Chapter 12 Congenital Anomalies of the Female Reproductive Tract
INTRODUCTION
In contrast, when congenital anomalies involve either ovaries or external genitalia, the müllerian-derived structures are often normal. In these cases, identifiable aberrations of the external genitalia are often noted in early infancy, with delays in secondary sexual characteristics occurring in later life. specific congenital anomalies of the reproductive tract can be difficult to diagnose because of the wide variety of clinical presentations. Once an accurate diagnosis is rendered, many treatment options exist and are often individualized for a given malformation. For infants born with ambiguous genitalia, a multidisciplinary approach is often necessary. Refinements in surgical and medical techniques have enabled many women with these disorders to have normal satisfying sexual relations. Advances in reproductive technologies have resulted in improved fertility and obstetrical outcomes. Indeed, assisted reproductive technologies now allow many women with congenital anomalies of the reproductive tract to conceive and deliver healthy babies.1
EMBRYOLOGY OF THE FEMALE REPRODUCTIVE TRACT
In the normal female embryo, a series of well-orchestrated, complex interactions is required for proper differentiation of the müllerian ducts and urogenital sinus. Although originating from different germ layers, the fates of the müllerian ducts (mesoderm) and urogenital sinus (endoderm) are interconnected as they differentiate to form the female reproductive tract. The müllerian ducts are the primordial anlage of the internal female reproductive organs and differentiate to form the fallopian tubes, uterus, cervix, and superior aspect of the vagina.1 When the dynamic processes of differentiation, migration, fusion, and canalization are interrupted, a wide spectrum of congenital anomalies of the reproductive tract can result. The anatomic malformations can range from agenesis of the uterus and vagina to duplication of the reproductive organs. Disruption of local mesoderm development and its contiguous somites can account for associated urologic and axial skeletal abnormalities, respectively, that quite often coexist.
Development of the Fallopian Tubes, Uterus, and Uterine Cervix
Two sets of paired genital ducts, paramesonephric (müllerian) and mesonephric (Wolffian), are present in female and male embryos at 6 weeks of development (Fig. 12-1). We use the two terms that refer to each duct somewhat interchangeably. For example, although most embryology books use the term paramesonephric duct, most clinicians use the term müllerian. Development of the mesonephric ducts precedes that of the paramesonephric ducts, and for a short period, the mesonephric ducts drain the contents of the primitive mesonephric kidney into the cloaca.2 A key gene for the development of paramesonephric and mesonephric ducts is PAX2. Mutations in this gene will result in impaired duct and renal development in both sexes.
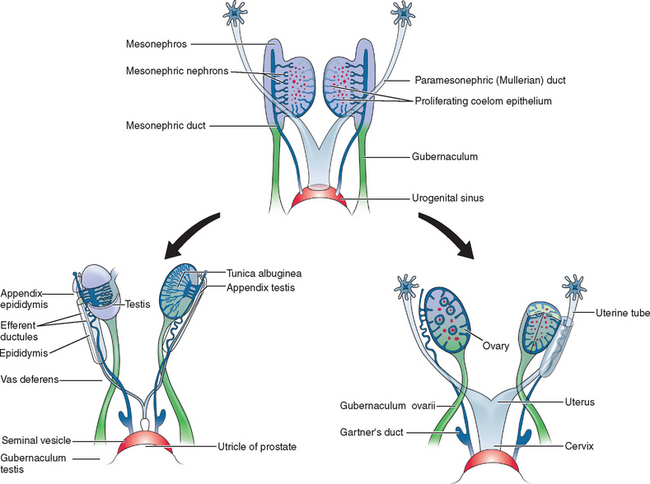
In the female, the mesonephric ducts will degenerate in the absence of testosterone, and the paramesonephric ducts develop because of the absence of anti-müllerian (AMH).2–4 At the same time, the paramesonephric ducts, which originated as longitudinal invaginations of coelomic epithelium, begin to elongate bidirectionally along the anterolateral aspects of the gonads.2 The regressing mesonephric ducts provide an ideal template for the elongating paramesonephric ducts. This primordial connection explains the frequent associations seen later between paramesonephric duct anomalies and urinary system malformations. In the male the paramesonephric ducts will regress under the influence of antimüllerian hormone secreted by the Sertoli cells of the testes. Persistence of the paramesonephric ducts in the male is seen with a mutation of the gene coding for antimüllerian hormone or its receptor.
As the paramesonephric ducts elongate at week 9, three recognizable regions can be identified: the cranial, horizontal, and caudal aspects. Each of these regions will have a distinct fate.2,5 The funnel-shaped cranial regions open directly into the early peritoneal cavity and will form the fimbria of the fallopian tubes. The paramesonephric ducts at this level are lateral to the mesonephric ducts (see Fig. 12-1).
The paired horizontal segments migrate lateral to the mesonephric ducts, then cross ventrally and extend caudomedially to form the remainder of the fallopian tubes. The caudal regions come into contact with their contralateral component at the median plane in the future pelvis and fuse, forming a single Y-shaped tubular structure known as the uterovaginal primordium. The uterovaginal primordium consists of a uterine and vaginal region.2,5 The uterine region gives rise to the uterus; correspondingly, the vaginal component will develop into the superior aspect of the vagina.2,3,5
The uterus has a bicornuate appearance at this stage, but its anatomic configuration will continue to develop through the processes of fusion and subsequent canalization. Uterine septum canalization or regression is mediated by apoptosis, which is regulated by the bcl-2 gene.6 Fusion was thought to occur from a caudal to cranial direction. However, anomalies found in postnatal life, such as a double cervix and vagina but a normal uterus, suggests that fusion may occur initially at the level of the internal uterine isthmus and then proceed in both directions.3
By week 12, the uterine fundus assumes its mature shape. The uterine endometrium is derived from the lining of the fused paramesonephric (müllerian) ducts, and the endometrial stroma and myometrium are derived from adjacent mesenchyme.4,7 This entire process is completed by week 22 of development and results in a uterine cervix and uterus with a single uterine cavity.2
Development of the Vagina and Hymen
Normal vaginal development requires the fusion of two different embryologic structures, the mesodermal paramesonephric (müllerian) ducts and the endodermal urogenital sinus. After formation of the Y-shaped uterovaginal primordium, the caudal tip of the uterovaginal primordium inserts into the dorsal wall of the urogenital sinus (see Fig. 12-1). This event creates an elevation called the müllerian or sinus tubercle. The sinus tubercle induces the formation of the sinovaginal bulbs at its distal aspect. As paired endodermal evaginations, the sinovaginal bulbs extend as a solid core from the urogenital sinus to the caudal aspect of the uterovaginal primordium and then fuse to form the vaginal plate.2,7,8
The vaginal plate and the sinovaginal bulbs restructure the urogenital sinus from its original long, tubular structure into a flat vestibule. This restructuring is responsible for positioning the female urethra at the primitive perineum. Meanwhile, the vaginal plate canalizes caudal to cephalad; when this process is complete at week 20, the vaginal canal is formed. The precise boundary between the uterovaginal primordium and urogenital sinus, as well as their individual contributions to the vagina, has not been established. Some authorities maintain that the superior one third of the vaginal epithelium is derived from the uterovaginal primordium and the inferior two thirds from the uterogenital sinus. However, most other experts consider the entire vaginal lining to be derived from the vaginal plate of the uterogenital sinus.2,7 The vaginal fibromuscular wall develops from the surrounding mesenchyme.
The hymen is formed by expansion of the caudal aspect of the vagina with the subsequent invagination of the posterior wall of the urogenital sinus.2,7 It serves to separate the vaginal lumen from the urogenital sinus cavity until late in fetal development. It ruptures perinatally, with the remnants remaining as a thin mucous membrane.8
Development of the Ovary
Chromosomal and genetic sex is determined at fertilization when the X ovum is fertilized by either an X-bearing or Y-bearing sperm. The XX or XY chromosomal complement of the embryo provides the genes coding for a group of transcription factors—Wilms’ tumor suppressor gene (WT1), DAX1, and steroidogenic factor (SF-1)—that orchestrate gonadal differentiation into either an ovary or testis. WT1 is also important for renal development. Renal and gonadal agenesis will occur with a mutation in the WT1 gene. SF1 and DAX1 gene mutations are associated with gonadal dysgenesis and impaired development of the adrenal cortex. DAX1, an X-linked molecule, suppresses testicular differentiation. Therefore, patients with a mosaic karyotype such as 45,XO/46,XY may have sufficient expression of DAX1 to suppress testicular development. The same concept applies to XXY Klinefelter’s syndrome. Sex determination of the gonads depends on a number of complex molecular events that direct the interactions of the genes encoding these factors.9
Just as the Y chromosome influences the development of the male gonad and phenotype, the X chromosome similarly influences ovarian differentiation and phenotype. The presence of two X chromosomes is crucial to maintenance of normal ovarian function after differentiation. Ovaries of 45,X females demonstrate accelerated oocyte atresia.10 Autosomal genes also influence ovarian development.11
By week 6, the primordial germ cells have integrated into the mesenchyme to become part of the primary sex cords. As the primary sex cords regress, the primordial germ cells then become incorporated into the secondary sex cords, which connect to the ovarian epithelium. At week 12, the ovarian cortex can be identified. Approximately 1 week later, the secondary cords fragment, leaving the primordial follicles.10 The primordial follicle is composed of a primordial germ cell surrounded by one layer of sex cord-derived follicular cells; it is now known as the oogonium. The oogonia undergo active mitosis and initiate meiosis during fetal development. Many degenerate before birth, and only about 2 million will remain after birth; they will enlarge to become primary oocytes.
Development of the External Female Genitalia
The external genitalia of female and male are similar at the indifferent stage of development between weeks 4 and 7 (Fig. 12-2). Distinguishing sexual characteristics begin to appear at week 9, although full differentiation is not achieved until week 12.2 Mesenchyme at the cranial aspect of the cloacal membrane proliferates, forming the genital tubercle. The genital tubercle elongates to form the phallus and later, the clitoris. The genital folds and the labioscrotal swellings develop bilaterally along the cloacal membrane.
