Chapter 7 Congenital Aortic Stenosis
Congenital Aortic Regurgitation
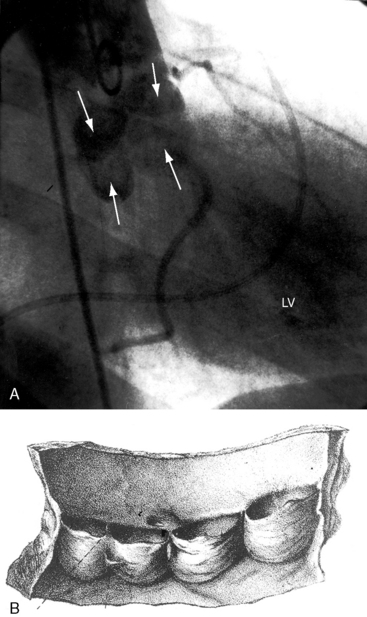
Clinical evaluation of congenital obstruction to left ventricular outflow seeks to establish the presence and degree of obstruction and the level and morphologic type.1–6 Five varieties of congenitally abnormal aortic valves are based on the number and types of cusps and commissures (Box 7-1).6 A unicuspid aortic valve is either acommissural7 or unicommissural.6 A unicuspid acommissural valve is characterized by a single leaflet with a central orifice that is usually stenotic but can be stenotic and regurgitant.7 Traces of three rudimentary commissures do not divide the valve (Figure 7-1A, left upper).6 This type of congenitally stenotic semilunar valve is found in the pulmonary location (see Chapter 11), but rarely in the aortic location.6 A unicommissural unicuspid valve is characterized by a single commissural attachment to the aorta (Figures 7-1A, left middle, and 7-2A).2,6 The single (unicuspid) leaflet originates from a single commissural attachment, proceeds across the aortic orifice without making contact with the aortic wall, bends on itself, and returns to reinsert at the same attachment site from which it originated.2,6 Remnants of rudimentary raphes are occasionally present.6 Viewed from above, the orifice resembles an exclamation point (see Figure 7-1A, left middle).6 The typical unicommissural valve is intrinsically stenotic, but if the free edge is sufficiently redundant and the single commissure is not fused, obstruction is initially absent but subsequently appears when mobility is reduced by fibrosis and calcification.2
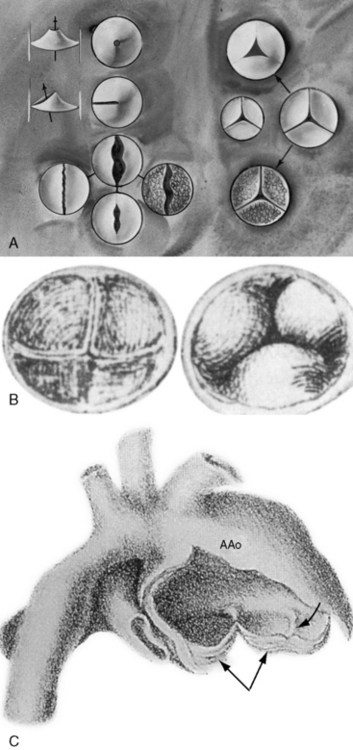
Figure 7-1 A, Illustrations of the various types of aortic valve stenosis. Figure on the left illustrates three types of congenitally abnormal aortic valves. The upper drawing shows a unicuspid acommissural valve. The middle drawing shows a unicuspid unicommissural valve with an eccentric orifice. The lower group of four drawings shows a functionally normal bicuspid aortic valve (upper center), a fibrocalcific stenotic bicuspid aortic valve (center right), a bicuspid aortic that is inherently stenotic because the free edges are not longer than the annular diameter (center left), and a bicuspid aortic valve that is inherently stenotic because of failure of commissural separation (lower center). Illustrations on the right show a normal trileaflet aortic valve with equal cusps and equal commissures (center right). A congenitally hypoplastic trifleaflet aortic valve (center left) is paired beside the normal trileaflet aortic valve (center left). Acquired aortic valve stenosis from fibrosis and calcification without commissural fusion (right lower) or from rheumatic fusion of commissures (upper) is shown. A dysplastic trileaflet valve is not shown. B, Congenital aortic cuspal inequality as represented by Leonardo da Vinci circa 1513.16 His legend read: “Figures of the cusps (aorti) of the gateway which the left ventricle possesses when it closes itself.” On the left is a trileaflet aortic valve as seen from above. On the right is a closed trileaflet aortic valve as seen from below. C, Dilation of the ascending aorta (AAo) above a congenitally bicuspid aortic valve (paired arrows) with a false raphe (curved arrow).
(From Maude Abbott. Atlas of congenital cardiac disease. Montreal: Osler Library, McGill University; 1936. Reproduced with permission.)
A bicuspid aortic valve is the most common congenital anomaly to which that structure is subject (see Figure 7-1A, left lower group)1,2,6 and is the most common gross morphologic congenital abnormality of the heart or great arteries in adults.8 Estimated frequency in the general population has been reported as 0.5% to 0.6%9 and 0.9% to 1.36%,10 with an overall prevalence rate in the United States of approximately four million.4,11 The male:female ratio is 2:1. The bicuspid aortic valve is a genetic disorder, with a transmission pattern that suggests autosomal dominant inheritance.10,12 A low prevalence rate of bicuspid aortic valve is found in African Americans.13 Acquired calcification of a congenitally bicuspid aortic valve accounts for approximately half of surgical cases of isolated aortic stenosis in adults.11,14 Hypercholesterolemia is an atherosclerotic risk factor for the development of calcification.15
The bicuspid aortic valve was first identified in the early 16th century by Leonardo da Vinci in his remarkable Anatomical, Physiological, and Embryological Drawings, released by Dover Publications in a facsimile edition.16 Normal aortic valves are composed of a connective tissue framework of interstitial cells and a matrix covered by endothelial cells. During valvulogenesis, extracellular matrix proteins direct cell differentiation and cusp formation. Differentiation of mesenchymal cells into mature aortic valve cells correlates with the expression of the matrix protein fibrillin-1, which is deficient in bicuspid aortic valve tissue. Three morphologic types of a bicuspid aortic valve based on cusp size are characterized by two cusps of equal size, two cusps of unequal size, and a conjoined cusp twice the size of its nonconjoined mate.11 Three morphologic types based on commissural fusion are characterized by fusion of the right and left coronary cusps (most common), fusion of the right and noncoronary cusps, and fusion of the left and noncoronary cusps (least common).8 A false raphe can be well-formed, fenestrated, calcified, or absent.11,17 If the free edges of the bicuspid leaflets are sufficiently long, if the cusps are thin and mobile, and if the commissures are not fused, the valve is functionally normal—unobstructed—which is the usual condition at birth (see Figure 7-1A, left lower).4–6 Conversely, if the commissures are congenitally fused, or if the free edges of the cusps are not longer than the diameter of the aortic ring, the valve is inherently obstructed (see Figure 7-1A, left lower). Fusion of the right and left coronary cusps is strongly associated with coarctation of the aorta.18 Fusion of the right and noncoronary cusps is associated with valve pathology.18 Sclerosis of a bicuspid aortic valve begins as early as the second decade, and calcification as early as the fourth decade.19 The fibrocalcific process is more rapid in bicuspid aortic valves with cusps of unequal size because of maldistribution of tension during diastolic closure.19 Rarely, a congenital bicuspid aortic valve is stenotic because of myxoid dysplasia.20 Bicuspid aortic and bicuspid pulmonary valves coexist in the Syrian hamster,21 but not in human subjects.
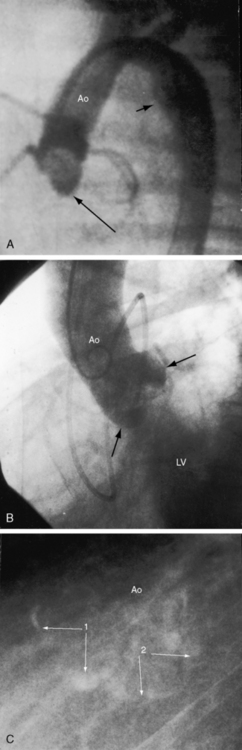
A functionally normal bicuspid aortic valve can continue to function normally throughout a long lifetime,4 but more often than not, fibrocalcific thickening or acquired commissural fusion decreases mobility and renders the valve stenotic (see Figures 7-1A, left lower group, and 7-2C).1,4,11 Thomas Peacock22 recognized this tendency in his On Malformations of the Human Heart (1858). Abnormal mechanical stress is an important factor in promotion of fibrosis and calcification of a bicuspid bicommissural aortic valve (see Figure 7-2C).23 An important consequence of a functionally normal bicuspid aortic valve is progressive regurgitation, which may be accelerated by infective endocarditis to which a bicuspid valve is highly susceptible (see subsequent discussion). Rarely, a severely incompetent bicuspid aortic valve becomes stenotic with virtual loss of regurgitation.24
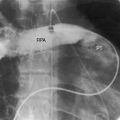
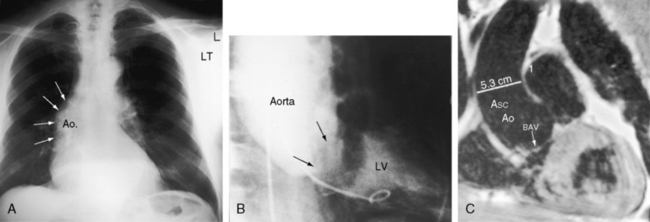
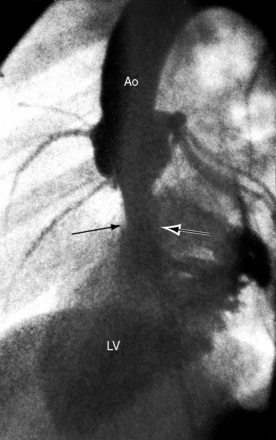
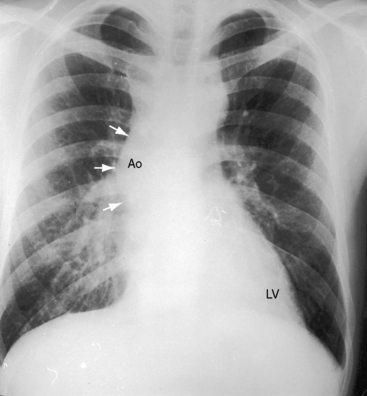
Dilation of the ascending aorta is consistently associated with a congenitally bicuspid aortic valve (Figures 7-1C, 7-3, and 7-4),25–27 but the term poststenotic dilation is a misnomer because the ascending aorta is dilated whether the bicuspid valve is stenotic, incompetent, or functionally normal.28 Dilation is the result of an inherent medial abnormality that expresses itself as an ascending aortic aneurysm with chronic aortic regurgitation (see Figure 7-4) or dramatically as a dissecting aneurysm (Figure 7-5).28–32 A decrease in ascending aortic elasticity is independent of dilation.33,34
Trileaflet aortic valves are congenitally abnormal when three cusps and three commissures are miniaturized within a small aortic ring35 or when an aortic valve is the site of myxoid dysplasia.20 In a hydraulically ideal aortic valve with three equal cusps, diastolic force is equally distributed among the three cusps and their sinus attachments. Cuspal inequality is a common variation of normal in trileaflet aortic valves but results in unequal distribution of diastolic force (see Figure 7-1B).5,36 The fibrocalcific process of aging proceeds more rapidly in the cusp or cusps that bear the greatest hemodynamic stress.11,36 Accordingly, cuspal inequality in a normal trileaflet aortic valve enhances the aging process, converting a functionally normal trileaflet valve into fibrocalcific aortic stenosis.5,36 Similarly, the tendency for a bicuspid aortic valve to become fibrocalcific (see Figure 7-2C) is related in part to the mechanical stress inherent in bicuspid cuspal inequality (see Figure 7-2B).23 The quadricuspid aortic valve, first reported in 1862, can function normally or cause incompetence (Figures 7-35 and 7-42)37 but is rarely stenotic.6 Six-cuspid semilunar valves sporadically occur with truncus arteriosus (see Chapter 28).6
Fixed subaortic stenosis is the second most common variety of congenital obstruction to left ventricular outflow6,38 and accounts for 15% to 20% of all types of congenital aortic stenosis.39 It occurs in isolation (Figure 7-6) or with other congenital cardiac defects.40,41 Nonfixed muscular subaortic stenosis can coexist with severe aortic valve stenosis and can account for as much as half the pressure gradient.40–42
This chapter deals with two principal varieties of fixed subaortic stenosis in hearts that are otherwise devoid of congenital heart disease. The first variety is characterized by a thin fibrous crescent-shaped membrane located immediately beneath the aortic valve (see Figure 7-6A).6,40 The membrane is occasionally relatively thick and forms a fibrous or fibromuscular collar that extends across an otherwise normal left ventricular outflow tract43 and inserts onto the anterior mitral leaflet. This form of fixed subaortic stenosis occurs in human hearts and in dogs, pigs, and cows.44 The aortic root is not dilated.28 Aortic regurgitation is associated with malformed leaflets that are damaged by the proximity of the subvalvular membrane or fibromuscular collar and by the impact of the eccentric systolic jet (see Figure 7-6B).6 Tubular subaortic stenosis is a less common variety of fixed obstruction to left ventricular outflow and is represented by a fibromuscular channel that occupies several centimeters within the outflow tract (Figure 7-7).40,41,45 A layer of fibrous tissue extends onto the ventricular surface of the anterior mitral leaflet. The aortic cusps show fibrous thickening as with discrete subaortic stenosis. The aortic root is not dilated (see Figure 7-7).28,45
Fixed subaortic stenosis as just defined is not present during intrauterine cardiac morphogenesis, is therefore not con genitus, and accordingly is uncommon if not rare in neonates and infants.41 The disorder becomes manifest after the first year of life and then changes in both severity and morphology. In contrast to rapid progression in infants and children, fixed subaortic stenosis in adults progresses slowly.40,41,45,46 Aortic regurgitation is common but is usually mild and nonprogressive (see Figure 7-6B).
