[level-membership-for-pediatrics-category]Chapter 132
Congenital Anomalies of Bone
Regardless of origin, abnormal limb development tends to fall into patterns that can be recognized clinically and radiographically (Fig. 132-1). Most classification systems of congenital malformations are based on osseous structures, but it is well recognized that anomalous conditions of surrounding soft tissues are certain to be present. Although one deficiency is usually dominant, dysplasia often involves the entire limb.
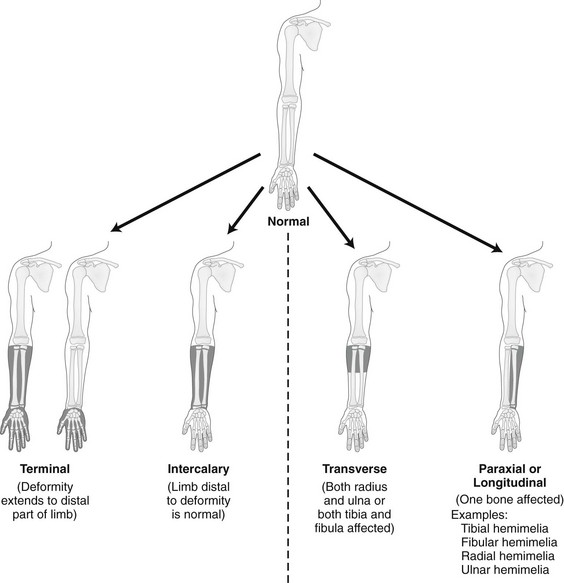
Figure 132-1 Skeletal deformities of the limbs.
The shaded, blackened areas indicate deficient parts.
Congenital deficiencies are more common than acquired amputations in children. Frantz and O’Rahilly developed a system of classification that is still commonly used to evaluate congenital anomalies of the extremities.1 Each malformation is defined by the part that is deficient. For example, the fibula is deficient in fibular hemimelia. The abnormality is considered terminal if the deformity extends to the distal aspect of the extremity; it is intercalary if the limb distal to the deformity is normal. For example, in fibular hemimelia, if the foot is abnormal, the deficiency is terminal, and if the foot is normal, it is intercalary. Likewise, a defect may be classified as longitudinal (paraxial) or transverse. The malformation is paraxial if only the fibular side is affected. If both the tibia and the fibula are affected, the deformity is considered transverse.
The cause of reduction malformations is known in only a small number of cases. A few abnormalities are heritable, but most are sporadic. Environmental factors are implicated infrequently. Medications such as thalidomide induced malformations that were reduction deformities, ranging from severe amelias to mild muscular hypoplasia.2 Most malformations were on the radial or tibial side, and the upper limbs were affected more often than were the lower extremities. Infrequently, excess deformities such as polydactyly were observed.
Extremity Deficiencies
Proximal Femoral Focal Deficiency
Etiology: Proximal femoral focal deficiency (PFFD) refers to abnormalities that range from mild shortening and hypoplasia of the femur to severe deficiency of the bone and dysplasia of the acetabulum. The defect is thought to be a result of altered proliferation and maturation of the chondrocytes of the proximal femoral physis in utero, which in turn results in underdevelopment of the ipsilateral acetabulum. A child with PFFD usually presents in infancy with a short extremity and an unstable hip.3 A well-formed acetabulum implies the presence of a femoral head, which might be cartilaginous and therefore is not apparent radiographically early in life. Most cases are sporadic; however, the combination of bilateral femoral deficiencies and abnormal facies, known as the femoral hypoplasia–unusual facies syndrome, is thought to be an autosomal-dominant disorder.
PFFD should be distinguished from developmental dysplasia of the hip, infantile coxa vara, and congenitally short femur. In the setting of developmental dysplasia of the hip, proximal femoral neck and head hypoplasia may be present. The subtrochanteric region of the femur is normal and distal congenital anomalies are absent, and the deformities at the level of the acetabulum are more severe than the deformities at the level of the femoral head and neck. Persons with infantile coxa vara have a normal subtrochanteric region and distal bones, and the femur is normal length, accounting for bowing. Unlike PFFD, infantile coxa vara is discovered after weight bearing occurs.4 A congenitally short femur may be unilateral or bilateral and often is associated with reduction defects elsewhere in the same limb. Most cases are sporadic, but several external factors have been implicated in more complex deformities. Known causes include drugs (such as thalidomide), trauma, irradiation, infection, and focal ischemia.5 Unlike patients with PFFD, patients with a congenitally short femur have a stable hip.
Imaging: The most commonly used classification for PFFD was proposed by Aitken (Fig. 132-2).6 The least severe type is class A, which refers to an adequate or only mildly dysplastic proximal femur and acetabulum. The femoral head is present but is separated from the shortened distal femoral segment. With age, a fibrous connection between the head and the distal femoral segment ossifies—usually, not completely. A subtrochanteric varus deformity invariably is present. Class C deformities have a residual acetabulum present (Fig. 132-3). In the most severe form, class D, most of the femur and the ipsilateral acetabulum are absent.
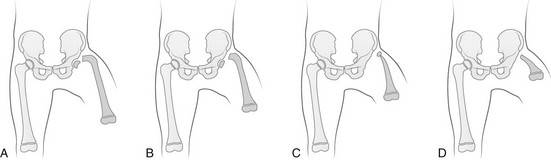
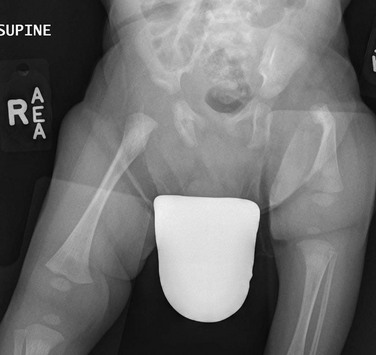
Figure 132-3 Aiken type 3 proximal femoral focal deficiency of the left hip in a 4-week-old boy.
The femur is substantially short and the left acetabulum is severely dysplastic. Mild developmental dysplasia of the right acetabulum is present.
PFFD is associated with other ipsilateral deformities, including fibular hemimelia (in more than 50% of affected children), shortening of the tibia, equinovalgus deformity of the foot (more often than equinovarus deformity), and deficiency of the lateral rays of the foot. PFFD is bilateral in 15% of affected children.7
Sonography can be used to delineate radiographically inapparent structures at an early age. If a cartilaginous femoral head and its connection to the femoral shaft can be shown, the stability of the hip joint is likely greater than is implied from radiographs. Similar to sonography, magnetic resonance imaging (MRI) is useful for defining the anatomy of the hip joint and associated deformities of the limb,8 allowing for appropriate early classification of deformity. Although the acetabulum might appear developed, even the least severe forms of PFFD are associated with acetabular deficiency. In contrast to the predominantly anterior deficiency of developmental acetabular dysplasia, the acetabulum in PFFD usually has an insufficient posterior wall. Most of the muscles about the affected hip are hypoplastic when compared with the contralateral normal side, with the exception of the sartorius muscle, which often is hypertrophied. This situation results in the characteristic flexion deformity of the hip and knee. MRI also defines the anatomy of soft tissues that can guide surgical exposure and preparation of the limb stump for fitting of a prosthesis.
Treatment and Follow-up: Therapy for children with PFFD is based on the severity of the deficiency and projection of growth and final limb length discrepancy at maturity. Objectives for treatment are to maximize the length of the extremity, to promote stability of the hip, knee, and ankle, and to optimize anatomic alignment. Mild deformities may not require surgery. Stabilization of the upper femoral defect is controversial and usually is indicated if a deformity is progressive. Children with severe deformities benefit from amputation of the foot and fusion of the knee. Rotationplasty of the tibia, in which the foot is rotated 180 degrees (so that the toes face posteriorly and the ankle now serves as a knee joint), allows the ankle and foot to control a distal prosthesis.9 Intact sensory feedback from the foot provides proprioceptive control of the knee. Thus it is imperative to assess the morphology of the proximal femur and acetabulum, the cartilaginous epiphyses of the knee, and the supporting soft tissue structures. For example, instability of the knee can result from absence of the anterior cruciate ligament. In the rare case of bilateral PFFD, amputation is contraindicated and therapy is based on extension prostheses that enhance the child’s height.
