[level-membership-for-pathology-category]
Congenital and Developmental Disorders of the Gastrointestinal Tract
Dale Huff
Pierre Russo
Molecular Mechanisms of Gastrointestinal Development
Recent advances in understanding of the molecular controls of gut development have flowed from studies of a number of vertebrate and invertebrate models, including Caenorhabditis elegans, Drosophila, sea urchins, zebrafish, and mice. These studies have provided insights into the genetic mechanisms that direct formation and modeling of the gastrointestinal (GI) tract, highlighted the importance of endodermal-mesenchymal interactions, and demonstrated the high degree of phylogenetic conservation of these mechanisms.
Gut development is controlled by a number of intercellular signaling pathways, such as the bone morphogenetic protein (BMP), Notch, Homeobox, Hedgehog, and Wnt pathways. A discussion of these studies is beyond the scope of this chapter, and several excellent reviews are available.1–4 Early development of the endoderm depends on molecular signaling pathways such as that of Wnt pathway, which acts by stabilizing β-catenin, allowing it to translocate to the nucleus to activate transcription genes. Ablation of β-catenin in the notochord and primitive streak abrogates endoderm formation.5 In Drosophila and in the mouse, there are regional differences in the specific expression of Homeobox (Hox) genes along the gut axis.2,4 For example, hindgut defects in mice can be linked to defective expression of Hoxd-13.4
Another family of signaling genes critical in cellular cross-talk is the Hedgehog (Hh) family, which appears to be essential to anterior-posterior, dorsal-ventral, and radial patterning. Knockout and transgenic mouse models of various hedgehog components result in a variety of malformed phenotypes, ranging from esophageal atresia (EA) to persistent cloaca.6 Vertebrate homologs of Hh exist in three forms: sonic (Shh), Indian (Ihh) and Desert (Dhh), which have different but overlapping expression patterns. For example, Shh−/− mutant embryos die in utero and have overgrown duodenal villi resulting in occlusion, analogous to duodenal stenosis in humans. Selective postnatal blocking of Hh signaling resulted in a wasting and runted phenotype characterized by diarrhea with disorganized intestinal villi, hyperplastic crypts, and enterocyte vacuolization.6
Epigenetic factors may also contribute to phenotypic development as well as disease susceptibility in genetically identical individuals. For example, different degrees of methylation of CpG groups in the agouti mouse, which can vary according to maternal intake of B group vitamins, may result in variation in coat colors.7 Epigenetic factors such as diet and the development of the microbiome appear to play a major role, especially in postnatal development of the GI tract.4
Embryology and Anatomic Development of the GI tract
The development of the GI tract proceeds through three major overlapping steps: formation of the gut tube during blastogenesis, differentiation of the specific segments of the digestive tract and its accessory organs during organogenesis, and histogenesis of the individual organs with their specialized cell types.2 Major developmental milestones are outlined in Table 8.1. The first two steps take place during the embryonic period, which begins on the day of fertilization and ends on the 56th postconceptual day (week 8). The developing human is more susceptible to teratogenetic agents during the embryonic period than at any other period of development. The fetal period, which begins on postconceptual day 57 and ends at birth, is characterized by the final stages of rotation and fixation and by continued elongation and histogenesis of the GI tract. By 15 to 20 weeks’ gestation, the fetal gut essentially resembles that of the newborn.1 An overview of these basic processes, especially pertaining to the GI tract, is presented in Table 8.2. The pattern of congenital anomalies of the GI tract varies depending on the developmental period from which they arise (Table 8.3).
Table 8.1
Developmental Milestones
| Event | Time of First Expression (Weeks after Conception) |
| Gastrulation | 3 |
| Gut tube largely closed | 4 |
| Liver and pancreas buds Growth of intestines into cord |
4 7 |
| Intestinal villus formation | 8 |
| Retraction of intestines into abdominal cavity | 10 |
| Organ formation complete | 12 |
| Parietal cells detectable, pancreatic islets appear, bile secretion, intestinal enzymes detectable | 12 |
| Swallowing detectable | 16 and 17 |
| Mature motility | 36 |
Data from Montgomery RK, Mulberg AE, Grand RJ. Development of the human gastrointestinal tract: twenty years of progress. Gastroenterology. 1999;116:702-731.
Table 8.2
Overview of Gastrointestinal Development in the First 10 Weeks after Conception
| Feature | Embryo | Fetus | ||||||||
| Blastogenesis | Organogenesis | |||||||||
| week | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Bilaminar disc; endoderm | X | |||||||||
| Yolk sac; connecting stalk | X | |||||||||
| Trilaminar disc | X | |||||||||
| Early: Midline developmental field; induction of gastrointestinal patterning | X | |||||||||
| Late: Primitive foregut, midgut, hindgut; respiratory and hepatic primordia | X | |||||||||
| Cylindrical embryo; definitive tubular gastrointestinal tract; umbilical cord | X | |||||||||
| Beginning intestinal rotation | X | |||||||||
| Remodeling, growth, histogenesis; anus completed | X | X | X | X | ||||||
| Return of bowel to abdomen; final rotation and fixation | X | |||||||||
| Sexual differentiation of perineum | X | |||||||||
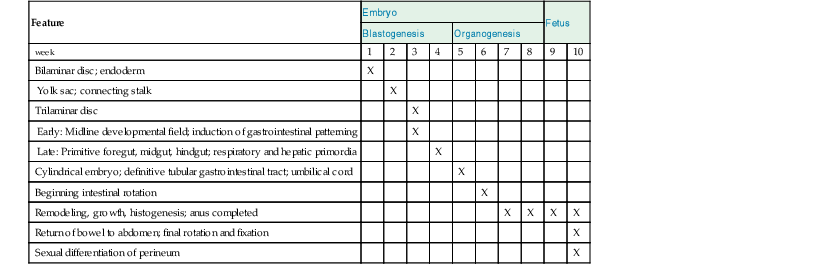
Table 8.3
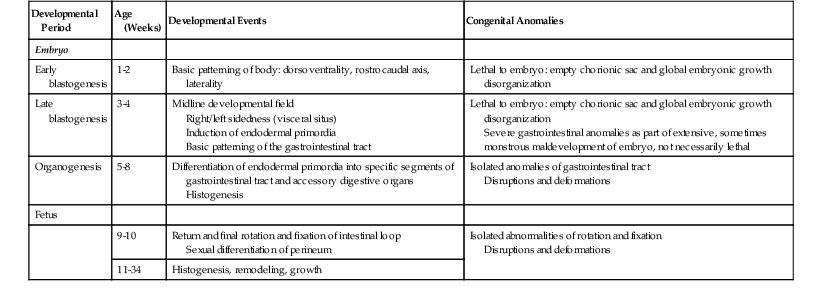
Patterns of Congenital Anomalies Arising during Various Developmental Periods
| Developmental Period | Age (Weeks) | Developmental Events | Congenital Anomalies |
| Embryo | |||
| Early blastogenesis | 1-2 | Basic patterning of body: dorsoventrality, rostrocaudal axis, laterality | Lethal to embryo: empty chorionic sac and global embryonic growth disorganization |
| Late blastogenesis | 3-4 | Midline developmental field Right/left sidedness (visceral situs) Induction of endodermal primordia Basic patterning of the gastrointestinal tract |
Lethal to embryo: empty chorionic sac and global embryonic growth disorganization Severe gastrointestinal anomalies as part of extensive, sometimes monstrous maldevelopment of embryo, not necessarily lethal |
| Organogenesis | 5-8 | Differentiation of endodermal primordia into specific segments of gastrointestinal tract and accessory digestive organs Histogenesis |
Isolated anomalies of gastrointestinal tract Disruptions and deformations |
| Fetus | |||
| 9-10 | Return and final rotation and fixation of intestinal loop Sexual differentiation of perineum |
Isolated abnormalities of rotation and fixation Disruptions and deformations |
|
| 11-34 | Histogenesis, remodeling, growth |
From Huff DS. Developmental anatomy and anomalies of the gastrointestinal tract, with involvement in major malformative syndromes. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. New York: Springer; 2004:3-37.
Blastogenesis extends from fertilization to day 28. During the first half of blastogenesis, the bilaminar disc and the basic body plan of dorsoventrality, rostrocaudal axis, and laterality are established. During the second half of blastogenesis, the midline developmental field directs the process of gastrulation, which establishes all three germ layers (endoderm, mesoderm, and ectoderm). The mammalian digestive system is derived from each of these layers—the epithelial lining from the endoderm, the muscle layers and supportive elements from the mesoderm, and the neurons of the enteric nervous system from the ectoderm. It is during this period that the basic plan of the GI tract is established through the inductive influences of the notochord, primitive streak, emerging mesoderm, and other anatomic components of the midline developmental field on the primitive endoderm. These inductive influences predetermine the sites of the specific segments of the GI tract and the primordia of its accessory organs of digestion. For example, at the end of gastrulation in mice, further patterning of the endoderm is determined by regional expression of factors such as Sox2 and Hhex in the anterior endoderm and Cdx2 in the posterior endoderm.3
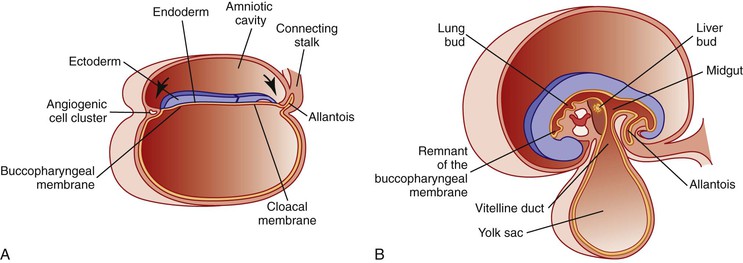
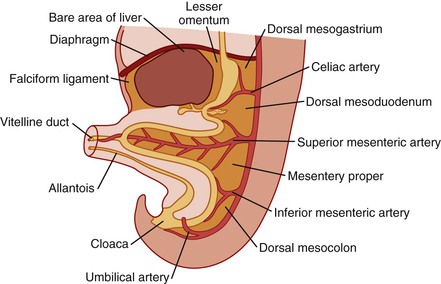
Simultaneously, during the third and fourth weeks of development, cephalocaudad and lateral folding of the embryo converts the trilaminar germ disc into an elongated cylinder. The primitive gut is at that point somewhat arbitrarily divided into three major segments: a cranial foregut, a midgut open to the yolk sac via the vitelline duct, and a hindgut (Fig. 8.1). Each of these segments will give rise to specialized regions of the gut and, as in the case of the foregut, to other organs such as the thyroid, lungs, liver, and pancreas. The blood supply to the primitive gut is derived from the vitelline arteries of the yolk sac. The celiac artery, superior mesenteric artery (SMA), and inferior mesenteric artery vascularize the abdominal foregut, midgut, and hindgut, respectively, and by convention determine the boundaries of each (Fig. 8.2).