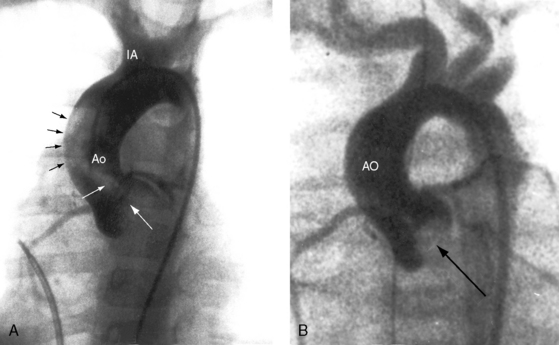
Supravalvular aortic stenosis is the least common variety of congenital obstruction to left ventricular outflow. The most frequent type is a localized segmental hourglass deformity immediately above the aortic sinuses with medial thickening and fibrous intimal proliferation (Figure 7-8A).6 The size of the aorta distal to the obstruction is normal or reduced. The sinuses of Valsalva are enlarged.47 Localized supravalvular aortic stenosis is occasionally caused by a fibrous membrane with a central opening.6 An uncommon variety is represented by tubular hypoplasia of the ascending aorta beginning above the sinuses of Valsalva and associated with narrowing of the orifices of the brachiocephalic arteries (Figure 7-8B).6,48 The aortic leaflets are usually thickened and adherent to the supravalvular stenosing ridge,6,47 are occasionally dysplastic, and may fuse to a coronary ostium.6 The aortic valve abnormalities usually cause no more than mild regurgitation.6
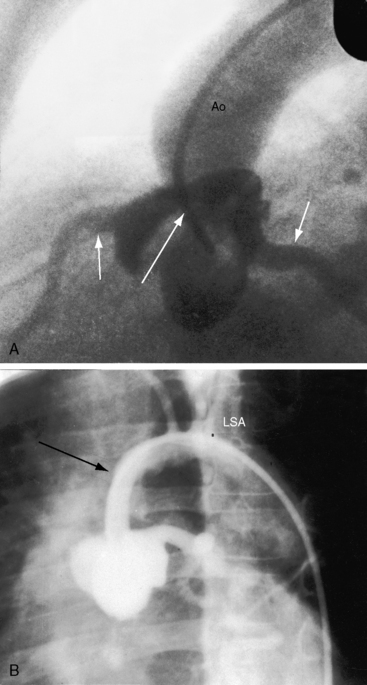
Three additional features of supravalvular aortic stenosis include: (1) the anatomy of the extramural coronary arteries; (2) the condition of the aortic leaflets and aortic sinuses; and (3) the association with Williams syndrome. Obstruction of a coronary ostium can be caused by adherence of a distorted aortic leaflet to the supravalvular stenotic ridge, by aortic medial proliferation, and by the supravalvular ridge itself, which can impede diastolic flow into an ostium.49 Because the coronary ostia are proximal to the supravalvular obstruction, the coronary arteries are exposed to elevated left ventricular systolic pressure6 and become thick-walled and dilated (see Figure 7-8). Coronary artery aneurysms have been described.50 and hypertension is a risk factor for premature atherosclerosis.6
In 1961, Williams, Barratt-Boyes, and Lowe51 described the association of supravalvular aortic stenosis with distinctive elfin facies and mental retardation. In 1962, Beuren, Apitz, and Harmjanz52 expanded the syndrome to include pulmonary artery stenosis. Williams syndrome or Williams-Beuren syndrome now includes elfin facies, mental retardation, small stature, infantile hypercalcemia, supravalvular aortic stenosis, pulmonary artery stenosis, and important vascular abnormalities, especially in adults.53,54 Renal abnormalities occur in nearly half of afflicted patients and are represented by renal artery stenosis, segmental scarring, cystic dysplasia, nephrocalcinosis, marked asymmetry in kidney size, solitary kidney, and pelvic kidney.53,55 Systemic hypertension is not necessarily related to the renovascular abnormalities56 but instead is related to stiffness of arterial walls.57 A generalized arteriopathy is characterized by medial thickening and luminal narrowing of systemic and pulmonary arteries.58 Long segment narrowing of the aorta may occur with or without localized coarctation.56 Involvement of cerebral arteries is responsible for strokes. Tortuous retinal arteries similar to those that accompany coarctation of the aorta (see Chapter 8, Figure 8-8) were described in the original report of Williams, Barratt-Boyes, and Lowe,51 and in 1985, the observation was confirmed.56
The physiologic consequences of congenital aortic valve stenosis are reflected in the response of the left ventricle to increased afterload. An adaptive increase in left ventricular mass in the immature heart is chiefly the result of hyperplasia (replication) of cardiomyocytes. In contrast, the increase in left ventricular mass in the mature heart is the result of hypertrophy (an increase in cell size) of terminally differentiated cardiomyocytes.59 The afterloaded immature heart is capable of capillary replication that is proportional to cardiomyocyte replication, so capillary density remains normal and coronary flow reserve remains normal.59,60 The increase in left ventricular mass characterized by myocyte hyperplasia with proportionate growth in the microvascular bed sets the stage for low left ventricular systolic wall stress and supernormal ejection performance.59 The left ventricle thickens concentrically and the cavity size is normal or small,59 so distensibility decreases. A greater force of left atrial contraction generates the end-diastolic fiber length necessary for left ventricular performance appropriate for the increased afterload without an increase in end-diastolic volume or left atrial mean pressure. The normal trileaflet aortic valve and its annulus increase in anatomic cross-sectional area with age even after maturity.61 The normal trileaflet physiologic orifice, which is the cross-sectional area defined by the leaflets in systole, is flow-dependent, varying directly with the volume and rate of left ventricular ejection. In the resting state, less than half the cross-sectional area of a normal trileaflet aortic valve is used during ejection, so a large reserve is available during high-flow states.
The physiologic consequences of congenital aortic stenosis take into account the morphology of the obstruction, the degree of obstruction, the age of onset, and whether or not the obstruction is progressive. A functionally normal bicuspid aortic valve awaits adulthood to become stenotic as its leaflets thicken and calcify. An stenotic aortic valve in infants and young children tends to generate an increasingly higher gradient in response to the progressive increase in transaortic flow that accompanies the normal age-related increase in body mass.62 In Williams syndrome, progressive supravalvular aortic stenosis results from inadequate growth of the sinotubular junction.63 Fixed subaortic stenosis is not present in utero but usually begins after the first year of life and undergoes a progressive decrease in cross-sectional area (see previous discussion).41,46 The time course in adults is much slower.
The subendocardium of the left ventricle in aortic stenosis is vulnerable to ischemia because of a selective decrease in perfusion.49 In neonates and infants with severe aortic stenosis, subendocardial ischemia is responsible for papillary muscle infarction with mitral regurgitation49 and for endocardial fibrosis and reduced cavity size that depress left ventricular contractility.64 A progressive rise in left ventricular filling pressure, in left atrial mean pressure, and in pulmonary arterial pressure provokes right ventricular failure.65 Supravalvular aortic stenosis incurs the additional impediment of compromised coronary perfusion (see previous discussion).
The physiologic response of the neonate to severe aortic stenosis is best understood in light of the fetal circulation. Intrauterine left ventricular volume is low because pulmonary blood flow is virtually nil. When lungs expand at birth, pulmonary blood flow commences and a severely obstructed, thick-walled left ventricle with reduced cavity size suddenly receives a sizable increment in volume. Left ventricular filling pressure rises steeply, left atrial pressure rises in parallel, and a left-to-right shunt is established across a stretched valve-incompetent foramen ovale. If a small left ventricular cavity is beset with endocardial fibrosis or fibroelastosis, the hemodynamic consequences are correspondingly worse.64 Vasoreactive pulmonary arterioles constrict, pulmonary artery pressure rises, and pressure overload is imposed on an already volume-overloaded right ventricle. Temporary patency of the ductus arteriosus diverts right ventricular blood into the aorta and delays the onset of symptoms, but when the ductus closes, that advantage is lost because the entire right ventricular output enters the pulmonary circulation and the left side of the heart.
The response to dynamic exercise mild aortic stenosis is normal, but when stenosis is severe, left ventricular stroke volume is blunted at each level of graded isotonic stress.66–68 In congenital aortic valve stenosis, the augmented flow and increased left ventricular systolic pressure induced by exercise result in a larger computed aortic valve area, which implies that the stenotic valve is sufficiently mobile to open more widely when stressed.66 Activation of canine left ventricular baroreceptors in response to an increase in left ventricular pressure or stretch induces hypotension from skeletal muscle vasodilation. Reflex vasodilation and bradycardia induced by activation of left ventricular baroreceptors during isotonic exercise are responsible for hypotension and exertional syncope.69
History
Congenital aortic valve stenosis is considerably more common in males, with a gender ratio of approximately 4:1.4,39,70 Male prevalence is less common in supravalvular aortic stenosis, depending in part on genetic transmission. Discrete and tunnel subaortic stenosis both have a distinct female prevalence.38 In the Newfoundland dog, genetically transmitted subaortic stenosis has an equal gender ratio.44
When congenital aortic stenosis is present at birth, the murmur is also present because the anatomic and physiologic conditions required to generate the murmur exist.41,46 An exception is fixed subaortic stenosis, which is not present at birth, and an additional qualification relates to specific types of congenitally malformed aortic valves.6 As a rule, the fewer the cusps and commissures, the greater the likelihood of an intrinsically stenotic valve and of a murmur in the newborn.6 Intrinsically stenotic valves are usually unicuspid unicommissural or bicuspid,6 but when a bicuspid aortic valve is functionally normal,4,6 the onset of the murmur awaits the development of fibrocalcific thickening in adulthood.1,4,6,39 Stenosis of a unicommissural aortic valve occasionally follows a protracted course similar to that of a bicuspid aortic valve.2,14
An impression of murmur intensity (loudness) can be inferred in the history with determination of how readily a murmur was heard during follow-up examinations. A loud murmur is likely to be heard even in uncooperative infants, whereas the soft murmur of mild obstruction is likely to be overlooked or mistaken for a normal murmur, even in cooperative infants and older children (see Chapter 2).
Growth and development are affected in patients with Williams syndrome (see subsequent discussion). Symptoms associated with aortic stenosis, especially in children, may be absent even in the presence of severe obstruction,39 and progression from mild to severe is not necessarily accompanied by symptomatic deterioration.71 Effort dyspnea and fatigue reflect an inadequate increment in cardiac output and an increase in left ventricular end-diastolic pressure. Effort syncope arouses suspicion of aortic stenosis in an acyanotic patient with a prominent cardiac murmur dating from infancy or childhood. Syncope depends on the degree of obstruction, not on its morphologic type,70 and is the result of reflex vasodilation and bradycardia induced by activation of left ventricular baroreceptors during isotonic exercise (see previous discussion).69 Syncope can be recurrent,39 and sudden death looms as a threat,70 although the risk in children is small compared with adults. Subtle cerebral symptoms consist of mild giddiness, faintness, or lightheadedness with effort. Syncope-induced hypotension may provoke electrical ventricular instability in adults with atherosclerotic coronary artery disease but not in young patients with aortic stenosis and normal coronary arteries.49,72
A potential disparity exists between the oxygen requirements of an hypertrophied left ventricle and coronary blood flow and flow reserve. The disparity is aggravated by acquired coronary artery disease in adults with fibrocalcific bicuspid aortic stenosis and in children with coronary artery abnormalities that accompany supravalvular aortic stenosis.73 Angina pectoris in young patients is a symptom of congenital aortic stenosis that arrests attention.