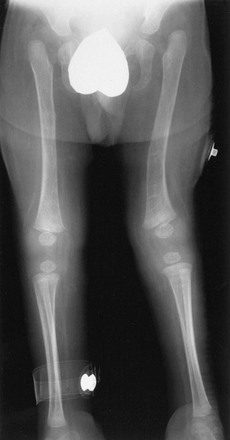
Fibular Hemimelia
Etiology: Fibular hemimelia is the most common hemimelia and the most common congenital anomaly of the fibula. It also is referred to as congenital short tibia with absent fibula syndrome. Unilateral absence is more frequent than bilateral deformity. A band of strong connective tissue may replace all or most of the fibula.10 Fibular hemimelia is associated with a short, bowed tibia, absence of lateral rays of the foot, tarsal abnormalities (particularly coalitions), and femoral shortening or deficiency in 15% of patients. Caskey and Lester11 reported a clubfoot deformity in 16% of patients with fibular hemimelia. Other associations include small, subluxed, or dislocated patellae, hypoplastic femoral condyles, and ligamentous deficiencies of the knee.
Imaging: Fibular hemimelia ranges from mild deficiency of the proximal end of the bone to complete absence accompanied by multiple malformations of neighboring structures (Fig. 132-4). MRI can document associated abnormalities, particularly if surgery is contemplated, including associated ligamentous deficiencies of the knee.
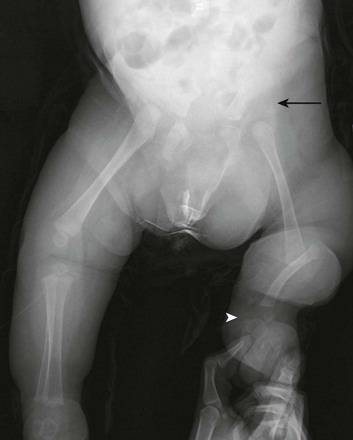
Treatment and Follow-up: The talipes equinovalgus deformity of the foot and severe shortening of the limb associated with fibular hemimelia result in poor function of the limb. Children with extensive abnormalities involving the fibula, ipsilateral tibia, femur, and foot benefit the most from early amputation of the foot and often the more proximal structures.12 If the deformity is less severe, lengthening of the affected side, realignment of the tibiotalar articulation, and epiphysiodesis of the contralateral limb often are undertaken. Attempts at lengthening a severely dysplastic limb often are unsatisfactory.
Tibial Hemimelia
Etiology: Most cases of tibial hemimelia are sporadic, although reported forms (particularly bilateral deformities) display an autosomal-dominant inheritance pattern. Jones et al.13 classified the spectrum of tibial dysplasia into four groups. In type I, which is the most severe form, the tibia is not recognized radiographically at birth. Lack of ossification of the distal femoral epiphysis implies that no proximal tibia is present. The least severe form, type IV, includes congenital tibial diastasis. In children with this form, the tibia is short and diverges from the fibula at the ankle. The talus is displaced proximally. No normal distal tibial articulation surface is present.
Imaging: Radiography of tibial hemimelia (Fig. 132-5) ranges from absence of a portion of the distal tibia to complete absence with multiple malformations of neighboring structures. MRI can document associated abnormalities, including associated muscular and ligamentous deficiencies, which is important if surgery is contemplated. MRI may identify a cartilaginous tibial remnant that may not be identified readily on radiography.
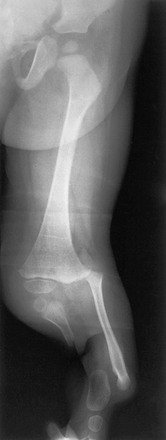
Treatment and Follow-up: Treatment of tibial hemimelia varies with the severity of the deformity. A knee disarticulation is recommended for complete tibial absence. Brown pioneered centralization of the fibula with a Syme amputation of the foot in children with complete tibial hemimelia.14 This approach produces adequate results in the rare cases in which the quadriceps muscles are well developed. When the quadriceps mechanism is abnormal, the patella often is absent. MRI can be used to assess deficiencies preoperatively, allowing the clinician to discern who will function better with primary knee disarticulation (most patients) rather than knee reconstruction.
Radial Deficiency (Radial Dysplasia, Radial Clubhand)
Etiology: Conditions associated with radial deficiency are listed in Box 132-1.15 Usually the entire limb is involved to some degree, which often results in joint dysfunction of the shoulder, elbow, wrist, carpus, and small joints of the hand. Associated muscular deficiencies also are proportionate to the degree of skeletal abnormality. Neurovascular abnormalities include an absent radial artery and superficial radial nerve.
Whether radial deficiency is isolated or is associated with a syndrome, the primary malformation is likely the result of a vascular abnormality in the embryo that occurred before differentiation of mesenchyme into muscle and bone.16 Most cases are sporadic, but some autosomal-dominant and autosomal-recessive inheritance patterns are reported. Other cases are likely the result of various environmental insults from viruses, chemicals, radiation, and drugs during limb bud development.
Imaging: Congenital radial deficiency ranges from hypoplasia of the thumb to various degrees of radial hypoplasia. Complete absence is the most common longitudinal deficiency. This deformity usually is accompanied by radial and volar deviation of the hand, which results in part from unopposed pull by the flexor carpi radialis and brachioradialis muscles.
In most cases of radial aplasia, the forearm is bowed to the radial side and the distal ulna is prominent (Fig. 132-6). The forearm is short (usually approximately two thirds the length of the normal contralateral side) and remains proportionately so throughout growth. Often, only the capitate, hamate, and triquetral bones and the metacarpals and phalanges of the four ulnar rays are present and normal. The trapezium, scaphoid, and first ray often are deformed or absent. If a remnant of the radius is present proximally, it usually is fused to the ulna, which is curved, shortened, and thickened. Bilateral deficiency occurs in approximately 50% of affected children.15 The degree of deformity of the hand is related to the severity of deficiency of the forearm.
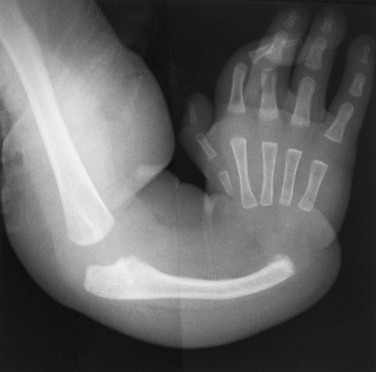
Treatment and Follow-up: Treatment of a longitudinal radial deficiency begins with serial casting and splinting intended to improve radial deviation by stretching the soft tissues. This treatment often is followed by surgical centralization of the hand over the ulna to improve function and appearance.17 Thumb reconstruction and pollicization of the index finger often are indicated.
Ulnar Deficiency (Ulnar Dysplasia, Ulnar Clubhand)
Etiology: Deficiency that occurs along the ulnar or postaxial border of the upper extremity is known as ulnar deficiency or ulnar clubhand. It is a relatively uncommon disorder that occurs much less frequently than radial deficiency (radial clubhand). Approximately 48% of cases have anomalies of the contralateral limb.18 Most cases are sporadic, but associations with many other syndromes have been reported, most commonly Brachmann-de Lange syndrome.
Imaging: In persons with ulnar deficiency, coexisting abnormalities almost always are present in the carpal, metacarpal, and phalangeal rays (e-Fig. 132-7). The pisiform is always absent, and the hamate frequently is not detected. Syndactyly, carpal fusion, and radiohumeral fusion are frequent. The forearm is shortened and bowed, with a concavity to the ulnar side. The hand is deviated in an ulnar direction, and elbow abnormalities are common. Hypoplasia of the shoulder girdle and the upper arm can coexist with ulnar deficiencies. Anomalies of the contralateral upper limb and the lower limbs (such as PFFD) have been reported.
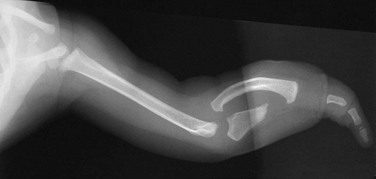
Treatment and Follow-up: Treatment for ulnar deficiency includes early correction of ulnar deviation of the hand, which is achieved with serial casting. Surgical treatment is reserved for cases with significant limitation of function.19 Forearm instability and associated hand deformities must be addressed.
Generalized Anomalies
Etiology: Constriction bands are thought to result from intrauterine rupture of the amnion and subsequent mechanical constriction of fetal limbs.20 Various body parts become entangled in the amnion as it separates from the chorion. In some instances, adjacent structures are pulled together by the bands and ultimately become fused, producing a soft tissue syndactyly. Bony syndactyly is very rare. The earlier the amnion ruptures, the more severe are the malformations. Limb abnormalities range from slight soft tissue grooves to transverse intrauterine amputations. Acrosyndactyly, craniofacial and visceral anomalies, and fetal death are part of the spectrum of congenital constriction band syndrome. Sporadic congenital constriction band syndrome is estimated to occur at an incidence of 1 : 1200 to 1 : 15,000 births.
