arachnoid cyst (Fig. 31-46) and mega cisterna magna (Fig. 31-47) are associated with a fully formed cerebellum. Retrocerebellar arachnoid cysts cause anterior displacement of the fourth ventricle and the cerebellum. The mega cisterna magna consists of enlarged retrocerebellar CSF spaces with no mass effect.
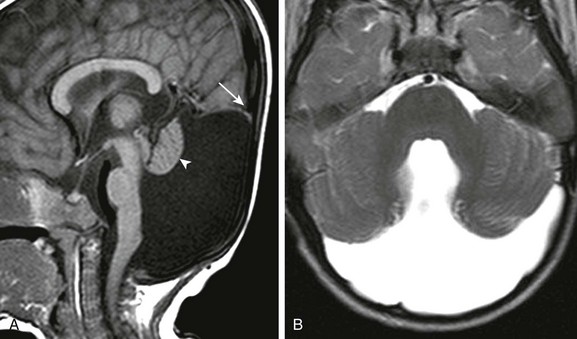
Figure 31-44 Dandy-Walker malformation.
A, A sagittal T1-weighted image shows that the cerebellum (arrowhead) is hypoplastic and the torcula (arrow) is elevated. Note the hypoplastic pons. B, An axial T2-weighted image shows absence of the inferior vermis; the large retrocerebellar fluid collection communicates with the dilated fourth ventricle.
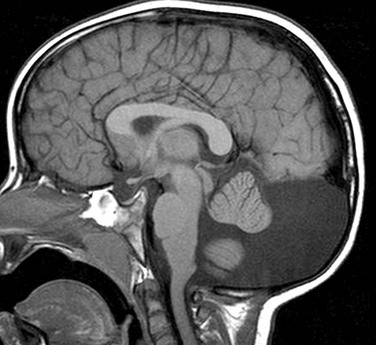
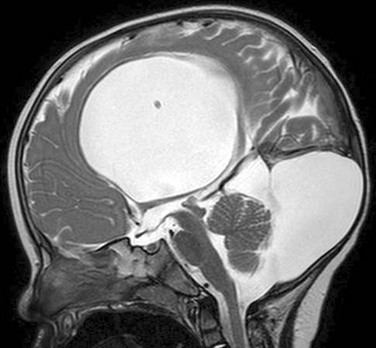
Figure 31-45 Dandy-Walker variant; a continuum in the spectrum.
Cerebellar Dysplasia
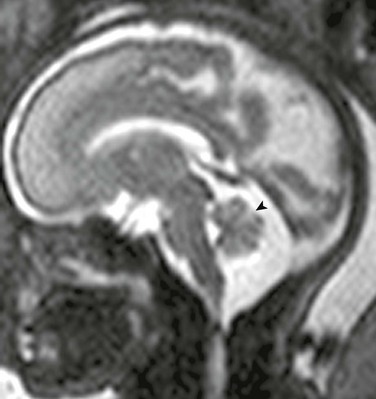
Cerebellar dysplasia may be isolated or associated with other anomalies. The affected cerebellar tissue may be hypoplastic (Fig. 31-48) or hyperplastic (Fig. 31-49).
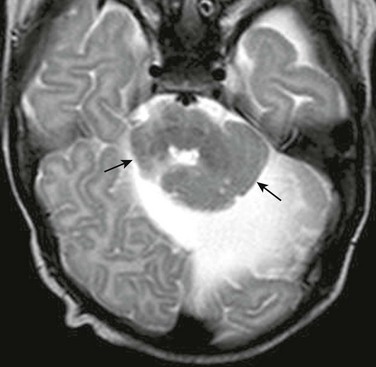
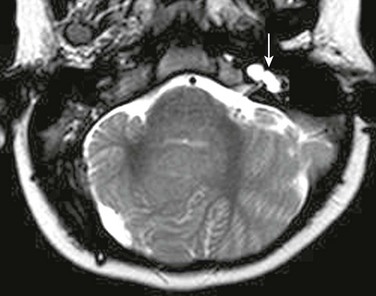
Rhombencephalosynapsis
Imaging: The cerebellar hemispheres are fused (Fig. 31-50). Associated brainstem abnormalities include fusion of the colliculi, atresia or dilatation of the fourth ventricle, and abnormalities of lower cranial nerves and the medulla. The corpus callosum may be hypoplastic or absent, and failure of formation of diencephalic structures with thalamic fusion, hydrocephalus, and malformations of cortical development may exist.
References
1. Tortori-Donati, P, Rossi, A. Congenital malformations in the neonate. In: Rutherford M, ed. MRI of the neonatal brain. London: WB Saunders; 2002:225–249.
2. Norman MG, McGillivray BC, Kalousek DK, et al, eds. Congenital malformations of the brain. pathological, embryological, clinical, radiological and genetic aspects. New York: Oxford University Press, 1995.