Inappropriate diaphoresis increases with the onset of congestive heart failure, especially in neonates.64,74 In infants with severe aortic stenosis, mitral regurgitation from papillary muscle infarction adds to the hemodynamic burden.72
Bicuspid aortic stenosis and subaortic stenosis are genetically transmitted (see previous discussion).44,75–77 Familial and nonfamilial types of supravalvular aortic stenosis are the basis of the following classification: (1) familial with normal facies and normal intelligence; (2) nonfamilial with normal facies and normal intelligence; and (3) nonfamilial with Williams syndrome,51,78 which results from mutation or deletion of the elastin gene located at chromosome 7q11.23.79–82 Pulmonary artery stenosis in Williams syndrome tends to improve with time, and the supravalvular aortic stenosis is progressive because of growth failure of the sinotubular junction, which may be associated with obstruction of coronary artery ostia.63 Supravalvular aortic stenosis has been experimentally produced in newborn rabbits with administration of maternal vitamin D during gestation83 and has occurred in human offspring when infantile hypercalcemia resulted from administration of vitamin D during pregnancy. The history should therefore include questions regarding maternal vitamin D ingestion.
Infective endocarditis is a potential risk in all types of congenital aortic stenosis,6,39 but the bicuspid aortic valve is especially vulnerable, whether functionally normal, stenotic, or incompetent, an observation made by William Osler84 more than a century ago. Dissecting aneurysm of the ascending aorta may dramatically interrupt the clinical course of bicuspid aortic stenosis (see Figure 7-5).85 Gastrointestinal bleeding associated with aortic stenosis, sometimes called Heyde’s syndrome, occurs in older adults.86 Angiodysplasia has been used to designate the offending lesions that tend to be present in the ascending colon but may be distributed throughout the gastrointestinal tract.86 Aortic stenosis is not thought to cause the lesions but is thought to increase the likelihood that the lesions will bleed.
Physical Appearance
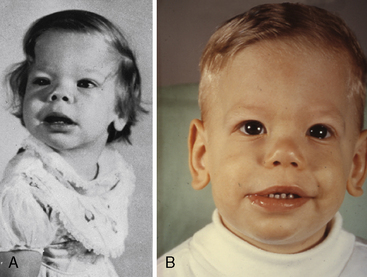
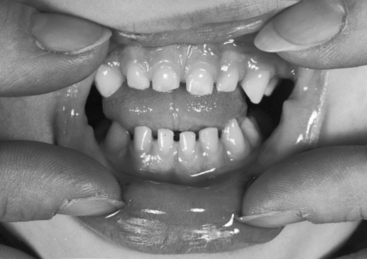
Williams syndrome (nonfamilial supravalvular aortic stenosis) is characterized by peculiar facies, short stature, and mental retardation.3,51,52 The chin is small (hypoplastic mandible), the mouth is large, the lips are patulous, the nose is blunt and upturned, the eyes are widely set with occasional internal strabismus, the cheeks are baggy (Figure 7-9), the teeth are malformed, and the bite is abnormal (malocclusion; Figure 7-10). Flat molar regions accentuate the prominence of a wide mouth with full lips, small jaw, and long philtrum.54 The brow is broad with prominent supraorbital ridges. The nasal tip is broad, and the nacres are anteverted. Adults with Williams syndrome are relatively short and tend to have lordosis, kyphoscoliosis, and joint abnormalities of the lower limbs that result in a stiff awkward gait.54 Friendly temperaments and deep, somewhat metallic voices further emphasize the similarities. XO Turner’s syndrome (see Chapter 8) represents another distinctive physical appearance that coexists with a bicuspid aortic valve.87 Congenital heart disease with Turner’s syndrome has been known since the initial description by Morgagni, and is coupled with different patterns of X monosomies.88 Abnormal karyotypes consist of 45 X mosaicism and X structural abnormalities.89 Patients with severe dysmorphic features have a significantly higher prevalence rate of congenital heart disease,89 and patients with 45 X Turner’s syndrome have the highest prevalence rate.89 X structural abnormalities are associated with an increased prevalence rate of bicuspid aortic valve,89 but with X deletion, the incidence rate of congenital heart disease is not increased.88 In Noonan’s syndrome (Turner’s phenotype with normal genotype), obstruction to left ventricular outflow is the result of hypertrophic obstructive cardiomyopathy (Figure 7-11).
Arterial Pulse
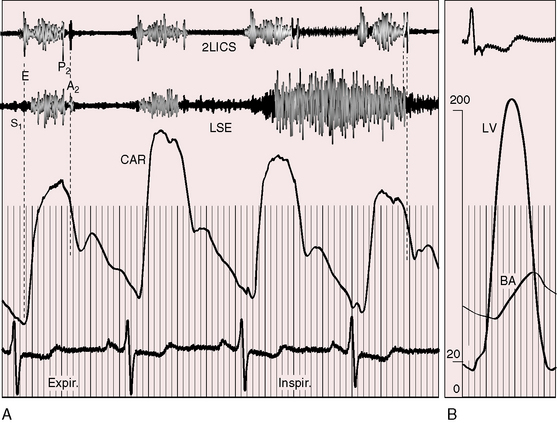
The object of the following paragraphs is to consider the aid in the diagnosis of aortic stenosis that may be supplied by a study of the pulse (Graham Steell, The Lancet, November 1894). Fixed obstruction to left ventricular outflow distinctively alters the arterial pulse.3,90 The pulse pressure is small, the rate of rise is slow, the peak is sustained, and the decline is gentle (Figures 7-12 and 7-13). This typical configuration is not as frequent in children as in adults with equivalent aortic stenosis.90,91 Children with severe obstruction may have a brachial arterial pulse that is interpreted as normal, although palpation of the carotid artery improves accuracy.91,92 A bisferiens pulse (twin peaked) implies coexisting aortic regurgitation.90
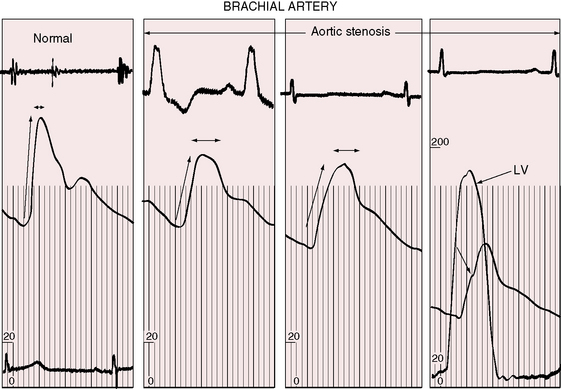
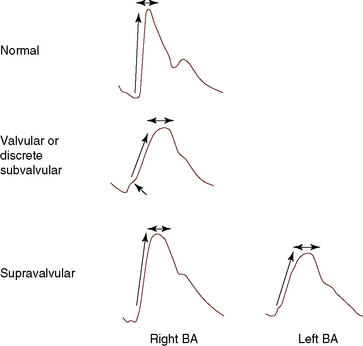
The right and left brachial and carotid arterial pulses are symmetric in valvular or fixed subvalvular aortic stenosis, but in supravalvular aortic stenosis, the rate of rise, the systolic pressure, and the pulse pressure are greater in the right brachial artery and in the right carotid artery (see Figure 7-13)93,94 because the hourglass deformity directs the high-velocity jet toward the right wall of the aorta and the Coanda effect (affinity of a jet stream for adherence to a wall) carries the jet into the innominate artery.93–95 Experimental observations with an aortic arch model showed that kinetic energy developed in a jet stream under conditions simulating supravalvular aortic stenosis is sufficient to account for the clinically observed differences in arterial pressure.93 Selective narrowing of the origins of the left carotid and left subclavian arteries (see Figure 7-8B) is an uncommon cause of asymmetric pulses.95 In healthy adults, the systolic pressure in the right arm is 10 to 15 mm Hg higher than in the left arm.90 Simultaneous determination of blood pressure in both arms minimizes these differences, and the technique of palpation shown in Figure 7-14 is useful in the clinical comparison of the right and left brachial pulses.3,90
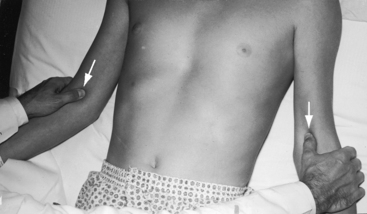
In valvular and fixed subaortic aortic stenosis, arterial thrills or shudders are common in the suprasternal notch and over the carotid and subclavian arteries.3,90,92 In supravalvular aortic stenosis, the thrill is distinctly more pronounced over the right carotid artery, which exhibits an increased pulsation.
Jugular Venous Pulse
Directing attention to the amplitude of the jugular venous A wave in subjects with isolated obstruction to left ventricular outflow is seemingly paradoxical.3,90 However, left ventricular hypertrophy serves to decrease right ventricular distensibility, so the right atrium contracts with greater force and the amplitude of the jugular venous A wave increases in the absence of pulmonary hypertension (Figure 7-15).3,90,96
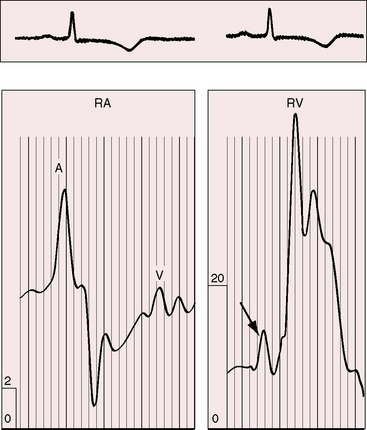
Precordial Movement and Palpation
Neonates with severe aortic valve stenosis have a prominent right ventricular impulse because of pulmonary hypertension and a left-to-right shunt at the atrial level.97 With these exceptions, the characteristic precordial impulse is left ventricular, varying from normal in mild aortic stenosis to the strong sustained impulse generated by an hypertrophied left ventricle of severe aortic stenosis.3,90 Presystolic distention is in response to the increased force of left atrial contraction (Figure 7-16), which is evidence that the aortic stenosis is hemodynamically appreciable.98 Dilation of the ascending aorta rarely transmits an impulse because the rate of ejection is blunted by the stenotic aortic valve. If an ascending aortic impulse occurs at all, it is likely to do so in patients with mild obstruction and an aortic aneurysm (see Figure 7-4).85
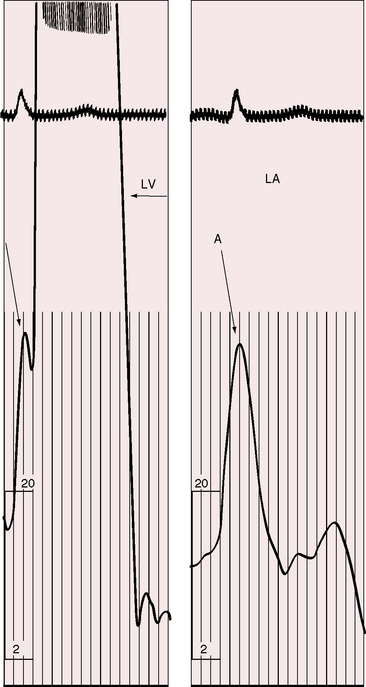
Systolic thrills are trivial or absent when aortic stenosis is mild or when severe aortic stenosis occurs with left ventricular failure. The thrill radiates upward and to the right, is maximal in the second right intercostal space, and is detected in the suprasternal notch and over both carotid arteries.90 The thrill in infants is sometimes maximal to the left of the sternum, inviting a mistaken diagnosis of ventricular septal defect. Even in older children, the thrill is occasionally more pronounced in the second or third left interspace, although radiation is still upward and to the right.90 In supravalvular aortic stenosis, the thrill is especially prominent below the right clavicle and on the right side of the neck.
Auscultation
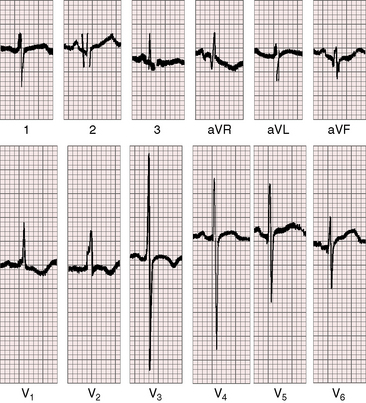
Normal splitting of the first heart sound must be distinguished from a first heart sound followed by an aortic ejection sound, which has considerable diagnostic significance.3,90 The ejection sound is separated from the first heart sound by a distinct interval, is louder and higher pitched than the first sound, and is heard best at the cardiac apex (Figure 7-17A). An aortic ejection sound is a valuable auscultatory sign of the level but not the degree of aortic stenosis.99 The sound coincides with abrupt cephalad movement of a mobile dome-shaped stenotic valve (see Figure 7-3B).17,99 The ejection sound may be difficult to hear in infants and disappears in adults when fibrocalcific changes impair valve mobility (see Figure 7-2C). Because the ejection sound is valvular in origin, it does not occur in fixed subaortic stenosis (Figure 7-17B) or in supravalvular aortic stenosis (Figure 7-18).