An alternative explanation is the intrinsic theory, which suggests that an inherent defect leads to bands during limb embryogenesis.20
Imaging: Radiography may underestimate the degree of soft tissue compromise, and associated anomalous vascular anatomy may not be recognized. Typical conventional radiographic findings include a tethered appearance of fingers with a characteristic tight distal waist with soft tissue syndactyly (Fig. 132-8). MRI can be used to evaluate the depth of the constriction band, the degree of resultant lymphedema, and the integrity of the involved musculature.
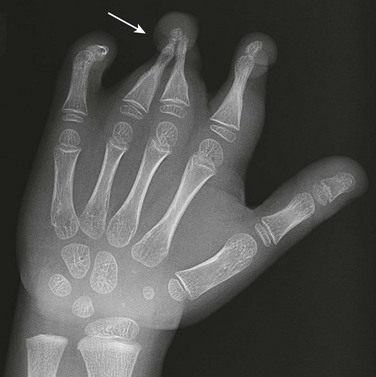
Treatment and Follow-up: Surgical treatment usually is cosmetic, although it may be needed to relieve massive edema distal to a constriction band or to improve neurovascular function. Intervention for patients with acrosyndactyly may result in partial correction of the deformity and improved longitudinal growth.
Arthrogryposis
Etiology: Arthrogryposis is a descriptive term used loosely to refer to conditions that result in congenital contractures involving two or more joints, such as those that limit intrauterine movement.21 The earlier in gestation that movement is limited and the longer the restriction lasts, the more severe will be the contractures at birth. Normal in utero motion is necessary for normal development of joints.
Arthrogryposis may result from abnormal muscles (such as myopathies), abnormal nerve function or innervation, abnormal connective tissues, or mechanical limitation of movement (such as oligohydramnios or multiple fetuses). Other possible causes include genetic defects, mutagenic agents, mitotic abnormalities, and toxic chemicals or drugs (Box 132-2).
Imaging: Contractures that are present at birth often are symmetric and most commonly affect distal parts of limbs. Clubfoot deformities often are present. The lower extremities are usually involved, but more than half of patients have upper limb abnormalities as well. Rigid joints and hypotonia contribute to frequent perinatal fractures. Soft tissue dimples may be seen over the affected joints, and muscle mass is decreased in affected limbs (e-Fig. 132-9). Affected muscles are partially or completely replaced by fatty or fibrous tissue. In many children, a scoliosis eventually develops. MRI can be used to assess joint integrity when surgery to maximize joint function is planned.
Treatment and Follow-up: The long-term prognosis for most children with arthrogryposis is good. However, children with associated central nervous system abnormalities usually have a limited life span. A neuromuscular workup is recommended for all children with arthrogryposis. Early therapy, including manipulation to increase range of motion, as well as surgery, is advocated for best results.22
Fusion Deformities
Etiology: Tarsal coalition is a congenital failure of segmentation of the primitive mesenchyme that results in the union of two or more tarsal bones. A preadolescent or adolescent child with pain in the midfoot and hindfoot that is associated with lack of motion in the subtalar region should be suspected of having a tarsal coalition. Prevalence in the U.S. population is approximately 1% or less. At least 50% of patients with coalitions have bilateral findings.23 An autosomal-dominant inheritance pattern with a high level of penetrance is reported, although the coalition need not involve the same joint. Although it typically presents during the second decade of life, tarsal coalition may not manifest until adulthood.
Tarsal coalition is defined on the basis of anatomic location and completeness of ossification. A complete ossific bar forms a synostosis, a cartilaginous bar forms a synchondrosis, and a fibrous union is termed a syndesmosis. The most common fusions are talocalcaneal and calcaneonavicular. The middle facet is the most common location for a talocalcaneal coalition. Talocalcaneal coalitions often may extend posteriorly, with involvement of the junction of the middle and posterior facet. Talonavicular (e-Fig. 132-10) and calcaneocuboid coalitions are much less common. Synostoses can occur between other bones of the foot but frequently are associated with other limb abnormalities, such as fibular hemimelia or short femur, or disorders such as Apert syndrome.
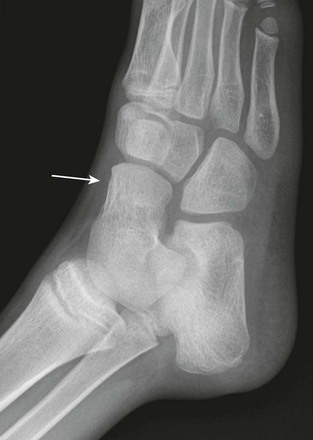
e-Figure 132-10 Talonavicular coalition (arrow).
Imaging: A 45-degree lateral oblique radiograph of the foot directly shows a calcaneonavicular coalition. The secondary radiographic features of calcaneonavicular coalitions visible on a lateral radiograph include pes planus, anteater sign (Fig. 132-11), and talar beak sign (e-Fig. 132-12). A reverse anteater sign is seen on the anteroposterior (AP) view of the foot and is defined as diminished AP height of the lateral portion of the navicular (compared with the medial navicular), with the navicular extending far lateral with respect to the talar head.
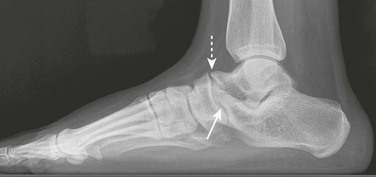
Figure 132-11 The anteater sign in a child with a calcaneonavicular coalition (arrow) and talar beak (dotted arrow).
Elongation of the anterior calcaneus resembling the nose of an anteater is present.
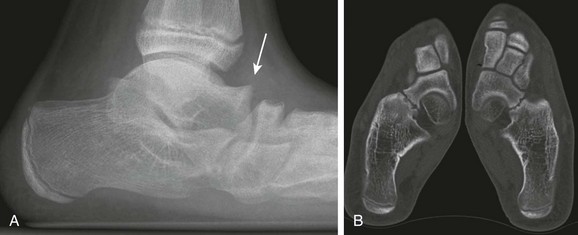
e-Figure 132-12 Talonavicular coalition.
A, A lateral radiograph demonstrates a talar beak (arrow) at the distal end of a shortened talar neck in a child with a talonavicular coalition. B, Computed tomography reconstructions demonstrate bilateral talonavicular coalitions.
Radiographic features of talocalcaneal coalitions include the C sign (Fig. 132-13, A), talar beak, an ovoid, enlarged sustentaculum tali (see Fig. 132-13, A), and broadened lateral process of the talus.
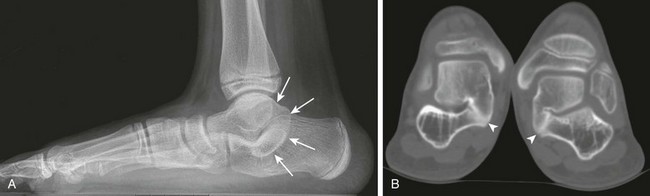
Figure 132-13 Talocalcaneal coalition.
A, A lateral radiograph demonstrates the C sign (arrows), ovoid, elongated sustentaculum tali, and pes planus. B, Computed tomography with coronal reformats in a different patient demonstrates bilateral middle facet subtalar coalitions (arrowheads).
The talar beak associated with tarsal coalitions is a dorsal extension of the talar head (see e-Fig. 132-12). A talar beak should not be confused with a talar ridge. The talar ridge represents bony proliferation related to the insertion of the anterior tibiotalar capsule and is located along the more proximal dorsal talar neck rather than at the distal talar head.24
A ball-and-socket articulation of the distal tibia and talus is an uncommon association with tarsal coalition. However, this configuration at the ankle joint is nonspecific and may be associated with congenital long bone deficiencies, as well as tarsal coalitions.25
Computed tomography (CT) and MRI help to define the nature and cross-sectional area of a fusion. CT is more cost-effective if the clinical suspicion for coalition is high (Fig. 132-13, B). If other causes of pain and limited hindfoot mobility are considered, then MRI may be indicated. MRI may directly show bony, cartilaginous, and fibrous unions. Bone marrow edema often is noted adjacent to the fused articulation (Fig. 132-14).