3. Barkovich, AJ, Kuzniecky, RI, Jackson, GD, et al. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65(12):1873–1887.
4. Naidich, TP, Altman, NR, Braffman, BH, et al. Cephaloceles and related malformations. AJNR Am J Neuroradiol. 1992;13(2):655–690.
5. Van der Put, NM, van Straaten, HW, Trijbels, FJ, et al. Folate, homocysteine and neural tube defects: an overview. Exp Biol Med (Maywood). 2001;226(4):243–270.
6. McLone, DG, Knepper, PA. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15(2):1–12.
7. Naidich, TP, McLone, DG, Fulling, KH. The Chiari II malformation. Part IV. The hindbrain deformity. Neuroradiology. 1983;25(4):179–197.
8. Herweh, C, Akbar, M, Wengenroth, M, et al. DTI of commissural fibers in patients with Chiari II-malformation. Neuroimage. 2009;44(2):306–311.
9. Castillo, M, Quencer, RM, Dominguez, R. Chiari III malformation: imaging features. AJNR Am J Neuroradiol. 1992;13(1):107–113.
10. Rollins, NK, Joglar, J, Perlman, J. Co-existent holoprosencephaly and Chiari II malformation. AJNR Am J Neuroradiol. 1999;20(9):1678–1681.
11. Mittelbronn, M, Beschorner, R, Capper, D, et al. Coincidence of semilobar holoprosencephaly and Chiari II malformation: correlation of prenatal diagnostics and neuropathologic findings. J Child Neurol. 2006;21(5):426–429.
12. Nagai, T, Aruga, J, Minowa, O, et al. Zic2 regulates the kinetics of neurulation. Proc Natl Acad Sci U S A. 2000;97(4):1618–1623.
13. Solomon BD, Gropman A, Muenke M. Holoprosencephaly overview. In: Pagon RA, Bird TD, Dolan CR, et al., eds. GeneReviews. Seattle, WA: 1993; University of Washington. Available at http://www.ncbi.nlm.nih.gov/books/NBK1291/. Accessed September 17, 2012.
14. Hahn, JS, Barnes, PD. Neuroimaging advances in holoprosencephaly: refining the spectrum of the midline malformation. Am J Med Genet Part C Semin Med Genet. 2010;154C:120–132.
15. Plawner, LL, Delgado, MR, et al. Neuroanatomy of holoprosencephaly as predictor of function: beyond the face predicting the brain. Neurology. 2002;59:1058–1066.
16. Rollins, NK. Semilobar holoprosencephaly as seen with diffusion tensor imaging and fiber tracking. AJNR Am J Neuroradiol. 2002;26(8):2148–2152.
17. Barkovich, AJ, Simon, EM, Clegg, NJ, et al. Analysis of the cerebral cortex in holoprosencpehlay with attention to the Sylvian fissures. AJNR Am J Neuroradiol. 2002;23:143–150.
18. Barkovich, AJ, Quint, DJ. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol. 1993;14(2):431–440.
19. Lewis, AJ, Simon, EM, Barkovich, AJ, et al. Middle interhemispheric variant of holoprosencephaly: a distinct cliniconeuroradiologic subtype. Neurology. 2002;59(12):1860–1865.
20. Kelberman, D, Dattani, MT. Septo-optic dysplasia: novel insights into the aetiology. Horm Res. 2008;69(5):257–265.
21. Paul, LK, Brown, WS, Adolphs, R, et al. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8(4):287–299.
22. Raybaud, C. The corpus callosum, the other great forebrain commissures, and the septum pellucidum: anatomy, development, and malformation. Neuroradiology. 2010;52(6):447–477.
23. Hanna, RM, Marsh, SE, Swistun, D, et al. Neurology. 2011;76(4):373–382.
24. Wahl, M, Barkovich, AJ, Mukherjee, P. Diffusion imaging and tractography of congenital brain malformations. Pediatr Radiol. 2010;40(1):59–67.
25. Rollins, NK. Clinical applications of diffusion tensor imaging and tractography in children. Pediatr Radiol. 2007;37(8):769–780.
26. Mochida, GH. Genetics and biology of microcephaly and lissencephaly. Semin in Pediatr Neurol. 2009;16(3):120–126.
27. Guerrini, R, Marini, C. Genetic malformations of cortical development. Exp Brain Res. 2006;173:322–333.
28. Sato, N, Yagishita, A, Oba, H, et al. Hemimegalencephaly: a study of abnormalities occurring outside the involved hemisphere. AJNR Am J Neuroradiol. 2007;28:678–682.
29. Bernasconi, A, Bernasconi, N, Bernhardt, BC, et al. Advances in MRI for “cryptogenic” epilepsies. Nat Rev Neurol. 2011;7:99–108.