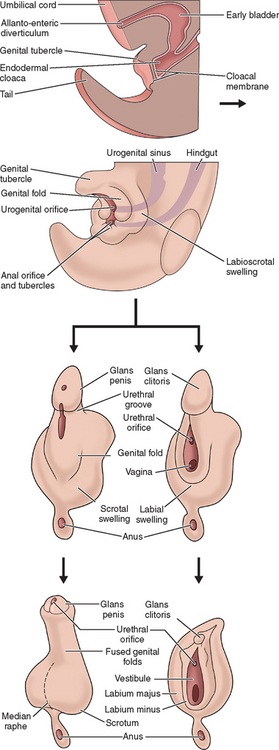
At the completion of week 6, the urorectal septum fuses with the cloacal membrane, dividing the membrane into anal (dorsal) and urogenital (ventral) aspects.2,8 The urogenital membrane lies in the floor of the urogenital groove and is bound by the genital folds. Approximately 1 week later, the membranes rupture to form the anus and urogenital orifices, respectively. The urogenital orifice in the female is the primordial urethra and vaginal vestibule.
The genital folds meet posteriorly, fuse, and form the labia minora frenulum. Their unfused anterior counterparts become the labia minora (minus) proper. The labioscrotal folds also fuse posteriorly and form the posterior labial commissure.2 When they fuse anteriorly, the anterior labial commissure and mons pubis are formed. However, most of the labioscrotal folds remain unfused and form the labia majora.
CLASSIFICATION OF CONGENITAL ANOMALIES OF THE INTERNAL REPRODUCTIVE TRACT
Müllerian anomalies have been reported dating as far back as the 16th century.12 The epidemiology of these anomalies has been vastly outpaced by the remarkable technical achievements used in their diagnosis and treatment. The actual incidences of these anomalies are not well understood. Indeed, reports widely vary, with most authors reporting an incidence ranging from 1:200 to 1:600 in fertile women.13–15
The genetics of various congenital anomalies of the reproductive tract are quite complex and beyond the scope of this chapter. Briefly, most cases occur sporadically. In familial cases, many anomalies appear to be multifactorial. Associations with other modes of inheritance also exist and include autosomal dominant and autosomal recessive patterns of inheritance as well as X-linked disorders. Müllerian anomalies can also represent a component of a multiple malformation syndrome.5,16,17,
CLASSIFICATION SYSTEM FOR REPRODUCTIVE TRACT CONGENITAL ANOMALIES
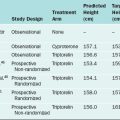
In 1988, the American Society for Reproductive Medicine (ASRM), formerly known as the American Fertility Society (AFS), published their classification scheme for female reproductive tract anomalies (Fig. 12-3).18 Their classification represents the most widely accepted method of categorizing these abnormalities and has the major advantages of allowing for standardization of reporting methods and providing a template for long-term studies of reproductive outcomes for each anomaly. The system is organized into seven classes according to the major developmental failure and separates the defects into groups having similar clinical manifestations. It also includes a class characterizing uterine abnormalities related to in utero DES exposure. Although vaginal anomalies are not included, the scheme allows for their incorporation.

Figure 12-3 Diagram from the American Fertility Society Classification of Müllerian Abnormalities
(American Fertility Society, Fertil Steril 49:944–955, 1998.)
Class I: Agenesis or Hypoplasia: Segmental or Complete
Agenesis or hypoplasia may involve the vagina, cervix, fundus, fallopian tubes, and/or any combination. The Mayer-Rokitansky-Küster-Hauser syndrome is the most common example in this category.
Class V: Septate Uterus: Complete or Partial
Either a complete or partial midline septum is present within a single uterus.
PATHOGENESIS AND DIAGNOSIS OF DEVELOPMENTAL ANOMALIES OF THE REPRODUCTIVE TRACT
Vaginal Agenesis
Description and Pathogenesis
Vaginal agenesis is the most common defect involving the vagina and uterus and is characterized by the absence or hypoplasia of the uterus and proximal vagina and, in some cases, the fallopian tubes. Frequently, this malformation is accompanied by urinary tract anomalies. Vaginal agenesis occurs when the sinovaginal bulbs fail to develop because the vaginal plate cannot form in the absence of the sinovaginal bulbs.2 It occurs in an estimated 1:5000 newborn females.19 Multiple variants have been reported, with some exhibiting complicated associated anomalies. Although the uterus is usually absent, 7% to 10% of cases will have a normal, albeit obstructed, uterus or a rudimentary uterus with functional endometrium.20 In the presence of active endometrium, the patient may experience cyclical pain.
Vaginal agenesis can be partial or complete. Partial vaginal agenesis is less commonly seen and is characterized by a normal uterus with a small vaginal pouch located distal to the cervix. Complete vaginal agenesis, known as the Mayer-Rokitansky-Küster-Hauser syndrome, is more common. In 90% to 95% of reported Mayer-Rokitansky-Küster-Hauser cases, the uterus is also absent.20–22 The fallopian tubes are normal and the ovaries demonstrate normal endocrine function.
Associated anomalies often coexist with vaginal agenesis. The incidence of associated urologic abnormalities varies from 15% to 40%; skeletal anomalies such as congenital fusion or absence of vertebra occur in approximately 12% to 50%.20,23 An association between the Mayer-Rokitansky-Küster-Hauser syndrome and the Klippel-Feil syndrome has been reported. Components of the Klippel-Feil syndrome include congenital fusion of the cervical spine, short neck, a low posterior hairline, and limited range of motion in the cervical spine.24 The Mayer-Rokitansky-Küster-Hauser is also associated with the MURCS syndrome. MURCS syndrome is characterized by aplasia of the müllerian ducts and renal system as well as cervicothoracic somite dysplasia.25,26
Patients with vaginal agenesis are 46,XX. Most cases occur sporadically, although approximately 4% of reported cases are clustered in families.20,21 Abnormalities in the expression of homeobox (Hox) genes are associated with müllerian duct agenesis. It has been reported that increased exposure to galactose is responsible for abnormal vaginal development based on the finding that vaginal agenesis has been reported to be associated with variants of the galactose 1-phosphate uridyl transferase enzyme.27–29 Other authorities have speculated that mutations in either the antimüllerian hormone or müllerian inhibitory substance gene or its receptor gene are responsible for this disorder.4 In a recent report, a de novo translocation in a young woman with Mayer-Rokitansky-Küster-Hauser syndrome is discussed, suggesting that this break point may be involved in midmüllerian differentiation.30
Diagnosis of Vaginal Agenesis
Vaginal agenesis is usually diagnosed at puberty when young adolescents present with primary amenorrhea. Indeed, vaginal agenesis is the second most common cause of primary amenorrhea in adolescents.20,21 Normal growth and development and the presence of age-appropriate secondary sexual characteristics are all evident. The external genitalia have a normal appearance, although visual inspection can often reveal a patulous urethra.30,31 The appearance of the vagina can be diverse; it can be completely absent or can be present as a short, blinding-ending pouch or as a vaginal dimple. The appearance of the vaginal dimple can range from a slight indentation to up to 5 to 6 cm in length. A uterus is not palpated on rectal examination.
Pelvic ultrasonography can support the clinical findings of an absent uterus or uterine remnant and the presence of normal ovaries. Magnetic resonance imaging (MRI) is extremely useful for those cases in which ultrasound findings are not definitive; lack of visualization of the vagina and a uterus in a technically adequate study indicates agenesis or hypoplasia of these structures.32–34 Laparoscopy is not usually indicated unless the diagnosis cannot be determined by other studies or if there is concern over the presence of a functioning uterus or rudimentary uterine tissue.
Unicornuate Uterus
Description and Pathogenesis
A unicornuate uterus occurs when one of the paired müllerian ducts fails to elongate while the other develops normally. This anomaly accounts for approximately 13% of all müllerian duct abnormalities.35 These ASRM class II uterine defects are structurally quite diverse. The unicornuate uterus may occur in isolation but is frequently accompanied by a rudimentary horn.35,36 Associated urologic anomalies frequently (44%) occur, especially when the rudimentary horn is obstructed. These anomalies include ipsilateral renal agenesis (67%), horseshoe kidneys, or ipsilateral pelvic kidney (15%).35,36
Obstetric outcomes are generally poor, although normal pregnancies can occur. Because these anomalies are uncommon, most reports do not classify reproductive outcomes according to the various malformation subclasses.35 For the entire unicornuate class, the spontaneous abortion rate is 51%, the preterm birth rate is 15%, and the fetal survival rate is approximately 40%.37 Common obstetrical complications include malpresentation, intrauterine growth retardation, and preterm birth.37–41
Diagnosis of Unicornuate Uterus
Although a hysterosalpingogram (HSG) is useful in diagnosing a unicornuate uterus, it cannot detect a noncommunicating rudimentary horn. MRI can reliably identify a noncommunicating horn and is characteristically not opacified when the endometrium is absent.32 High-resolution ultrasound evaluation can usually identify rudimentary horns and is more reliable than laparoscopy in determining whether the horn is communicating. Indeed, laparoscopy is rarely indicated as part of the diagnostic evaluation of an obstructed, noncommunicating horn. Additional studies should include an intravenous pyelogram (IVP) or renal ultrasound to evaluate for ipsilateral renal agenesis, a horseshoe kidney, or an ipsilateral pelvic kidney.35,42
Uterus Didelphys
Pathogenesis
Either unilateral or bilateral duplication of the müllerian ducts can result in uterus didelphys. This anomaly is characterized by the presence of two separate, normal-sized uteri and cervices that are fused at the lower uterine segment. Vaginal duplication is frequently a component, and an associated vaginal septum is often seen. A longitudinal (horizontal) septum extends either completely or partially from the cervices to the introitus. A complete vaginal septum with a sagittal orientation occurs in approximately 75% of cases; some have occasional obstructing transverse septum.43–45
Uncommonly, patients can present with an obstructed unilateral vagina. This abnormality is frequently associated with ipsilateral renal and ureter agenesis and is a component of the Wunderlich-Herlyn-Werner syndrome.46,47
Reported reproductive outcomes are slightly better for uterus didelphys than for the unicornuate uterus. Fertility is not compromised, but the spontaneous abortion rate is high (40%). However, when the pregnancies are carried to term, few obstetrical difficulties occur.35,37,38
Diagnosis of Uterus Didelphys
Uterus didelphys with obstructed unilateral vagina can be diagnosed early and accurately. The clinical presentation is similar to that in patients having a unicornuate uterus and noncommunicating, functional horn.35,38 Ipsilateral renal agenesis frequently accompanies both these conditions as well.46,47 On pelvic examination, one cervix is identified, and a paravaginal cystic-type structure can usually be palpated that represents the noncommunicating second vagina.