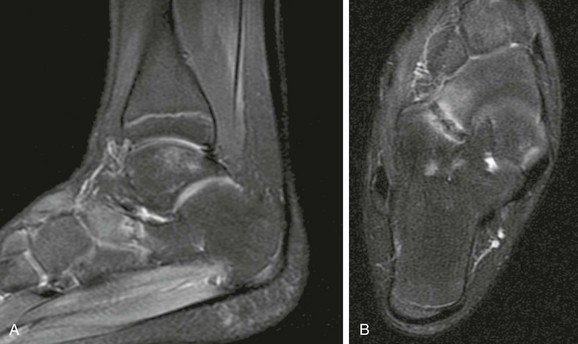
Treatment and Follow-up: Casting and nonsteroidal antiinflammatory medications often are the initial treatment for symptomatic children. Steroid injections and physical therapy also may be used. If symptoms are not relieved and no degenerative changes have occurred, then surgical resection of the abnormal tarsal bridging often is attempted.23 Regrowth of a calcaneonavicular coalition can be halted by interposition of the extensor digitorum brevis tendon. Fat frequently is interposed after resection of a subtalar coalition. Subtalar fusion or triple arthrodesis may be indicated in refractory cases.
Carpal Fusion
Isolated fusion of the carpal bones is a normal variant that is seen in approximately 0.1% of the white population and approximately 1.6% of the African American population (e-Fig. 132-15).26 Carpal fusion is seen most frequently between the lunate and triquetrum. Fusion between bones of the proximal and distal carpal rows usually is associated with a syndrome. Fusions also may be acquired, as in juvenile idiopathic arthritis or after trauma.
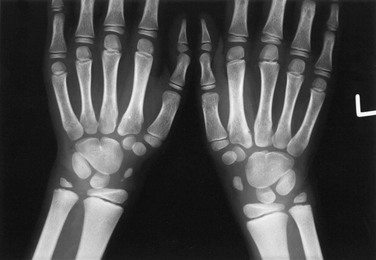
e-Figure 132-15 Fusion of the hamate and capitate bones bilaterally.
Syndactyly
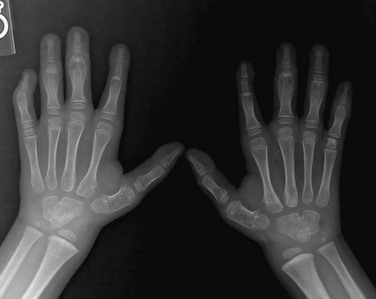
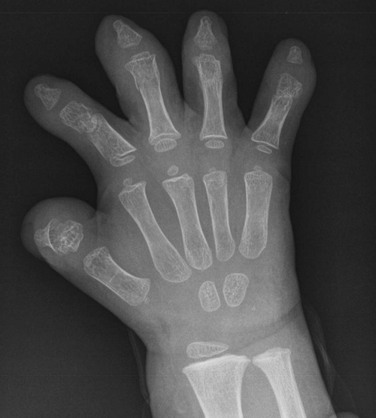
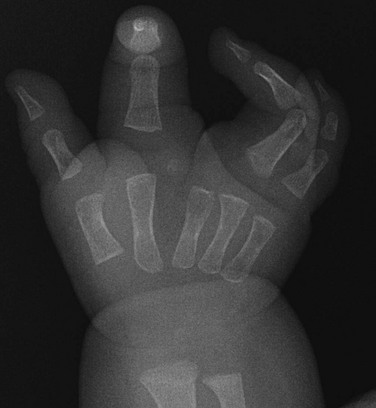
Fusion between adjacent digits resulting from intrauterine failure to separate is termed syndactyly (also referred to as “webbed toes” or “webbed fingers”). These fusions may involve only soft tissue (simple syndactyly), or they also may involve bone (complex syndactyly). If the entire length of the digit is involved, syndactyly is termed complete, and if only a partial length is bridged, it is incomplete. Fusion may be unilateral or bilateral and usually involves the second, third, and fourth digits. Syndactyly may be isolated or may be associated with congenital disorders such as Poland, Apert (Fig. 132-16), and Carpenter syndromes. Poland syndrome includes symbrachydactyly (brachydactyly and syndactyly) and ipsilateral chest wall anomalies, most commonly pectoralis muscle hypoplasia or aplasia. Apert syndrome includes syndactyly, craniosynostosis, and facial bone anomalies. Carpenter syndrome includes polysyndactyly, craniosynostosis, and facial bone anomalies. Syndactyly is thought to have an autosomal-dominant inheritance pattern.27 Surgery is performed early in life to improve function and appearance when the hand is involved. Surgery is rarely indicated for the foot.
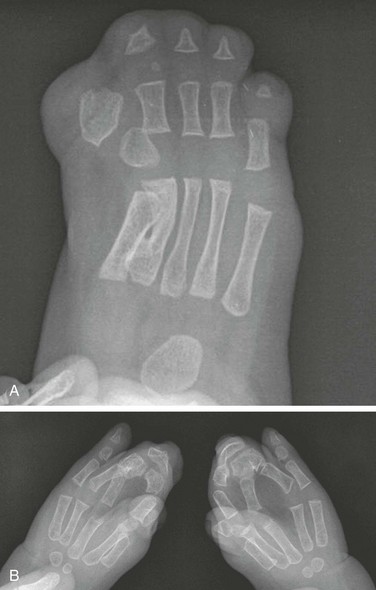
Figure 132-16 A 1-year-old child with Apert syndrome.
A, A frontal view of the foot shows syndactyly of the digits. Hypoplasia of many bones of the foot is seen with absent middle phalanges. The first and second metatarsals are partially fused. B, A frontal radiograph of both hands of the same child shows syndactyly, synostosis, and symphalangism bilaterally.
Symphalangism
Symphalangism is an uncommon autosomal-dominant disorder of fusion of the interphalangeal joints of the hands and feet (Fig. 132-17). Although proximal interphalangeal joint fusion is more common, fusion also can be more distal.28 The anomaly is usually bilateral, and the little finger and little toe are affected most often. Fusion may not be recognized radiographically until later in childhood. Despite the radiographic appearance of the digits, seldom is disability or loss of function of the hand reported. Symphalangism may be associated with other skeletal abnormalities, various digit deformities (e.g., brachydactyly, clinodactyly, and camptodactyly), radioulnar fusion, hip dislocation, tarsal coalition, and spinal anomalies.
Radioulnar Synostosis
Etiology: Congenital radioulnar synostosis anomaly is due to failure of longitudinal segmentation between the radius and the ulna.29 In utero, cartilaginous anlagen of the humerus, radius, and ulna are connected. Normal separation progresses from the distal end of the forearm to the proximal end. An insult to segmentation during early in utero development can lead to bony or fibrous synostosis. Radioulnar synostosis is associated with disorders such as Apert and Carpenter syndromes, as well as with other upper extremity abnormalities such as polydactyly, syndactyly, and carpal coalition.
Imaging: Osseous coalition is best seen on AP radiographs (Fig. 132-18). Radioulnar synostosis may lead to secondary radial head dysplasia (see Fig. 132-18, B) because of the lack of articulation between the radial head and capitellum needed for normal development. Posterior dislocation of the radial head also may be seen (e-Fig. 132-19).
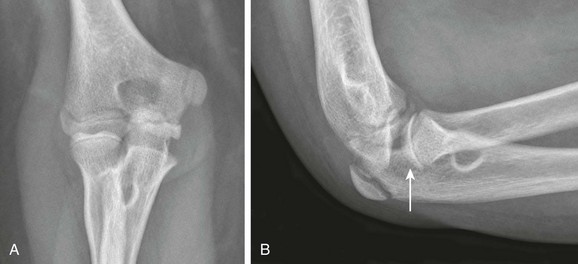
Figure 132-18 Radioulnar synostosis in a 12-year-old boy.
A, Anteroposterior radiographs demonstrate osseous synostosis. B, A lateral radiograph demonstrates a convex-appearing radial head (arrow) related to secondary radial head dysplasia.
Treatment and Follow-up: Surgical treatment usually is reserved for patients with a severe pronation deformity or symptomatic radial head subluxation, particularly when it involves the dominant hand. A both bone extraarticular derotational osteotomy through the forearm to improve supination may be performed.30 Resection of the proximal synostosis is complex and may not lead to improved forearm and hand function.