30. Blümcke, I, Thom, M, Aronica, E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52(1):158–174.
31. Guerrini, R, Filippi, T. Topical review: neuronal migration disorders, genetics, and epiloptogenesis. J Child Neurol. 2005;20(4):287–299.
32. Barkovich, AJ, Gressens, P, Evrard, P. Formation, maturation, and disorders of brain neocortex. AJNR Am J Neuroradiol. 1992;13(2):423–446.
33. Barkovich, AJ, Kjos, BO. Gray matter heterotopias: MR characteristics and correlation with developmental and neurological manifestations. Radiology. 1992;182(2):483–489.
34. Wynshaw-Boris, A. Lissencephaly and LIS1: insights into the molecular mechanisms of neuronal migration and development. Clin Genet. 2007;72:296–304.
35. Friocourt, G, Marcorelles, P, Saugier-Veber, P, et al. Role of cytoskeletal abnormalities in the neuropathology and pathophysiology of type I lissencephaly. Acta Neuropathol. 2011;121(2):149–170.
36. Cardoso, C, Leventer, RJ, Dowling, JJ, et al. Clinical and molecular basis of classical lissencephaly: mutations in the LIS1 gene. Hum Mutat. 2002;19:4–15.
37. Dobyns, WB, Truwit, CL, Ross, ME, et al. Differences in the gyral pattern distinguish chromosome 17-linked and X- linked lissencephaly. Neurology. 1999;53:270–277.
38. Barkovich, AJ, Koch, TK, Carrol, CL. The spectrum of lissencephaly: report of ten patients analyzed by magnetic resonance imaging. Ann Neurol. 1991;30(2):139–146.
39. Rollins, N, Reyes, T, Chia, J. Diffusion tensor imaging in lissencephaly. AJNR Am J Neuroradiol. 2005;26(6):1583–1586.
40. Barkovich, AJ, Guerrini, R, Battaglia, G, et al. Band heterotopia: correlation of outcome with magnetic resonance imaging parameters. Ann Neurol. 1994;36:609–617.
41. Sparks S, Quijano S, Harper A, et al. Congenital muscular dystrophy overview. In: Pagon RA, Bird TD, Dolan CR, et al., eds. GeneReviews. Seattle, WA: 1993; University of Washington. Available at http://www.ncbi.nlm.nih.gov/books/NBK1291/. Accessed September 17, 2012.
42. Barkovich, AJ. Neuroimaging manifestations and classification of congenital muscular dystrophies. AJNR Am J Neuroradiol. 1998;19:1389–1396.
43. Parrini, E, Ferrari, AR, Dorn, T, et al. Bilateral frontoparietal polymicrogyria, Lennox-Gastaut syndrome, and GPR56 gene mutations. Epilepsia. 2009;50(6):1344–1353.
44. Robin, NH, Taylor, CJ, McDonald-McGinn, DM, et al. Polymicrogyria and deletion 22q11.2 syndrome: window to the etiology of a common cortical malformation. Am J Med Genet A. 2006;140:2416–2425.
45. Barkovich, AJ. MRI analysis of sulcation morphology in polymicrogyria. Epilepsia. 2010;51(suppl 1):17–22.
46. Barkovich, A. Bilateral symmetrical polymicrogyria. AJNR Am J Neuroradiol. 1999;20(10):1814–1821.
47. Barkovich, AJ, Kjos, BO. Schizencephaly: correlation of clinical findings with MR characteristics. AJNR Am J Neuroradiol. 1992;13(1):85–94.
48. Barkovich, AJ, Millen, KJ, Dobyns, WB. A developmental and genetic classification for midbrain-hindbrain malformations. Brain. 2009;132(12):3199–3230.
49. Spampinato, MV, Kraas, J, Maria, BL, et al. Absence of decussation of the superior cerebellar peduncles in patients with Joubert syndrome. Am J Med Genet A. 2008;146A(11):1389–1394.
50. Brancati, F, Dallapiccola, B, Valente, EM. Joubert syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20.
51. Poretti, A, Boltshauser, E, Loenneker, T, et al. Diffusion tensor imaging in Joubert syndrome. AJNR Am J Neuroradiol. 2007;28(10):1929–1933.
52. Chan, WM, Traboulsi, EI, Arthur, B, et al. Horizontal gaze palsy with progressive scoliosis can result from compound heterozygous mutations in ROBO3. J Med Genet. 2006;43(3):e11.
53. Mori, H, Fujishiro, T, Hayashi, N, et al. Partially uncrossed pyramidal tracts shown by tractography in horizontal gaze palsy and scoliosis. AJR Am J Roentgenol. 2005;184(suppl 3):S4–S6.
54. Jissendi-Tchofo, P, Doherty, D, McGillivray, G, et al. Pontine tegmental cap dysplasia: MR imaging and diffusion tensor images features of impaired axonal navigation. AJNR Am J Neuroradiol. 2009;30(1):113–119.