Preoperative diagnostic studies are similar to those used for the unicornuate uterus and include HSG, pelvic ultrasound or MRI, and an IVP or renal ultrasound to either confirm or exclude associated urinary tract anomalies.33,34,42
Bicornuate Uterus
Pathogenesis
Müllerian ducts that incompletely fuse at the level of the uterine fundus result in the bicornuate uterus malformation. The lower uterus and cervix are completely fused, but the incompletely fused ducts elongate and develop into two separate uterine horns. Their respective endometrial cavities communicate, although an intervening muscular uterine septum is present. A single-chamber cervix and vagina are present. The subclassification of this anomaly is contingent on whether the separation is complete or partial. Complete variants include the uterus bicornis unicollis, in which the uterine cavity division extends to the internal os, and the uterus bicornis bicollis, in which the division extends to the external os. Partial bicornuate uteri are characterized by a uterine division that is confined to the fundal region. In this anomaly, the uterine cavity division corresponds to a visible sagittal groove on the external fundal uterine surface. The depth of the groove and the extent of the separation are subject to the length of the incompletely fused müllerian ducts.35,48
Usually the bicornuate uterus is an incidental finding. Women with this uterine malformation usually have no difficulty becoming pregnant, and approximately 60% can expect to deliver a viable infant.36 However, they can present with late abortion or premature labor. This observation may be related to whether the bicornuate uterus is partial or complete. According to one study, women with partial bicornuate uterus experienced a spontaneous abortion rate of 28% and preterm delivery rate of 20%. These rates contrasted with a spontaneous abortion rate of 66% and a higher rate of preterm deliveries in women diagnosed with a complete bicornuate uterus.35
Diagnosis of Bicornuate Uterus
Evaluation begins with an ultrasound performed during the luteal phase of the menstrual cycle when the endometrial echo complex is better identified.42 Ultrasound based on the angle between the two cornu cannot accurately distinguish the bicornuate uterus from the septate uterus, and MRI must be performed to establish this diagnosis. HSG, usually the cornerstone in diagnosing most uterine structural anomalies, cannot reliably distinguish between these two entities because they can display similar uterine cavity images.49,50
Septate Uterus
Pathogenesis
There are three types of septa: complete, partial, and segmental. The complete septum extends from the fundus to the internal os, dividing the endometrial cavity. The partial septum is located at the fundus; the segmental septum, also located at the fundus, is noncontiguous, allowing for partial communication between the endometrial cavities.51,52
The fertility of women with this type of anomaly is not significantly compromised. However, the septate uterus is associated with the poorest reproductive outcomes of all the müllerian duct anomalies. Spontaneous abortion rates of 67% have been reported, and the live birth rate ranges from 15% to 28%; obstetric complications such as incompetent cervix, premature labor, and abnormal presentation are common.53–56 Hysteroscopic resection of the septum increases the live birth rate to as high as 75%.
Diagnosis of the Septate Uterus
HSG is an essential study in diagnosing and planning surgery for the septate uterus. It can assess the presence of a two-chambered uterus, determine the length and thickness of the septum, and permit concomitant assessment of tubal patency. However, HSG is limited since it cannot distinguish the septate from the bicornuate uterus or detect minor septal defects.50 MRI provides excellent tissue characterization and can reliably differentiate between septate uterus and bicornuate uterus.56 Transvaginal ultrasound can also be useful in identifying this abnormality. In one study, transvaginal ultrasound had a sensitivity of 100% and a specificity of 80% in detecting the septate uterus.57 Additionally, if three-dimensional ultrasound is available, the reported accuracy was 92% for the diagnosis of septate uterus.
Arcuate Uterus
Description
The arcuate uterus has a small (<1.5 cm) septate projection located at the fundal aspect of the uterine cavity. The external contour of the uterus is convex or flat. This congenital anomaly is the most common uterine abnormality detected using HSG.58,59 Classifying the arcuate uterus has been challenging. Earlier classification systems considered it a mild form of the bicornuate uterus and the ASRM classification places this anomaly in a separate class.18 Some speculate that the arcuate uterus represents a normal variant because it is not associated with an increased risk of spontaneous abortion or other pregnancy complications. Interestingly data on outcome after resection of the septum in a septate uterus demonstrates that leaving approximately 1 cm of septum will not affect pregnancy outcomes.
Diagnosis of Arcuate Uterus
There is limited data regarding the diagnosis, management, and reproductive outcomes for the arcuate uterus. Using HSG, a single uterine cavity with a saddle-shaped fundus can be identified. MRI shows a normal fundal contour with a minimal indentation.58,59 Renal ultrasound and IVP may be performed to exclude any associated urinary tract anomalies, although these studies generally are not part of a standard evaluation. Management is similar to the septate uterus with only selected patients fulfilling criteria for poor reproductive performance recommended for surgical correction.
Diethylstilbestrol-Related Anomalies
Pathogenesis
DES is a synthetic estrogen prescribed from the late 1940s to the 1970s to prevent recurrent pregnancy loss, premature delivery, and other obstetric problems. In the early 1970s it was found to be a teratogen and its use during pregnancy was banned.60 It is now known that there is a strong association between in utero DES exposure and an increased risk of developing vaginal adenocarcinoma later in life. Additional studies revealed that in utero DES exposure was also associated with structural abnormalities of the developing uterus, cervix, and vagina. Although some women exposed to DES in utero are in their midthirties, the majority of these women are now postmenopausal or approaching the end of their reproductive years.
Uterine anomalies include a T-shaped endometrial cavity, widened lower uterine segment, midfundal constrictions, endometrial filling defects, and uterine hypoplasia.61,62 DES-associated structural abnormalities of the cervix also have been identified but are less frequent than those of the uterus. These abnormalities include cervical hypoplasia, anterior ridge or collar (cock’s comb cervix), and pseudopolyps. Vaginal adenosis and vaginal constrictions have also been associated with DES.61,62 Interestingly, uterine anomalies similar to those related to DES exposure have been reported in women without in utero DES exposure.
Although there is no convincing evidence that in utero DES exposure has an unfavorable impact on fertility, there is considerable evidence to indicate that many patients with DES exposure will have poor obstetrical outcomes. Complications include an increased risk of spontaneous abortions, ectopic pregnancies, and cervical incompetence.63 However, it remains uncertain whether the anomalies themselves or some subclinical abnormality of these women’s reproductive tracts are responsible for these associations.
Diagnosis of DES-Related Anomalies
These anomalies of the uterine cavity and endocervix are diagnosed exclusively by HSG. MRI and ultrasound are uncommonly required.49,50 Abnormalities of the vagina and ectocervix are diagnosed by direct visualization at the time of speculum examination. Cervical changes often indicate coexisting uterine anomalies. In one study, 86% of women with a history of in utero DES exposure that had cervical changes also had concomitant uterine abnormalities.61 No associations between DES-related anomalies and urinary-renal system anomalies have been identified, and this would preclude the need for imaging of the renal system.
Transverse Vaginal Septum
Pathogenesis
A transverse vaginal septum is one of the rarest congenital anomalies of the female reproductive tract, with a frequency of 1 in 70,000 females.64,65 This malformation arises either from incomplete vertical fusion between the caudal aspect of the sinus tubercle and sinovaginal bulbs or because the vaginal plate fails to canalize. The septum divides the vagina into two segments, thereby reducing its functional length. It can be located at nearly all levels in the vagina and may be perforate or imperforate. The majority of septa (46%) are located at the superior aspect, at the putative junction between the vaginal plate and caudal aspect of the uterovaginal primordium. The next most common site is the midvagina (40%), followed by the inferior vagina (14%).66
Urologic defects are only occasionally seen with a transverse vaginal septum. Instead this anomaly is often associated with other structural anomalies, including imperforate anus, bicornuate uterus, coarctation of the aorta, atrial septal defect, and malformations of the lumbar spine.65 Although there are no indications that a transverse vaginal septum is genetic, an autosomal disorder associated with a transverse vaginal septum and hydromucocolpos has been discovered in the Amish community.65
Diagnosis of Transverse Vaginal Septum
Fetus, Neonate, and Infant
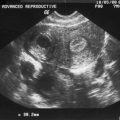
Before puberty, a transverse vaginal septum can sometimes manifest as hydromucocolpos. In rare cases, a hydromucocolpos can be diagnosed in utero during a third-trimester transabdominal ultrasound, which reveals fetal abdominal distension secondary to an abdominal or pelvic mass.67
Diagnosis of hydromucocolpos in the neonate and infant can be challenging. Indeed, unless significant hydromucocolpos is evident, a transverse vaginal septum is rarely diagnosed. In these cases a large mass is palpated in the lower abdomen, but unlike an imperforate hymen, the obstruction is well within the vagina so that an introital bulge will not be visible. Although rare, profuse amounts of fluid can collect in the vagina superior to the obstructing septum, resulting in a mass effect that compresses the surrounding organs. The compression may cause serious consequences if not promptly diagnosed and treated.68 Initial studies include an abdominal-pelvic ultrasound. Proper imaging studies can frequently eliminate the need for laparoscopy or laparotomy. MRI is useful in depicting pelvic anatomy and determining the thickness of the vaginal septum and should also be performed.65
Young Adolescent
The hymen is generally open and there are no bulging membranes on Valsalva. When the septum is incomplete, presenting complaints often include a foul-smelling vaginal discharge, dyspareunia secondary to a short vagina, and infertility. Because an incomplete transvaginal septum allows menstrual flow to escape periodically, a hematocolpos and hematometrium often develop over time. For patients who become pregnant, a transverse vaginal septum can cause soft tissue dystocia.64
Imperforate Hymen
Pathogenesis
This anomaly is the most common obstructive condition of the female reproductive tract. The embryonic defect that gives rise to the imperforate hymen occurs when the inferior aspect of the vaginal plate fails to perforate (open). It appears to be an isolated condition, although there are some reports of familial occurrences.69
Diagnosis of Imperforate Hymen
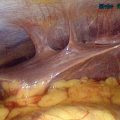
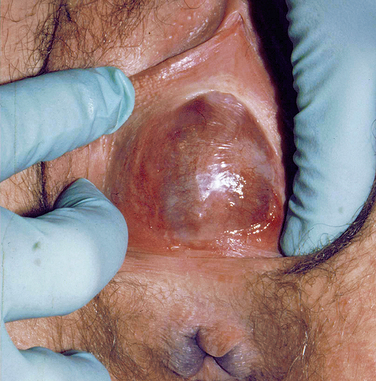
At puberty, the hymen can then again become distended from menstrual-associated hematocolpos.70 Often the patient complains of back and cyclic abdominal pain, pain on defecation, difficulty with micturition, and amenorrhea. Examination of the external genitalia reveals a bulging, blue mass at the introitus, and rectoabdominal palpitation confirms a distended vagina (Fig. 12-4). In these cases, retrograde menstrual flow into the abdominal cavity can predispose the patient to endometriosis.