Organogenesis extends from day 29 to day 56 (weeks 5 through 8). Suddenly, during the fifth week, the entire tubular GI tract, its major divisions, and its accessory organs of digestion, having been predetermined during blastogenesis, emerge from the imprinted primordium of the primitive endodermal tube. The abdominal portion of the foregut is divided into the esophagus, stomach, and proximal duodenum. The common origin of the trachea and esophagus from the foregut results in various types of fistulas if separation is incomplete. The hepatic diverticulum arises from the proximal duodenum, its cephalic portion budding into the transverse septum (precursor of the diaphragm) to become the liver, and its caudal portion giving rise to the gallbladder and the extrahepatic biliary tree. Dorsal and ventral pancreatic buds also emerge from the proximal duodenum.
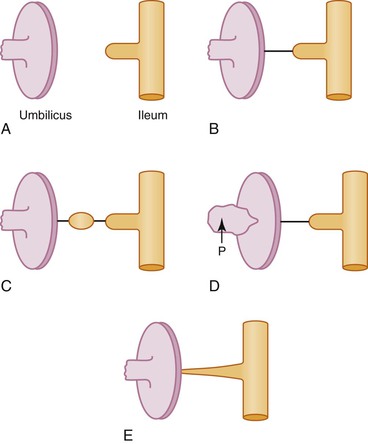
It is generally agreed and illustrated in most embryology texts that further development and growth of the gut involves a counterclockwise rotation of 270 degrees during development (Fig. 8.3). However, the exact timing of these events is still subject to disagreement.8 The initial event appears to be herniation of the developing gut into the stalk of the yolk sac during the sixth week, likely because elongation of the midgut proceeds much faster than growth of the embryo and because the concomitant rapid growth of the liver displaces the intestines from the abdominal cavity. As the midgut elongates, it rotates 90 degrees counterclockwise (as viewed from the front of the embryo) around the axis of the SMA, so that the cranial limb (“prearterial” in relation to the SMA) moves to the embryo’s right and the caudal limb (“postarterial”) moves to the embryo’s left (see Fig. 8.3, A and B). Continued elongation, especially of the prearterial segment, results in a series of folds called the jejunoileal loops, the identity of which Keibel believed was retained in the adult.9 The postarterial loop, most of which will form the colon, remains relatively straight. At approximately 10 weeks of life, the intestines return to the abdominal cavity. The factors responsible for this step are unclear, but decrease in the relative growth of the liver and rapid expansion of the abdominal cavity have been suggested. As the gut returns, there is a further anticlockwise, 180-degree rotation, which, added to the previous rotation, makes a total of 270 degrees (see Fig. 8.3, C). Alternatively, some authors have suggested that the gut rotates 180 degrees in the cord followed by a 90-degree rotation during return.8
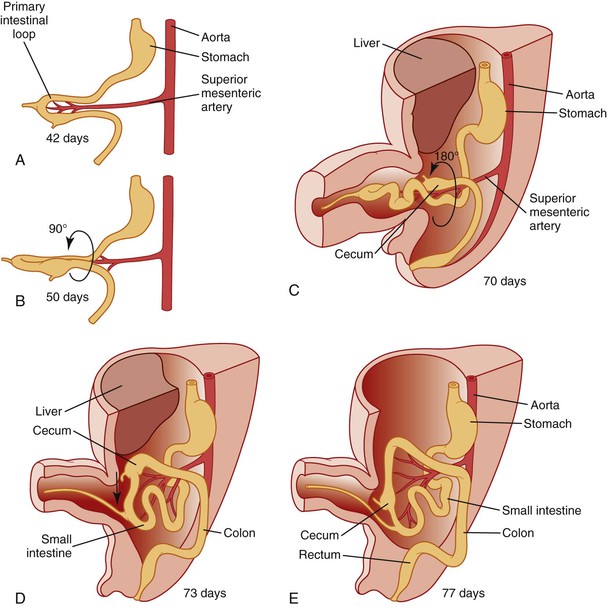
As a net result of this 270-degree rotation, the third portion of the duodenum passes horizontally caudal and dorsal to the artery, and the proximal anchoring point comes to lie near the final position of the ligament of Treitz to the left of the artery. The SMA hangs over the ventral wall of the third portion of the duodenum. As the distal limb then rapidly returns, it swings ventral and rostral to the proximal loop, and the cecum comes to lie in the right abdomen near the liver (see Fig. 8.3, D). Rotation is completed by the 10th week, and fixation continues throughout fetal life as the mesenteries become adherent to the parietal peritoneum. The cecum and liver then separate by unknown mechanisms, and the increasing distance becomes occupied by the lengthening ascending colon, with the final position of the liver being the right upper quadrant and that of the cecum being the right lower quadrant (see Fig. 8.3, E). This separation is referred to, probably incorrectly, as “cecal descent.” See Estrada10 for an extensive review and Kluth and colleagues11 for a recent reevaluation of these events.
Mucosal histogenesis transforms the primitive undifferentiated epithelium of the gut tube into the specific epithelia of the final differentiated segments of the digestive tract. Although histogenesis begins in the late embryonic period, most of the histologic transformation occurs during fetal life. It begins with a transient phase of epithelial proliferation. The proliferating epithelium completely occludes the lumen of the duodenum, significantly narrows the lumen of the esophagus, and may mildly narrow the lumen of the cardia, pylorus, upper jejunum, and distal ileum. These proliferations may be accompanied by the transient formation of multiple antimesenteric diverticula in the duodenum, upper jejunum, and distal ileum. Some instances of congenital atresia, stenosis, or diverticula may be the result of abnormalities in the formation or resolution of the proliferative phase.
Esophagus
Ciliated columnar epithelium covers the epithelial surface of the midesophagus at 10 weeks and spreads to both ends by the 11th week (Fig. 8.4). Stratified squamous epithelium begins to replace the ciliated columnar epithelium in the midesophagus at 16 weeks and spreads proximally and distally to cover the entire esophagus by birth, except for the proximal esophagus, where islands of ciliated columnar epithelium may persist, disappearing shortly after birth.12–14 Intestinal goblet cells in the distal esophagus have been rarely observed in the neonate and fetus, although positive staining with acidic mucins at the squamocolumnar junction in this age group is common.15 Residual embryonic cells that are induced to proliferate after damage to the squamous epithelium have been postulated to be the source of Barrett metaplasia.16
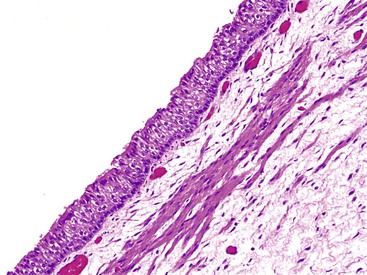
Pancreatic acinar tissue has been observed in young children at the gastroesophageal junction, independent of Barrett esophagus, esophagitis, or gastritis.17 The superficial cardiac glands of the lamina propria appear during the 13th week. The submucosal mucous glands appear in the 27th week. The circular muscle layer is present at 8 weeks and the longitudinal layer at approximately 13 weeks, followed by the muscularis mucosae.
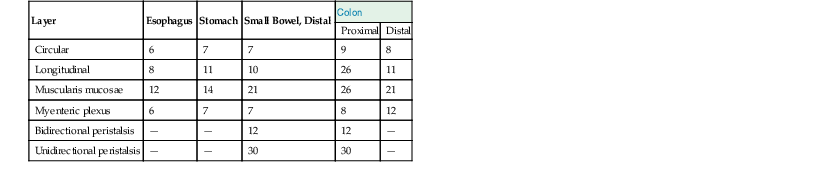
The waves of differentiation begin in the esophagus, propagating caudally, and at the anorectal junction, propagating cranially; the two meet at the ileocecal junction. Ganglion cells become recognizable by 8 weeks. Their density peaks between 16 and 20 weeks, decreases with increasing gestational age until 30 weeks, then becomes constant.18 The timing of development of the muscle layers and myenteric plexuses in the various segments of the GI tract is outlined in Table 8.4.
