Chapter 531 Congenital Anomalies and Dysgenesis of the Kidneys
Renal Agenesis
Renal agenesis, or absent kidney development, can occur secondary to a defect of the wolffian duct, ureteric bud, or metanephric blastema. Unilateral renal agenesis has an incidence of 1/450-1,000 births. Unilateral renal agenesis often is discovered during the course of an evaluation for other congenital anomalies (VATER syndrome; e.g., Chapter 311). Its incidence is increased in newborns with a single umbilical artery. In true agenesis, the ureter and the ipsilateral bladder hemitrigone are absent. The contralateral kidney undergoes compensatory hypertrophy, to some degree prenatally but primarily after birth. Approximately 15% of these children have contralateral vesicoureteral reflux, and most males have an ipsilateral absent vas deferens because the wolffian duct is absent. Because the wolffian and müllerian ducts are contiguous, müllerian abnormalities in girls also are common. The Mayer-Rokitansky-Kuster-Hauser syndrome is a group of associated findings that includes unilateral renal agenesis or ectopia, ipsilateral müllerian defects, and vaginal agenesis (Chapter 548).
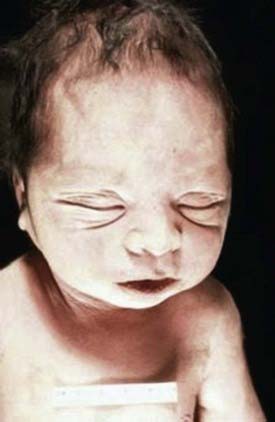
Bilateral renal agenesis is incompatible with extrauterine life and produces the Potter syndrome. Death occurs shortly after birth from pulmonary hypoplasia. The newborn has a characteristic facial appearance, termed Potter facies (Fig. 531-1). The eyes are widely separated with epicanthic folds, the ears are low set, the nose is broad and compressed flat, the chin is receding, and there are limb anomalies. Bilateral renal agenesis should be suspected when maternal ultrasonography demonstrates oligohydramnios, nonvisualization of the bladder, and absent kidneys. The incidence of this disorder is 1/3,000 births, with a male predominance, and represents 20% of newborns with the Potter phenotype. Other common causes of neonatal renal failure associated with the Potter phenotype include cystic renal dysplasia and obstructive uropathy. Less-common causes are autosomal recessive polycystic kidney disease (infantile), renal hypoplasia, and medullary dysplasia. Neonates with bilateral renal agenesis die of pulmonary insufficiency from pulmonary hypoplasia rather than renal failure (Chapter 95).
Renal Dysgenesis: Dysplasia, Hypoplasia, and Cystic Anomalies
Renal dysgenesis refers to maldevelopment of the kidney that affects its size, shape, or structure. The 3 principal types of dysgenesis are dysplastic, hypoplastic, and cystic. Although dysplasia always is accompanied by a decreased number of nephrons (hypoplasia), the converse is not true: Hypoplasia can occur in isolation. When both conditions are present, the term hypodysplasia is preferred. The term dysplasia is technically a histologic diagnosis and refers to focal, diffuse, or segmentally arranged primitive structures, specifically primitive ductal structures, resulting from abnormal metanephric differentiation. Nonrenal elements, such as cartilage, also may be present. The condition can affect all or only part of the kidney. If cysts are present, the condition is termed cystic dysplasia. If the entire kidney is dysplastic with a preponderance of cysts, the kidney is referred to as a multicystic dysplastic kidney (MCDK) (Fig. 531-2).
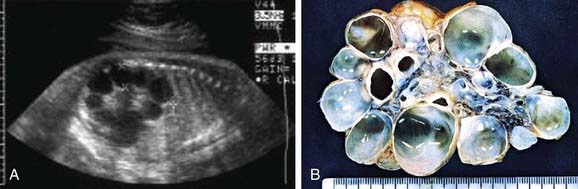
Figure 531-2 A, Prenatal sonogram demonstrating multicystic dysplastic kidney. B, Surgical specimen.
MCDK is a congenital condition in which the kidney is replaced by cysts and does not function; it can result from ureteral atresia. Kidney size is highly variable. The incidence is approximately 1/2,000. Some clinicians assume incorrectly that the terms multicystic kidney and polycystic kidney are synonymous. However, polycystic kidney disease is an inherited disorder that may be autosomal recessive or autosomal dominant and affects both kidneys (Chapter 515). MCDK usually is unilateral and generally is not inherited. Bilateral MCDKs are incompatible with life.
MCDK is the most common cause of an abdominal mass in the newborn, but the vast majority are nonpalpable at birth. In most cases it is discovered incidentally during prenatal sonography. In some patients, the cysts are identified prenatally, but the cysts regress in utero and no kidney is identified on imaging at birth. Contralateral hydronephrosis is present in 5-10% of patients. Sonography shows the characteristic appearance of a kidney replaced by multiple cysts of varying sizes that do not communicate, and no identifiable parenchyma is present; in most cases the diagnosis should be confirmed with a renal scan, which should demonstrate nonfunction. In some patients, usually boys, a small nonobstructing ureterocele is present in the bladder (Chapter 534). Although 15% have contralateral vesicoureteral reflux, obtaining a voiding cystourethrogram is optional, unless there is significant contralateral hydronephrosis. Management is controversial. Complete cyst regression occurs in nearly half of MCDKs by age 7 years. The risk of associated hypertension is 0.2-1.2%, and the risk of Wilms tumor arising from an MCDK is approximately 1/333. Because neoplasms arise from the stromal rather than the cystic component, even if the cysts regress completely, the likelihood that the kidney could develop a neoplasm is not altered.