Vaginal Atresia
Pathogenesis
This anomaly results when the urogenital sinus fails to contribute to the inferior portion of the vagina.2,8 The müllerian structures are usually normal, but the lower portion of the vagina is completely replaced by fibrous tissue. Vaginal atresia can clinically mimic vaginal agenesis and an imperforate hymen.
Most cases of vaginal atresia occur sporadically. However, vaginal atresia has also been described as a component of an autosomal recessive syndrome that includes a multitude of other anomalies, including middle ear ossicle anomalies and renal dysgenesis.71,72
Diagnosis of Vaginal Atresia
Abdominal ultrasound reveals the presence of ovaries, uterus, cervix, and an obstructed, blind-ending superior vagina. This is in contradistinction to vaginal agenesis, in which the uterus and cervix are absent and only the ovaries are seen.73 MRI can aid in detecting the presence of a cervix, which distinguishes this anomaly from cervical agenesis. Additionally, MRI can determine the presence of a functioning endometrial cavity.74 Obviously, these patients are not candidates for HSG studies. The karyotype is 46,XX and endocrine studies are normal.
CONGENITAL ABNORMALITIES OF THE EXTERNAL GENITALIA
Ambiguity of the external genitalia is the result of defects during the processes of sexual determination and differentiation (Table 12-1).9 Four major groups account for most disorders resulting in ambiguous genitalia; they include female pseudohermaphroditism, male pseudohermaphroditism, gonadal differentiation disorders, and malformation syndromes. The discovery of ambiguous genitalia in an infant requires special investigative studies to determine the presence of life-threatening conditions. Multispecialty consultations are indicated. An important issue to be addressed at this time includes determining the sex of rearing. In older patients presenting with ambiguity of sexual development, quality of life issues and reproductive potential are the major concerns.
Congenital Adrenal Hyperplasia
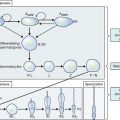
The most frequent metabolic cause of ambiguous genitalia in the newborn is female pseudohermaphroditism due to CAH.75,76 CAH represents a group of autosomal recessive disorders that includes enzymatic defects resulting in impaired adrenal steroidogenesis (Fig. 12-5).75–77 All forms of CAH have in common these endocrine irregularities: diminished cortisol production, increased corticotropin levels, adrenal hyperplasia, and overproduction of intermediate steroids. The metabolic intermediates are typically shunted to other enzymatic pathways and result in the production of androgenic steroids. It is these steroids that cause virilization of the female fetus in utero. The extent of virilization will depend on androgen levels, but often includes varying degrees of posterior labial fusion, clitorimegaly, and vaginal introitus abnormalities. When CAH occurs in males, genital enlargement and hyperpigmentation of the skin are often seen.
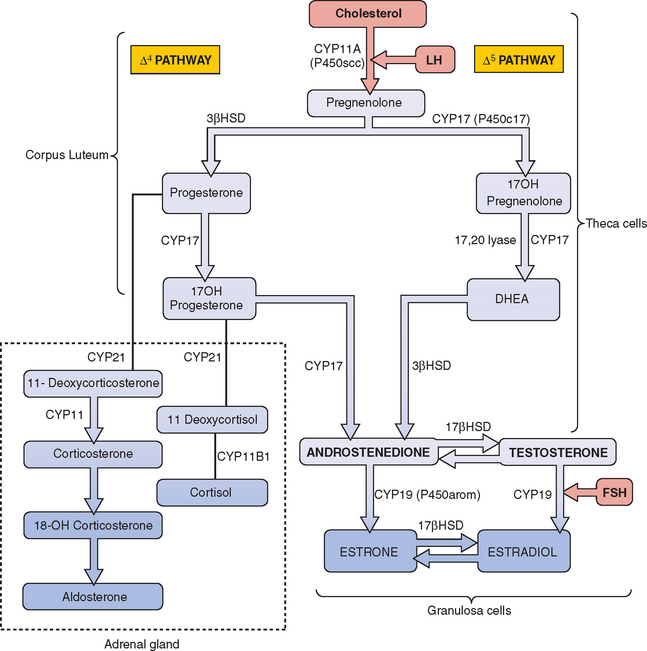
21-Hydroxylase Deficiency: Classic Salt-Losing and Simple Virilizing Variants
The most common form of CAH is a deficiency of cytochrome P450 21-hydroxylase; this is responsible for 90% to 95% of these cases.78 Cytochrome P450 21-hydroxylase deficiency is categorized into classic and nonclassic types. In the classic form, the condition presents at birth. It is divided into two subtypes based on the severity of the enzymatic defect. Seventy-five percent of these infants will have the severe salt-losing form, in which aldosterone production is significantly impaired.78 The remainder of infants with classic 21-hydroxylase deficiency have the simple virilizing form, in which aldosterone production is adequate and there is no salt wasting.
The incidence of classic CAH varies among different ethnic groups and occurs in 1:15,000 live births worldwide.78,79 Nonclassic 21-hydroxylase deficiency is characterized by a partial enzymatic defect; this type is associated with neither ambiguous genitalia nor salt wasting. It is usually detected later in life and is often confused with polycystic ovary syndrome.80
Pathogenesis
The enzyme 21-hydroxylase is a cytochrome P450 protein, designated CYP21, and functions in both cortisol and aldosterone synthesis. The CYP21 gene is localized to chromosome 6p21.3 and encodes this enzyme. Either a mutation or partial deletion in this gene can result in markedly reduced or absent enzyme activity.78,79 In classic 21-hydroxylase deficiency, both alleles of the CYP21 gene are affected and three major endocrine regulatory systems are disrupted: the hypothalamic-pituitary-adrenal axis, renin-angiotensin-aldosterone axis, and hypothalamic-pituitary-gonadal axis. As a consequence, cortisol, aldosterone, and gonadal sex steroid production is altered.
Excess androgen production in utero results in abnormal clitoral growth and virilization of the urogenital structures. Newborn females with either the salt-losing or simple virilizing forms of cytochrome P450 21-hydroxylase deficiency have ambiguous genitalia with appearances that can range from mild clitorimegaly to fully masculinized penile urethra. Affected male infants generally have no genital malformations at birth, but continued androgen excess results in accelerated body growth. In classic salt-losing 21-hydroxylase deficiency, infants are unable to retain sodium and excrete potassium from the renal tubules. The infant will often present with hyponatremia, hyperkalemia, hypotension, and metabolic acidosis. Importantly, this condition can lead to cardiovascular collapse.78,79
Diagnosis
The accurate diagnosis of 21-hydroxylase deficiency is based on observations from the physical examination and laboratory studies. Laboratory tests reveal the following abnormalities: decreased serum cortisol and aldosterone levels (in salt-losing form), increased serum 17-hydroxyprogesterone and increased urinary pregnanetriol, a metabolite of 17-hydroxyprogesterone. Salt-losing 21-hydroxylase deficiency is also accompanied by decreased serum sodium, increased potassium, and increased plasma renin activity. The diagnosis of cytochrome P450 21-hydroxylase deficiency can also be rendered using allele-specific polymerase chain reaction in affected infants.81
11β-Hydroxylase Deficiency
Defects in 11β-hydroxylase are the second most common cause of all CAH-associated ambiguous genitalia, representing 5% to 8% of these cases. The clinical presentation can be quite varied, with symptoms occasionally overlapping with those seen in 21-hydroxylase deficiency. Excessive virilization of genitalia is often seen in 46,XX females and gender-appropriate masculinization in 46,XY males.82
Pathogenesis
The CYP11B1 gene, located on chromosome 8q21, encodes for the enzyme 11β-hydroxylase.82,83 Mutations in the gene result in impaired conversion of 11-deoxycortisol to cortisol and 11-deoxycorticosterone to corticosterone. The intermediate corticoids are shunted to form androgenic steroids. Accumulation of mineralocorticoids results in hypernatremia, hypokalemia, plasma volume expansion, and hypertension.
Diagnosis
The diagnosis of 11β-hydroxylase deficiency can be established by the clinical observation of ambiguous genitalia in the female along with laboratory abnormalities. The following abnormalities are commonly identified: increased serum sodium, decreased serum potassium, decreased serum cortisol, increased serum 11-deoxycortisol, increased serum desoxycorticosterone, increased 24-hour urinary 17-ketosteroids, decreased plasma renin activity, and increased ratio of tetrahydrocompounds (11-deoxycortisol to cortisol).84
3β-Hydroxysteroid Dehydrogenase Deficiency
Deficiency of 3β-hydroxysteroid dehydrogenase (3βHSD) accounts for less than 5% of all CAH cases.85 It is the only form of CAH associated with genital ambiguity in both males and females. In most cases the adrenal insufficiency is quite severe. There is reduced synthesis of all three groups of adrenal corticosteroids, with elevated serum levels of pregnenolone, 17-hydroxypregnenolone, and dehydroepiandrosterone (DHEA). A variety of phenotypic manifestations can present in infants as well as older individuals. Production of the more potent androgens is blocked, and DHEA is the major intermediate that accumulates. DHEA is a weak androgen, and it is associated with mild virilization in female infants and undervirilization of newborn males. Newborns of both sexes may have salt-wasting because of deficiencies in aldosterone and other mineralocorticoids.85
Pathogenesis
The HSD3B2 gene is located on chromosome 1 and encodes for two isoforms of the 3βHSD enzyme, Type I and Type II. Type I, located in the liver and peripheral tissue, is not associated with mutations affecting enzymatic activity of the enzyme. Type II is located in the adrenal glands and gonads and is associated with a number of mutations affecting enzymatic activity. Patients with the salt-losing disorder have no functional 3βHSD Type II activity; those with the nonsalt-wasting variant will demonstrate partial enzyme activity.86,87
Diagnosis
In infants with the salt-wasting form, early diagnosis and replacement of glucocorticoids and/or mineralocorticoids are essential to avoiding adrenal crisis and death. Affected female neonates can appear normal or have clitorimegaly and labial fusion. Most newborn males will have incompletely masculinized genitalia with varying degrees of hypospadias.85,86 The testes often descend and can be palpated in the inguinal canal or labioscrotal folds. Signs of adrenal insufficiency may also be present.