Table 8.4
Histogenesis of the Muscular Coats and Myenteric Plexus
| Layer | Esophagus | Stomach | Small Bowel, Distal | Colon | |
| Proximal | Distal | ||||
| Circular | 6 | 7 | 7 | 9 | 8 |
| Longitudinal | 8 | 11 | 10 | 26 | 11 |
| Muscularis mucosae | 12 | 14 | 21 | 26 | 21 |
| Myenteric plexus | 6 | 7 | 7 | 8 | 12 |
| Bidirectional peristalsis | — | — | 12 | 12 | — |
| Unidirectional peristalsis | — | — | 30 | 30 | — |
Stomach
During the fifth week of life, differential growth of the dorsal wall of the stomach results in formation of the greater curvature. Subsequent rotation of the stomach 90 degrees along a craniocaudal axis during the seventh week, followed by fixation of the second part of the duodenum to the dorsal body wall, forms the lesser sac of the peritoneal cavity. Prenatal ultrasound examinations have shown that the stomach continues to grow in a linear fashion from 13 to 39 weeks. Studies of the development of the mouse stomach have established that epithelial stem cells of the gastric pits reside in the neck region, producing different cell populations that move either upward or downward.19,20
Intestinal villi appear in the cardia and pylorus, where they are normally abundant by 30 weeks (Fig. 8.5). They disappear by birth. Intestinal metaplasia of cardiac or pyloric epithelium may represent a dedifferentiation to the normal fetal condition. The cardiac mucosa is thought to arise from undifferentiated gastric mucosa and not from esophageal metaplasia.21 The development of the gastric glands occurs early during fetal life. Glandular pits are formed during the 11th to 12th weeks of fetal life, along with emergence of the first cells of the parietal lineage. By 15 to 17 weeks of gestation, the fetal gastric glands are essentially similar to an adult’s, with compartmentalization into foveolus, isthmus, neck, and base containing the various phenotypically differentiated cell types.1
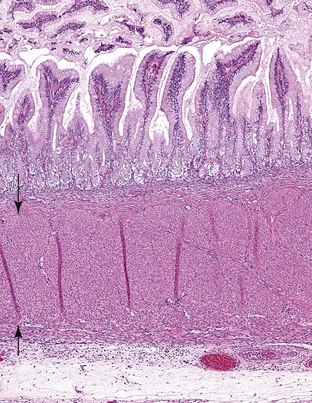
Further development of the stomach involves thickening of the glandular region with proliferation and maturation of the chief cells, which are relatively fewer in the neonatal stomach than in the adult stomach and which do not produce pepsin in the newborn.22 Gastric pH is relatively high in the neonate; it becomes comparable to that of adults by the age of 2 years. This may result in part from buffering by amniotic fluid but also from a relative lack of gastrin, levels of which increase during the first few postnatal months.22
Small and Large Intestine
Rearrangement of the endodermal epithelium resulting from elongation of the gut tube, rather than epithelial proliferation as previously thought, leads to temporary occlusion of the lumen by the end of the sixth week.23 Defects in subsequent recanalization of the lumen can result in stenoses or duplications of the digestive tract. As the lumen expands, the epithelium undergoes folding, which will eventually lead to the formation of villi. Mesenchymal cells grow toward the lumen to form early villi, and this process is orchestrated by an elaborate endodermal-mesodermal cross-talk under the control of signaling pathways including the BMP, Hedgehog, platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), and Wnt pathways and the mesenchymal transcription factors FoxL1, FoxF1, and FoxF2.3
Villi and crypts appear first in the duodenum in the 8th week, spread to the middle of the small intestine by the 9th week, and reach the distal ileum by the 12th week. The early intestinal mucosa consists of stratified epithelium; columnar epithelium appears gradually, first at the apices and then along the sides of villi. By the 10th week, only intervillus epithelium remains stratified.2 By 9 to 10 weeks, absorptive cells of the proximal intestine display a brush border with an array of microvilli.24 Eosinophilic globules frequently can be observed within the fetal intestinal epithelium (Fig. 8.6); these have been referred to as “thanatosomes,” and seem to reflect apoptotic activity.25
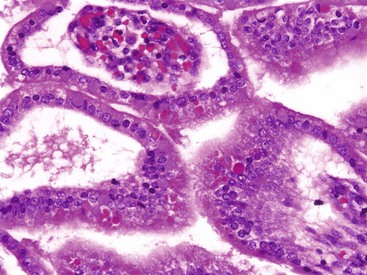
Both Wnt and BMP signaling pathways appear to be involved in the formation of crypts.3 These crypts contain the stem cells that will serve as the source of the epithelial cells, which are renewed every 4 days.4 Cytologic differentiation of the crypts begins with the appearance of goblet cells in the eighth week, followed by Paneth cells and enteroendocrine cells in the ninth week. Clusters of enteroendocrine cells occurring on top of villi in the duodenum and upper jejunum during the 20th week of gestation have been described as Segi’s cap.26
The Notch pathway plays a critical role in epithelial differentiation by regulating the specification of absorptive versus secretory lineages, which is controlled by differences in expression of factors such as Hes1, Atoh1, and Neurog3.3 Atoh1 (also called Math1) is a basic loop-helix-loop transcription factor that appears to be a key regulator of the development of secretory cells (goblet, Paneth, and enteroendocrine cells), whereas absorptive cells appear to be Math1 independent.27 Neonates have been reported with an absence of gut secretory cells.28 Patients with mutations in a gene called neurogenin 3 (i.e., NEUROG3) have presented with malabsorptive diarrhea and a complete absence of enteroendocrine cells.29 The time of appearance and electronmicroscopic features of 13 enteroendocrine cells have been described in human embryos between 9 and 22 weeks’ gestation.30 Brunner glands appear in the proximal duodenum in the 12th week. The histologic appearance of the small intestine resembles that of a newborn by 20 weeks.31
The enteric nervous system results from contributions from the sympathetic system, growing along the arterial supply, and from the parasympathetic system, with branches of the vagal nerve innervating the upper GI tract; the pelvic splanchnic nerves innervate the descending colon and rectum. Postganglionic neurons are derived from neural crest cells, which appear in the fetal gut at approximately 8 weeks’ gestation and migrate in a craniocaudal direction; they may be detected in the rectum by 12 weeks.2 The development of the enteric nervous system is beyond the scope of this chapter, and recent references should be consulted.32–36
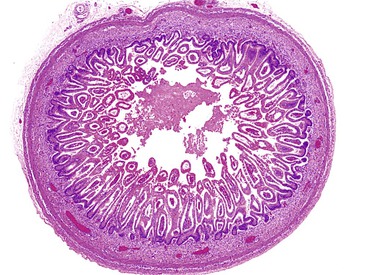
Similar to the intestinal epithelium, the colonic mucosa consists of a stratified epithelium beginning at about 8 weeks. At approximately the 10th week, villi with developing crypts cover the surface of the large intestine and persist until the 28th week. Therefore, intestinal villi are normally seen in the embryo not only in the small intestine but also in the cardia, pylorus, and colon (Fig. 8.7). The intervillous surface epithelium differentiates into a single layer containing goblet cells by the 13th to 16th week. After birth, a 100-fold increase in the number of intestinal crypts occur, along with an expansion of crypt cells.22 An outline of the histogenesis of the muscular coats and myenteric plexus is presented in Table 8.4.
The development of the mucosal lymphoid system is outlined in Table 8.5. The fetal mucosal lymphoid system has the capacity to respond to an abnormal intrauterine antigenic stimulus with the formation of germinal centers and plasma cells possibly as early as 20 weeks. The neonate responds to the antigenic stimulus of colonization at birth with the formation of germinal centers and plasma cells 2 to 4 weeks after birth.
Table 8.5
Histogenesis of Mucosal Lymphoid Tissue
| Week | Feature |
| Intrauterine | |
| 7 | Intraepithelial lymphocytes. |
| 10 | T cells with surface recognition |
| 12 | PHA-responsive lymphocytes |
| 14 | Lymphocytes with PHA cytotoxicity and ability to mediate graft-vs-host response |
| 17-20 | Mast cells in small intestine |
| 21-24 | Mast cells in large intestine; solitary lymphoid follicle in distal ileum, appendix, and colon |
| 24 | Peyer patches in distal ileum |
| 40 | Solitary lymphoid follicles in duodenum, rectum, and possibly stomach |
| Postnatal | |
| 2-4 | Germinal centers and plasma cells |
PHA, Phytohemagglutinin.
From Huff DS. Developmental anatomy and anomalies of the gastrointestinal tract, with involvement in major malformative syndromes. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. New York: Springer; 2004:3-37.
Congenital Anomalies of the GI Tract
General Aspects
The causes of anomalies of the GI tract include chromosomal abnormalities (numerical and structural); single gene defects; maternal diseases, especially diabetes; and maternal exposure to drugs, especially hydantoin (pyloric stenosis, duodenal and anal atresia). Causes of disruptions include inherited37 and noninherited maternal and fetal thrombophilic diseases, intrauterine hypoxic/ischemic events, intrauterine infection including varicella,38 iatrogenic vascular disruptions,39 and maternal exposure to vasoactive drugs.40 Other diseases of the embryo, such as cystic fibrosis and epidermolysis bullosa, underlie some GI anomalies. Deformations are limited to abnormal shapes of the liver and abnormal rotation and fixation associated with defects of the diaphragm, body wall, and umbilicus. The cause of most anomalies is unknown. Anomalies that arise after completion of organogenesis are often disruptions or deformations; otherwise, the causes are not specific to any developmental period.