55. Barth, PG, Majoie, CB, Caan, MW, et al. Pontine tegmental cap dysplasia: a novel brain malformation with a defect in axonal guidance. Brain. 2007;130(9):2258–2266.
56. Shaffer, N, Martin, B, Loth, F. Cerebrospinal fluid hydrodynamics in type I Chiari malformation. Neurol Res. 2011;33(3):247–260.
57. Tubbs, RS, Beckman, J, Naftel, RP, et al. Institutional experience with 500 cases of surgically treated pediatric Chiari malformation Type I. J Neurosurg Pediatr. 2011;7(3):248–256.
58. Patel, S, Barkovich, AJ. Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol. 2002;23(7):1074–1087.
59. Aldinger, KA, Lehmann, OJ, Hudgins, L, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009;41(9):1037–1042.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 31
Congenital Brain Malformations
With advances in magnetic resonance imaging (MRI) and molecular biology and the availability of mutant mouse models of human cortical malformations, many malformations of brain development have been reclassified.1–3 Given the large number of congenital human brain malformations, the complexity of the molecular genetics, and the degree of anatomic variability, an in-depth discussion of some malformations is beyond the scope of this chapter. This chapter addresses malformations seen most often in clinical practice and those malformations that are less common but are distinctive and have a profound impact of early childhood development. The malformations presented here are grouped according to presumed dominant defect in embryologic or fetal development. Diffusion tensor imaging (DTI) is included in the diagnosis of malformations in which aberrant white matter tracts are a dominant feature.
A rudimentary description of relevant embryology is needed. Around 26 to 28 days after conception, the process of neurulation, in which the lateral edges of the neural plate elevate into neural folds, takes place.1 The folds then fuse medially to form the neural tube. At the cranial end of the neural tube, the primitive brain vesicles form and include the prosencephalon, mesencephalon, and rhombencephalon.1,2,4 The prosencephalon divides into the telencephalon, which gives rise to the cerebral hemispheres and the corpus striatum, and the diencephalon, which gives rise to the thalami and the hypothalamus. The cerebral peduncles and midbrain arise from the mesenchephalon. The rhombencephalon gives rise to the metencephalon, which forms the pons and cerebellum, and the myelencephalon, from which the medulla arises. Malformations of brain development may result from chromosomal aberrations, single gene mutations, teratogenic infections and agents, or ischemia.1,3 However, in 70% of malformations, the etiology is identifiable.
Defects of Neurulation
Complete failure of closure of the cranial end of the neuropore results in anencephaly, in which the forebrain, skull, and scalp are absent. Affected patients die soon after birth and postnatal imaging is not usually indicated. Less severe disorders of neural tube closure result in meningoceles and encephaloceles, which are protrusions of the meninges or brain, respectively, through a congenital defect of the skull and dura; the latter occurs in about 1 per 5000 live births.4 Encephaloceles tend to be midline. In the countries of the Western hemisphere, a predominance of posterior cephaloceles is seen, whereas in Asian children, encephaloceles tend to be anterior.1 Frontal encephaloceles include interorbital frontal, nasofrontal, nasoethmoidal, and naso-orbital lesions. Interorbital frontal encephaloceles protrude through a defect in frontal bone (Fig. 31-1). Nasofrontal encephaloceles involve the region of nasal bridge, the floor of the anterior cranial fossa, or both (Fig. 31-2). Nasofrontal encephaloceles are also referred to as nasal gliomas, although they are not neoplastic. These masses of neuroglial tissue are categorized as extranasal (60%), intranasal (30%), or mixed (10%).1,2,4 When telangiectasias occur on the skin overlaying an external nasal glioma, the lesion may be mistaken for a hemangioma. Often, hypertelorism is present with a broad nasal bridge. Intranasal glioma presents as an intranasal mass; biopsy should be avoided prior to imaging because of the risk of meningitis. With nasoethmoidal encephaloceles, frontal bone is intact. Neural tissue bulges into the ethmoid sinus through a defect in the floor of the anterior cranial fossa, and the nasal septum defines the posterior margin. A defect in the medial orbital wall results in a naso-orbital encephalocele, which protrudes into the orbit and produces unilateral exophthalmos. Other facial anomalies seen with anterior encephaloceles include a bifid nasal tip or complete midline splitting of the nose in an uncommon malformation known as frontonasal dysplasia (e-Fig. 31-3). Morning Glory syndrome includes midline facial defects, callosal agenesis, frontal encephaloceles, and characteristic eye anomalies.
Figure 31-1 Frontal encephalocele.
A sagittal T1 image shows agenesis of the corpus callosum associated with encephalocele at the glabella.
Figure 31-2 Incidental nasofrontal encephalocele found on head computed tomography done for trauma.