Other Congenital Malformations
Clinodactyly is a curvature of the finger in a radial or ulnar direction (in the mediolateral plane) away from the axis of joint flexion and extension. Although it may involve any digit, clinodactyly most often refers to radial curvature of the distal interphalangeal joint of the fifth digit (Fig. 132-20).31 Clinodactyly is associated with a short middle phalanx and usually is bilateral. It may be seen in the normal population as a sporadic variant but also can be inherited in an autosomal-dominant pattern. It has been described in numerous syndromes (most commonly Down syndrome), in bone dysplasias, after trauma, and in many miscellaneous conditions. The curvature deformity may occur in the setting of a normal phalanx, as a result of physeal growth disturbance, or related to a longitudinal epiphyseal bracket deformity. Treatment is undertaken for cosmesis or for excessive scissoring of the digits.
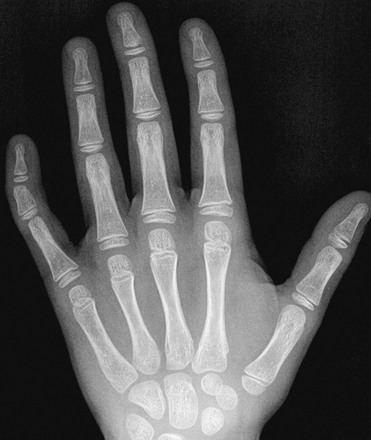
Camptodactyly
Camptodactyly refers to a congenital or acquired flexion contracture of the finger. It usually is located at the proximal interphalangeal joint of the fifth finger but also can involve the second through fourth digits (Fig. 132-21).31 Occasionally the distal interphalangeal joints may be affected. The cause remains unknown but may relate to abnormal insertion of the lumbrical muscles or the flexor digitorum superficialis tendon. Similar to clinodactyly, camptodactyly may be sporadic or may show an autosomal-dominant inheritance pattern. Occasionally it is associated with a chromosomal abnormality, a bony dysplasia, or another syndrome. Treatment includes bracing or, infrequently, surgical release.
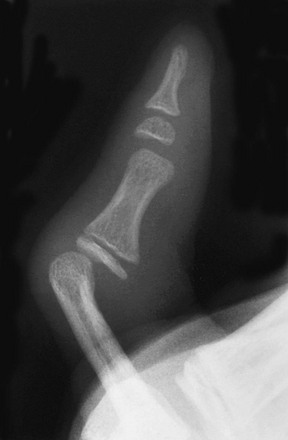
Kirner Deformity
Palmar bending of the distal phalanx of the fifth digit is termed Kirner deformity.31 It usually is an isolated abnormality that is bilateral and symmetric. The long axis of the distal phalanx is bent toward the palm, and the epiphysis is normally oriented (Fig. 132-22). The physis is widened. With skeletal maturation, the physis fuses on an angle. The distal phalanx exhibits a permanent palmar curvature. Similar to the other digit deformities, Kirner deformity can be a sporadic or an autosomal-dominant inherited anomaly.
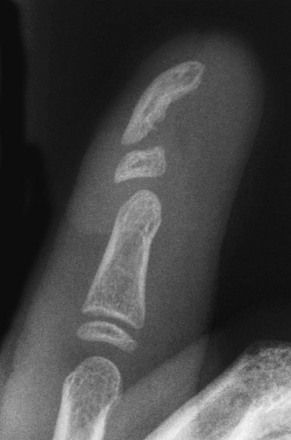
Brachydactyly
Brachydactyly is a descriptive term that describes hypoplasia or complete aplasia of phalanges of the hands or feet (e-Fig. 132-23). Morphologically the affected bone may appear normal but just abnormally shortened compared with the other digits. The most commonly affected level is the middle phalanges. Brachydactyly may coexist with other processes such as longitudinal epiphyseal bracket and symphalangism. Brachydactyly may be sporadic or may be inherited in an autosomal-dominant fashion. Brachydactyly also may be seen with syndromes such as Apert and Poland syndromes.
Longitudinal Epiphyseal Bracket (Delta Phalanx)
Etiology: Longitudinal epiphyseal bracket is a congenital deformity that affects the short tubular bones that normally develop a proximal epiphyseal ossification center (e.g., phalanges, first metacarpal, and first metatarsal). The deformity may be sporadic or may occur in association with conditions such as Rubinstein-Taybi syndrome and fibrodysplasia ossificans progressiva. The cause is likely incomplete development of the primary ossification center of the bone during early fetal growth. The growth disturbance relates to the longitudinally oriented cartilaginous bracket, which causes growth to follow a “C-shaped” curve.32
Imaging: The involved bone is trapezoid or triangular in shape (Fig. 132-24). The diaphyseal–metaphyseal osseous unit is bracketed along the longitudinal side by a physis and an epiphysis. The physis is arclike (see Fig. 132-24, B), extending from the medial proximal surface along the longitudinal margin of the bone to the distal medial surface, and is similar in configuration to a bracket. The thumb usually is affected. Other anomalies of the digits frequently are associated with the longitudinal epiphyseal bracket.
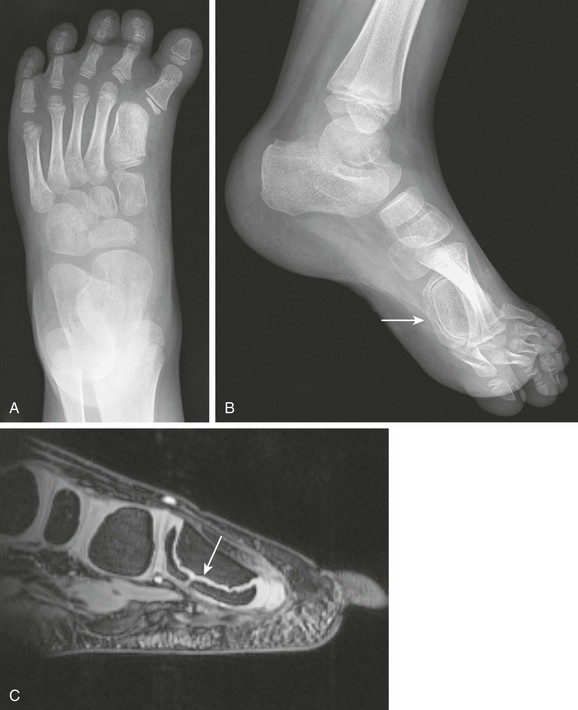
Figure 132-24 A longitudinal epiphyseal bracket.
A, A frontal radiograph of the foot of a 4-year-old girl. The first metatarsal is short and widened and has a shorter medial than lateral length, forming a trapezoidal shape. B, A lateral radiograph of the same child shows the C-shaped epiphysis and physis of the first metatarsal (arrow). C, A sagittal gradient echo image shows a high-signal intensity C-shaped physis (arrow) and adjacent epiphysis.
Split Hand-Foot Malformations
Etiology: Split hand-foot malformation (SHFM) refers to the congenital splitting of the hands or feet into two halves. SHFM is attributed to hypoplasia or aplasia of the phalanges, metacarpals, or metatarsals of one or more fingers or toes. The deformity also is referred to as ectrodactyly, cleft hand or cleft foot, or lobster or crab claw deformity.
SHFM may be isolated or associated with congenital constriction band syndrome, or, most commonly, it may occur as a component of the ectrodactyly–ectodermal dysplasia–cleft lip/palate syndrome.34 More than 75 syndromes have been associated with SHFM. Ectrodactyly–ectodermal dysplasia–cleft lip/palate syndrome usually has an autosomal-dominant inheritance pattern with incomplete penetrance and variable expression, although autosomal-recessive forms have been described. SHFM has been detected on prenatal ultrasonography, which can guide counseling and reconstructive efforts.
Imaging: SHFM is highly variable in presentation. It can range from mild digital changes to the most severe monodactyly with only the fifth digit remaining. Two distinctive clinical forms have been described—typical and atypical forms. The typical form has an autosomal-dominant inheritance pattern and involves the lack of phalanges and metacarpals, resulting in a deep V-shaped cleft with the two halves resembling a lobster claw (Fig. 132-25). The much less frequent atypical form is sporadic and results in a much wider cleft, which forms a U-shaped central defect. The atypical form rarely involves the feet. Vascular disruption has been implicated as the cause. In most forms the digits frequently curve in toward the cleft, and a reduction in the number of digits may occur. Syndactyly or synostosis often is present.