Esophagus
Short Esophagus
A congenital short esophagus is a rare anomaly that is associated with intrathoracic development of the stomach. It may be difficult to distinguish from the more common congenital hiatal hernia. Features that favor the diagnosis of a congenital short esophagus include early identification of the intrathoracic stomach during the second trimester and the consistent absence of an abdominal stomach bubble on antenatal ultrasound.41 Furthermore, in the congenital short esophagus, the intrathoracic stomach is supplied by segmental arteries from the descending thoracic aorta, rather than by intrathoracic extensions of the gastric artery, as observed in hiatal hernia.14 Congenital hiatal hernia, by contrast, appears to develop later in gestation and is caused by defective development of the lumbar part of the diaphragm. The differences between these conditions are of more than academic interest, because the outcome of repair of a congenital short esophagus is more guarded than that of hiatal hernia.41
Esophageal Atresia and Tracheoesophageal Fistula
Atresias and stenoses may occur at any site along the tract, but some sites are more commonly involved than others (Table 8.6). Several disorders associated with multiple atresias or stenoses of the GI tract are listed in Table 8.7. Key distinguishing features of the various types of congenital atresias and stenoses of the GI tract are outlined in Table 8.8. EA with or without tracheoesophageal fistula (TEF) occurs in approximately 1 of every 3000 live births. There is a slight male predominance. A history of polyhydramnios is found in a majority of patients with atresia. EA without a fistula is associated with a small stomach42 and absence of a GI gas pattern. A fistula from the distal esophageal segment allows passage of gastric content into the respiratory tract, causing respiratory symptoms. Approximately 50% of patients have associated congenital anomalies, of which the VATER/VACTERL association (consisting of a combination of vertebral anomalies, anal atresia, cardiac defect, tracheoesophageal fistula, renal and limb anomalies) is the most common.43 Conditions associated with EA and TEF are listed in Table 8.9. Familial forms not associated with hereditary syndromes are probably multifactorial.
Table 8.6
Occurrence of Atresia and Stenosis of the Gastrointestinal Tract
| No. Per Live Births* | % of All Intestinal Atresia | |
| Esophagus | 1 : 3000 | |
| Stomach | Rare | |
| Duodenum | 1 : 1500 | 50 |
| Jejunum | 1 : 2000 | 20 |
| Ileum | 1 : 2000 | 25 |
| Colon | Rare | 5 |
| Rectum anus | 1 : 5000 | |
| Multiple | 15 |
* Number per live births vary widely from series to series.
Data from Huff DS. Developmental anatomy and anomalies of the gastrointestinal tract, with involvement in major malformative syndromes. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. New York: Springer; 2004:3-37.
Table 8.7
Disorders with Multiple Gastrointestinal Atresias or Stenoses

Table 8.8
Congenital Atresias and Stenoses of the Gastrointestinal Tract
| Location | Incidence | Key Clinical Features | Key Pathologic Features |
| Esophagus | Atresia 1 : 3000 Stenosis 1 : 30,000 |
>50% associated anomalies; VACTERL association most common Choking and respiratory distress first day of life Absence of normal gastrointestinal gas pattern Stenoses may manifest later in life |
Atresias associated with tracheoesophageal fistula in 85% of cases Stenosis in middle third usually membranous Stenosis in distal third usually due to tracheobronchial remnant |
| Stomach | 1 : 100,000 | Variable; birth to childhood Prenatal history of polyhydramnios Nonbilious vomiting; single large gastric bubble “Single bubble” sign on radiographs in cases of complete obstruction |
Some cases may be associated with epidermolysis bullosa, aplasia cutis, or due to pancreatic heterotopias or adenomyoma |
| Idiopathic hypertrophic pyloric stenosis | 1 : 500 | Projectile vomiting in first weeks of life Palpable epigastric mass Associated with several multiple congenital anomalies syndromes |
Concentric hyperplasia and hypertrophy of pyloric muscularis propria |
| Duodenum | 1 : 1500 | Most common site of intestinal stenoses Nonbilious vomiting if proximal to ampulla; bilious if distal “Double-bubble” gas pattern |
Atresias are usually membranous 50% of stenoses are associated with other anomalies, including annular pancreas, or malrotation |
| Jejunum/ileum | 1 : 1500 | 85% single; 15% multiple Rare familial forms Abdominal distention; gas-filled loops Meconium ileus is presenting feature in 20% of cases of cystic fibrosis |
95% are atresias; 5% are stenoses 50% of atresias are type III with associated mesenteric defect |
| Anorectal | 1 : 4000 | 60% associated with other malformations Imperforate anus with abnormal perineum Failure to pass meconium with cutaneous, vesicular, urethral, or vaginal (females) fistulas |
High atresias more common in males; low atresias more common in females Most severe form is persistent cloaca |
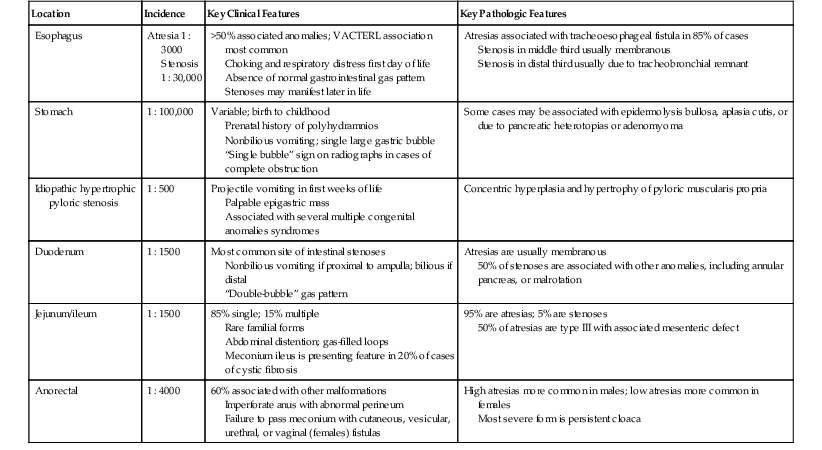
Table 8.9
Disorders with Esophageal Atresia and Tracheoesophageal Fistula
| Single gene disorders |

CHARGE, Coloboma, heart defect, atresia choanae, retarded growth and development, genital abnormality, and ear abnormality; VACTERL, vertebral anomalies, anal atresia, cardiac defect, tracheoesophageal fistula, renal and limb anomalies)
Data from de Jong EM, Felix JF, de Klein A, Tibboel D. Etiology of esophageal atresia and tracheoesophageal fistula: “mind the gap.” Curr Gastroenterol Rep. 2010;12:215-222.
Gross44 and Swenson and colleagues45 proposed the most commonly used classifications of EA and TEF. Figure 8.8 schematically depicts five common variations found in many current publications. The most common form, comprising approximately 85% of cases, consists of a blind-ending proximal pouch with a fistula from the trachea to the distal portion (Fig. 8.9). The tracheal ostium of a fistula to the distal esophageal segment is often at the carina but may be higher in the trachea. Likewise, the ostium of a fistula to the proximal esophageal pouch is often in the upper trachea but is sometimes lower. The length of the distal segment varies. In some cases, it is a short stump barely visible above the diaphragm, and in some it is subdiaphragmatic.46 The distance between the upper esophageal pouch and the distal esophageal segment may be short or long, depending on these variations. Short gap lesions are more easily and successfully repaired than long gap lesions. A fistula from the upper esophageal pouch or the distal esophageal segment may arise from one or both bronchi forming a bronchoesophageal fistula (or fistulas). The trachea may be absent, in which case the distal trachea or both main bronchi arise from the esophagus. An esophageal stenosis may be present at the site of a TEF without EA.
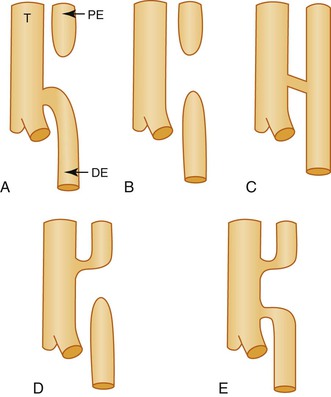
The association with a multitude of genetic defects, particularly the VATER/VACTERL association, suggests that multiple genetic pathways may be involved, of which mutations and deletions of the FOX gene cluster on chromosome 16q24 appear to be the most signficant.43,47 Adriamycin administered intraperitoneally to pregnant rodents results in esophageal anomalies and tracheoesophageal fistulas in about 50% of the pups. In that model, there appeared to be interference with the division of the esophagotracheal tube into ventral and dorsal components.48 No such association has been reported in humans. The Hedgehog pathway may also play a role. Shh−/− mice have severe TEF.48 The occasional coexistence of EA and TEF with foregut duplications and bronchopulmonary foregut malformations suggests a common pathogenesis.
The prognosis is poorer for babies who have low birth weight and associated anomalies, especially cardiac abnormalities. Postoperatively, there is a risk of anastomotic leaks leading to pneumonitis and mediastinitis.49 Gastroesophageal reflux, resulting from abnormal peristalsis, is common, occurring in as much as 50% of children.50 Motility defects after successful surgical repair have been attributed to histologic abnormalities of the Auerbach plexus in the distal esophagus and stomach.51 The proximal portion of the distal esophageal segment may lack a muscularis propria. Tracheomalacia and other tracheal anomalies are also common, occurring in 75% of patients, and may, in some cases, be severe enough to require tracheostomy.50 Ectopic gastric mucosa has been reported at the site of repair of an atresia. Chronic esophagitis and gastric metaplasia are late complications.52
Congenital esophageal stenosis has an incidence of approximately 1 : 50,000 live births and is caused by a congenital malformation of the esophageal wall due to a membranous diaphragm or web,53 intramural tumors such as pancreatic heterotopias,54 adenomyomas or leiomyomas,55,56 or cartilage nodules suggestive of tracheobronchial remnants.56 Membranous stenosis is most frequent in the middle third of the esophagus14,49 (Fig. 8.10), and tracheobronchial remnants are most common in the distal portion. In one third of cases, these are associated with other anomalies, including TEF.49,57 Concomitant atresias of other segments of the GI tract have been reported. Some patients with Hirschsprung disease have congenital GI atresias and stenoses including EA.58 Associations with congenital pyloric stenosis,59 duodenal atresia,60 and biliary and anorectal atresia61 have been reported. An autosomal recessive syndrome called multiple GI anomalies syndrome, consisting of atresias of the esophagus, duodenum, extrahepatic biliary tree, and anorectal area together with hypoplasia of the pancreas, malrotation of the intestines, and hypospadius, has been described.62, 63 Associated malformations of the respiratory tract include communicating64 and noncommunicating bronchopulmonary foregut malformations,65 abnormal pulmonary lobation, horseshoe lung,66 pulmonary hypoplasia, and tracheobronchial malacia.67 Unrecognized associated malformations may be the source of persistent symptoms after successful surgery. The typical symptoms, signs, and imaging features of gastric, duodenal, and jejunal atresia may be masked by a more proximal EA.