A, Axial computed tomography shows a focally expansile lesion within the right ethmoid sinus (arrow). B, A high-resolution coronal T2 image shows the clinically occult defect in the right cribriform plate through which has herniated the frontal cortex (arrow).
e-Figure 31-3 Median facial cleft.
A, Shaded surface rendering from computed tomography. The frontal encephalocele and median facial cleft are skin covered; marked hypertelorism and a broad nasal bridge are seen. B, Axial computed tomography shows the large defect in the frontal bone; an interhemispheric lipoma (arrow) associated with absence of the corpus callosum is seen.
Basal encephaloceles result from defects in the sphenoid bone (Fig. 31-4); the encephalocele herniates into the posterior nasopharynx anterior to the dorsum sella and may contain pituitary tissue, optic nerves, branches of the circle of Willis, or all (Fig. 31-5). Encephaloceles in the parietal bone range from large deforming “towering” lesions to small meningoceles (Fig. 31-6). Atretic encephaloceles or meningoceles present as small subcutaneous fibrofatty masses, which are often painful to palpation (Fig. 31-7). Occipital encephaloceles usually contain dysplastic cerebellar tissue alone or with the cerebral cortex (Fig. 31-8). Rarely, the brainstem may be within the encephalocele; this malformation is lethal.
Figure 31-4 Basal encephalocele.
A coronal reformatted computed tomography image shows the floor of the anterior cranial fossa is deficient, the nasal turbinates are disorganized, and marked hypertelorism exists.
Figure 31-5 Basal encephalocele.
A, A sagittal T1-weighted image shows callosal agenesis with a tiny lipoma (arrow). A large defect in the basisphenoid is seen. Note the apparent absence of the pituitary, floor of the third ventricle, and optic pathways. B, A high-resolution sagittal T2-weighted image shows the pituitary-hypothalamic structures (arrow) and optic pathways are contained within the encephalocele.
Figure 31-6 Parietal encephalocele.
A, A coronal T2-weighted image shows extremely dysmorphic hemisphere and diencephalic structures contained within the massive parietal encephalocele. B, Coronal maximum-intensity projection from two-dimensional magnetic resonance venogram shows dysplastic dural sinuses (arrowheads) contained within the encephalocele.
Figure 31-7 Atretic parietal meningocele.
A, A sagittal T1-weighted image shows the small meningocele (arrow) over the parietal vertex. Focal expansion of the posterior interhemispheric subarachnoid space is shown (arrowhead). B, A sagittal image from a gadolinium-enhanced magnetic resonance venogram shows a persistent falcine sinus (arrow) draining the deep medullary venous system; no straight sinus is present.
Figure 31-8 Occipital encephalocele.
A, A sagittal T1-weighted image shows an occipital encephalocele containing cerebrospinal fluid and dysplastic cerebellum. A beaked tectal plate and kinking of the cervicomedullary brainstem (arrow), similar to that seen in the Chiari II malformation, are present. B, A sagittal T1-weighted image in a different patient shows a large cephalocele containing both cerebellar tissue and brainstem (arrow); the patient died after closure of the defect.
Conditions and syndromes associated with encephaloceles include trisomy 13 and 18, amniotic band syndrome, Meckel-Gruber syndrome, dyssegmental dwarfism, Knobloch syndrome, Walker-Warburg (type II lissencephaly) syndrome, cryptophthalmos, and Voss syndrome. The prognosis depends on the severity of associated brain anomalies and the amount of dysplastic brain contained within the encephalocele.1,2,4
Imaging: High-resolution MRI is performed soon after birth to define contents of the encephalocele, the severity of the malformation, and the frequent associated anomalies. These include callosal agenesis, anomalies of cortical formation, and variable anomalies of the cerebellum, diencephalon, and brainstem. Although dysplastic nonfunctional neural tissue is usually resected during closure of the encephalocele, major dural venous sinuses are preserved. Therefore, magnetic resonance venography is essential for diagnosing large occipital and midline parietal encephaloceles that may contain dural venous sinuses. Basal encephaloceles containing optic chiasm or nerves or pituitary tissue cannot be closed without sacrificing these structures. Affected patients are at risk for meningitis and cerebrospinal fluid (CSF) leakage into the nasophayrnx. The typical intracranial manifestations of atretic parietal encephaloceles include posterior tenting of the tectal plate, a persistent falcine sinus with or without atresia of the straight sinus, and expansion of posterior interhemispheric CSF. Hydrocephalus is common after closure of large parietal and occipital encephaloceles.