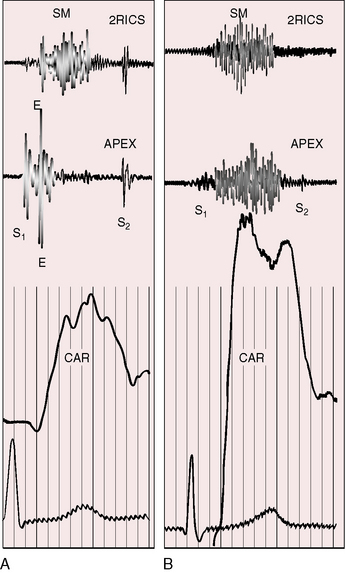
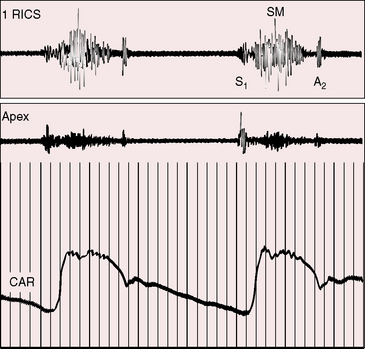
The aortic stenotic murmur is the prototype of the left-sided midsystolic murmur, beginning after the first heart sound or with the ejection sound, rising in crescendo to a systolic peak, and then declining in decrescendo to end before the aortic component of the second sound (Figures 7-17 through 7-19).3,90 The murmur is harsh, rough, and grunting, especially when loud, and may have an early systolic peak and a short duration, a relatively late peak and a prolonged duration, or all gradations in between, but the shape remains symmetric (see Figure 7-19).3,90 Intensity varies from bare audibility to grade 6/6, decreases in the presence of left ventricular failure, and may alternate in a fashion analogous to pulsus alternans.59,90 Although configuration, length, and loudness do not necessarily correspond to the degree of obstruction, some conclusions can be drawn (see Figure 7-19).100 The longer and louder the murmur, and the later its symmetric systolic peak, the greater the likelihood of severe stenosis. The shorter and softer the murmur and the earlier its symmetric peak, the greater the likelihood of mild stenosis or of a functionally normal bicuspid aortic valve. Irrespective of length, the murmur remains symmetric. A late symmetric peak reflects prolonged ejection and increased severity (see Figure 7-19), but severity does not change the shape of the aortic stenotic murmur, as in congenital pulmonary valve stenosis in which the murmur becomes progressively asymmetric as severity increases (see Chapter 11).
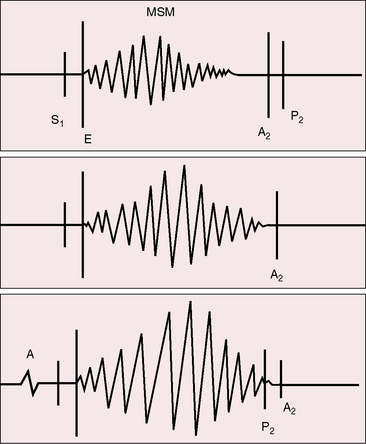
The typical murmur of congenital aortic valve stenosis is maximal in the second right interspace with radiation upward, to the right, and into the neck, reflecting the upward and rightward direction of the high velocity jet within the ascending aorta. Very loud murmurs are occasionally transmitted to the shoulders and elbows. In infants with aortic valve stenosis, the murmur may be maximal to the left of the sternum and mistaken for the murmur of ventricular septal defect, but with the passage of time, maximum intensity shifts to the right base.3 Maximum intensity in tubular subaortic stenosis is likely to be at the mid left sternal edge.45
When aortic stenosis is supravalvular, the jet may be as distal as the innominate artery, so the murmur is most prominent in the first right intercostal space (see Figure 7-18) and in the right side of the neck.90 A saccular aneurysm near the orifice of the innominate artery was ascribed to the jet impact. Systolic murmurs in the axillae and back are likely to be caused by coexisting stenosis of the pulmonary artery and its branches.52,90 Normal supraclavicular systolic murmurs in children can be mistaken for aortic stenosis,90 but supraclavicular murmurs are softer below the clavicles and decrease or vanish with hyperextension of the shoulders (see Chapter 2).90,101
An aortic stenotic midsystolic murmur at the apex must be distinguished from the holosystolic murmur of mitral regurgitation. Clear audibility of the aortic component of the second heart sound in the presence of a prominent apical murmur generally means that the murmur ends before aortic closure and is therefore midsystolic.90 An apical holosystolic murmur is likely to envelope the aortic component second sound, which is then inaudible. When the aortic valve is fibrocalcific and immobile, the closure sound is soft or inaudible at the base and at the apex, so an accurate auscultatory assessment of murmur length is difficult or impossible. However, an aortic stenotic murmur is louder in the beat after the compensatory pause that follows a premature ventricular contraction, whereas the murmur of mitral regurgitation does not amplify in the postpremature beat.102
The second heart sound should be analyzed regarding the intensity of the aortic component and the presence, degree, and type of splitting.3,90 The mobile valve that generates an ejection sound during its abrupt cephalad movement generates a well-preserved aortic component of the second sound during its abrupt caudal movement, even in the presence of appreciable obstruction (see Figure 7-17A).103 In subvalvular or supravalvular aortic stenosis, the aortic component of the second sound is normal, diminished, or absent, depending on the degree of stenosis (see Figures 7-17 and 7-18).
Splitting of the second heart sound is not necessarily related to severity.3,90 A delay in the aortic component results from prolonged left ventricular ejection from an increase in duration of left ventricular systole and a delay in the aortic incisura.104 The incisura is delayed because of the time required for the elevated left ventricular systolic pressure to fall below the level of the low aortic root pressure, at which point the incisura is inscribed.104 Inspiratory splitting of the second heart sound means that the duration of left ventricular ejection is not prolonged and aortic stenosis is mild (see Figure 7-19). Paradoxical splitting or reversed sequence of semilunar valve closure (Figures 7-19 and 7-20) means that the duration of left ventricular ejection is prolonged and aortic stenosis is severe. Left ventricular systolic pressure is then at or near its maximum of 250 mm Hg in older children and adults, so the approximate gradient is the difference between 250 mm Hg and the cuff brachial arterial systolic blood pressure.3,90 Reversed splitting may appear after exercise. Because paradoxical splitting is difficult to detect and because prominent inspiratory splitting is confined to mild obstruction, it follows that most patients with aortic stenosis have a second sound that is single or closely split through a wide range of severity.
A soft early diastolic murmur of aortic regurgitation occurs in approximately 50% of cases of fixed subvalvular stenosis because of the high incidence rate of abnormalities of the aortic valve.6 An aortic regurgitation murmur may be present with mild bicuspid aortic stenosis or with a functionally normal bicuspid aortic valve because the free edges of the bicuspid leaflets must be greater than the diameter if the aortic annulus to permit unobstructed flow (see Figure 7-1A). Aortic regurgitation is least likely to accompany supravalvular aortic stenosis but may do so when the cusps are malformed.6
A fourth heart sound (see Figure 7-19) is the auscultatory counterpart of presystolic distention of the left ventricle (see Figure 7-16), although the low frequency sound may be soft or absent despite the presence of a presystolic impulse. These signs imply an increased force of left atrial contraction required by the hypertrophied left ventricle to achieve an end-diastolic fiber length appropriate for greater contractile force (see Figure 7-16). Potain recognized this tendency in left ventricular hypertrophy when he wrote that the wall of the ventricle is placed under tension precisely at the moment that this (the added sound) occurs.98 Accordingly, the fourth heart sound is a feature of hemodynamically significant aortic stenosis.98 However, after age 40 years, the fourth heart sound is not a reliable sign of severity because it is heard in healthy adults.98
Electrocardiogram
Left ventricular hypertrophy in the electrocardiogram can occur with no more than mild to moderate congenital aortic stenosis (Figure 7-21),105,106 and sudden death occasionally occurs, with severe aortic stenosis and a normal or near normal electrocardiogram.107 Severity can progress without a change in the electrocardiogram, which if initially normal, remains so. Although these points are noteworthy, they should not detract from the value derived from careful interpretation of the scalar tracing.108
P waves are usually normal (Figures 7-21 and 7-22). Broad notched P waves are exceptional and are likely to indicate significant coexisting mitral regurgitation.106 The QRS axis is usually normal irrespective of severity (see Figures 7-21 and 7-22).106 Depolarization is clockwise, so Q waves appear in leads 3 and aVF (see Figures 7-21 and 7-22). In severe congenital aortic stenosis, electrocardiographic evidence of subendocardial ischemia or infarction can occur even in infants (Figure 7-23).109 The electrocardiogram in supravalvular aortic stenosis can reflect the ischemia of coronary ostial obstruction.
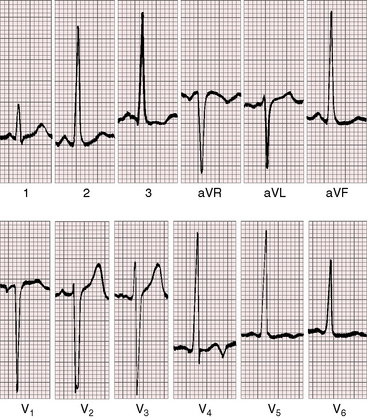
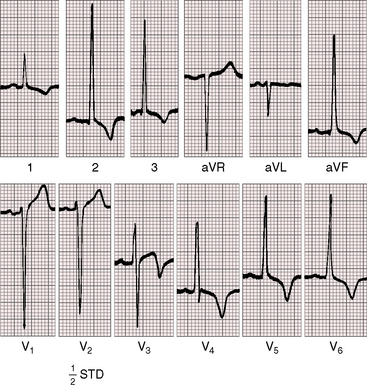
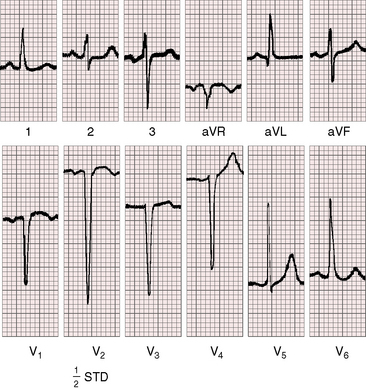
Left ventricular hypertrophy is manifested by tall R waves in leads 2 and aVF, deep S waves in lead V1, tall R waves in V5-6, and deeply inverted asymmetric T waves (see Figures 7-21 through 7-23).106 The T waves usually point in a direction opposite the QRS complex (wide QRS-T angle; see Figure 7-22). Exercise stress tests can provoke ST-T wave abnormalities when the resting electrocardiogram is normal,68 and digitalis glycosides induce or exaggerate the ST-T patterns of left ventricular hypertrophy. Electrocardiographic evidence of right ventricular hypertrophy is reserved for neonates with pinpoint aortic valve stenosis, pulmonary hypertension, a small left ventricular cavity, and a left-to-right shunt through a patent foramen ovale (Figure 7-24). Right ventricular hypertrophy occurs when mild to moderate supravalvular aortic stenosis is accompanied by severe pulmonary artery stenosis.52