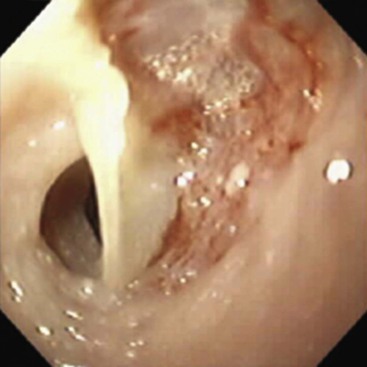
Esophageal Cysts and Duplications
The GI malformations referred to as duplications represent an array of lesions with a confusing nomenclature. Other names used include dorsal enteric remnants, posterior mediastinal cysts, dorsal enteric cysts, enterocysts, enterogenous cysts, neurenteric cysts, persistent neurenteric canal, diverticula, giant diverticula, and thoracic duplications of the intestine. Much of the confusion results from the fact that these cysts may be lined by any type of epithelium derived from the alimentary or respiratory tract. A useful and simple classification proposed by Dimmick and Hardwick68 divides them into two groups: (1) duplications and (2) neurenteric (dorsal enteric) cysts. Neurenteric remnant is a more appropriate term than neurenteric cyst, because a cyst is only one of several malformations included in this spectrum.
Duplications are cystic or tubular replicas of a segment of the GI tract. They are directly contiguous with the segment with which they are associated. Anomalies of adjacent vertebrae, spinal cord, or dorsal body wall are not present. Any segment from the mouth to the anus may be involved (Table 8.10). Cystic duplications, especially noncommunicating ones, may form an expanding intramural mass and cause stenosis or atresia with signs and symptoms of obstruction appropriate to the site. Upper esophageal duplications often produce respiratory obstruction.
Table 8.10
Distributions of Gastrointestinal Duplications*
| Site | % |
| Esophageal | 20 |
| Thoracoabdominal | 4 |
| Gastric | 7 |
| Duodenal | 5 |
| Jejunoileal | 44 |
| Colonic | 15 |
| Rectal | 5 |
| TOTAL | 100 |
* May include neurenteric cysts.
Data from Bond SJ, Groff DB. Gastrointestinal duplications. In: O’Neill JA, Rowe MI, Grosfeld JL, eds. Pediatric Surgery. St. Louis: Mosby; 1998:1257-1267.
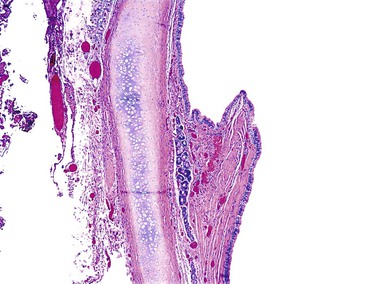
Cystic duplications are partially intramural or are attached to the wall and often share muscularis propria with the involved segment. The lumen of the cyst may communicate with the normal lumen. Histologic diagnostic criteria include (1) attachment to the esophagus, (2) enclosure by two muscle layers, and (3) lining by epithelium.57 Typically, the wall of the cyst is composed of all layers of the normal GI tract, including inner and outer layers of muscularis propria and myenteric and submucosal plexuses (Fig. 8.11). The mucosa is that of the segment with which the duplication is associated, but heterotopias of any other GI mucosa may be present. Cysts containing various epithelial types have also been referred to as enteric cysts (Fig. 8.12). Gastric heterotopias are most common in esophageal and small intestinal duplications but also may be seen in those of the anorectum. A summary of the key distinguishing characteristics of congenital intestinal cysts and duplications is presented in Table 8.11.
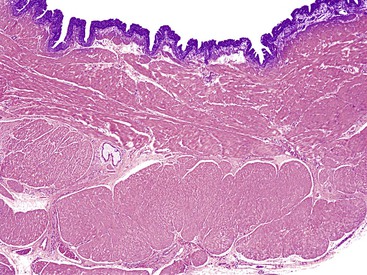
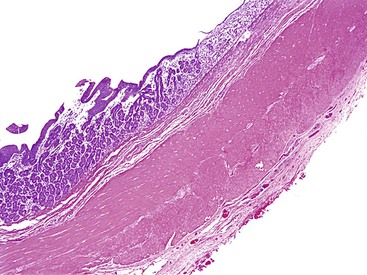
Table 8.11
Congenital Intestinal Cysts and Diverticula
| Anomaly | Embryologic Origin | Key Clinical Features | Key Pathological Features |
| Duplication cyst | Unknown | May be cystic or tubular 40% occur in terminal ileum Attached to dorsal or mesenteric side of intestinal segment May be intramural |
Composed of normal intestinal muscle layers with nerve plexuses Epithelial lining may be stratified squamous, ciliated columnar, gastric or enteric |
| Neurenteric remnant (enteric cyst) | Persistence of neurenteric canal | Posterior cyst, sinus, or fistula extending to or involving spinal cord Most common in cervical or lumbar area Vertebral anomalies in 50% Overlying cutaneous hyperpigmentation or hypertrichosis |
Composed of normal layers of gastrointestinal tract in association with neuroglial tissue |
| Tracheobronchial foregut duplication cyst | Incomplete separation of lung bud from foregut | Anterior mediastinum | Wall may contain mucous glands or cartilage Epithelial lining is ciliated columnar |
| Intestinal diverticulum | Remnant of omphalomesenteric duct | Located on antimesenteric portion of intestinal segment Most frequent in terminal ileum (Meckel diverticulum) |
Composed of all layers of the intestinal wall Mucosa enteric with frequent heterotopias |
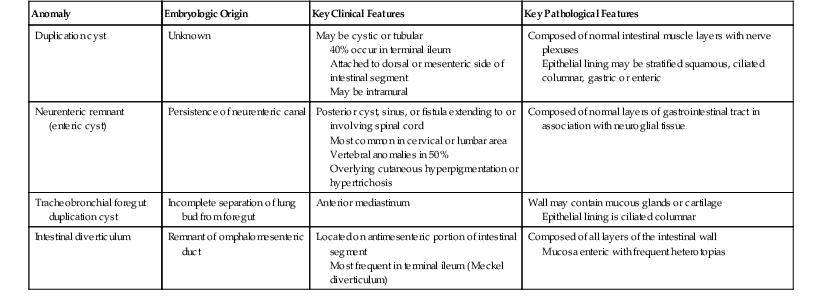
Tracheobronchial foregut duplications result from incomplete separation of the primitive lung bud from the foregut during the fourth to ninth week of gestation. They are located anteriorly and are usually lined by ciliated columnar epithelium; the wall may contain mucous glands and cartilage (Fig. 8.13). The rare esophageal duplications with intramural cartilaginous heterotopias may be confused with bronchogenic cysts. Bronchogenic cysts are attached to or adjacent to the bronchial tree, not the esophagus. The wall mimics bronchus, not esophagus, and lacks esophageal muscularis propria with myenteric and submucosal plexuses. The lumen is lined with well-differentiated respiratory mucosa and is less likely than in esophageal duplications to have extensive stratified squamous, gastric, or intestinal mucosa. Despite these differences, esophageal duplications may sometimes be difficult to differentiate from bronchogenic cysts.
Neurenteric Remnants
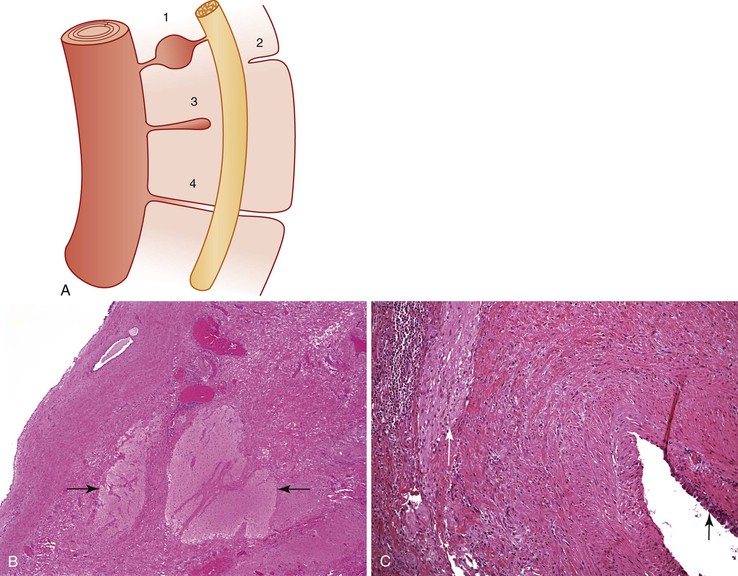
Neurenteric remnants, sometimes called the split notochord syndrome,69 include diverticula, fistulas, cysts, and fibrous cords that originate from the dorsal midline of the GI tract, extend in a cranial direction, and attach to or pass through the vertebral column and spinal cord cranial to their enteric origins; they may continue to the skin of the dorsal midline overlying the involved vertebra (Fig. 8.14, A). They may be located at any level but are most common in the cervicothoracic and lumbosacral areas.70 They tend to be slow-growing masses with a significant rate of recurrence after surgery. They are frequently associated with other features of spinal dysraphism, such as split cord malformations, and with vertebral anomalies.71 The walls of the fistulas, diverticula, and cysts are composed of all the normal layers of the gastrointestinal tract. Neuroglial and, less likely, leptomeningeal tissues are found in and around the lesions close to or involving the vertebra and spinal cord (see Fig. 8.14, B). The mucosa may be indigenous to any segment of the GI tract, and several types may coexist. The most severe forms are part of malformation complexes that are lethal to the fetus or neonate and are diagnosed during a perinatal autopsy. Symptoms and signs in living patients relate to the GI tract, central nervous system, or midline of the back. GI manifestations are similar to those of duplications described earlier and include respiratory distress, obstruction of the GI tract, obstruction of hepatobiliary and pancreatic ducts, and peptic ulceration with GI hemorrhage or perforation. Central nervous manifestations include abnormal function of the spinal cord associated with its malformations or compression, infectious meningitis associated with fistulas from the GI tract or the dorsal cutaneous surface to the meninges, and chemical meningitis due to perforation of an intraspinal cyst.