Congenital Radial Head Dislocation
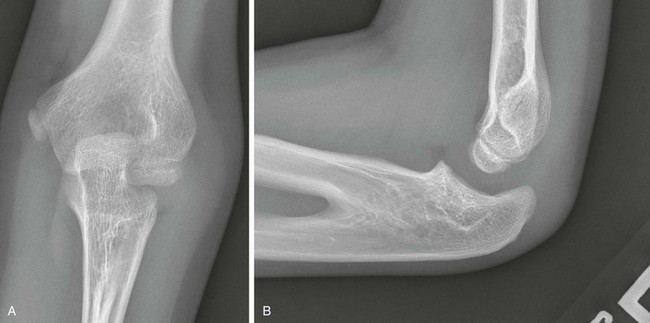
Etiology: Congenital radial head dislocation is the most commonly identified congenital abnormality of the elbow. More than half of cases are associated with other conditions, such as anomalies of the lower extremities, scoliosis, and various syndromes (e.g., Klippel-Feil).35 Whether it is an isolated abnormality or is associated with other conditions, congenital radial head dislocation is believed to have an autosomal-dominant inheritance pattern. The deformity can be unilateral or bilateral.
Imaging: Congenital radial head dislocation is associated with a small, underdeveloped forearm, a flattened and hypoplastic capitellum, and a short ulna. The proximal ulnar diaphysis often may have a posterior apex bowing (Fig. 132-26, B). The radial head is dysplastic with an elongated, thinned, and convex articular surface instead of the normal concave shape. The radial head is most commonly dislocated posteriorly (see Fig. 132-26). The degree of radial head dysplasia is correlated with the severity of dislocation. Radiographs should be carefully evaluated for concomitant radioulnar synostosis.
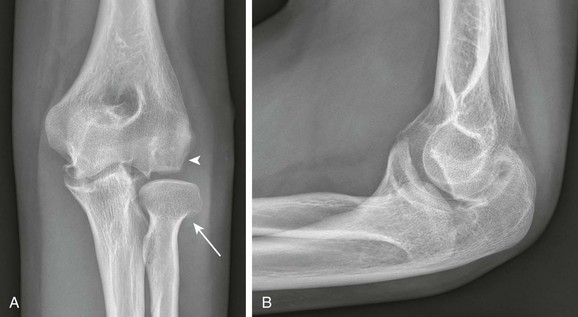
Figure 132-26 Congenital radial head dislocation in a 12-year-old girl.
Anteroposterior (A) and lateral (B) radiographs show a dysplastic convex-shaped radial head (arrow) with posterior dislocation as well as posterior bowing of the proximal ulnar diaphysis. Note the presence of a capitellar osteochondral lesion (arrowhead) and medial compartmental osteoarthritis, which likely is related to abnormal elbow biomechanics.
Treatment and Follow-up: Asymptomatic dislocations are not treated. If the child has pain, the radial head can be excised. However, excision of the radial head frequently is accompanied by pain from abnormal mechanics at the wrist. Recent work suggests that open reduction of the proximal radius at an early age may be beneficial.
Tibial Bowing Deformities
Etiology: Posteromedial congenital bowing affects the tibia, fibula, and soft tissues of the lower extremity. The origin and pathogenesis remain uncertain and may be related to abnormal early embryologic development rather than to abnormal fetal positioning or intrauterine fracture. Fetal vascular insufficiency also may play a role. After examination of a fetus with congenital bowing, De Maio and associates suggested that compressive events associated with amnion rupture may be responsible.36 As the child grows, tibial bowing and shortening often partially resolve.
Posteromedial congenital bowing should be differentiated from congenital tibial dysplasia (CTD). Other terms for CTD include anterolateral tibia bowing or congenital pseudoarthrosis of the tibia. It is a rare anomaly but does occur in a small number of patients with neurofibromatosis type 1 (NF1). Approximately 20% to 50% of patients with CTD do not have NF1.37
Imaging: With posteromedial congenital bowing, the tibia and the fibula show marked posteromedial bowing at the middle or distal one third of the shaft (Fig. 132-27). Rarely is the bowing lateral. At birth, the foot is held in a calcaneovalgus (dorsiflexed) position.
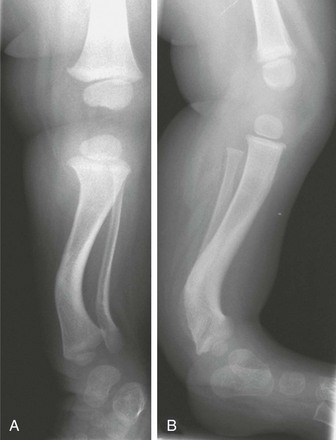
Figure 132-27 A 1-year-old girl with congenital posteromedial bowing.
A, On the frontal radiograph, medial bowing is seen at the junction of the middle and distal one third of the tibia. The fibula is similarly bowed. B, The posterior component of the bowing is seen on the lateral radiograph.
Radiographic changes seen with CTD are variable and include anterolateral bowing of the tibia, fracture, pseudarthrosis, hourglasslike constriction of the midshaft of the tibia, cystic changes usually at the junction of the upper and middle thirds of the tibia, sclerosis that narrows the medullary canal, and, infrequently, involvement of the fibula alone (Fig. 132-28).
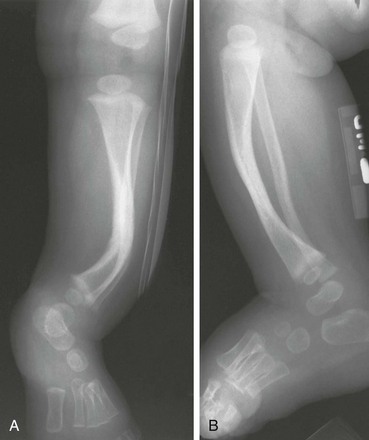
Treatment and Follow-up: Treatment for posteromedial congenital bowing of the tibia is conservative; for example, corrective casts or splints may be used to hold the foot and support the leg during growth and remodeling. If a severe persistent deformity continues beyond toddler age, a leg length discrepancy, usually on the order of 3 to 7 cm, may result that requires osteotomies and lengthening procedures. Occasionally, epiphysiodesis of the contralateral side may be needed.
References
1. Frantz, C, O’Rahilly, R. Congenital limb deficiencies. J Bone Joint Surg Am. 1961;43:1202–1224.
2. Lenz, W. Malformations caused by drugs in pregnancy. Am J Dis Child. 1966;112:99–106.
3. Schatz, SL, Kopts, SE. Proximal femoral focal deficiency. AJR Am J Roentgenol. 1978;131:289–295.
4. Amstutz, HC. Developmental (infantile) coxa vara—a distinct entity: report of two patients with previously normal roentgenograms. Clin Orthop Relat Res. 1970;72:242–247.
5. Hamanishi, C. Congenital short femur: clinical, genetic and epidemiological comparison of the naturally occurring condition with that caused by thalidomide. J Bone Joint Surg Br. 1980;62:307–320.
6. Proximal femoral focal deficiency—definition, classification, and management. In: Aitken G, ed. Proximal femoral focal deficiency: a congenital anomaly. Washington, DC: National Academy of Sciences, 1969.
7. Anton, CG, Applegate, KE, Kuivila, TE, et al. Proximal femoral focal deficiency (PFFD): more than an abnormal hip. Semin Musculoskelet Radiol. 1999;3:215–226.
8. Biko, DM, Davidson, R, Pena, A, et al. Proximal focal femoral deficiency: evaluation by MR imaging. Pediatr Radiol. 2012;42:50–56.
9. VanNes, C. Rotation-plasty for congenital defects of the femur: making use of the ankle of the shortened limb to control the knee joint of a prosthesis. J Bone Joint Surg Br. 1950;32:12–16.
10. Laor, T, Jaramillo, D, Hoffer, FA, et al. MR imaging in congenital lower limb deformities. Pediatr Radiol. 1996;26:381–387.
11. Caskey, PM, Lester, EL. Association of fibular hemimelia and clubfoot. J Pediatr Orthop. 2002;22:522–525.
12. McCarthy, JJ, Glancy, GL, Chang, FM, et al. Fibular hemimelia: comparison of outcome measurements after amputation and lengthening. J Bone Joint Surg Am. 2000;82-A(12):1732–1735.
13. Jones, D, Barnes, J, Lloyd-Roberts, GC. Congenital aplasia and dysplasia of the tibia with intact fibula: classification and management. J Bone Joint Surg Br. 1978;60:31–39.
14. Simmons, ED, Jr., Ginsburg, GM, Hall, JE. Brown’s procedure for congenital absence of the tibia revisited. J Pediatr Orthop. 1996;16:85–89.
15. Lourie, GM, Lins, RE. Radial longitudinal deficiency: a review and update. Hand Clin. 1998;14:85–99.
16. Van Allen, MI, Hoyme, HE, Jones, KL. Vascular pathogenesis of limb defects. I. Radial artery anatomy in radial aplasia. J Pediatr. 1982;101:832–838.