Chiari II Malformations
Clinical Presentation: Chiari II malformations are the intracranial manifestations of posterior dysraphic defects such as myelomeningoceles. Neural tube defects have been reported with multiple chromosomal abnormalities, including trisomies 18, 13, and 9; triploidy; unbalanced translocations and deletions; and in Turner, DiGeorge, and velocardiofacial syndromes. The genes in the region of 22q11 have been implicated in the development of neural tube defects, although neural tube defects and the associated Chiari II malformation are probably caused by a combination of genetic polymorphisms and environmental factors, including dietary folate intake and maternal folate metabolism.5
The most commonly accepted unifying theory for the Chiari II malformation suggests that the failure of neural tube closure prevents the transient closure of the central canal, which is essential for the distension of the primitive ventricular system;6 this results in the premature fusion of the mesenchymal components that form the calvarium, whereas the hindbrain manifestations result from leakage of CSF through the neural tube defect. The Chiari II malformation is associated with a wide range of brain malformations;1,2,7,8 DTI shows disordered axonal migration, suggesting that the malformation is not explained by mechanical alterations caused by faulty CSF dynamics alone.8 The Chiari II malformation is characterized by mesodermal dysplasia, small and dysplastic lower cranial nerve ganglia, deficient tentorium cerebelli, hypoplastic and dysmorphic cerebellum, and thickened basal meninges.6 As in holoprosencephaly (HPE), these anomalies are attributable to a defective or deficient mesenchyme, which presumably deprives the skull base, hindbrain, and rhombencephalon of normal inductive effects.7 Clinical problems related specifically to the malformed hindbrain include apnea, aspiration, feeding difficulties, and recurrent respiratory infections.
Imaging: The calvarial manifestations of the Chiari II malformation include a bifid frontal bone and luckenschadel, or lacunar, skull. The latter is a manifestation of the mesodermal dysplasia of the membranous skull and is caused by nonossified fibrous bone in the inner and outer tables of the skull, resulting in the apparent cranial scalloping (Fig. 31-9); the affected cranium ossifies and appears normal by 6 months of age. Luckenschadel is not the result of increased intracranial pressure and is not synonymous with “the beaten copper skull.”
Figure 31-9 Luchenschadel.
A, A skull radiograph shows a lacelike appearance to the cranium. B, Axial computed tomography shows scalloping of the inner and outer tables of the calvarium.
The intracranial stigmata of the Chiari II malformation are complex and variable; the hallmarks of the malformation are infratentorial. In the most severe forms, the foramen magnum is enlarged and the posterior cranial fossa is constricted, with effacement of CSF spaces; the cerebellum wraps around the ventral aspect of the brainstem (see Fig. 31-9), and the clivus and petrous ridges are concave. MRI shows caudal displacement of a dysplastic brainstem and a hypoplastic cerebellum into the upper cervical canal with “kinking” of the cervicomedullary brainstem (Fig. 31-10). The fourth ventricle is effaced and caudally displaced. The torcula and transverse sinuses are low lying, which presents a potential surgical hazard during decompressive suboccipital craniectomy, which may be performed for relief of symptomatic hindbrain compression. A “beaked” tectal plate is present, and a variably thickened massa intermedia may be so thick that the thalami appear virtually fused with partial atresia of the third ventricle.1,7 Common supratentorial abnormities include callosal dysgenesis, neuronal migration anomalies, and hydrocephalus. After ventricular shunt placement, the cerebral hemispheres drop away from the inner table of the skull, allowing interdigitations of the cortex across the midline under the hypoplastic falx and projection of the superior cerebellar vermis superiorly across a widened tentorial incisura. These findings are nonspecific manifestations seen after CSF diversion of severe congenital obstructive hydrocephalus. DTI of Chiari II malformations associated with more severe degrees of cerebellar hypoplasia shows absence of the dorsal transverse fibers at the level of the pons with preservation of the corticospinal tracts and mediolateral lemniscus fibers (Fig. 31-11). The cingulum may be anomalous, crossing the midline above the corpus callosum (Fig. 31-12). These alterations in axonal migration evident by DTI are difficult to appreciate with conventional MRI.
Figure 31-10 Chiari II malformation.
A, Axial computed tomography showing a constricted posterior fossa, the fourth ventricle is small and low lying, and a paucity of cerebrospinal fluid exists. B, An axial T2-weighted image through the posterior fossa shows cerebellar tissue wrapping around the brainstem.
Figure 31-11 Chiari II malformation.
A, A sagittal T1-weighted image shows a small cerebellum, the fourth ventricle caudally displaced, and a kink at the cervicomedullary junction (arrow). The brainstem is well-formed. B, A sagittal T1-weighted image in another patient shows that the cerebellum and brainstem are more hypoplastic and the tectal plate is distorted (arrowhead). Note the large massa intermedia (arrow) and hydrocephalus. C, Sagittal T1-weighted image in another patient shows the brainstem is hypoplastic with a beaked tectal plate. The cerebellum is dysplastic and caudally herniated. The fourth ventricle is inapparent.
Figure 31-12 Diffusion tensor imaging in Chiari II malformation.