Stomach
Congenital agastria is exceptionally rare; one case documented an esophagoduodenal junction with microscopic evidence of gastric mucosa.72 Agastria can be part of the complex of anomalies in acephalic-acardiac fetuses. This condition is believed to be caused by an interruption in the normal vascular supply, as in the arterial disruption sequence in monozygotic twins with placental artery–artery shunts, resulting in absence of the head and heart, upper limbs, and foregut derivatives.73 Microgastria is a rare malformation of the stomach that is almost always associated with multiple other congenital anomalies, including limb-reduction anomalies and the VACTERL association.74–78 Children may present with a history of failure to thrive or severe gastroesophageal reflux. Nonvizualisation of the stomach on prenatal ultrasound studies may enable early diagnosis.79 Isolated microgastria is extremely rare; case reports document successful treatment by gastric augmentation.80,81 Patients can present with asplenia, hepatic symmetry, and intestinal malrotation. Microgastria is believed to result from interference with elongation of the posterior wall of the stomach, which during the fifth week grows faster than the anterior wall, forming the greater curvature. Dextroposition usually occurs in visceral situs inversus as part of asplenia–bilateral right-sidedness (Ivemark syndrome). In more than 50% of cases, the liver is symmetric, with the gallbladder, stomach, pancreas, and duodenum on the right side.73 Reviews on the clinical and genetic aspects of laterality syndromes and on the genetic control of left-right symmetry in vertebrate development are available.82–84 There are reports of isolated dextrogastria.85,86
Gastric Atresia and Stenosis
Atresia and stenosis of the stomach are rare. Fewer than 1% of all cases of atresia and stenosis of the esophagogastrointestinal tract are gastric.87 Most involve the prepyloric antrum or the pylorus. The body is rarely involved. Most are membranous. Segmental stenosis of the body may result in the hour glass stomach. Pyloric atresia associated with epidermolysis bullosa (EB-PA) results from healing of a circumferential mucosal injury by exuberant granulation tissue which fuses the apposed mucosal surfaces, occluding the narrow pyloric channel, and matures into fibrous tissue.88,89 It is caused by mutations in the ITGA6 and ITGB4 genes, which code for the corresponding units of α6β4 integrin, an adhesion molecule expressed in a variety of epithelia, including the epidermis and GI tract, where it forms an integral part of the hemidesmosomes.90 Rarely, a large intramural pyloric pancreatic heterotopia or adenomyoma causes pyloric obstruction.91
More than 50% of patients with gastric atresia have a history of polyhydramnios. Newborns with atresia or severe stenosis are seen with persistent nonbilious vomiting after their first feeding. Excessive salivation may mimic that of EA. The first stools are less voluminous than usual, and subsequent stools decrease in volume and then disappear. A single large gastric gas bubble and absence of an intestinal gas pattern are present at birth. Patients with mild stenosis may have less severe symptoms at birth, resulting in delayed diagnosis. The symptoms may be delayed for several years, making differentiation between congenital and acquired stenosis difficult. Most isolated, nonsyndromic cases are sporadic, but some are familial with an autosomal recessive inheritance pattern. A few are associated with some of the disorders of multiple GI atresias and stenoses listed in Table 8.7. Pyloric atresia with atresia of the small intestine and colon is a recognized association.92
Infantile hypertrophic pyloric stenosis (IHPS) is a clinical-pathologic entity distinct from congenital pyloric atresias and stenoses. Some reports may have applied this name to the congenital pyloric atresias and stenoses discussed earlier, causing confusion between the two.93 IHPS arises postnatally and is the most common surgical condition causing emesis in infancy. The incidence ranges from approximately 0.5 to 4 per 1000 live births; the differences in incidence probably relate largely to differences in ascertainment.94 Intrauterine and neonatal imaging studies detect no pyloric abnormalities in babies who subsequently develop IHPS, and the lesion has rarely been diagnosed in a fetus. The pathology is characterized by concentric hyperplasia, hypertrophy, fibrosis, and elastosis of the pyloric muscularis, especially the circular layer, sometimes with mucosal erosions and inflammation. The clinical presentation is characterized by the onset of projectile vomiting at 3 to 5 weeks of age and visible gastric peristaltic waves. A palpable epigastric mass with the size and consistency of an olive and a distinctive appearance on ultrasound is usually present. It is more often familial and has a higher male-to-female ratio than congenital lesions. Five genetic loci have been associated with IHPS, including the gene coding for nitric oxide synthase (NOS), IHPS1, which is located on chromosome 12q24 (review by Ranells and associates94). Abnormalities of NOS-containing neurons have been demonstrated in IHPS.95 IHPS has also been described in a number of syndromes, including Cornelia de Lange, Apert, trisomy 21, and trisomy 18 syndromes.
Gastric Duplication
Foregut duplications account for approximately 35% of all duplications of the GI tract, with gastric duplications accounting for approximately 7% (see Table 8.10). Gastric duplications involve the greater curvature. They usually do not communicate with the gastric lumen. Gastric duplication cysts are most often lined by gastric or enteric-type epithelium, although respiratory epithelium may be observed.96 Associated anomalies, including duplications in other parts of the GI tract, are common.97
Neonatal Gastric Perforation
Neonatal gastric perforation can be categorized as spontaneous or traumatic. Spontaneous cases are associated with asphyxia, very low birth weight, necrotizing enterocolitis, steroid use, or an idiopathic cause. There also appears to be an association with other GI malformations such as midgut volvulus and intestinal atresia.98 Truly idiopathic cases are rare and appear to be associated with prematurity99 and with infection of the gastric wall by invasive Candida or Staphylococcus.100 An abnormal distribution of pacemaker cells in the gastric wall, resulting in hypomotility, has been postulated in some cases.101 Traumatic cases are usually the result of gastric intubation. Infants are seen with abdominal distention, respiratory distress, and pneumoperitoneum.102
Small and Large Intestine
Hernias and Abdominal Wall Defects
Hernias represent a heterogeneous group of disorders that frequently involve the GI tract (Table 8.12). Only the first three types are discussed here; they are the ones most frequently confused with each other and are particularly relevant to the developing intestinal tract. Studies have suggested an increasing prevalence of omphalocele and gastroschisis in the United States and elsewhere, a significant finding given the relatively high mortality rate of these conditions.103
Table 8.12
Types of Hernias in Children

Data from Tovar JA. Hernias. In: Walker AW, Goulet O, Kleinman RE, et al., eds. Pediatric Gastrointestinal Disease: Pathophysiology, Diagnosis, Management. 4th ed. Hamilton, Ont.: BC Decker, 2004:573-588.
Omphalocele (Exomphalos)
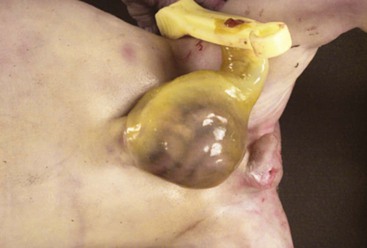
An omphalocele is a defect of the anterior abdominal wall at the insertion of the umbilical cord. It occurs in 1 of every 5,000 to 20,000 live births, but the incidence is probably higher if stillbirths and aborted fetuses are included.104 The umbilical cord at the site the defect is replaced by a sac, formed of amnion and peritoneum, which contains intestinal loops and sometimes parts of the liver (Fig. 8.15). The size of the defect can range from a few centimeters to involvement of a major portion of the anterior abdominal wall. Muscle, fascia, and skin are absent at the site of the defect.105 It is associated most characteristically with the Beckwith-Wiedemann syndrome but also with various trisomies as well as numerous single-gene defects.106,107 The cause is unknown but likely involves failure of abdominal wall closure when the intestines return to the abdominal cavity. An association with maternal smoking during pregnancy has been found.108, 109 Most cases are now diagnosed on prenatal ultrasonography. The prognosis depends on associated malformations. Successful surgical treatment usually involves excising the membrane and covering the exposed viscera in a Silastic bag with progressive reintegration of the contents into the abdominal cavity.104 Intestinal necrosis resulting in short bowel syndrome is a major complication.