17. Bayne, LG, Klug, MS. Long-term review of the surgical treatment of radial deficiencies. J Hand Surg Am. 1987;12:169–179.
18. Swanson, AB, Tada, K, Yonenobu, K. Ulnar ray deficiency: its various manifestations. J Hand Surg Am. 1984;9:658–664.
19. Manske, PR, Goldfarb, CA. Congenital failure of formation of the upper limb. Hand Clin. 2009;25:157–170.
20. Kawamura, K, Chung, KC. Constriction band syndrome. Hand Clin. 2009;25:257–264.
21. Swinyard, CA, Bleck, EE. The etiology of arthrogryposis (multiple congenital contractures). Clin Orthop. 1985;194:15–29.
22. Shapiro, F, Specht, L. The diagnosis and orthopaedic treatment of childhood spinal muscular atrophy, peripheral neuropathy, Friedreich ataxia, and arthrogryposis. J Bone Joint Surg Am. 1993;75:1699–1714.
23. Zaw, H, Calder, JD. Tarsal coalitions. Foot Ankle Clin. 2010;15(2):349–364.
24. Crim, J. Imaging of tarsal coalition. Radiol Clin North Am. 2008;46:1017–1026.
25. Takakura, Y, Tanaka, Y, Kumai, T, et al. Development of the ball-and-socket ankle as assessed by radiography and arthrography: a long-term follow-up report. J Bone Joint Surg Br. 1999;81(6):1001–1004.
26. Poznanski, AK, Holt, JF. The carpals in congenital malformation syndromes. AJR Am J Roentgenol. 1971;112(3):443–459.
27. Tonkin, MA, Failure of differentiation part I: syndactyly. Hand Clin, 2009;25:171–193.
28. Letts, M, Davidson, D, Beaule, P, Symphalangism in children: case report and review of the literature. Clin Orthop Relat Res, 1999;366:178–185.
29. Elliot, AM, Kibria, L, Reed, MH. The developmental spectrum of proximal radioulnar synostosis. Skeletal Radiol. 2010;39:49–54.
30. Hung, NN. Derotational osteotomy of the proximal radius and the distal ulna for congenital radioulnar synostosis. J Child Orthop. 2008;2:481–489.
31. Poznanski, AK, Pratt, GB, Manson, G, et al. Clinodactyly, camptodactyly, Kirner’s deformity, and other crooked fingers. Radiology. 1969;93:573–582.
32. Mahboub, S, Davidson, R. MR imaging in longitudinal epiphyseal bracket in children. Pediatr Radiol. 1999;29:259–261.
33. Light, TR, Ogden, JA. The longitudinal epiphyseal bracket: implications for surgical correction. J Pediatr Orthop. 1981;1:299–305.
34. Arbues, J, Galindo, A, Puente, JM, et al. Typical isolated ectrodactyly of hands and feet: early antenatal diagnosis. J Matern Fetal Neonatal Med. 2005;17:299–301.
35. Sachar, K, Mih, AD. Congenital radial head dislocations. Hand Clin. 1998;14:39–47.
36. De Maio, F, Corsi, A, Roggini, M, et al. Congenital unilateral posteromedial bowing of the tibia and fibula: insights regarding pathogenesis from prenatal pathology: a case report. J Bone Joint Surg Am. 2005;87(7):1601–1605.
37. Stevenson, DA, Viskochil, DH, Schorry, EK, et al. The use of anterolateral bowing of the lower leg in the diagnostic criteria for neurofibromatosis type 1. Genet Med. 2007;9(7):409–412.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 132
Congenital Anomalies of Bone
Regardless of origin, abnormal limb development tends to fall into patterns that can be recognized clinically and radiographically (Fig. 132-1). Most classification systems of congenital malformations are based on osseous structures, but it is well recognized that anomalous conditions of surrounding soft tissues are certain to be present. Although one deficiency is usually dominant, dysplasia often involves the entire limb.
Figure 132-1 Skeletal deformities of the limbs.
The shaded, blackened areas indicate deficient parts.
Congenital deficiencies are more common than acquired amputations in children. Frantz and O’Rahilly developed a system of classification that is still commonly used to evaluate congenital anomalies of the extremities.1 Each malformation is defined by the part that is deficient. For example, the fibula is deficient in fibular hemimelia. The abnormality is considered terminal if the deformity extends to the distal aspect of the extremity; it is intercalary if the limb distal to the deformity is normal. For example, in fibular hemimelia, if the foot is abnormal, the deficiency is terminal, and if the foot is normal, it is intercalary. Likewise, a defect may be classified as longitudinal (paraxial) or transverse. The malformation is paraxial if only the fibular side is affected. If both the tibia and the fibula are affected, the deformity is considered transverse.
The cause of reduction malformations is known in only a small number of cases. A few abnormalities are heritable, but most are sporadic. Environmental factors are implicated infrequently. Medications such as thalidomide induced malformations that were reduction deformities, ranging from severe amelias to mild muscular hypoplasia.2 Most malformations were on the radial or tibial side, and the upper limbs were affected more often than were the lower extremities. Infrequently, excess deformities such as polydactyly were observed.
Extremity Deficiencies
Proximal Femoral Focal Deficiency
Etiology: Proximal femoral focal deficiency (PFFD) refers to abnormalities that range from mild shortening and hypoplasia of the femur to severe deficiency of the bone and dysplasia of the acetabulum. The defect is thought to be a result of altered proliferation and maturation of the chondrocytes of the proximal femoral physis in utero, which in turn results in underdevelopment of the ipsilateral acetabulum. A child with PFFD usually presents in infancy with a short extremity and an unstable hip.3 A well-formed acetabulum implies the presence of a femoral head, which might be cartilaginous and therefore is not apparent radiographically early in life. Most cases are sporadic; however, the combination of bilateral femoral deficiencies and abnormal facies, known as the femoral hypoplasia–unusual facies syndrome, is thought to be an autosomal-dominant disorder.
PFFD should be distinguished from developmental dysplasia of the hip, infantile coxa vara, and congenitally short femur. In the setting of developmental dysplasia of the hip, proximal femoral neck and head hypoplasia may be present. The subtrochanteric region of the femur is normal and distal congenital anomalies are absent, and the deformities at the level of the acetabulum are more severe than the deformities at the level of the femoral head and neck. Persons with infantile coxa vara have a normal subtrochanteric region and distal bones, and the femur is normal length, accounting for bowing. Unlike PFFD, infantile coxa vara is discovered after weight bearing occurs.4 A congenitally short femur may be unilateral or bilateral and often is associated with reduction defects elsewhere in the same limb. Most cases are sporadic, but several external factors have been implicated in more complex deformities. Known causes include drugs (such as thalidomide), trauma, irradiation, infection, and focal ischemia.5 Unlike patients with PFFD, patients with a congenitally short femur have a stable hip.
Imaging: The most commonly used classification for PFFD was proposed by Aitken (Fig. 132-2).6 The least severe type is class A, which refers to an adequate or only mildly dysplastic proximal femur and acetabulum. The femoral head is present but is separated from the shortened distal femoral segment. With age, a fibrous connection between the head and the distal femoral segment ossifies—usually, not completely. A subtrochanteric varus deformity invariably is present. Class C deformities have a residual acetabulum present (Fig. 132-3). In the most severe form, class D, most of the femur and the ipsilateral acetabulum are absent.
Figure 132-3 Aiken type 3 proximal femoral focal deficiency of the left hip in a 4-week-old boy.
The femur is substantially short and the left acetabulum is severely dysplastic. Mild developmental dysplasia of the right acetabulum is present.
PFFD is associated with other ipsilateral deformities, including fibular hemimelia (in more than 50% of affected children), shortening of the tibia, equinovalgus deformity of the foot (more often than equinovarus deformity), and deficiency of the lateral rays of the foot. PFFD is bilateral in 15% of affected children.7
Sonography can be used to delineate radiographically inapparent structures at an early age. If a cartilaginous femoral head and its connection to the femoral shaft can be shown, the stability of the hip joint is likely greater than is implied from radiographs. Similar to sonography, magnetic resonance imaging (MRI) is useful for defining the anatomy of the hip joint and associated deformities of the limb,8 allowing for appropriate early classification of deformity. Although the acetabulum might appear developed, even the least severe forms of PFFD are associated with acetabular deficiency. In contrast to the predominantly anterior deficiency of developmental acetabular dysplasia, the acetabulum in PFFD usually has an insufficient posterior wall. Most of the muscles about the affected hip are hypoplastic when compared with the contralateral normal side, with the exception of the sartorius muscle, which often is hypertrophied. This situation results in the characteristic flexion deformity of the hip and knee. MRI also defines the anatomy of soft tissues that can guide surgical exposure and preparation of the limb stump for fitting of a prosthesis.