A, Diffusion tractography from a normal patient shows paired cingula (arrows), which are association fibers of the outer limbic system, above the corpus callosum. The green orientation indicates the fibers are oriented back-to-front. By definition, association fibers do not cross the midline. B, In a patient with Chiari II malformation, diffusion tractography shows the left cingulum (arrow) is medially displaced. C, In another patient with Chiari II malformation, the right cingulum is absent, and the left cingulum (arrow) crosses the midline.
Chari III malformations are high cervical or low occipital encephaloceles.9 The term Chiari IV malformation describes severe cerebellar hypoplasia in association with a neural tube defect. A better description of affected patients would be Chiari II malformation with cerebellar hypoplasia or aplasia (e-Fig. 31-13). Defects of neurulation have also been reported to coexist in patients with HPE; the latter malformation is considered a disorder of differentiation of the dorsal neural plate.10,11 The coexistence of these malformations, traditionally considered disparate in embryologic timing and insult, may be explained, in part, by mutations in genes implicated in both developmental pathways.12,13
Disorders of Differentiation of Dorsal Neural Plate
Clinical Presentation: HPE is the most common anomaly affecting the ventral forebrain, resulting from a primary defect in patterning and induction of the basal forebrain expressed around the fifth to sixth week of gestation, and occurs in 1 in 250 embryos and 1 in 8300 to 16,000 live births.1,13,14 HPE is thought to be caused by a deficient or defective prechordal mesoderm, with failure of induction or abnormal fusion of normally paired and separate neo-cortex, caudates, and claustrum. The consequences of the inductive failure of the median and paramedian structures are most pronounced in the ventral forebrain and decline in severity from rostral to caudal and from medial to lateral. HPE is caused by a combination of genetic polymorphisms and environmental factors; the incidence of HPE is increased 200-fold in maternal diabetes mellitus. HPE is notable for its genetic heterogeneity. Mutations in single genes result in syndromic forms of HPE, triploidies of entire chromosomes (e.g., 13 and 18), deletions or duplications of regions of a chromosome, and copy number variants. At least 12 HPE loci that play some role in the midline of the developing nervous system have been identified, including Shh, Otx2, Emx, Pax, Nkx-2.2, and some POU domain genes.13 Although 24% to 45% of affected live-born individuals have chromosomal abnormalities, no known correlation exists between the type or severity of the holoprosencephalic defect and the specific mutation. In addition to controlling prosencephalic separation and differentiation, the inductive effects of the prechordal mesoderm also influence the corpus striatum, thalami, eyes, face, and cerebral vasculature.13 The spectrum of associated facial anomalies range from flattening of the nasal bridge, hypotelorism without metopic synostosis, a single central maxillary incisor, cleft lip or palate to facial clefting, and cyclopia with a central proboscis.15 More severe facial anomalies are seen with more severe variants of HPE, but mild facial anomalies may occur in the absence of brain anomalies. Clinical problems associated with HPE include developmental delay, seizures, hypothalamic and brainstem dysfunction with swallowing and respiratory problems, thermal instability, pituitary dysfunction, and erratic sleeping patterns.13,15
Imaging: The hallmark of HPE is incomplete separation of the forebrain, absence of the anterior interhemispheric falx, and fusion of central gray nuclei. The septum pellucidum is absent. Considerable topographic variation exists in HPE, which is most often characterized as alobar, semilobar, or lobar.13–19 The malformation represents a continuum, ranging from overt hypotelorism, with severe facial anomalies, to mild hypotelorism, with subtle fusion of basal forebrain and central gray nuclei. Hydrocephalus may be present.
Alobar Holoprosencephaly
In this most severe form of HPE, separation between the cerebral hemispheres is completely lacking, with absence of the falx. The corpus callosum and septum pellucidum are absent, the central gray nuclei are fused, and the rudimentary single ventricle has a “U” configuration, which may communicate with a dorsal cyst (Fig. 31-14).1,14,17
Semilobar Holoprosencephaly
Relative preservation of the lateral and posterior cerebrum and the splenium exists in semilobar HPE. The posterior interhemispheric fissure and falx are present, whereas the hypoplastic frontal lobe is undivided. (Fig. 31-15).1,13,16 Lack of formation of the frontal lobes results in an anterior position of the Sylvian fissures, termed a “wide Sylvian angle” by Barkovich et al.17 The globus pallidi are absent or hypoplastic, and the caudate nuclei are fused, resulting in obliteration of or lack of formation of the septal region. The posterior limbs of the internal capsules are ventral to partially or totally fused thalami. The hippocampus is virtually always present, although usually incompletely or abnormally developed. A dorsal cyst may be present (e-Fig. 31-16).
Figure 31-15 Semilobar holoprosencephaly.
A, A sagittal T1-weighted image shows underdeveloped frontal lobes with midface hypoplasia. The splenium (arrow) of the corpus callosum is formed. B, An axial T2-weighted image shows failure of separation of the frontal lobes; the caudate and thalami are fused. Arrows indicate the anomalous anterior position of the Sylvian fissures. C, Diffusion tractography. The brain is viewed from the inferior aspect; arrows indicate the optic pathways. The red fibers are midline extensions of anomalous association fibers.