Close monitoring of daily weights, electrolytes, fluid status, and plasma renin activity is indicated.88 In some cases, plasma renin activity increases in the absence of aldosterone deficiency and salt losing. For these patients, fludrocortisone acetate improves renin activity and hormonal control. Therapy is initiated as soon as possible. In infants, hydrocortisone therapy may reverse the virilizing effects of the excess androgens.
Male Pseudohermaphroditism
Male pseudohermaphrodites are genetic 46,XY males; they have testes but exhibit incompletely masculinized external genitalia. Discovery of this condition generates a number of diagnostic and therapeutic challenges for the clinician. Unlike female pseudohermaphroditism, in which most cases are caused by CAH and exogenous androgens, a constellation of disorders can result in male pseudohermaphroditism and in many cases a definitive diagnosis cannot be rendered.87
Defects in Androgen Action: 5-α-Reductase Type 2 Deficiency
Many individuals with 5-ARD virilize during early adolescence, and this disorder has been historically referred to as “penis at 12.” In the past, these individuals were often reared as females. At puberty, when the genitalia underwent masculinization due to an increase in type 1 enzyme activity, they then changed gender.89 Obviously, the major issue with 5-α-reductase type 2 deficiency is determining the sex of rearing.
Pathogenesis
The defect in androgen action is related to mutations in the 5-α-reductase type 2 enzyme, which converts testosterone to the more physiologically active form, dihydroxytestosterone (DHT) in androgen-dependent tissues. The enzyme is encoded by the SRD5A2 (or 5α-RD2) gene on chromosome 5. More than 40 mutations have been reported in all five exons of this gene. Although most mutations are amino acid substitutions, other mutations, including complete deletions, nonsense, and/or splice site mutations, have been reported.90 Masculinization of external genitalia and differentiation of the urogenital sinus depends on DHT. Incomplete masculinization occurs as the result of decreased conversion of testosterone to the active DHT. Because testosterone levels are normal, dependent Wolffian duct structures are not affected. Antimüllerian hormone is normally produced and therefore all müllerian duct development is arrested.
Diagnosis
The diagnosis of 5-ARD is often rendered at birth. However, near-identical phenotypes can occur in androgen insensitivity syndrome. Preliminary laboratory testing reveals normal or slightly increased serum testosterone levels with decreased DHT. On stimulation by human chorionic gonadotropin (hCG), the characteristic lab finding is an increased testosterone to DHT ratio (>20). Cultured genital skin fibroblasts are used to measure 5-α-reductase type 2 enzyme activity by assaying the conversion of testosterone to DHT.89 In androgen insensitivity syndromes the ratio of testosterone to DHT is not increased.
Androgen Insensitivity Syndrome
There are two forms of androgen insensitivity syndrome, complete and partial; each depends on the amount of residual androgen receptor function. Complete androgen insensitivity syndrome is characterized by the failure of Wolffian duct-derived structures to develop. There are no ambiguous genitalia, although features of testicular feminization, such as a normal labia, clitoris, and a vaginal introitus, are evident. Testes can occasionally be palpated in the inguinal region; when this occurs in a phenotypic female, it is imperative to determine the contents,90 because 7% of newborn females with an inguinal hernia and no cervix by palpation or vaginoscopy are either male pseudohermaphrodites, usually secondary to androgen insensitivity syndrome, or have mixed gonadal dysgenesis.91 In partial androgen insensitivity syndrome, ambiguous genitalia with varying degrees of Wolffian development and labioscrotal fusion are found.
Pathogenesis
The androgen action defect is due to a mutation in the androgen receptor gene with impaired binding of testosterone and DHT in androgen-dependent target organs. The androgen receptor gene has a loss-of-function mutation localized to Xq11-13. The typical postreceptor events that mediate androgen effects on target tissues do not occur, despite normal levels of androgen synthesis, and prenatal undervirilization of external genitalia occurs.92 More than 250 mutations have been described, including complete and partial gene deletions, point mutations, and small insertions/deletions.93 As expected, an assortment of functional defects can arise from any of these mutations. Some defects result in complete loss of cell surface receptors, whereas others exhibit incomplete protein synthesis due to modifications in substrate binding affinity. Binding affinity alterations can contribute to a loss of signal transmission, despite the presence of normal cell surface receptors. In complete androgen insensitivity syndrome, the genotypes are consistent with most phenotypic presentations. However, the same is not true for partial androgen insensitivity syndrome.93 Normal antimüllerian hormone levels result in müllerian duct regression. However, Wolffian duct structures fail to develop properly due to diminished to absent testosterone and DHT binding.
Diagnosis
Screening hormone levels are within normal limits. However, the hCG stimulation study reveals an increase in both testosterone and DHT. Diagnostic confirmation can be rendered either by mutational analysis of the androgen receptor gene, which detects greater than 95% of the complete and partial androgen insensitivity syndrome mutations or by measuring androgen receptor binding in scrotal skin, which should be reduced.91 Sonography can be useful in identifying the presence of any müllerian structures. When present, the diagnoses of complete and partial androgen insensitivity syndrome are excluded.
Leydig Cell Hypoplasia or Agenesis
Leydig cell hypoplasia is an autosomal recessive disorder in which Leydig cell differentiation and testosterone production are impaired. The estimated incidence of Leydig cell hypoplasia is 1:1,000,000 and preliminary laboratory findings usually reveal markedly reduced serum testosterone and elevated LH levels.94 The phenotype depends on the extent of intrauterine testosterone secretion, and affected males can present with a wide variety of external genital abnormalities.
Pathogenesis
In most cases of Leydig cell hypoplasia, the androgen action defect is due to point mutations in exon 11 of the LH receptor gene.94,95 Testosterone synthesis by the fetal testes is diminished, and masculinization of the external genitalia is altered. Sertoli cells are present, producing antimüllerian hormone, and müllerian duct structures regress. Type I Leydig cell hypoplasia is the most severe form of this disorder and involves inactivating mutations in the LH receptor gene that prevent signal transduction.96 This break in the signal pathway results in significant decreases in testosterone production by the fetal testes. The androgen deficiency is milder in type II Leydig cell hypoplasia, in which mutations in the LH receptor gene only partially inactivate signal transduction.94,95
Diagnosis
Hormone levels reveal decreased serum testosterone, decreased DHT, and increased serum LH. In the hCG stimulation study, serum levels of testosterone and DHT do not increase. On rectal examination, a uterus cannot be palpated. Abdominal ultrasound usually reveals intra-abdominal gonads. Confirmation of the diagnosis is rendered by testicular biopsy, which reveals absence or marked hypoplasia of Leydig cells, presence of Sertoli cells, and arrest of spermatogenesis in the seminiferous tubules.93
Congenital Lipoid Adrenal Hyperplasia
Congenital lipoid adrenal hyperplasia is a rare, severe form of CAH that is fatal in two thirds of affected neonates. There is complete deficiency of all adrenal steroid hormones, and neonates present with female external genitalia and marked salt-wasting. Symptoms of severe adrenal insufficiency, including failure to thrive, vomiting, diarrhea, hyponatremia, and hypokalemia, present at birth.97
Pathogenesis and Diagnosis
Congenital lipoid adrenal hyperplasia is associated with mutations in two different genes. Mutations in the gene encoding steroidogenic acute regulatory protein (StAR) are responsible for most cases of this disorder. The StAR protein mediates rapid entry of cholesterol into the mitochondria and is an acute regulator of steroidogenesis.98 Mutations involving the CPY11A gene that encodes for the cholesterol side chain cleavage enzyme 20,22-desmolase are less common causes of this disorder.98 Preliminary laboratory testing on infants reveals decreased glucocorticoids, mineralocorticoids, testosterone, and DHT along with decreased sodium and potassium. Most newborns do not survive infancy, and the ones that do require hormone replacement.97
17-α-Hydroxylase Deficiency
Deficiency of 17-α-hydroxylase is characterized by reduced to absent gonadal and adrenal sex hormones with increased synthesis of mineralocorticoid precursors. In newborn males, ambiguous genitalia are evident, whereas sexual infantilism is present in newborn females. Varying degrees of hypertension and hypokalemia accompany this disorder. In the female, 17-α-hydroxylase deficiency is often diagnosed in the young adolescent during an evaluation for delayed puberty, absent secondary sexual characteristics, or primary amenorrhea, although some cases are diagnosed at birth.99
Pathogenesis
Mutations in the CYP17 gene can manifest as 17-α-hydroxylase deficiency, 17,20-lyase deficiency, or a combination of the two.99,100 In 17-α-hydroxylase deficiency, decreased cortisol levels stimulate corticotropin and although steroid production is increased, it is blocked proximal to the 17-α-hydroxylase step. There is compensatory accumulation of 17-deoxysteroids, including pregnenolone, progesterone, deoxycorticosterone, and corticosterone. Diminished androgen production results in hypogonadism. The mineralocorticoid activity of deoxycorticosterone causes hypernatremia and sodium retention, plasma volume expansion, and hypertension. Hypokalemia and subsequent decreased serum aldosterone and plasma renin activity are usually evident.99
Diagnosis
Markedly increased serum levels of 11-deoxycorticosterone and corticosterone confirm the diagnosis. Pregnenolone and progesterone levels are slightly increased. Serum levels of the following steroids are very low or absent: 17-α-hydroxypregnenolone, 17-α hydroxyprogesterone, 11-deoxycortisol, DHEA, androstenedione, and testosterone. Serum and urinary estrogens are decreased and corticotropin, FSH, and LH levels are all increased. The urinary metabolites 17-α-hydroxycorticosteroid and 17-ketosteroid are reduced or absent. Prenatal diagnosis of affected infants is possible by measuring adrenal steroid levels in amniotic fluid.99
17,20-Desmolase (Lyase) Deficiency
In 17,20-desmolase (lyase) deficiency, the biosynthetic pathways that convert 21-carbon steroids, 17-hydroxypregnenolone, and 17-hydroxyprogesterone to 19-carbon steroids, DHEAS, and androstenedione, respectively, are disrupted.100 Diminished to absent levels of androgens, testosterone, and estradiol also occur. There can be either partial or complete blockages of these pathways, and presenting symptoms will differ depending on the enzyme block.
Pathogenesis and diagnosis
As stated previously, the CYP17 gene encodes the 17-α-hydroxylase 17,20-lyase complex, and defects in the gene can result in 17,20-desmolase (lyase) deficiency.99,100 Laboratory tests diagnostic of 17,20-desmolase deficiency include increased serum 17-hydroxypregnenolone, increased 17-hydroxyprogesterone, normal FSH, increased LH, decreased testosterone (estradiol), decreased DHEA, decreased androstene-dione, and normal to slightly increased pregnenolone and progesterone.