Anomalies in Shape and Position
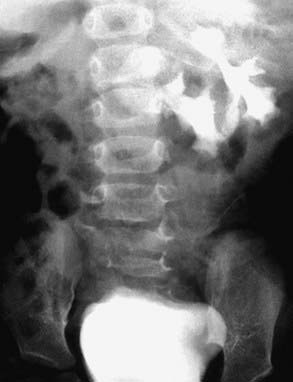
During renal development the kidneys normally ascend from the pelvis into their normal position behind the ribs. The normal process of ascent and rotation of the kidney may be incomplete, resulting in renal ectopia or nonrotation. The ectopic kidney may be in a pelvic, iliac, thoracic, or contralateral position. If the ectopia is bilateral, in 90% of persons the two kidneys fuse. The incidence of renal ectopia is approximately 1 in 900 (Fig. 531-3).
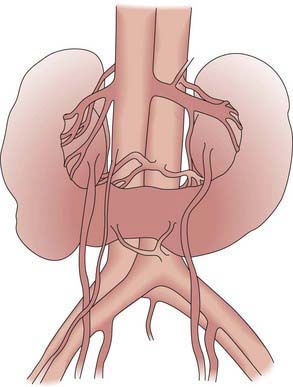
Renal fusion anomalies are more common. The lower poles of the kidneys can fuse in the midline, resulting in a horseshoe kidney (Fig. 531-4); the fused portion is termed the isthmus and may be thick functioning parenchyma or a thin fibrous strand. Horseshoe kidneys occur in 1/400-500 births but are seen in 7% of patients with Turner syndrome. Horseshoe kidney is one of the many renal anomalies that occur in 30% of patients with Turner syndrome (Chapter 580). Wilms tumors are 4 times more common in children with horseshoe kidneys than in the general population. Stone disease and hydronephrosis secondary to ureteropelvic junction obstruction are other potential late complications. The incidence of MCDK affecting one of the 2 sides of a horseshoe kidney also appears to be increased. With crossed fused ectopia, one kidney crosses over to the other side and the parenchyma of the 2 kidneys is fused. Renal function usually is normal. In the most common finding, the left kidney may cross over and fuse with the lower pole of the right kidney. The insertion of the ureter to the bladder does not change, and the adrenal glands remain in their normal positions. The clinical significance of this anomaly is that if renal surgery is necessary, the blood supply is variable and can make partial nephrectomy more difficult.
Cain JE, Di Giovanni V, Smeeton J, et al. Genetics of renal hypoplasia: insights into the mechanisms controlling nephron endowment. Pediatr Res. 2010;68(2):91-98.
Cambio AJ, Evans CP, Kurzrock EA. Non-surgical management of multicystic dysplastic kidney. BJU Int. 2008;101:804-808.
Glodny B, Petersen J, Hofmann KJ, et al. Kidney fusion anomalies revisited: clinical and radiological analysis of 209 cases of crossed fused ectopia and horseshoe kidney. BJU Int. 2009;103:224-235.
Leung VYF, Rasalkar DD, Liu JX, et al. Dynamic ultrasound study on urinary bladder in infants with antenatally detected fetal hydronephrosis. Pediatr Res. 2010;67(4):440-443.
Narchi H. Risk of hypertension with multicystic kidney disease: a systematic review. Arch Dis Child. 2005;90:921-924.
Narchi H. Risk of Wilms’ tumour with multicystic kidney disease: a systematic review. Arch Dis Child. 2005;90:147-149.
Park JM. Normal development of the urogenital system. In Wein AJ, Kavoussi LR, Novick AC, et al, editors: Campbell-Walsh urology, ed 9, Philadelphia: Saunders, 2007. 3121–2148
Schreuder MF, Langemeijer ME, Bökenkamp A, et al. Hypertension and microalbuminuria in children with congenital solitary kidneys. J Paediatr Child Health. 2008;44:363-368.
Schreuder MF, Westland R, van Wijk JA. Unilateral multicystic dysplastic kidney: a meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol Dial Transplant. 2009;24:1810-1818.
Spira EM, Jacobi C, Frankenschmidt A, et al. Sonographic long-term study: paediatric growth charts for single kidneys. Arch Dis Child. 2009;94:693-698.
Vu KH, Van Dyck M, Daniels H, et al. Renal outcome of children with one functioning kidney from birth. A study of 99 patients and a review of the literature. Eur J Pediatr. 2008;167:885-890.
Weinstein A, Goodman TR, Iragorri S. Simple multicystic dysplastic kidney disease: end points for subspecialty follow-up. Pediatr Nephrol. 2008;23:111-116.