Treatment and Follow-up: Therapy for children with PFFD is based on the severity of the deficiency and projection of growth and final limb length discrepancy at maturity. Objectives for treatment are to maximize the length of the extremity, to promote stability of the hip, knee, and ankle, and to optimize anatomic alignment. Mild deformities may not require surgery. Stabilization of the upper femoral defect is controversial and usually is indicated if a deformity is progressive. Children with severe deformities benefit from amputation of the foot and fusion of the knee. Rotationplasty of the tibia, in which the foot is rotated 180 degrees (so that the toes face posteriorly and the ankle now serves as a knee joint), allows the ankle and foot to control a distal prosthesis.9 Intact sensory feedback from the foot provides proprioceptive control of the knee. Thus it is imperative to assess the morphology of the proximal femur and acetabulum, the cartilaginous epiphyses of the knee, and the supporting soft tissue structures. For example, instability of the knee can result from absence of the anterior cruciate ligament. In the rare case of bilateral PFFD, amputation is contraindicated and therapy is based on extension prostheses that enhance the child’s height.
Fibular Hemimelia
Etiology: Fibular hemimelia is the most common hemimelia and the most common congenital anomaly of the fibula. It also is referred to as congenital short tibia with absent fibula syndrome. Unilateral absence is more frequent than bilateral deformity. A band of strong connective tissue may replace all or most of the fibula.10 Fibular hemimelia is associated with a short, bowed tibia, absence of lateral rays of the foot, tarsal abnormalities (particularly coalitions), and femoral shortening or deficiency in 15% of patients. Caskey and Lester11 reported a clubfoot deformity in 16% of patients with fibular hemimelia. Other associations include small, subluxed, or dislocated patellae, hypoplastic femoral condyles, and ligamentous deficiencies of the knee.
Imaging: Fibular hemimelia ranges from mild deficiency of the proximal end of the bone to complete absence accompanied by multiple malformations of neighboring structures (Fig. 132-4). MRI can document associated abnormalities, particularly if surgery is contemplated, including associated ligamentous deficiencies of the knee.
Treatment and Follow-up: The talipes equinovalgus deformity of the foot and severe shortening of the limb associated with fibular hemimelia result in poor function of the limb. Children with extensive abnormalities involving the fibula, ipsilateral tibia, femur, and foot benefit the most from early amputation of the foot and often the more proximal structures.12 If the deformity is less severe, lengthening of the affected side, realignment of the tibiotalar articulation, and epiphysiodesis of the contralateral limb often are undertaken. Attempts at lengthening a severely dysplastic limb often are unsatisfactory.
Tibial Hemimelia
Etiology: Most cases of tibial hemimelia are sporadic, although reported forms (particularly bilateral deformities) display an autosomal-dominant inheritance pattern. Jones et al.13 classified the spectrum of tibial dysplasia into four groups. In type I, which is the most severe form, the tibia is not recognized radiographically at birth. Lack of ossification of the distal femoral epiphysis implies that no proximal tibia is present. The least severe form, type IV, includes congenital tibial diastasis. In children with this form, the tibia is short and diverges from the fibula at the ankle. The talus is displaced proximally. No normal distal tibial articulation surface is present.
Imaging: Radiography of tibial hemimelia (Fig. 132-5) ranges from absence of a portion of the distal tibia to complete absence with multiple malformations of neighboring structures. MRI can document associated abnormalities, including associated muscular and ligamentous deficiencies, which is important if surgery is contemplated. MRI may identify a cartilaginous tibial remnant that may not be identified readily on radiography.
Treatment and Follow-up: Treatment of tibial hemimelia varies with the severity of the deformity. A knee disarticulation is recommended for complete tibial absence. Brown pioneered centralization of the fibula with a Syme amputation of the foot in children with complete tibial hemimelia.14 This approach produces adequate results in the rare cases in which the quadriceps muscles are well developed. When the quadriceps mechanism is abnormal, the patella often is absent. MRI can be used to assess deficiencies preoperatively, allowing the clinician to discern who will function better with primary knee disarticulation (most patients) rather than knee reconstruction.
Radial Deficiency (Radial Dysplasia, Radial Clubhand)
Etiology: Conditions associated with radial deficiency are listed in Box 132-1.15 Usually the entire limb is involved to some degree, which often results in joint dysfunction of the shoulder, elbow, wrist, carpus, and small joints of the hand. Associated muscular deficiencies also are proportionate to the degree of skeletal abnormality. Neurovascular abnormalities include an absent radial artery and superficial radial nerve.
Whether radial deficiency is isolated or is associated with a syndrome, the primary malformation is likely the result of a vascular abnormality in the embryo that occurred before differentiation of mesenchyme into muscle and bone.16 Most cases are sporadic, but some autosomal-dominant and autosomal-recessive inheritance patterns are reported. Other cases are likely the result of various environmental insults from viruses, chemicals, radiation, and drugs during limb bud development.
Imaging: Congenital radial deficiency ranges from hypoplasia of the thumb to various degrees of radial hypoplasia. Complete absence is the most common longitudinal deficiency. This deformity usually is accompanied by radial and volar deviation of the hand, which results in part from unopposed pull by the flexor carpi radialis and brachioradialis muscles.
In most cases of radial aplasia, the forearm is bowed to the radial side and the distal ulna is prominent (Fig. 132-6). The forearm is short (usually approximately two thirds the length of the normal contralateral side) and remains proportionately so throughout growth. Often, only the capitate, hamate, and triquetral bones and the metacarpals and phalanges of the four ulnar rays are present and normal. The trapezium, scaphoid, and first ray often are deformed or absent. If a remnant of the radius is present proximally, it usually is fused to the ulna, which is curved, shortened, and thickened. Bilateral deficiency occurs in approximately 50% of affected children.15 The degree of deformity of the hand is related to the severity of deficiency of the forearm.
Treatment and Follow-up: Treatment of a longitudinal radial deficiency begins with serial casting and splinting intended to improve radial deviation by stretching the soft tissues. This treatment often is followed by surgical centralization of the hand over the ulna to improve function and appearance.17 Thumb reconstruction and pollicization of the index finger often are indicated.
Ulnar Deficiency (Ulnar Dysplasia, Ulnar Clubhand)
Etiology: Deficiency that occurs along the ulnar or postaxial border of the upper extremity is known as ulnar deficiency or ulnar clubhand. It is a relatively uncommon disorder that occurs much less frequently than radial deficiency (radial clubhand). Approximately 48% of cases have anomalies of the contralateral limb.18 Most cases are sporadic, but associations with many other syndromes have been reported, most commonly Brachmann-de Lange syndrome.
Imaging: In persons with ulnar deficiency, coexisting abnormalities almost always are present in the carpal, metacarpal, and phalangeal rays (e-Fig. 132-7). The pisiform is always absent, and the hamate frequently is not detected. Syndactyly, carpal fusion, and radiohumeral fusion are frequent. The forearm is shortened and bowed, with a concavity to the ulnar side. The hand is deviated in an ulnar direction, and elbow abnormalities are common. Hypoplasia of the shoulder girdle and the upper arm can coexist with ulnar deficiencies. Anomalies of the contralateral upper limb and the lower limbs (such as PFFD) have been reported.
Treatment and Follow-up: Treatment for ulnar deficiency includes early correction of ulnar deviation of the hand, which is achieved with serial casting. Surgical treatment is reserved for cases with significant limitation of function.19 Forearm instability and associated hand deformities must be addressed.
Generalized Anomalies
Etiology: Constriction bands are thought to result from intrauterine rupture of the amnion and subsequent mechanical constriction of fetal limbs.20 Various body parts become entangled in the amnion as it separates from the chorion. In some instances, adjacent structures are pulled together by the bands and ultimately become fused, producing a soft tissue syndactyly. Bony syndactyly is very rare. The earlier the amnion ruptures, the more severe are the malformations. Limb abnormalities range from slight soft tissue grooves to transverse intrauterine amputations. Acrosyndactyly, craniofacial and visceral anomalies, and fetal death are part of the spectrum of congenital constriction band syndrome. Sporadic congenital constriction band syndrome is estimated to occur at an incidence of 1 : 1200 to 1 : 15,000 births.
An alternative explanation is the intrinsic theory, which suggests that an inherent defect leads to bands during limb embryogenesis.20