17β-Hydroxysteroid Dehydrogenase Deficiency
The diagnosis of this disorder is often rendered at puberty in genetic males. These males have either been raised as females and present with primary amenorrhea and hirsutism or have been raised as males and present with gynecomastia and incomplete male genital development.101 In affected males, the virilization, including phallic enlargement and male secondary sexual characteristics at puberty, is very similar to that seen in 5-AHD. Affected individuals are infertile.
Pathogenesis
The enzyme 17β-hydroxysteroid dehydrogenase, or 17-ketosteriod reductase, catalyzes the conversion of androstenedione to testosterone in the testes. Mutations in the 17-βHSD type 3 isozyme gene (HSD17B3), located on chromosome 9q22, result in deficient testosterone production. Most of these mutations are missense/nonsense.101 No significant correlation between the genotype and phenotype has been determined.
Diagnosis
Characteristic laboratory findings include increased ratio of androstenedione to testosterone, with serum levels revealing increased androstenedione and decreased testosterone. Prenatal diagnosis is available in the kindred of affected patients if causal mutations have been characterized.101
Persistent Müllerian Duct Syndrome
Persistent müllerian duct syndrome is characterized by the presence of completely developed müllerian duct derivatives in an otherwise normally masculinized 46,XY male. Fewer than 200 cases have been reported.102 Failure of müllerian duct regression results in the presence of internal female reproductive organs in affected males, who also have varying degrees of Wolffian duct structures. Ambiguous genitalia are not often associated with this disorder, although unilateral or bilateral cryptorchidism may be present.102
Gonadal Differentiation Disorders
Gonadal Dysgenesis
Gonadal dysgenesis is a descriptive term that refers to a broad category of individuals born with female genitalia, müllerian duct structures, and dysgenetic or streak gonads. The term is often used in association with Turner’s syndrome, although additional forms of gonadal dysgenesis have been described. Patients with Turner’s syndrome will not only have dysgenic ovaries, but other associated findings as well, including short stature, shield chest, and coarctation of the aorta. Chromosomal analysis reveals either an abnormality or absence of an X chromosome. Turner’s syndrome accounts for approximately 50% of patients with gonadal dysgenesis.103,104
Pure Gonadal Dysgenesis
In pure gonadal dysgenesis, the gonads are dysgenetic, although the internal reproductive organs and external genitalia are normal. Occasionally, hypoplastic genitalia occur. Most cases are 46,XY, although some are 46,XX. The condition can occur sporadically or be inherited as either an autosomal recessive or X-linked trait in some cases of XY gonadal dysgenesis. In Swyer syndrome, 46,XY females present as phenotypic females, achieve average height, and exhibit none of the physical stigmata associated with Turner’s syndrome. FSH levels become elevated in early adolescence because “streak” gonads do not produce steroid hormones or inhibin. Microscopically, the dysgenetic gonads are often composed of ovarian stroma and fibrous tissue with no evidence of primordial follicles. The diagnosis is usually rendered during adolescence when these patients are evaluated for primary amenorrhea. However, in a recent report an adolescent with Swyer syndrome presenting with secondary amenorrhea is discussed.105 Patients with pure XY gonadal dysgenesis require gonadectomy because of the high risk for developing gonadoblastomas, which may transform into dysgerminomas or other germ cell neoplasms.106
Mixed Gonadal Dysgenesis
Most patients with mixed gonadal dysgenesis have a mosaic karyotype, 45,X/46,XY. The condition is characterized by asymmetric gonadal development and persistence of müllerian duct structures.107 Either an abnormal testis or a germ cell neoplasm is identified along with a contralateral dysgenetic or absent gonad. Incomplete müllerian duct development is the result of a functional deficit imposed by the abnormal testis. As indicated previously, gonadectomy is indicated.
Diagnosis of Gonadal Dysgenesis
Ovarian dysgenesis is often first suspected in a young adolescent who fails to achieve normal secondary sexual milestones, such as adequate breast development and menstruation. Ultrasonography can confirm the presence of the dysgenetic ovaries and often reveals small atrophic structures. Although rare, steroid-producing gonadoblastomas can develop in the dysgenetic gonads and may resemble normal ovaries on ultrasound.106 Other useful imaging studies include MRI and computed tomography scan. Both 46,XX and 46,XY patients benefit from exogenous estrogens and are potential candidates for assisted reproductive technology, particularly donor oocytes.
True Hermaphroditism
True hermaphroditism is the rarest of all the intersex disorders. Individuals have both ovarian and testicular tissue, although the gonad combinations vary. Gonad combinations can include one ovary and one testis, two ovotestes, one ovotestis and one ovary, or one ovotestis and one testis. More than 50% of these individuals are 46,XX; one third can be either chimeric 46,XX/46,XY or mosaic 46,XY/47,XXY or 45,X/46,XY. Very few true hermaphrodites are 46,XY.107 The amount of functional testicular tissue determines how the internal reproductive tract will differentiate.
Congenital Abnormalities of the Ovaries
Congenital anomalies of the ovaries include agenesis, supernumerary ovaries, and gonadal dysgenesis. These anomalies are all quite rare. Ovarian agenesis or dysgenesis is most often associated with a 46,XY karyotype and sexual infantilism, although cases involving 46,XX karyotype have been reported.108 Indeed, Gorgojo and colleagues recently reported a 46,XX female with gonadal agenesis and Mayer-Rokitansky-Küster-Hauser syndrome.109
Supernumerary ovaries occur when ovarian tissue has been separated during development from the normally located ovary.110 Such ovaries have been found in the omentum or retroperitoneum, and their location corresponds to the migration path of the primordial germ cells. Only 15 cases have been reported, and in 5 of those (33%) cases, primary neoplasms were identified arising in the supernumerary tissue.110,111 This condition is distinguished from accessory ovaries. In accessory ovaries, the additional ovarian tissue is connected to the normally positioned ovary. It has been estimated that accessory ovaries occur in about 1 in 93,000 patients.110
Malformation Syndromes Associated with Genital Ambiguity
Denys-Drash syndrome is associated with XY gonadal dysgenesis and mutations of the WT1 gene. In addition to dysgenetic testis and hypoplastic müllerian duct derivatives, these patients have focal or diffuse mesangial sclerosis of the kidneys. Approximately 75% will develop Wilms’ tumor in the first decade of life.112
Camphomelic dysplasia is a lethal bony dysplasia associated with a male to female sex reversal in two-thirds of affected males. Approximately 70% of these patients present with ambiguous genitalia. Camphomelic dysplasia is inherited as an autosomal dominant trait, and the condition is associated with a heterozygous loss of function of the SOX9 coding region.113
Smith-Lemli-Opitz syndrome type I is characterized by microcephaly, mental retardation, ptosis, micrognathia, polydactyly, and micropenis. Approximately 75% of XY patients will have abnormalities of the external genitalia that vary from hypospadias to complete failure of masculinization. This syndrome is caused by a deficiency of 7-dehydrocholesterol reductase, an enzyme required in the biosynthesis of cholesterol from acetate. It is inherited as an autosomal recessive disorder.113
Antley-Bixler syndrome is a rare disorder characterized by craniosynostosis, multiple joint contractures, including radiohumeral synostosis, and ambiguous genitalia. It is associated with abnormal steroid metabolism resulting from a deficiency of lanosteral 14-α-demethylase required for cholesterol synthesis and is inherited as an autosomal dominant trait.114
Another malformation syndrome associated with ambiguous genitalia is the Meckel-Gruber syndrome. This syndrome is inherited in an autosomal recessive manner and is characterized by polycystic kidneys, occipital encephalocele, polydactyly, cleft palate, and eye abnormalities. The gene responsible for this condition has not been identified.115
Finally, two associations that have ambiguous genitalia as one component include the CHARGE and VATER associations. The CHARGE association is characterized by coloboma, heart disease, atresia choanae, retarded growth, genital hypoplasia, and ear defects. The cause of this heterogeneous condition is unknown, although a familial inheritance pattern is occasionally reported. The VATER association is characterized by vertebral, anal, tracheoesophageal, and renal anomalies. Inheritance type is unknown. These patients present with ambiguous genitalia as part of other cloacal abnormalities.116
Diagnostic Evaluation of Ambiguous Genitalia in the Infant
Evaluation of an infant with ambiguous genitalia requires a thorough maternal history, physical examination, a karyotype, laboratory testing and, in some cases, radiographic studies. The results of these preliminary studies should determine whether the infant is a female or male pseudohermaphrodite, has a gonadal differentiation disorder, or has a malformation syndrome. Subsequent studies should be individualized to the patient and will depend on the initial findings.
COUNSELING
The diagnosis of any congenital anomaly involving the reproductive system will have profound implications on the patient’s sexual development and future fertility. In many cases, particularly those involving gender ambiguity, the abnormalities are often detected and diagnosed in infancy. When any deviation from normal involves the external genitalia in the neonate, gender assignments should be delayed until necessary studies have been completed. In the interim, the parents are to be reassured that they have a “healthy baby,” but that the external genitalia have not completed development. They should be informed that additional testing should clarify the sex of rearing. Most clinicians recommend referring to the infant as the “baby” until gender assignment can be rendered. Determining the sex of rearing must be made with the parents, taking into account cultural and religious beliefs, as well as fertility issues. Once diagnosed, effective communication with family members is critical. In addition to providing details to the family on the infant’s condition, the physician should also supply information regarding support groups.117
Gender assignment should be rendered as soon as test results are completed. Once assignment is made, steps should be made to reinforce the gender identity. Aside from patients with 5-ARD, reversing gender identity after the first 2 years of life will have serious consequences. The physician should make every effort—including surgery, hormonal manipulation, and psychological support—to ensure that the infant’s genital structures conform to the patient’s own sexual identity.117 This task is sometimes difficult in infancy because one can only speculate on the newborn’s gender identity. Indeed, there has been a movement in recent years to address the problems of surgery timing in these cases. This problem is of particular concern in patients with CAH. A number of interest groups believe that genitoplasty should not be conducted without the informed consent of the involved patient. Requirements for such consent would require rethinking when such surgeries should be performed and would obviate surgery being performed in infancy. If this approach is adopted, problems still exist as to the sex of rearing, because the first 2 years of life appear to be critical in reinforcing gender identity.