Lobar Holoprosencephaly
In this mildest form of HPE, hypoplasia of the frontal poles is present, along with agenesis of the septal pellucidum and incomplete separation of the basal forebrain, which is best depicted with high-resolution coronal images. The posterior frontal, parietal, and occipital lobes are more normally formed.1,14,17 The callosal body and splenium are preserved (Fig. 31-17).
Syntelencephaly (Middle Frontal Variant)
The middle frontal variant is an unusual variant of lobar HPE characterized by separation of the frontal and occipital poles, fusion of the middle portions of the cerebral hemispheres (Fig. 31-18), preservation of portions of the commissural fibers of the corpus callosum, and neuronal migration anomalies; thalamic fusion is variable.18,19
Figure 31-18 Middle frontal variant of holoprosencephaly.
A, A sagittal T1-weighted image shows that the callosal genu (arrow) is formed; the callosal body appears formed but is hypoplastic. Partial fusion of the frontal lobes is seen. B, An axial T2-weighted image shows fusion of cortex and white matter across the midline in the middle frontal region.
Septo-optic Dysplasia
Clinical Presentation: Septo-optic dysplasia (SOD) is considered along the continuum of disorders of ventral forebrain differentiation.20 Patients typically present in early childhood with nystagmus, optic nerve atrophy, short stature because of growth hormone deficiency, or panhypopituitarism without diabetes insipidus. Affected patients are at risk for Addisonion crisis from subclinical adrenal insufficiency, which may become clinically apparent only during illness or severe stress. Most cases of isolated SOD are sporadic.
Imaging: SOD is characterized by absence of the septum pellucidum and variable optic nerve hypoplasia (e-Fig. 31-19). The neurohypophysis is often ectopic or may be absent, in which case the pituitary stalk may be interrupted. However, agenesis of the septum pellucidum may be isolated with normal optic pathways and intact neuroendocrine function. Absence of the septal leaves results in downward displacement of the fornices into the third ventricle. SOD is associated with multiple malformations, such as schizencephaly, neuronal migration anomalies, and neural tube defects.
e-Figure 31-19 Variable appearance of septal agenesis.
A, A sagittal T1-weighted image shows that the genu and rostral portions of the corpus callosum are hypoplastic (arrow). An ectopic neurohypophysis (arrowhead) along the floor of the third ventricle is present. The optic pathways are hypoplastic. B, Septal agenesis with hydrocephalus has been caused by aqueductal stenosis. A normal hyperintense neurohypophysis (arrowhead) exists within the sella turcica. The optic pathways are normal. The fornices (arrow) are caudally displaced. C, A coronal T2-weighted image better shows the caudal displacement of the fornices (arrow) into the third ventricle.
Callosal Agenesis or Dysgenesis
Complete or partial malformation of the corpus callosum has an incidence of 1 in 4000.21 Normal corpus callosal development requires a precise sequence of cellular proliferation and migration, axonal growth and guidance, and midline glial development and patterning. Disruptions in this sequence may result from gene mutations, genetic polymorphisms, intrauterine infections, ischemia, and toxins.21
The telencephalic commissures normally develop in a predictable sequence.1,21 The anterior commissure (AC) forms around 55 days after conception, arising from the primitive hippocampal formations and ultimately connecting the anterior temporal lobes. Approximately 3 weeks later, the initial interhemispheric migration of pioneering callosal axons occurs, aided by the “glial sling,” which consists of primitive subependymal glial cells. The “glial wedge” is composed of radial glial cells that guide callosal axons across the midline and then repel the axons away from the midline into the contralateral hemisphere.1,21 After crossing the midline, callosal axons grow into the contralateral hemisphere; their ultimate destination mirror-images their region of origin and is within the same cortical layer from which the axon arose.21,22 The crossing axons follow a rostrocaudal gradient with the callosal rostrum forming before the splenium; formation of the splenium is complete by around 85 days after conception. Many of the crossing axons undergo programmed cell death after reaching their final destination; this apoptosis commences during the second trimester, with pruning of axonal axons continuing after birth.1,22
Imaging: The corpus callosum is the largest of the three telencephalic commissures, which include the anterior and hippocampal commissures. The AC is a variably-sized tract embedded in the cranial aspect of the lamina terminalis, which demarcates the anterior wall of the third ventricle and connects the anterior temporal lobes. The hippocampal commissure is a thin sheet of white matter connecting the two crura of the fornices and is not routinely visualized on MRI of the normal brain. Absence of the corpus callosum, AC, and hippocampal commissure is described as “complete commissural agenesis,” although some portion of the corpus callosum may be preserved (Fig. 31-20).1
[/not-level-membership-for-pediatrics-category]