X-Ray
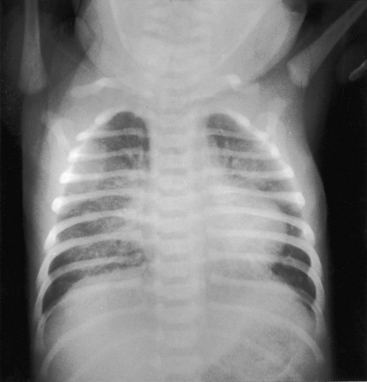
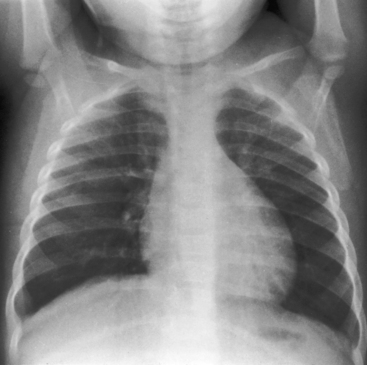
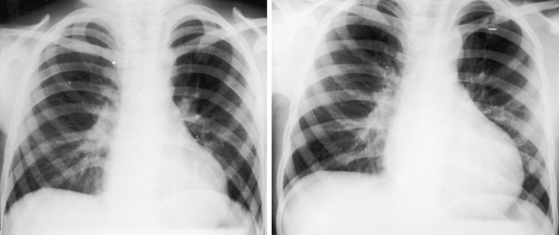
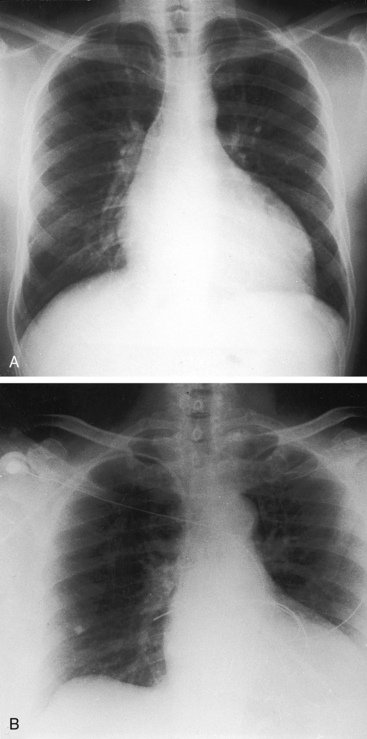
In neonates with severe aortic stenosis, pulmonary venous congestion coincides with an increase in left ventricular filling pressure (Figure 7-25). Dilation of the ascending aorta is an important radiologic sign of bicuspid aortic stenosis, but not of its severity, and may be the only abnormality in the x-ray when the stenosis is mild (Figure 7-26).110 The ascending aorta is normal in fixed subvalvular aortic stenosis (Figure 7-27),45 is normal to small in supravalvular aortic stenosis (Figure 7-28), and is distinctly undersized with hypoplasia of the ascending aorta (see Figure 7-8B) or with a hypoplastic aortic annulus and a miniature valve.45 Dilation of the ascending aorta in XO Turner’s syndrome reflects a medial abnormality that is present whether or not the aortic valve is bicuspid (see Chapter 8, Figure 8-6).87
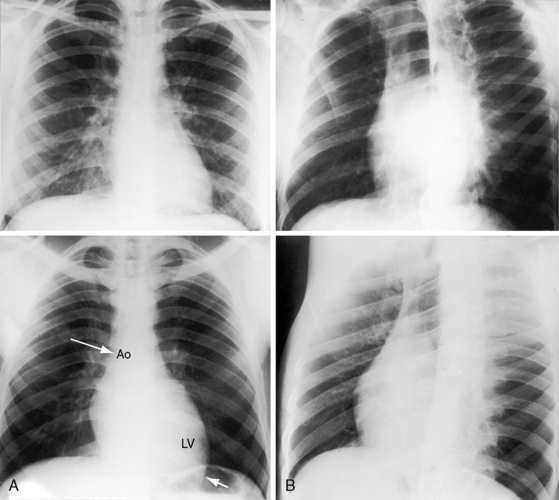
Calcification is presumptive evidence that the stenosis is valvular, and dense calcium is evidence of severity.1,39,110 A calcified bicuspid aortic valve can be recognized by the bulbous club-like configuration of a calcified raphe or by a circle or semicircle of calcium with the bulbous raphe pointing toward its center (see Figure 7-2C).111
The left ventricle attracts attention because of its shape rather than its size (Figures 7-27 through 7-29) and can remain normal-sized through a wide range of severity because the adaptive response is concentric hypertrophy with a normal or reduced cavity. The left ventricle enlarges downward and to the left and posterior so that in the frontal view the apex extends below the left hemidiaphragm (see Figures 7-27B and 7-29) and, in the left lateral projection, extends behind the inferior vena cava (see Figure 7-27B). Significant left ventricular enlargement is reserved for infants with severe aortic stenosis and congestive heart failure and for adults with chronic congestive heart failure, whether or not the aortic stenosis is severe. An increase in left atrial size in the lateral projection is a sign of severity.110
Echocardiogram
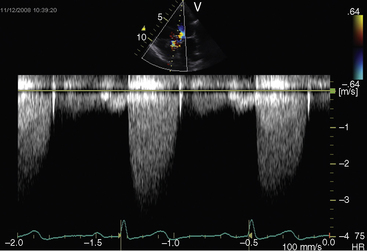
Echocardiography is used to identify the level, morphologic type, and severity of congenital aortic stenosis.112 The gradient is determined with continuous-wave Doppler scan (Figure 7-30), the aortic orifice size can be calculated, and the two-dimensionally targeted M mode can be used to measure septal/free wall thickness and left ventricular cavity size. Real-time imaging is used to determine the ejection fraction.
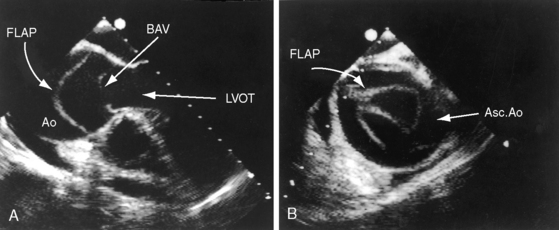
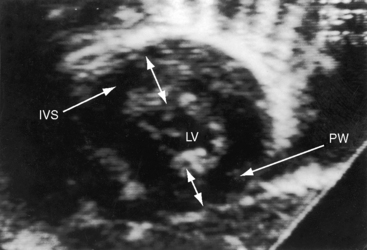
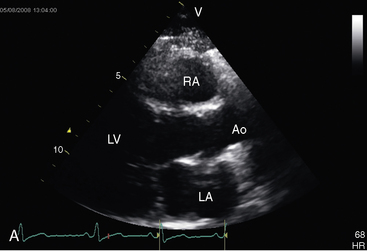
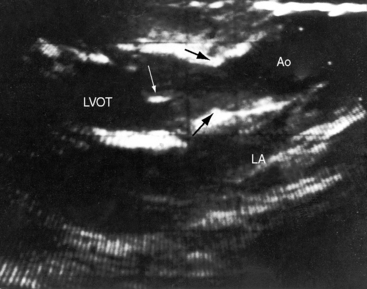
The parasternal long axis view is used to identify doming of a bicuspid aortic valve, and the parasternal short axis is used to identify a single diastolic closure line (Figure 7-31 and Video 7-1). The bicuspid valve can appear trileaflet when the false raphe is prominent, but error is avoided with imaging of the open valve that shows doming of two unequal leaflets with an elliptical orifice (see Figure 7-31) and an M mode that shows an eccentric diastolic closure line. The suprasternal notch view visualizes the dilated ascending aorta (Figure 7-32 and Video 7-2). Transesophageal echocardiography is used to confirm the bicuspid aortic valve (see Figure 7-31) and the aortic root size and to detect a dissecting aneurysm (see Figure 7-5). Infants with severe aortic valve stenosis usually have a unicommissural unicuspid valve that is less mobile and more echodense than a bicuspid valve and may be accompanied by increased reflectivity of an infarcted fibrotic mitral papillary muscle.
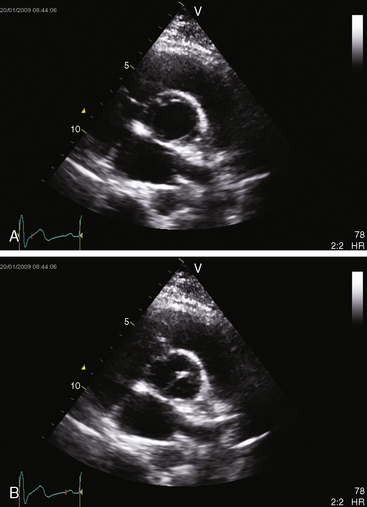
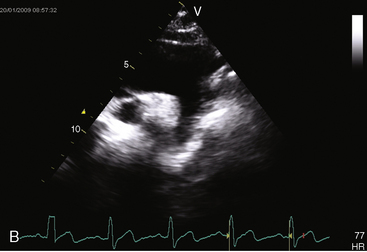
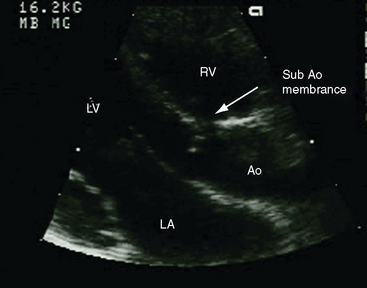
In the parasternal long-axis view, a subaortic membrane is imaged immediately beneath the aortic valve attached to the ventricular septum and anterior mitral leaflet (Figure 7-33 and Video 7-3). A subaortic fibromuscular collar or tunnel produces a long dense ridge of echoes that attach to the ventricular septum and extend onto the anterior mitral leaflet. The M mode in fixed subaortic stenosis reveals distinctive brisk early systolic closure of the aortic valve followed by marked fluttering.113
The hourglass deformity of supravalvular aortic stenosis is identified in the parasternal long-axis view just above the sinuses of Valsalva (Figure 7-34 and Video 7-4). Continuous-wave Doppler scan establishes the gradient.
Congenital aortic regurgitation
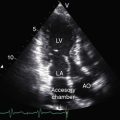
The classic features of chronic severe aortic regurgitation have been familiar to clinicians since Corrigan’s1141832 treatise, On Permanent Patency of the Mouth of the Aorta, or Inadequacy of the Aortic Valves. The remainder of this chapter deals with pure congenital aortic regurgitation when the regurgitant flow is through an incompetent aortic valve directly into the left ventricle (Boxes 7-2 and 7-3). Regurgitant flow that reaches the left ventricle through channels other than the aortic valve is dealt with briefly (see Boxes 7-2 and 7-3).
The causes of pure aortic regurgitation include abnormalities of the aortic valve, abnormalities of the aortic wall, and abnormalities that affect neither the valve nor the aortic wall.115,116 Abnormalities that primarily affect the aortic valve are the most common causes of pure aortic regurgitation.
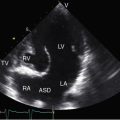
Pure congenital aortic regurgitation occurs with valves that are unicuspid unicommissural,2 bicuspid,117 tricuspid, or quadricuspid41,118 or that are devoid of one or more cusps119–121 and occurs with ventricular septal defect (see Chapter 17), aortic–left ventricular tunnel,122,123 and coronary artery to left ventricular fistula.124 The most common cause of pure congenital aortic valve regurgitation is the bicuspid aortic valve.4 Tricuspid aortic valves with cuspal inequality are seldom incompetent in the young but become incompetent in older patients in whom minor degrees of fibrocalcification exaggerate inherent cuspal inequality and interfere with leaflet coaptation.90 Isolated quadricuspid aortic valves are rare (Figure 7-35) and usually function normally in early life despite cuspal inequality.125,126 When quadricuspid aortic valves function abnormally, the physiologic derangement is almost always expressed as pure regurgitation (see Figure 7-35A).118,125,127 Unicuspid unicommissural valves rarely have sufficient redundancy of the free edge of the single leaflet to render the mechanism incompetent.2
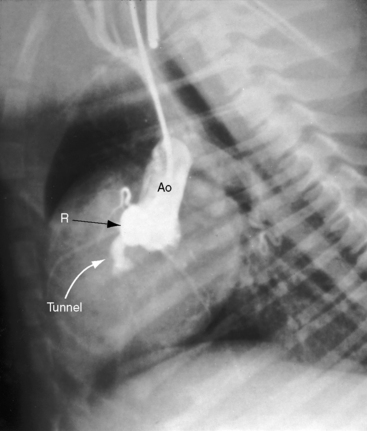
The foregoing types of congenital aortic regurgitation are characterized by regurgitant flow that is mild or absent in infancy and early childhood. However, three types of severe aortic regurgitation begin in the young. One or more cusps or the entire aortic valve mechanism can be congenitally absent,119,121,128–130 with deleterious effects that begin in utero and continue in neonates and infants.119–121,130 A second form of severe aortic regurgitation that originates in infancy is an aortic–left ventricular tunnel, a rare malformation characterized by a vascular channel that originates immediately above the right sinus of Valsalva, tunnels through the ventricular septum, terminates in the paramembranous or infundibular septum, and enters the left ventricle immediately below the right and noncoronary cusps.122 The aortic end of the tunnel is separated from the right coronary ostium (Figure 7-36 and Video 7-5). A third and also rare form of severe aortic regurgitation that begins in infancy is a large coronary artery to left ventricular fistula, fewer than 10% of which terminate in the left ventricle (see Chapter 22). Although congestive heart failure can develop in the neonate, the magnitude of flow is usually limited, so adult survival is the rule.131
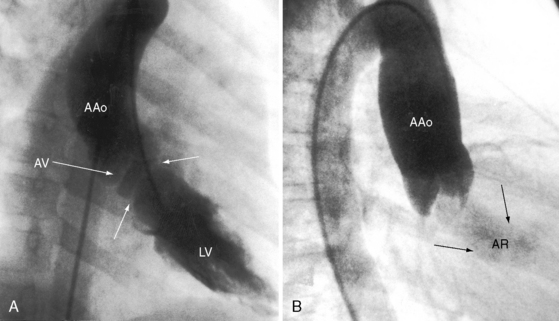
A functionally normal bicuspid aortic valve4 is associated with mild aortic regurgitation (Figure 7-37) because the free edges of bicuspid leaflets are longer than the diameter of the aortic ring to permit unobstructed systolic flow (see Figure 7-1A, left panel). Regurgitation may remain mild or gradually or acutely increase (Figure 7-38).4,132,133 Chronic severe bicuspid aortic regurgitation is caused by gradually increasing prolapse of the relatively redundant edge of the larger of the two bicuspid leaflets.132 Regurgitation is insidious, with the fully developed hemodynamic fault awaiting adulthood.117 However, infective endocarditis can convert a functionally normal congenital bicuspid aortic valve into the catastrophic hemodynamic fault of acute severe aortic regurgitation (see Figure 7-38B).4,132,133 Bicuspid aortic regurgitation is augmented by dilation and dissection of the aortic root caused by an inherent medial abnormality of the ascending aorta above a bicuspid aortic valve (see previous discussion and Figure 7-4).28,85,127
(Courtesy Dr. N. Sidney Moise, Cornell University College of Veterinary Medicine, Ithaca, New York.)