Gastroschisis (Laparoschisis)
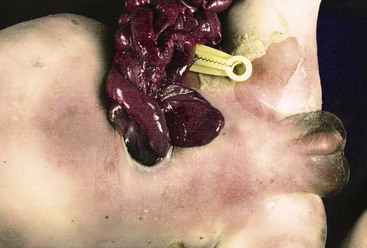
Gastroschisis results from a small (typically <5 cm) defect of the abdominal wall, just to the right of the umbilical cord insertion, that allows evisceration of bowel loops, stomach, and sometimes the gonads (Fig. 8.16). It is distinguished from an omphalocele by the absence of a sac covering the eviscerated contents and by a normal insertion of the cord. The condition has a prevalence of approximately 1 : 10,000 live births, a rate that has increased in the last 30 years and is probably higher if stillbirths and aborted fetuses are included.104 Its etiology is unknown; occlusion or disruption of the right omphalomesenteric artery has been proposed.110 Exchanges between the fetal internal environment and the amniotic fluid in utero results in fetal malnutrition and growth retardation and exposes the serosa of the loops to inflammation-generating substances, resulting in edema and thickening of the loops.104Associated malformations and abnormal karyotypes are less frequent than in infants with omphalocele. Most cases are detected by prenatal ultrasonography, and prenatal interventions such as repeated amniotic fluid exchange to remove inflammatory compounds in the amniotic fluid have been proposed.111 Elective cesarean section by 36 weeks’ gestation with early primary surgical repair appears to be the treatment of choice.112 Interruption of vascular flow in utero to a portion of the bowel may result in a focal atresia. An intraabdominal “compartment” syndrome may develop after surgical repair due to increased intraabdominal pressure and decreased vascular perfusion, leading to necrosis of variable lengths of bowel.104
Cloacal Exstrophy
Cloacal exstrophy is a very rare malformation that appears to result from rupture of the cloacal membrane and failure of descent of the urorectal septum.113 An omphalocele protrudes through a major defect in the abdominal wall and contains an exteriorized bladder and prolapsed ileum. Imperforate anus and many other abnormalities are present, including abnormal and ambiguous genitalia, separated pubic bones, and vertebral anomalies.
Congenital Atresias and Stenoses
Approximately 75% of all intestinal stenoses and 40% of all intestinal atresias are duodenal (see Table 8.6). Intrinsic duodenal atresias and stenoses most often involve the first and second portions (i.e., the foregut portion) of the duodenum, in close proximity to the entrances of the biliary and pancreatic ducts. This explains the association of duodenal atresia with hepatobiliary and pancreatic duct abnormalities.114 Approximately 75% of duodenal stenoses are immediately distal to the ampulla. The majority are membranous (Fig. 8.17). Those of the third and fourth portions are often located at the duodenojejunal junction and are associated with malrotation and midgut volvulus. Ladd bands, resulting from incomplete absorption of the cecal and ascending colon mesenteries, can cause external compression and stenosis of the duodenum; they are usually associated with associated with malrotation, preduodenal portal vein, and superior mesenteric artery syndrome.
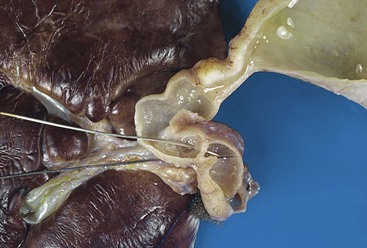
Polyhydramnios and prematurity are common in patients with duodenal atresia. Vomiting begins at birth; it is nonbilious if the obstruction is proximal to the ampulla of Vater or bilious if the obstruction is distal to the ampulla. Intrauterine bilious vomiting may be associated with ulcers of the umbilical cord, necrosis of adjacent umbilical vessels, or fatal fetal hemorrhage into the amniotic fluid. The presence of ulcers of the umbilical cord should lead to the suspicion of duodenal or proximal jejunal atresia/stenosis distal to the ampulla.115 The classic radiographic “double bubble” sign—a dilated, gas-filled duodenum proximal to the obstruction and separated by the pylorus from a dilated, gas-filled stomach—may not be seen if atresia of the esophagus or stomach is also present. The symptoms and signs of less severe stenoses may be mild and delayed, leading to delayed diagnosis and confusion with acquired obstruction. More than 50% of patients have additional congenital anomalies.116 These include annular pancreas in 33% and malrotation in 28%117 both of which may be familial.
Annular pancreas is a complete encirclement of the second part of the duodenum by pancreatic tissue (Fig. 8.18). It occurs with a frequency of 1 : 20,000 live births.118 When manifested in infancy, it is associated with other anomalies in 75% of cases, including trisomy 21, TEF, and cardiac anomalies.119 The age at presentation is determined by the severity of obstruction and by associated anomalies, with one third of cases manifesting in the neonatal period, one third during infancy, and one third later in life.118 Symptoms in older children and adults may include recurrent vomiting due to partial obstruction and pain due to gastritis and peptic ulcers.118 Peptic symptoms result from gastric overdistention caused by the partial obstruction, with consequent hypergastrinemia, hyperchlorhydria, and ulceration.120 The histology of the pancreas is unremarkable.
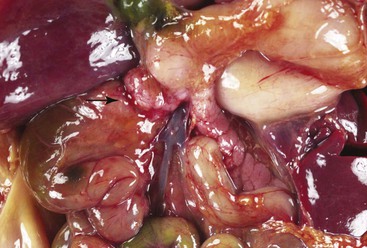
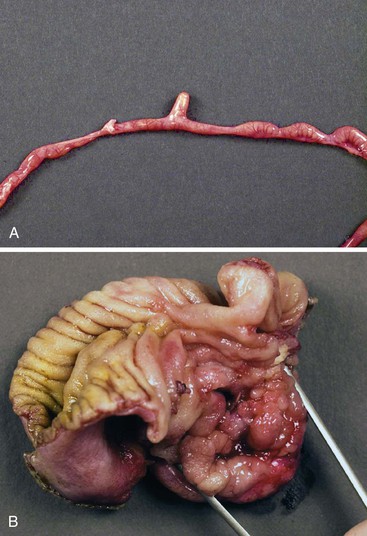
Any type of atresia or stenosis can occur in the jejunum and ileum. Approximately 95% of cases are atresias; 5% are stenoses. Atresias in which the mucosa and submucosa form an obstructing web or diaphragm (type I) account for 19% of cases; those in which the blind ends are connected by a cord (type II) account for 31%; and those with no connection between the blind ends and with a defect in the mesentery (type III) account for 46% (Fig. 8.19). Although the jejunum and ileum are about equally involved, approximately one third of atresias involve the proximal jejunum; one third, the distal ileum; and the remaining one third, all other segments. Approximately 85% are single, and 15% are multiple. These figures vary from study to study.117,121
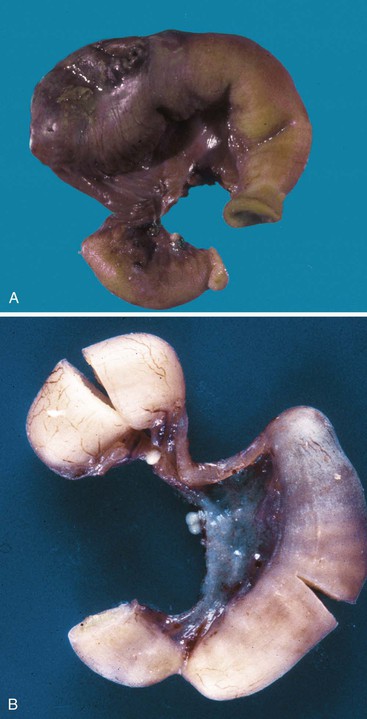
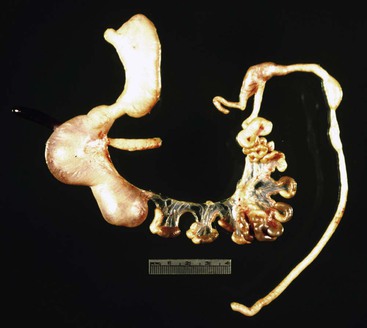
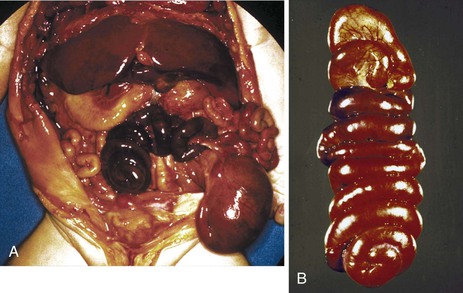
Two additional types are unique to the small intestine. The first is multiple atresias with a gross pathologic appearance that has been appropriately described as a string of sausages or string of beads (Fig. 8.20). The radiographic appearance has been described as a string of pearls.122 The total length of the small bowel is extremely short. This condition is familial and autosomal recessive in many cases. It has been associated in some instances with severe congenital immunodeficiency.123,124 The second unique type is the “apple peel” or “Christmas tree” atresia (Fig. 8.21). A long segment of jejunum and ileum is missing between the two blind ends, which are not connected by a fibrous chord, and there is a large defect in the mesentery. The dilated proximal blind end is similar to that of any atresia, but the distal blind segment is spiraled around retrograde mesenteric arteries from the ileocolic, right colic, or inferior mesenteric artery. This atresia is also often familial and autosomal recessive125 and is more frequently associated with other malformations than the usual types of jejunoileal atresia.
The clinical presentation of proximal jejunal atresia/stenosis is similar to that of duodenal atresia/stenosis distal to the ampulla. The more distal the obstruction. the less likely are polyhydramnios, immediate bilious vomiting at birth, and jaundice and the more likely are generalized abdominal distention and early obstipation. Radiographically, proximal lesions are characterized by a few dilated loops of bowel, with air-fluid levels, and distal lesions are characterized by numerous such loops. Associated congenital anomalies are less common than with atresia and stenoses in the esophagus, duodenum, rectum, and anus because many are caused by disruptions in fetuses who have had normal development during blastogenesis and organogenesis. There are many associated disorders.126 In white infants, the risk for cystic fibrosis is more than 210 times higher in those with jejunoileal atresia than in those without the atresia.127
Colonic atresia or stenosis is rare. It may be part of any multiple GI atresia syndrome (see Table 8.7). Atresia of the ileum128 or colon is seen in association with syndromic or nonsyndromic Hirschsprung disease.129 The clinical presentation is usually that of rapidly progressive abdominal distention. Failure to pass meconium is common. No meconium is found in the rectum, which instead contains mucus. Radiographs demonstrate numerous loops of distended bowel, some with air-fluid levels and with a low cutoff point. The presenting symptoms, signs, and radiographic changes of colonic atresia may be masked by an associated jejunoileal atresia.