The final aspect of counseling occurs when the chromosomal sex does not match the phenotypic sex, as seen in androgen insensitivity. The amount of information that should be disclosed is a matter of controversy. Some authors believe that although there are instances in which an accurate pathophysiologic explanation is appropriate, it may not be necessary to present detailed genetic and anatomic information. Some experts believe that the discussion should be tailored to the perceived need and psychological makeup of the patient.117 Others believe that this information should be disclosed in the course of genetic counseling, rather than being inadvertently divulged in later encounters in the patient’s medical record. These authorities recommend using gonads instead of testes or ovaries as well as providing an explanation as to why their removal is necessary. That explanation should include the nonfunctioning nature of the gonad as well as the potential that these gonads may develop into a neoplasia.
CONCLUSIONS
1 Amesse L, Pfaff-Amesse T. Surgical Management of Müllerian Duct Anomalies. Available at www.emedicine.com. Accessed 2006.
2 Moore KL, Persaud TVN. The urogenital system: The development of the genital system. In The Developing Human: Clinically Oriented Embryology, 6th ed., Philadelphia: WB Saunders; 1998:303.
3 Gidwani G, Falcone T, editors. Congenital Anomalies of the Female Genital Tract: Diagnosis and Management. Philadelphia: Lippincott Williams & Wilkins, 1999.
4 Lindenman E, Shepard MK, Pescovitz OH. Müllerian agenesis: An update. Obstet Gynecol. 1997;90:307-312.
5 Shulman LP, Elias S. Developmental abnormalities of the female reproductive tract: Pathogenesis and nosology. Adolesc Pediatr Gynecol. 1988;1:230-237.
6 Lee DM, Osathanondh R, Yeh J. Localization of Bcl-2 in the human fetal müllerian tract. Fertil Steril. 1998;70:135-140.
7 Persaud TN. Embryology of the female genital tract and gonads. In: Copeland IJ, Jarrell J, McGregor Y, editors. Textbook of Gynecology. Philadelphia: WB Saunders; 1993:321.
8 Acien P. Embryological observations on the female genital tract. Hum Reprod. 1992;7:437-445.
9 MacLaughlin DT, Donahue PK. Sex determination and differentiation. NEJM. 2004;350:367-378.
10 Voutilainen R. Differentiation of the fetal gonad. Hormone Res. 1992;38:66-71.
11 Rapport R. Disorders of the gonads. In: Behrman RE, Jenson HB, editors. Nelson Textbook of Pediatrics. 15th ed. Philadelphia: WB Saunders; 2005:1921-1946.
12 Steinmetz GP. Formation of artificial vagina. West J Surg. 1940;48:169-173.
13 Stampe Sorenson S. Estimated prevalence of müllerian anomalies. Acta Obstet Gynecol Scand. 1988;67:441-445.
14 Byne J, Nussbaum-Blask A, Taylor SW, et al. Prevalence of müllerian duct anomalies detected at ultrasound. Am J Med Gen. 2000;94:9-12.
15 Golan A, Langer R, Bukovsky I, Caspi E. Congenital anomalies of the müllerian system. Fertil Steril. 1989;51:747-755.
16 Verp MS, Simpson JL, Elias S, et al. Heritable aspects of uterine anomalies. I. Three familial aggregates with müllerian fusion anomalies. Fertil Steril. 1983;40:80-85.
17 Carson SA, Simpson JL, Malinak LR, et al. Heritable aspects of uterine anomalies. II. Genetic analysis of müllerian aplasia. Fertil Steril. 1983;40:86-90.
18 American Fertility Society. The American Fertility Society classification of adnexal adhesions, distal tubal occlusion, tubal occlusion secondary to tubal ligation, tubal pregnancies, müllerian anomalies and intrauterine adhesions. Fertil Steril. 1988;49:944-955.
19 Evans RN, Poland ML, Boving RE. Vaginal malformations. Am J Obstet Gynecol. 1981;141:910-920.
20 Rock JA. Surgery for anomalies of the müllerian ducts. In: Tompson JD, Rock JA, editors. Te Linde’s Operative Gynecology. 7th ed. Philadelphia: JB Lippincott; 1992:603.
21 Murray J, Gambrell RD. Complete and partial vaginal agenesis. J Reprod Med. 1979;22:101-105.
22 Griffin JE, Edwards C, Madden JD, et al. Congenital absence of the vagina. The Mayer-Rokitansky-Küster-Hauser Syndrome. Ann Intern Med. 1976;85:224-236.
23 Turunen A, Unnerus CE. Spinal changes in patients with congenital aplasia of the vagina. Acta Obstet Gynecol Scand. 1967;46:99-106.
24 Willemson WN. Combination of Mayer-Rokitansky-Küster and Klippel-Feil syndrome: A case report and review of the literature. Eur J Obstet Gynecol Reprod Biol. 1982;13:229-235.
25 Duncan PA, Shapiro LR, Stangel JJ, et al. The MURCS association: Müllerian duct aplasia, renal aplasia and cervicothoracic somite dysplasia. J Pediatr. 1979;95:399-402.
26 Lyons Jones K. MURCS association: Müllerian duct, renal and cervical vertebral defects. In: Lyons Jones K, editor. Smith’s Recognizable Patterns of Human Malformation. 5th ed. Philadelphia: WB Saunders; 1997:666.
27 Cramer DW, Ravnikar VA, Craighill M, et al. Müllerian aplasia associated with maternal deficiency of galactose-phosphate uridyl transferase. Fertil Steril. 1987;47:930-934.
28 Chen Y, Mattison DR, Feigenbaum L, et al. Reduction in oocyte number following prenatal exposure to a diet high in galactose. Science. 1981;214:1145-1147.
29 Aughton DJ. Müllerian duct abnormalities and galactosaemia heterozygosity: Report of a family. Clin Dysmorphol. 1993;2:55-61.
30 Amesse LS, Yen F, Weisskopf B, Hertweck SP. Vaginal uterine agenesis associated with amastia in a phenotypic female with a de novo 46,XX,t(8;13)(q22.1;q32.1) translocation. Clin Genet. 1999;55:493-495.
31 Petrozza JC, Gray MR, Davis AJ, Reindollar RH. Congenital absence of the uterus and vagina is not commonly transmitted as a dominant genetic trait: Outcomes of surrogate pregnancies. Fertil Steril. 1997;67:387.
32 Mitchell DG. Benign disease of the uterus and ovaries. Applications of magnetic resonance imaging. Radiol Clin North Am. 1992;30:777-787.
33 Mitchell DG, Outwater EK. Benign gynecologic disease: Applications of magnetic resonance imaging. Top Magnet Reson Imaging. 1995;7:26-43.
34 Doyle MB. Magnetic resonance imaging in müllerian fusion defects. J Reprod Med. 1992;37:33-38.
35 Heinonen P. Unicornuate uterus and rudimentary horn. Fertil Steril. 1997;68:224-230.
36 Rock JA, Schlaff WD. The obstetric consequences of uterovaginal anomalies. Fertil Steril. 1985;43:681-692.
37 Rolen AC, Choquette AJ, Semmens JP. Rudimentary uterine horn: Obstetric and gynecologic implications. Obstet Gynecol. 1966;27:806-813.
38 Raga F, Bauset C, Remohi J, et al. Reproductive impact of congenital müllerian anomalies. Hum Reprod. 1997;12:2277-2281.
39 Fedele L, Zamberletti D, Vercellini P, et al. Reproductive performance of women with unicornuate uterus. Fertil Steril. 1987;47:416-419.
40 Andrews MC, Jones HWJr. Impaired reproductive performances of the unicornuate uterus: Intrauterine growth retardation, infertility and recurrent abortion in five cases. Am J Obstet Gynecol. 1982;144:173-176.
41 Michalas SP. Outcome of pregnancy in women with uterine malformations. Int J Gynecol Obstet. 1991;35:215-219.
42 Forstner R, Hricak H. Congenital malformations of uterus and vagina. Radiol. 1994;34:397-404.
43 Tridenti G, Bruni V, Ghirardini G, et al. Double uterus with a blind hemivagina and ipsilateral renal agenesis: Clinical variants in three adolescent women: Case report and literature review. Adolesc Pediatr Gynecol. 1995;8:201-207.
44 Propst AM, Hill JA3rd. Anatomic factors associated with recurrent pregnancy loss. Semin Reprod Med. 2000;18:341-350.
45 Constantian HM. Ureteral ectopia, hydrocolpos, and uterus didelphys. JAMA. 1966;197:54-56.
46 Gilliland B, Dyck F. Uterus didelphys associated with unilateral imperforate vagina. Obstet Gynecol. 1976;48:5S-8S.
47 Erdogan E, Okan G, Daragenli O. Uterus didelphys with unilateral obstructed hemivagina and renal agenesis on the same side. Acta Obstet Gynecol Scand. 1992;71:76-77.
48 Heinonen PK, Saarikoski S, Pystynen P. Reproductive performance of women with uterine anomalies. An evaluation of 182 cases. Acta Obstet Gynecol Scand. 1982;61:157-162.
49 Carrington BM, Hricak H, Nuruddin RN, et al. Müllerian duct anomalies: MR imaging evaluation. Radiology. 1990;176:715-720.
50 Pellerito JS, McCarthy SM, Doyle MB, et al. Diagnosis of uterine anomalies: Hysterosalpingography. Radiology. 1992;183:795-800.
51 Strassmann EO. Fertility and unification of the double uterus. Fertil Steril. 1966;17:165-176.
52 Candiani GB, Fedele L, Zamberletti D, et al. Endometrial patterns in malformed uteri. Acta Eur Fertil. 1983;14:311-318.
53 Malik E, Berg C, Sterzik K, Stoz F, Rossmanith WG. Reproductive outcome of 32 patients with primary or secondary infertility and uterine pathology. Arch Gynecol Obstet. 2000;264:24-26.
54 Musich JR, Behrman SJ. Obstetric outcome before and after metroplasty in women with uterine anomalies. Obstet Gynecol. 1978;52:63-66.
55 Bennett MJ, Berry JV. Preterm labour and congenital malformations of the uterus. Ultrasound Med Biol. 1979;5:83-85.
56 Homer HA, Li TC, Cooke ID. The septate uterus: A review of management and reproductive outcomes. Fertil Steril. 2000;73:1-14.
57 Wu MH, Hsu CC, Huang KE. Detection of congenital müllerian duct anomalies using three-dimensional ultrasound. J Clin Ultrasound. 1997;25:487-492.
58 Maneschi F, Zupi E, Marconi D, et al. Hysteroscopically detected asymptomatic müllerian anomalies. Prevalence and reproductive implications. J Reprod Med. 1995;40:684-688.