The physiologic consequences of aortic regurgitation depend on the magnitude of regurgitant flow, its rate development, and the adaptive response of the left ventricle to volume overload. Fundamental differences between gradual progression culminate decades later in severe aortic regurgitation and the development of acute severe aortic regurgitation.133,134 Adaptation of the left ventricle to gradual chronic aortic regurgitation was addressed in 1858 by Peacock22: “This process is often so slow in its progress, that the ventricle accommodates itself to the additional exertion required; the disease becomes a source of manifest evil only after the lapse of many years.” Gradual development of severe aortic regurgitation permits an adaptive response of the left ventricle that is precluded if equivalent aortic regurgitation is suddenly incurred.133
The response of the left ventricle to a gradual increase in volume is typified by chronic severe aortic regurgitation incurred before the onset of depressed myocardial contractility (Table 7-1). The volume-overloaded ventricle achieves an adaptive increase in mass by increasing its internal dimensions (radius and base to apex) with a proportionate increase in septal/free wall thickness. The result is a magnified normal heart that is geometrically ideal for ejecting an augmented stroke volume against normal or reduced systemic resistance.59 The increase in ventricular mass is initially a desirable adaptive response that permits the heart to function normally at greater workloads.59 Left ventricular end-diastolic volume and fiber length increase; end-diastolic pressure remains normal. Stroke volume and ejection fraction increase, so effective cardiac output is maintained. The velocity of ejection increases, left ventricular and aortic systolic pressures rise, and aortic diastolic pressure falls, so the pulse pressure widens. Despite a considerable increase in stroke volume, the duration of left ventricular contraction remains normal because the velocity of ejection is increased and the pre-ejection period is shortened. With isotonic exercise, systemic systolic pressure rises, peripheral resistance falls, diastole shortens as heart rate increases, and regurgitant fraction per beat decreases. An appropriate increment in effective cardiac output is achieved without an increase in end-diastolic volume or end-diastolic pressure.135 Despite these favorable adaptations, left ventricular response to isotonic or isometric exercise is not necessarily normal, even in young asymptomatic patients.135,136
Table 7-1 Major Hemodynamic Features of Chronic and Acute Severe Aortic Regurgitation
| Chronic | Acute | |
|---|---|---|
| LV compliance | Increased | Not increased |
| Regurgitant volume | Increased | Increased |
| LV end-diastolic pressure | Normal | Markedly increased |
| LV ejection velocity (dp/dt) | Markedly increased | Not significantly increased |
| Aortic systolic pressure | Increased | Not increased |
| Aortic diastolic pressure | Markedly decreased | Not significantly decreased |
| Systemic arterial pulse pressure | Markedly increased | Not significantly increased |
| Ejection fraction | Increased | Not increased |
| Effective stroke volume | Normal | Decreased |
| Effective cardiac output | Normal | Decreased |
| Heart rate | Normal | Increased |
| Peripheral vascular resistance | Normal | Increased |
LV, Left ventricular; dp/dt.
Acute severe aortic regurgitation is characterized by a dramatic rise in left ventricular end-diastolic pressure in response to the sudden increase in volume imposed on a ventricle that is operating on the less compliant portion of its pressure-volume curve.134 As stroke volume falls, heart rate accelerates in a vain attempt to maintain cardiac output. Peripheral resistance rises in an equally vain attempt to maintain systemic arterial pressure.134 The velocity of ejection does not significantly increase, left ventricular and aortic systolic pressures are not significantly elevated, and aortic diastolic pressure cannot fall below the relatively high left ventricular end-diastolic pressure, so the systemic arterial pulse pressure remains normal or nearly so. The steep rise in left ventricular end-diastolic pressure exceeds left atrial pressure, prematurely closing the mitral valve before inscription of the P wave in the electrocardiogram.133 The mitral valve opens later than normal because of prolonged ejection from the acutely volume-loaded left ventricle.133 Premature closure and delayed opening of the mitral valve, together with obligatory tachycardia, reduce the time during which the mitral valve is open. The pulmonary arterial pressure rises in tandem with left atrial mean pressure, but competence of the mitral valve protects the left atrium from linear transmission of the high left ventricular end-diastolic pressure into the pulmonary venous bed.134 This protection is lost with the advent of mitral regurgitation, so left atrial pressure rises further and cardiac output reciprocally falls.134
History
Congenital aortic valve regurgitation predominates in males, a gender distribution that reflects the prevalence of bicuspid aortic valves.4 In asymptomatic infants and children, the soft diastolic murmur of bicuspid aortic regurgitation usually goes undetected. Patients with moderate to severe aortic regurgitation are often asymptomatic except for awareness of neck pulsations or awareness of left ventricular premature contractions when lying in the left lateral decubitus position.135 Vascular wall pain is occasionally experienced in the carotid and subclavian arteries and in the thoracic or abdominal aorta. The retrosternal chest pain of myocardial ischemia occasionally accompanies acute severe aortic regurgitation.137 Inappropriate diaphoresis sometimes begins long before the onset of congestive heart failure. When bicuspid valve infective endocarditis causes acute severe aortic regurgitation, cardiac failure is sudden and intractable.133
When aortic regurgitation is caused by an aortic–left ventricular tunnel, severe regurgitant flow and congestive heart failure develop early, but exceptional asymptomatic survivals have been reported in children and young adults.123 Congenital absence of one or more aortic cusps with severe regurgitation is manifest by in utero heart failure with nonimmune fetal hydrops.128 Exceptional childhood or postadolescent survivals have been reported.121
Physical Signs
In 1832, Dominic Corrigan114 described the visible arterial pulse associated with inadequacy of the aortic valve:
The arterial pulse in chronic severe aortic regurgitation is characterized by a rapid rate of rise, an increased pulse pressure (high systolic/low diastolic), a single or double peak (bisferiens), and a brisk collapse.90 The Quincke’s pulse (flushing and blanching of the digital capillary bed), palpable pulsations in the fingertips, and posteroanterior head movements are synchronous with systole and diastole.90 The arterial pulse in acute severe aortic regurgitation is characterized by an unimpressive rate of rise, a moderately increased pulse pressure (systolic normal, diastolic normal or slightly diminished), a single peak, and the pulsus alternans of left ventricular failure.133
Precordial palpation in chronic severe aortic regurgitation reveals a laterally displaced hyperdynamic left ventricular impulse that imparts a rocking motion to the chest. A systolic thrill and murmur are present in the suprasternal notch and over the carotid and subclavian arteries (Corrigan’s bruit de soufflet)114 because of high-velocity ejection. In acute severe aortic regurgitation, the location of the left ventricular impulse is normal or moderately displaced and is not hyperdynamic.
In chronic severe aortic regurgitation, the first heart sound is normal, the aortic component of the second sound is soft, and there is neither a third nor a fourth heart sound.138 There is a grade 3/6 right basal aortic midsystolic murmur, a long high-pitched aortic diastolic murmur at the mid left sternal border, and a presystolic Austin Flint murmur at the apex.90 An aortic ejection sound is absent because a severely incompetent bicuspid aortic valve does not dome.138 Peripheral arterial auscultatory signs include Traube’s pistol shot sounds and the systolic/diastolic Duroziez’s murmur over a partially compressed femoral artery.
In acute severe aortic regurgitation,133 the first heart sound is soft or absent and a fourth heart sound is precluded because of premature closure of the mitral valve. A third heart sound reflects left ventricular failure. The pulmonary component of the second sound is increased in response to elevated pulmonary artery pressure. A midsystolic aortic flow murmur is less than grade 3 and is followed by an aortic diastolic murmur that is short, medium-pitched, and disarmingly soft because of the relatively low velocity of regurgitant flow. Distinguishing systole from diastole is difficult because the first heart sound is soft or absent and the heart rate is rapid. A presystolic Austin Flint murmur is precluded because of premature closure of the mitral valve. No auscultatory signs are found over peripheral arteries.133
An aortic–left ventricular tunnel is accompanied by a systolic/diastolic to-and-fro murmur analogous to midsystolic/early diastolic murmur of chronic severe aortic regurgitation.123 Doppler scan interrogation of flow patterns within the ascending aorta and within the tunnel are used to identify diastolic flow from the tunnel into the left ventricle and systolic flow from left ventricle into the tunnel.139 The phasic flow is predominantly diastolic, which is consistent with the auscultatory observation that the diastolic murmur is loud and the systolic murmur is soft.123 Occasionally, only the diastolic murmur is audible.
X-Ray
In chronic severe bicuspid aortic regurgitation, the x-ray shows an enlarged convex left ventricle and a conspicuously dilated ascending aorta because pulsatile regurgitant flow aggregates the dilatory effect of an inherent medial abnormality (see Figure 7-38A).28 Pulmonary venous vascularity is normal because left ventricular filling pressure is not increased. In acute severe aortic regurgitation, the left ventricular silhouette is nearly normal, and pulmonary venous vascularity is increased because left ventricular filling pressure is elevated (see Figure 7-38B). In aortic regurgitation associated with aortic–left ventricular tunnel, the ascending aorta is consistently dilated and is sometimes aneurysmal.123,139
Echocardiogram
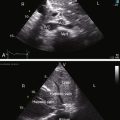
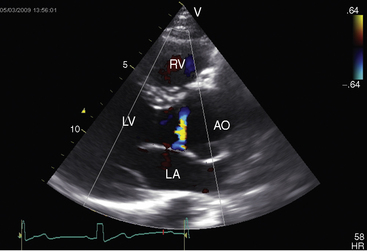
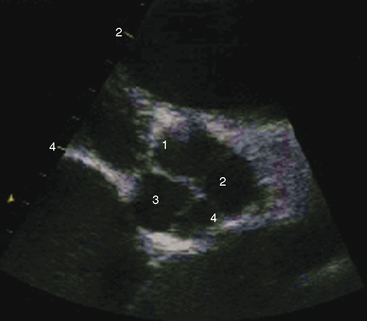
Echocardiography with Doppler scan interrogation and color flow imaging establishes the presence and degree of aortic regurgitation, determines aortic valve morphology, and defines the physiologic consequences of regurgitant flow. Mild aortic regurgitation of a functionally normal bicuspid aortic valve is identified with color flow imaging (see Figure 7-37B). The regurgitant jet of bicuspid aortic regurgitation is eccentric in the left ventricular outflow tract (Figure 7-39 and Video 7-6). Continuous-wave Doppler scan records the profile of regurgitant flow and is used to determine whether aortic stenosis coexists (Figure 7-40). The four cusps of a congenitally quadricuspid aortic valve are best recognized in a transesophageal echocardiogram (Figure 7-41 and Video 7-7).
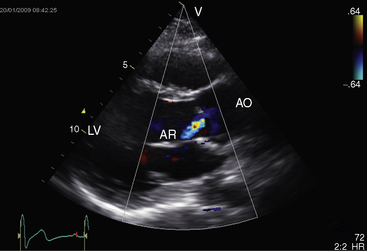
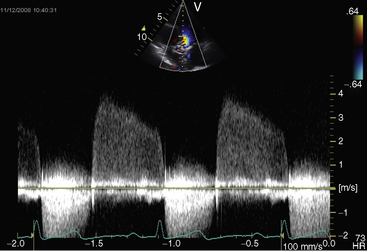
Figure 7-40 Continuous wave Doppler across a bicuspid aortic valve showing regurgitation in diastole.
An aortic–left ventricular tunnel can be identified as the cause of aortic regurgitation, and Doppler interrogation with color flow imaging permits study of phasic directional flow within the tunnel.122,124,139 An aortic–left ventricular tunnel arising from the left aortic sinus was diagnosed with echocardiography.
1 Campbell M. Calcific aortic stenosis and congenital bicuspid aortic valves. Br Heart J. 1968;30:606-616.
2 Falcone M.W., Roberts W.C., Morrow A.G., Perloff J.K. Congenital aortic stenosis resulting from a unicommisssural valve. Clinical and anatomic features in twenty-one adult patients. Circulation. 1971;44:272-280.
3 Perloff J. Clinical recognition of aortic stenosis the physical signs and differential diagnosis of the various forms of obstruction to left ventricular outflow. Prog Cardiovasc Dis. 1968;10:323-352.
4 Roberts W.C. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol. 1970;26:72-83.
5 Roberts W.C. The structure of the aortic valve in clinically isolated aortic stenosis: an autopsy study of 162 patients over 15 years of age. Circulation. 1970;42:91-97.
6 Roberts W.C. Valvular, subvalvular and supravalvular aortic stenosis: morphologic features. Cardiovasc Clin. 1973;5:97-126.
7 Roberts W.C., Ko J.M. Clinical and morphologic features of the congenitally unicuspid acommissural stenotic and regurgitant aortic valve. Cardiology. 2007;108:79-81.
8 Tzemos N., Therrien J., Yip J., et al. Outcomes in adults with bicuspid aortic valves. JAMA. 2008;300:1317-1325.
9 Movahed M.-R., Hepner A.D., Ahmadi-Kashani M. Echocardiographic prevalence of bicuspid aortic valve in the population. Heart Lung Circ. 2006;15:297-299.
10 Lewin M.B., Otto C.M. The bicuspid aortic valve: adverse outcomes from infancy to old age. Circulation. 2005;111:832-834.
11 Sabet H.Y., Edwards W.D., Tazelaar H.D., Daly R.C. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc. 1999;74:14-26.
12 Biner S., Rafique A.M., Ray I., Cuk O., Siegel R.J., Tolstrup K. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol. 2009;53:2288-2295.
13 Khan W., Milsevic M., Salciccioli L., Lazar J. Low prevalence of bicuspid aortic valve in African Americans. Am Heart J. 2008;156:e25.
14 Subramanian R., Olson L.J., Edwards W.D. Surgical pathology of pure aortic stenosis: a study of 374 cases. Mayo Clin Proc. 1984;59:683-690.
15 Chan K.L., Ghani M., Woodend K., Burwash I.G. Case-controlled study to assess risk factors for aortic stenosis in congenitally bicuspid aortic valve. Am J Cardiol. 2001;88:690-693.
16 Da Vinci L. Leonardo on the human body. Mineola, NY: Dover Publications; 1983.
17 Waider W., Craige E. First heart sound and ejection sounds. Echocardiographic and phonocardiographic correlation with valvular events. Am J Cardiol. 1975;35:346-356.
18 Fernandes S.M., Sanders S.P., Khairy P., et al. Morphology of bicuspid aortic valve in children and adolescents. J Am Coll Cardiol. 2004;44:1648-1651.
19 Beppu S., Suzuki S., Matsuda H., Ohmori F., Nagata S., Miyatake K. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol. 1993;71:322-327.
20 Davis G.L., Mcalister W.H., Friedenberg M.J. Congenital aortic stenosis due to failure of histogenesis of the aortic valve (myxoid dysplasia). Am J Roentgenol Radium Ther Nucl Med. 1965;95:621-628.
21 Sans-Coma V., Cardo M., Thiene G., Fernandez B., Arque J.M., Duran A.C. Bicuspid aortic and pulmonary valves in the Syrian hamster. Int J Cardiol. 1992;34:249-254.
22 Peacock T. On malformations of the human heart. London: John Churchill; 1858.
23 Song Z.-Z. Valve calcification and patients with bicuspid aortic valves. JAMA. 2009;301:935-936. author reply 936
24 Kalan J.M., Mcintosh C.L., Bonow R.O., Roberts W.C. Development of severe stenosis in a previously purely regurgitant, congenitally bicuspid aortic valve. Am J Cardiol. 1988;62:988-989.
25 Bonow R.O. Bicuspid aortic valves and dilated aortas: a critical review of the ACC/AHA practice guidelines recommendations. Am J Cardiol. 2008;102:111-114.
26 Basso C., Boschello M., Perrone C., et al. An echocardiographic survey of primary school children for bicuspid aortic valve. Am J Cardiol. 2004;93:661-663.
27 Beroukhim R.S., Kruzick T.L., Taylor A.L., Gao D., Yetman A.T. Progression of aortic dilation in children with a functionally normal bicuspid aortic valve. Am J Cardiol. 2006;98:828-830.
28 Niwa K., Perloff J.K., Bhuta S.M., et al. Structural abnormalities of great arterial walls in congenital heart disease: light and electron microscopic analyses. Circulation. 2001;103:393-400.
29 Silberbach M. Bicuspid aortic valve and thoracic aortic aneurysm: toward a unified theory. J Am Coll Cardiol. 2009;53:2296-2297.
30 Novaro G.M., Griffin B.P. Congenital bicuspid aortic valve and rate of ascending aortic dilatation. Am J Cardiol. 2004;93:525-526.
31 Novaro G.M., Tiong I.Y., Pearce G.L., Grimm R.A., Smedira N., Griffin B.P. Features and predictors of ascending aortic dilatation in association with a congenital bicuspid aortic valve. Am J Cardiol. 2003;92:99-101.
32 Gurvitz M., Chang R.-K., Drant S., Allada V. Frequency of aortic root dilation in children with a bicuspid aortic valve. Am J Cardiol. 2004;94:1337-1340.
33 Nistri S., Grande-Allen J., Noale M., et al. Aortic elasticity and size in bicuspid aortic valve syndrome. Eur Heart J. 2008;29:472-479.
34 Nemes A., Soliman O.I., Csanady M., Forster T. Aortic distensibility in patients with bicuspid aortic valves. Am J Cardiol. 2008;102:370.
35 Reeve R.Jr, Robinson S.J. Hypoplastic annulus—an unusual type of aortic stenosis: a report of three cases in children. Dis Chest. 1964;45:99-102.
36 Vollebergh F.E., Becker A.E. Minor congenital variations of cusp size in tricuspid aortic valves. Possible link with isolated aortic stenosis. Br Heart J. 1977;39:1006-1011.
37 Fratellone P., Berger M., Khan M., Bassiri-Tehrani M. Quadricuspid aortic valve diagnosed by echocardiography in two cases identical twins. Am J Cardiol. 2007;100:1490-1491.
38 Tentolouris K., Kontozoglou T., Trikas A., et al. Fixed subaortic stenosis revisited. congenital abnormalities in 72 new cases and review of the literature. Cardiology. 1999;92:4-10.
39 Campbell M. The natural history of congenital aortic stenosis. Br Heart J. 1968;30:514-526.
40 Choi J.Y., Sullivan I.D. Fixed subaortic stenosis: anatomical spectrum and nature of progression. Br Heart J. 1991;65:280-286.
41 Firpo C., Maitre Azcarate M.J., Quero Jimenez M., Saravalli O. Discrete subaortic stenosis (DSS) in childhood: a congenital or acquired disease? Follow-up in 65 patients. Eur Heart J. 1990;11:1033-1040.
42 Laskey W.K., Kussmaul W.G. Subvalvular gradients in patients with valvular aortic stenosis: prevalence, magnitude, and physiological importance. Circulation. 2001;104:1019-1022.
43 El Habbal M.H., Suliman R.F. The aortic root in subaortic stenosis. Am Heart J. 1989;117:1127-1132.
44 Pyle R.L., Patterson D.F., Chacko S. The genetics and pathology of discrete subaortic stenosis in the Newfoundland dog. Am Heart J. 1976;92:324-334.
45 Maron B.J., Redwood D.R., Roberts W.C., Henry W.L., Morrow A.G., Epstein S.E. Tunnel subaortic stenosis: left ventricular outflow tract obstruction produced by fibromuscular tubular narrowing. Circulation. 1976;54:404-416.
46 Freedom R.M., Fowler R.S., Duncan W.J. Rapid evolution from “normal” left ventricular outflow tract to fatal subaortic stenosis in infancy. Br Heart J. 1981;45:605-609.
47 Stamm C., Li J., Ho S.Y., Redington A.N., Anderson R.H. The aortic root in supravalvular aortic stenosis: the potential surgical relevance of morphologic findings. J Thorac Cardiovasc Surg. 1997;114:16-24.
48 Vaideeswar P., Shankar V., Deshpande J.R., Sivaraman A., Jain N. Pathology of the diffuse variant of supravalvar aortic stenosis. Cardiovasc Pathol. 2001;10:33-37.
49 Vincent W.R., Buckberg G.D., Hoffman J.I. Left ventricular subendocardial ischemia in severe valvar and supravalvar aortic stenosis. A common mechanism. Circulation. 1974;49:326-333.
50 Yilmaz A.T., Arslan M., Ozal E., Byngol H., Tatar H., Ozturk O.Y. Coronary artery aneurysm associated with adult supravalvular aortic stenosis. Ann Thorac Surg. 1996;62:1205-1207.
51 Williams J.C., Barratt-Boyes B.G., Lowe J.B. Supravalvular aortic stenosis. Circulation. 1961;24:1311-1318.
52 Beuren A.J., Apitz J., Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation. 1962;26:1235-1240.
53 Ingelfinger J.R., Newburger J.W. Spectrum of renal anomalies in patients with Williams syndrome. J Pediatr. 1991;119:771-773.
54 Morris C.A., Leonard C.O., Dilts C., Demsey S.A. Adults with Williams syndrome. Am J Med Genet Suppl. 1990;6:102-107.
55 Pober B.R., Lacro R.V., Rice C., Mandell V., Teele R.L. Renal findings in 40 individuals with Williams syndrome. Am J Med Genet. 1993;46:271-274.
56 Daniels S.R., Loggie J.M., Schwartz D.C., Strife J.L., Kaplan S. Systemic hypertension secondary to peripheral vascular anomalies in patients with Williams syndrome. J Pediatr. 1985;106:249-251.
57 Salaymeh K.J., Banerjee A. Evaluation of arterial stiffness in children with Williams syndrome: Does it play a role in evolving hypertension? Am Heart J. 2001;142:549-555.
58 Rein A.J., Preminger T.J., Perry S.B., Lock J.E., Sanders S.P. Generalized arteriopathy in Williams syndrome: an intravascular ultrasound study. J Am Coll Cardiol. 1993;21:1727-1730.
59 Perloff J.K. Normal myocardial growth and the development and regression of increased ventricular mass. In: Perloff J.K., Child J.S., editors. Congenital heart disease in adults. 2nd ed. Philadelphia: W.B. Saunders; 1998:346.
60 Rakusan K., Flanagan M.F., Geva T., Southern J., Van Praagh R. Morphometry of human coronary capillaries during normal growth and the effect of age in left ventricular pressure-overload hypertrophy. Circulation. 1992;86:38-46.
61 Krovetz L.J., Kurlinski J.P. Subendocardial blood flow in children with congenital aortic stenosis. Circulation. 1976;54:961-965.
62 El-Said G., Galioto F.M.Jr, Mullins C.E., Mcnamara D.G. Natural hemodynamic history of congenital aortic stenosis in childhood. Am J Cardiol. 1972;30:6-12.
63 Kim Y.M., Yoo S.J., Choi J.Y., Kim S.H., Bae E.J., Lee Y.T. Natural course of supravalvar aortic stenosis and peripheral pulmonary arterial stenosis in Williams’ syndrome. Cardiol Young. 1999;9:37-41.
64 Mocellin R., Sauer U., Simon B., Comazzi M., Sebening F., Buhlmeyer K. Reduced left ventricular size and endocardial fibroelastosis as correlates of mortality in newborns and young infants with severe aortic valve stenosis. Pediatr Cardiol. 1983;4:265-272.
65 Gould L., Venkataraman K., Goswami M., Demartino A., Gomprecht R.F. Right-sided heart failure in aortic stenosis. Am J Cardiol. 1973;31:381-383.
66 Bache R.J., Wang Y., Jorgensen C.R. Hemodynamic effects of exercise in isolated valvular aortic stenosis. Circulation. 1971;44:1003-1013.
67 Cyran S.E., James F.W., Daniels S., Mays W., Shukla R., Kaplan S. Comparison of the cardiac output and stroke volume response to upright exercise in children with valvular and subvalvular aortic stenosis. J Am Coll Cardiol. 1988;11:651-658.
68 Kveselis D.A., Rocchini A.P., Rosenthal A., et al. Hemodynamic determinants of exercise-induced ST-segment depression in children with valvar aortic stenosis. Am J Cardiol. 1985;55:1133-1139.
69 Mark A.L., Kioschos J.M., Abboud F.M., Heistad D.D., Schmid P.G. Abnormal vascular responses to exercise in patients with aortic stenosis. J Clin Invest. 1973;52:1138-1146.
70 Keane J.F., Driscoll D.J., Gersony W.M., et al. Second natural history study of congenital heart defects. Results of treatment of patients with aortic valvar stenosis. Circulation. 1993;87:I16-I27.
71 Friedman W.F., Modlinger J., Morgan J.R. Serial hemodynamic observations in asymptomatic children with valvar aortic stenosis. Circulation. 1971;43:91-97.
72 Moller J.H., Nakib A., Edwards J.E. Infarction of papillary muscles and mitral insufficiency associated with congenital aortic stenosis. Circulation. 1966;34:87-91.