59 Zanetti E, Ferrari LR, Rossi G. Classification and radiologic features of uterine malformations: Hysterosalpingographic study. Br J Radiol. 1978;51:161-170.
60 Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. NEJM. 1971;284:878-881.
61 Kaufman RH, Adam E, Binder GL, Gerthoffer E. Upper genital tract changes and pregnancy outcome in offspring exposed in utero to diethylstilbestrol. Am J Obstet Gynecol. 1980;137:299-308.
62 Goldberg JM, Falcone T. Effect of diethylstilbestrol on reproductive function. Fertil Steril. 1999;72:1-7.
63 Berger MJ, Goldstein DP. Impaired reproductive performance in DES-exposed women. Obstet Gynecol. 1980;55:25-27.
64 Suidan F, Azoury RS. The transverse vaginal septum: A clinicopathologic evaluation. Obstet Gynecol. 1979;54:278-283.
65 McKusick VA, Bauer L, Kopp CE, Scott RB. Hydrometrocolpos as a simply inherited malformation. JAMA. 1964;189:813-816.
66 Rock JA, Zacur HA, Dlugi AM, et al. Pregnancy success following surgical correction of imperforate hymen and complete transverse vaginal septum. Obstet Gynecol. 1982;59:448-451.
67 Banerjee AK, Clarke O, MacDonald LM. Sonographic detection of neonatal hydrometrocolpos. Br J Radiol. 1992;65:268-271.
68 Jones HW. Reconstruction of congenital uterovaginal anomalies. In: Rock JA, Murphy AA, Jones HW, editors. Female Reproductive Surgery. Baltimore: Williams & Wilkins; 1992:246.
69 Lim YH, Ng SP, Jamil MA. Imperforate hymen: Report of an unusual familial occurrence. J Obstet Gynaecol Res. 2003;29:399-401.
70 Laufer MR, Goldstein DP, Hendren H. Structural abnormalities of the female reproductive tract. In: Emans SJ, Laufer MR, Goldstein D, editors. Pediatric and Adolescent Gynecology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2005:334-416.
71 Winter JS, Kohn G, Mellman WJ, Wagner S. A familial syndrome of renal, genital and middle ear anomalies. J Pediatr. 1968;72:88-93.
72 Turner G. A second family with renal, genital and middle ear anomalies. J Pediatr. 1970;76:641.
73 Scanlan KA, Pozniak MA, Fagerholm M, Shapiro S. Value of transperineal sonography in the assessment of vaginal atresia. Am J Roentgenol. 1990;154:545-548.
74 Shatzkes DR, Haller JO, Velcek FT. Imaging of uterovaginal anomalies in the pediatric patient. Urol Radiol. 1991;13:58-66.
75 Miller WL. Congenital adrenal hyperplasias. Endocrinol Metab Clin North Am. 1991;20:721-749.
76 Pang S. Congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 1997;26:853-891.
77 Ritzen EM, Lajic S, Wedell A. How can molecular biology contribute to the management of congenital adrenal hyperplasia? Hormone Res. 2000;53(Suppl):34-37.
78 Cutler GBJr, Laue L. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. NEJM. 1990;323:1806-1813.
79 Migeon CJ, Donohoue PA. Congenital adrenal hyperplasia caused by 21-hydroxylase deficiency. Its molecular basis and its remaining therapeutic problems. Endocrinol Metab Clin North Am. 1991;20:277-296.
80 Azziz R, Dewailly D, Owerbach D. Clinical review 56: Nonclassic adrenal hyperplasia: Current Concepts. J Clin Endocrinol Metab. 1994;78:810-815.
81 Mercado AB, Wilson RC, Cheng KC, et al. Prenatal treatment and diagnosis of congenital adrenal hyperplasia owing to steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1995;80:2014-2020.
82 White PC, Speiser PW. Steriod 11-β-hydroxylase deficiency and related disorders. Endocrinol Metab Clin North Am. 1994;23:325-339.
83 New MI. Genetic disorders of adrenal hormone synthesis. Hormone Res. 1992;37(Suppl 3):22-33.
84 Zachman M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11-β hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab. 1983;56:222-229.
85 Marui S, Castro M, Latronico AC, et al. Mutations in the type II 3βa-hydroxysteroid dehydrogenase (HSD3B2) gene can cause premature pubarche in girls. Clin Endocrinol (Oxf). 2000;52:67-75.
86 Moisan AM, Ricketts ML, Tardy V, et al. New insight into the molecular basis of 3 β-hydroxysteroid dehydrogenase deficiency: Identification of eight mutations in the HSD3B2 gene: 11 patients from seven new families and comparison of the functional properties of 25 mutant enzymes. J Clin Endocrinol Metab. 1999;84:4410-4425.
87 Morel Y, Rey R, Teinturier C, et al. Aetiological diagnosis of male sex ambiguity: A collaborative study. Eur J Pediatr. 2002;161:49-59.
88 Merke DP, Cutier GB. New approaches to the treatment of congenital adrenal hypoplasia. JAMA. 1997;277:1073-1076.
89 Mendonca BB, Inacio M, Costa EM, et al. Male pseudohermaphroditism due to steroid 5 αa-reductase 2 deficiency. Diagnosis, psychological evaluation, and management. Medicine (Baltimore). 1996;75:64-76.
90 Kaufman M, Pinsky L, Bowin A, Au MW. Familial external genital ambiguity due to a transformation defect of androgen–receptor complexes that is expressed with 5 α- dihydrotestosterone and the synthetic androgen methyltrienolone. Am J Med Genet. 1984;18:493-507.
91 Imperato-McGinley J, Ip NY, Gautier T, et al. DNA linkage analysis and studies of the androgen receptor gene in a large kindred with complete androgen insensitivity. Am J Med Genet. 1990;36:104-108.
92 Gottlieb B, Beitel LK, Lumbroso R, et al. Update of the androgen receptor gene mutations database. Hum Mutat. 1999;14:103-114.
93 Quigley CA, DeBellis A, Marschke KB, et al. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr Rev. 1995;16:271-321.
94 Themmen AP, Verhoef-Post M. LH receptor defects. Semin Reprod Med. 2002;20:199-204.
95 Laue L, Wu SM, Kudo M, et al. Compound heterozygous mutations of the luteinizing hormone receptor gene in Leydig cell hypoplasia. Mol Endocrinol. 1996;10:987-997.
96 Martens JW, Lumbroso S, Verhoef-Post M, et al. Mutant luteinizing hormone receptors in a compound heterozygous patient with complete Leydig cell hypoplasia: Abnormal processing causes signaling deficiency. J Clin Endocrinol Metab. 2002;87:2506-2513.
97 Miller WL. Disorders of androgen biosynthesis. Semin Reprod Med. 2002;20:205-216.
98 Lin D, Sugawara T, Strauss JF3rd, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 1995;267:1828-1831.
99 Kater CE, Biglieri EG. Disorders of steroid 17 α-hydroxylase deficiency. Endocrinol Metab Clin North Am. 1994;23:341-357.
100 Forest MG, Lecornu M, de Peretti E. Familial male pseudohermaphroditism due to 17-20-desmolase deficiency. I. In vivo endocrine studies. J Clin Endocrinol Metab. 1980;50:826-833.
101 Andersson S, Geissler WM, Wu L, et al. Molecular genetics and pathophysiology of 17-β-hydroxysteroid dehydrogenase 3 deficiency. J Clin Endocrinol Metab. 1996;81:130-136.
102 Asthana S, Deo SV, Shukla NK, et al. Persistent müllerian duct syndrome presenting with bilateral intra-abdominal gonadal tumours and obstructive uropathy. Clin Oncol (R Coll Radiol). 2001;13:304-306.
103 Stratakis CA, Rennert OM. Turner’s syndrome: Molecular and cytogenics, dysmorphology, endocrine and other clinical manifestations and their management. Endocrinol. 1994;4:442-453.
104 Ogato T, Matsuo N. Turner’s syndrome and female sex chromosome aberrations: Deduction of the principal factors involved in the development of clinical features. Hum Genet. 1995;95:607-629.
105 Ester J, Pfaff-Amesse T, Gruber J, Amesse LS. Secondary amenorrhea: An unusual twist. J Pediatr Adolesc Gynecol. 2005;18:47-52.
106 Uehara S, Funato T, Yaegashi N, et al. SRY mutation and tumor formation on the gonads of XY pure gonadal dysgenesis patients. Cancer Genet Cytogenet. 1999;113:78-84.
107 Mendez JP, Ulloa-Aguirre A, Kofman-Alfaro S, et al. Mixed gonadal dysgenesis: Clinical, cytogenetic, endocrinological and histopathological findings in 16 patients. Am J Med Genet. 1993;46:263-267.
108 Queipo G, Zenteno JC, Pena R, et al. Molecular analysis in true hermaphroditism: Demonstration of low-level hidden mosaicism for Y-derived sequences in 46,XX cases. Hum Genet. 2002;111:278-283.
109 Gorgojo JJ, Almodover F, Lopez E, Donnay S. Gonadal agenesis 46,XX associated with the atypical form of Rokitansky syndrome. Fertil Steril. 2002;77:185-187.
110 Wharton LR. Two cases of supernumerary ovary and one of accessory ovary with an analysis of previously reported cases. Am J Obstet Gynecol. 1959;78:1101-1109.
111 Mercer LJ, Toub DB, Cibils LA. Tumors originating in supernumerary ovaries. A report of two cases. J Reprod Med. 1987;32:932-934.
112 Coppes MJ, Huff V, Pelletier J. Denys-Drash syndrome: Relating a clinical disorder to genetic alterations in the tumor suppressor gene WT1.. J Pediatr. 1993;123:673-678.
113 Aarskog D. Syndromes and genital dysmorpology. Hormone Res. 1992;38(Suppl 2):82-85.
114 Antley RM, Bixler D. X-trapezoidocephaly, midfacial hypoplasia and cartilage abnormalities with multiple synostoses and skeletal fractures. Birth Defects. 1975;11:397-401.
115 Kompanje EJ. Features described and illustrated in 1684 suggesting Meckel-Gruber syndrome. Pediatr Dev Pathol. 2003;6:595-598.
116 Warburg M. Update of sporadic microphthalmos and coloboma. Noninherited anomalies. Ophthalmic Paediatr Genet. 1992;13:111-122.
117 Meyer-Bahlburg HF, Migeon CJ, Berkovitz GD, et al. Attitudes of adult 46,XY intersex persons to clinical management policies. J Urol. 2004;171:1615-1619.