73 Conway E.E.Jr, Noonan J., Marion R.W., Steeg C.N. Myocardial infarction leading to sudden death in the Williams syndrome: report of three cases. J Pediatr. 1990;117:593-595.
74 Morgan C., Nadas A. Sweating and congestive heart failure. N Engl J Med. 1963;268:580-585.
75 Gale A.W., Cartmill T.B., Bernstein L. Familial subaortic membranous stenosis. Aust N Z J Med. 1974;4:576-581.
76 Richardson M.E., Menahem S., Wilkinson J.L. Familial fixed subaortic stenosis. Int J Cardiol. 1991;30:351-353.
77 Ensing G.J., Schmidt M.A., Hagler D.J., Michels V.V., Carter G.A., Feldt R.H. Spectrum of findings in a family with nonsyndromic autosomal dominant supravalvular aortic stenosis: A Doppler echocardiographic study. J Am Coll Cardiol. 1989;13:413-419.
78 Eisenberg R., Young D., Jacobson B., Boito A. Familial supravalvular aortic stenosis. Am J Dis Child. 1964;108:341-347.
79 Dridi S.M., Ghomrasseni S., Bonnet D., et al. Skin elastic fibers in Williams syndrome. Am J Med Genet. 1999;87:134-138.
80 Morris C.A. Genetic aspects of supravalvular aortic stenosis. Curr Opin Cardiol. 1998;13:214-219.
81 Urban Z., Michels V.V., Thibodeau S.N., et al. Isolated supravalvular aortic stenosis: functional haploinsufficiency of the elastin gene as a result of nonsense-mediated decay. Hum Genet. 2000;106:577-588.
82 Von Dadelszen P., Chitayat D., Winsor E.J., et al. De novo 46,XX,t(6;7)(q27;q11;23) associated with severe cardiovascular manifestations characteristic of supravalvular aortic stenosis and Williams syndrome. Am J Med Genet. 2000;90:270-275.
83 Friedman W.F., Roberts W.C. Vitamin D and the supravalvar aortic stenosis syndrome. The transplacental effects of vitamin D on the aorta of the rabbit. Circulation. 1966;34:77-86.
84 Osler W. The bicuspid condition of the aortic valves. Trans Assoc Am Physicians. 1886;1:185-192.
85 Larson E.W., Edwards W.D. Risk factors for aortic dissection: a necropsy study of 161 cases. Am J Cardiol. 1984;53:849-855.
86 Scheffer S.M., Leatherman L.L. Resolution of Heyde’s syndrome of aortic stenosis and gastrointestinal bleeding after aortic valve replacement. Ann Thorac Surg. 1986;42:477-480.
87 Miller M.J., Geffner M.E., Lippe B.M., et al. Echocardiography reveals a high incidence of bicuspid aortic valve in Turner syndrome. J Pediatr. 1983;102:47-50.
88 Prandstraller D., Mazzanti L., Picchio F.M., et al. Turner’s syndrome: cardiologic profile according to the different chromosomal patterns and long-term clinical follow-Up of 136 nonpreselected patients. Pediatr Cardiol. 1999;20:108-112.
89 Mazzanti L., Cacciari E. Congenital heart disease in patients with Turner’s syndrome. Italian Study Group for Turner Syndrome (ISGTS). J Pediatr. 1998;133:688-692.
90 Perloff J.K. Physical examination of the heart and circulation, 4th ed. Shelton, Connecticut: People’s Medical Publishing House; 2009.
91 Farrar J.F., Gray R.E. The pulse of aortic stenosis during childhood. Br Heart J. 1965;27:199-204.
92 Alpert J.S., Vieweg W.V., Hagan A.D. Incidence and morphology of carotid shudders in aortic valve disease. Am Heart J. 1976;92:435-440.
93 Goldstein R.E., Epstein S.E. Mechanism of elevated innominate artery pressures in supravalvular aortic stenosis. Circulation. 1970;42:23-29.
94 Lurie P., Mendelbaum I. Mechanism of brachial pulse asymmetry in congenital aortic stenotic lesions. Circulation. 1963;28:760.
95 Franch R., Oran E. Asymmetric arm and neck pulses: A clue to supravalvular aortic stenosis. Circulation. 1963;28:722.
96 Braunwald E., Goldblatt A., Aygen M.M., Rockoff S.D., Morrow A.G. Congenital aortic stenosis. I. Clinical and hemodynamic findings in 100 patients. II. Surgical treatment and the results of operation. Circulation. 1963;27:426-462.
97 Lakier J.B., Lewis A.B., Heymann M.A., Stanger P., Hoffman J.I., Rudolph A.M. Isolated aortic stenosis in the neonate. Natural history and hemodynamic considerations. Circulation. 1974;50:801-808.
98 Caulfield W.H., De Leon A.C.Jr, Perloff J.K., Steelman R.B. The clinical significance of the fourth heart sound in aortic stenosis. Am J Cardiol. 1971;28:179-182.
99 Epstein E.J., Criley J.M., Raftery E.B., Humphries J.O., Ross R.S. Cineradiographic studies of the early systolic click in aortic valve stenosis. Circulation. 1965;31:842-853.
100 Gamboa R., Hugenholtz P.G., Nadas A.S. Accuracy of the phonocardiogram in assessing severity of aortic and pulmonic stenosis. Circulation. 1964;30:35-46.
101 Nelson W.P., Hall R.J. The innocent supraclavicular arterial bruit—utility of shoulder maneuvers in its recognition. N Engl J Med. 1968;278:778.
102 Perloff J.K., Roberts W.C. The mitral apparatus. Functional anatomy of mitral regurgitation. Circulation. 1972;46:227-239.
103 Hirschfeld S., Liebman J., Borkat G., Bormuth C. Intracardiac pressure-sound correlates of echographic aortic valve closure. Circulation. 1977;55:602-604.
104 Kumar S., Luisada A.A. Mechanism of changes in the second heart sound in aortic stenosis. Am J Cardiol. 1971;28:162-167.
105 Cripps T., Leech G., Leatham A. Inappropriate left ventricular hypertrophy in minor aortic valve disease: a source of error in clinical assessment. Eur Heart J. 1987;8:895-901.
106 Hugenholtz P.G., Lees M.M., Nadas A.S. The scalar electrocardiogram, vectorcardiogram, and exercise electrocardiogram in the assessment of congenital aortic stenosis. Circulation. 1962;26:79-91.
107 Braverman I.B., Gibson S. The outlook for children with congenital aortic stenosis. Am Heart J. 1957;53:487-493.
108 Griep A.H. Pitfalls in the electrocardiographic diagnosis of left ventricular hypertrophy; a correlative study of 200 autopsied patients. Circulation. 1959;20:30-34.
109 Kangos J.J., Ferrer M.I., Franciosi R.A., Blanc W.A., Blumenthal S. Electrocardiographic changes associated with papillary muscle infarction in congenital heart disease. Am J Cardiol. 1969;23:801-809.
110 Rockoff S.D., Levine N.D., Austen W.G. Roentgenographic clues to the cardiac hemodynamics of aortic stenosis. Radiology. 1964;83:58-62.
111 Spindola-Franco H., Fish B.G., Dachman A., Grose R., Attai L. Recognition of bicuspid aortic valve by plain film calcification. AJR Am J Roentgenol. 1982;139:867-872.
112 Child J. Transthoracic and transesophageal echocardiographic imaging: anatomic and hemodynamic assessment. In: Congenital heart disease in adults. WB Saunders Company; 1998:91.
113 Sabbah H.N., Stein P.D. Mechanism of early systolic closure of the aortic valve in discrete membranous subaortic stenosis. Circulation. 1982;65:399-402.
114 Corrigan D. On permanent patency of the mouth of the aorta, or inadequacy of the aortic valves. In: Willus R.A., editor. Classics of cardiology: a collection of classic works on the heart and circulation with comprehensive biographic accounts of the authors. Malabar, FL: Robert E. Krieger Pub. Co, 1983.
115 Waller B., Howard J., Fess S. Pathology of aortic valve stenosis and pure aortic regurgitation. A clinical morphologic assessment—Part I. Clin Cardiol. 1994;17:85-92.
116 Waller B.F., Howard J., Fess S. Pathology of aortic valve stenosis and pure aortic regurgitation: a clinical morphologic assessment—Part II. Clin Cardiol. 1994;17:150-156.
117 Sadee A.S., Becker A.E., Verheul H.A., Bouma B., Hoedemaker G. Aortic valve regurgitation and the congenitally bicuspid aortic valve: a clinico-pathological correlation. Br Heart J. 1992;67:439-441.
118 Waller B.F., Taliercio C.P., Dickos D.K., Howard J., Adlam J.H., Jolly W. Rare or unusual causes of chronic, isolated, pure aortic regurgitation. Clin Cardiol. 1990;13:577-581.
119 Carvalho A.C., Andrade J.L., Lima V.C., Leal S.B., Martinez E.E., Buffolo E. Absence of an aortic valve cusp, a cause of severe aortic regurgitation in infancy. Pediatr Cardiol. 1992;13:122-124.
120 Issenberg H.J., Mathew R., Kim E.S., Bharati S. Congenital absence of the noncoronary aortic cusp. Am Heart J. 1987;113:400-402.
121 Lin A.E., Chin A.J. Absent aortic valve: a complex anomaly. Pediatr Cardiol. 1990;11:195-198.
122 Humes R.A., Hagler D.J., Julsrud P.R., Levy J.M., Feldt R.H., Schaff H.V. Aortico-left ventricular tunnel: Diagnosis based on two-dimensional echocardiography, color flow Doppler imaging, and magnetic resonance imaging. Mayo Clin Proc. 1986;61:901-907.
123 Levy M.J., Schachner A., Blieden L.C. Aortico-left ventricular tunnel: Collective review. J Thorac Cardiovasc Surg. 1982;84:102-109.
124 Wu J.R., Huang T.Y., Chen Y.F., Lin Y.T., Roan H.R. Aortico-left ventricular tunnel: Two-dimensional echocardiographic and angiocardiographic features. Am Heart J. 1989;117:697-699.
125 Fernicola D.J., Mann J.M., Roberts W.C. Congenitally quadricuspid aortic valve: Analysis of six necropsy patients. Am J Cardiol. 1989;63:136-138.
126 Hurwitz L.E., Roberts W.C. Quadricuspid semilunar valve. Am J Cardiol. 1973;31:623-626.
127 Olson L.J., Subramanian R., Edwards W.D. Surgical pathology of pure aortic insufficiency: A study of 225 cases. Mayo Clin Proc. 1984;59:835-841.
128 Bicoff J.P., Thompson W., Arbeiter H.I., Weinberg M.Jr, Agustsson M.H. Severe aortic stenosis in infancy. J Pediatr. 1963;63:161-164.
129 Hashimoto R., Miyamura H., Eguchi S. Congenital aortic regurgitation in a child with a tricuspid non-stenotic aortic valve. Br Heart J. 1984;51:358-360.
130 Tran Quang H., Smolinsky A., Neufeld H.N., Goor D.A. Dysplastic aortic valve with absence of aortic valve cusp: An unreported cause of congenital aortic insufficiency. J Thorac Cardiovasc Surg. 1986;91:471-472.
131 Starc T.J., Bowman F.O., Hordof A.J. Congestive heart failure in a newborn secondary to coronary artery-left ventricular fistula. Am J Cardiol. 1986;58:366-367.
132 Carter J.B., Sethi S., Lee G.B., Edwards J.E. Prolapse of semilunar cusps as causes of aortic insufficiency. Circulation. 1971;43:922-932.
133 Morganroth J., Perloff J.K., Zeldis S.M., Dunkman W.B. Acute severe aortic regurgitation. Pathophysiology, clinical recognition, and management. Ann Intern Med. 1977;87:223-232.
134 Welch G.H.Jr, Braunwald E., Sarnoff S.J. Hemodynamic effects of quantitatively varied experimental aortic regurgitation. Circ Res. 1957;5:546-551.
135 Bonow R.O., Rosing D.R., Mcintosh C.L., et al. The natural history of asymptomatic patients with aortic regurgitation and normal left ventricular function. Circulation. 1983;68:509-517.
136 Goforth D., James F.W., Kaplan S., Donner R., Mays W. Maximal exercise in children with aortic regurgitation: An adjunct to noninvasive assessment of disease severity. Am Heart J. 1984;108:1306-1311.
137 Saito S., Naik M.J., Westaby S. Ischemic pain in aortic regurgitation. Circulation. 2001;104:1984.
138 Sabbah H.N., Khaja F., Anbe D.T., Stein P.D. The aortic closure sound in pure aortic insufficiency. Circulation. 1977;56:859-863.
139 Fripp R.R., Werner J.C., Whitman V., Nordenberg A., Waldhausen J.A. Pulsed Doppler and two-dimensional echocardiographic findings in aortico-left ventricular tunnel. J Am Coll Cardiol. 1984;4:1012-1014.