Malrotation
Failure of the normal process of rotation and return to the abdomen of the intestinal loops may occur at any stage during the process. Normal rotation is probably a gradual, continuous process except for the return of the intestine to the abdomen and completion of the rotation, which are abrupt. Abnormalities of rotation do not correspond to any position encountered during normal rotation,11 and each case has a somewhat unique anatomy. Despite this, many authors find that use of a simplification of the thorough classification of Estrada10 facilitates description of these malformations. The three types frequently listed are nonrotation, mixed rotation, and reverse rotation. The position of the third portion of the duodenum relative to the SMA is crucial in differentiating these anomalies from normal intestine and from each other.
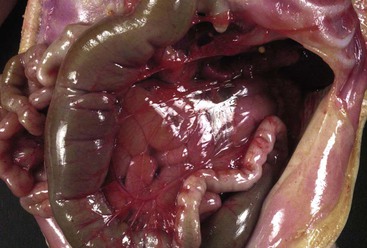
Complete failure of rotation (i.e., nonrotation) results in a right-sided jejunum and ileum, a low midabdominal cecum, and a colon residing in the left abdomen. Incomplete or mixed rotation, which is more common, results in the small bowel occupying mainly the right side of the abdomen, with the cecum usually residing in the right upper quadrant (Fig. 8.22). All cases are associated with a short mesenteric attachment and midgut volvulus. With mixed rotation, the volvulus may occur during the first few days of life, whereas in the other types, symptoms may be delayed or intermittent. The short mesenteric attachment can lead to twisting of the midgut around the pedicle of the SMA, with consequent bowel obstruction and vascular compromise. An unattached cecum, ascending colon, and hepatic flexure may be associated with Ladd bands that bind the hepatic flexure to the third portion of the duodenum and to the right abdominal wall, sometimes resulting in duodenal obstruction. Reverse rotation, the rarest type, occurs consequent to two errors of reentry of the bowel loops after the combined 270-degree counterclockwise rotation outside the abdominal cavity: (1) If the postarterial segment returns first, the small bowel will lie ventral to the colon and SMA, with the colon behind. This may result in compression of the mesenteric arterial flow.130 (2) If the prearterial segment returns first, the colon will occupy the right half of the abdomen, and the small bowel will lie in the left side. These cases can be associated with situs anomalies of other organs.131,132
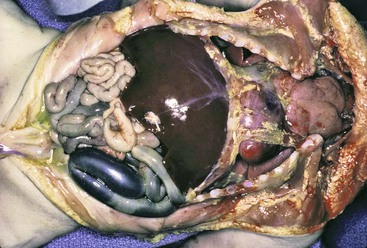
Abnormal fixation and abnormal mesenteries of various segments form internal hernias. Right and left mesocolic hernias are two common forms. An unusually long mesentery of an unfixed cecum and ascending colon attached along or to the right of the midline may be so redundant as to form an internal hernia pouch, called a right mesocolic hernia. If, as in the case of nonrotation, the small bowel is in the right side of the abdomen, it can become entrapped in the right mesocolic internal hernia, resulting in obstruction and strangulation. Similarly, an unusually long mesentery of an unfixed descending colon attached along the left side of the abdomen may be so redundant as to form a left mesocolic internal hernia (Fig. 8.23). If the small bowel is located in the left abdomen, it can become entrapped in the left mesocolic hernia, resulting in obstruction and strangulation.
Malrotation occurs in approximately 3 of every 10,000 live births and fetal deaths,133 and 60% to 90% have associated malformations.133–135
Duplications
As with foregut duplications, intestinal duplications are directly contiguous with the segment with which they are associated. They can occur anywhere along the GI tract, although the ileum is the most frequent site.131 Duplications are dorsal to the intestine, usually mesenteric, and sometimes intramural.136 Duodenal duplications involve the concave medial border of the second portion, adjacent to the hepatobiliary and pancreatic ductal systems. Cystic duodenal duplications may clinically mimic choledochal cysts, cysts of the pancreatic duct, or pancreatic pseudocysts. Jejunal, ileal, and colonic duplications may cause intussusception or volvulus.
Tubular duplications may be short, or they may involve entire segments, such as the entire esophagus and stomach combined or the distal ileum, cecum, entire colon, and anus. Most are attached along the dorsal or mesenteric border (Fig. 8.24). Some are attached to the lateral border and form parallel, side-by-side segments referred to as double-barrel duplications. Rarely, the duplication has a separate mesentery and blood supply and is called a loop duplication. Communications between the lumen of a tubular duplication and the normal lumen are common and may be located at the proximal or distal end of the duplication or at both ends. Multiple communications may occur. Double-barrel duplications of the colon, rectum, and anus are associated with duplications of part or all of the genitourinary tracts and external genitalia, forming symmetric or asymmetric right and left GI and genitourinary tracts and perineums. A tubular duplication with a proximal communication and a blind distal end forms an expanding mass that may obstruct the normal lumen or adjacent structures. Similarly, a cystic duplication may cause obstruction of the involved segment (Fig. 8.25).
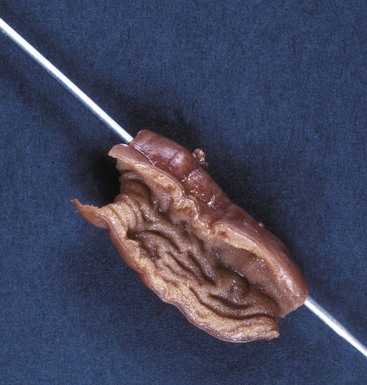
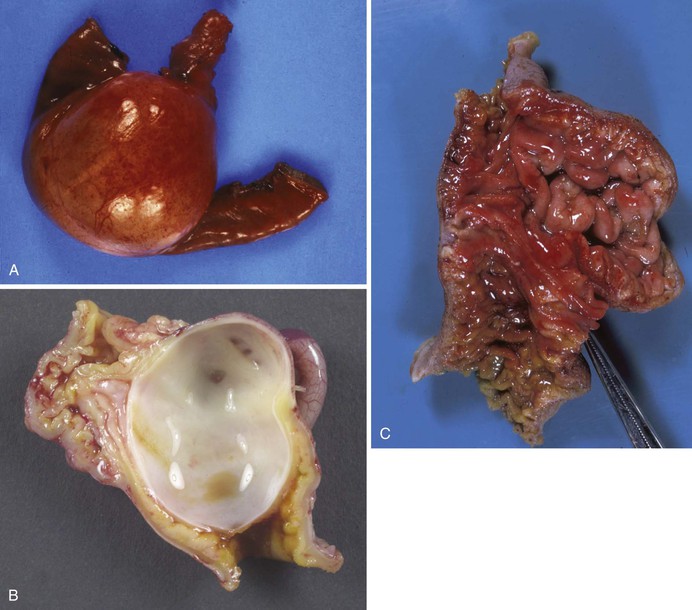
The wall of a duplication is usually thick, with well-formed muscle layers. The epithelial lining may consist of gastric, intestinal, or respiratory-type epithelium and may contain heterotopic pancreatic rests. Complete excision is the treatment of choice. The embryogenesis of duplications is not well understood. An abnormally exuberant proliferative phase, defective resolution of the proliferative phase, and persistence of the normal transient diverticula seen in embryos are some possibilities. Some may be caused by mild abnormalities of development of the notochord, neurenteric canal, or related structures of the midline developmental field and therefore may be a forme fruste of neurenteric cysts.
Congenital Diverticula, Including Omphalomesenteric Remnants and Meckel Diverticulum
GI diverticula can be divided into mesenteric and antimesenteric types. Mesenteric diverticula are thought to be primarily related to the neurenteric remnants and duplications discussed earlier. Antimesenteric diverticula are, with few exceptions, part of the spectrum of omphalomesenteric remnants that includes Meckel diverticulum and is summarized in Table 8.13 and Fig. 8.26.
Table 8.13
The Spectrum of Omphalomesenteric Remnants
| %* | |
| Meckel diverticulum | 90 |
| Tip unattached | — |
| Tip attached to umbilicus | — |
| Tip attached to mesentery | — |
| Solid cord | 5 |
| Fistula | 3 |
| Cyst | 1 |
| Intraabdominal within solid cord | — |
| In abdominal wall at umbilicus | — |
| Umbilical remnant | 1 |
| Umbilical polyp | — |
| Umbilical sinus | — |
| Total | 100 |
| Omphalomesenteric vessel remnants | — |
| From mesentery to umbilicus | — |
| From Meckel diverticulum to umbilicus | — |
| From Meckel diverticulum to mesentery | — |
* Percentages are estimates.
Data from Huff DS. Developmental anatomy and anomalies of the gastrointestinal tract, with involvement in major malformative syndromes. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. New York: Springer; 2004:3-37; Moses WR. Meckel’s diverticulum: a report of 2 unusual cases. N Engl J Med. 1947;182:251-253; Skandalakis JE, Gray SW, Ricketts R. The esophagus. In: Skandalakis JE, Gray SE, eds. Embryology for Surgeons. Baltimore: Williams & Wilkins; 1994:65-112; and Soderland S. Meckel’s diverticulum: a clinical and histological study. Acta Chir Scand. 1959;118(suppl):1-233.
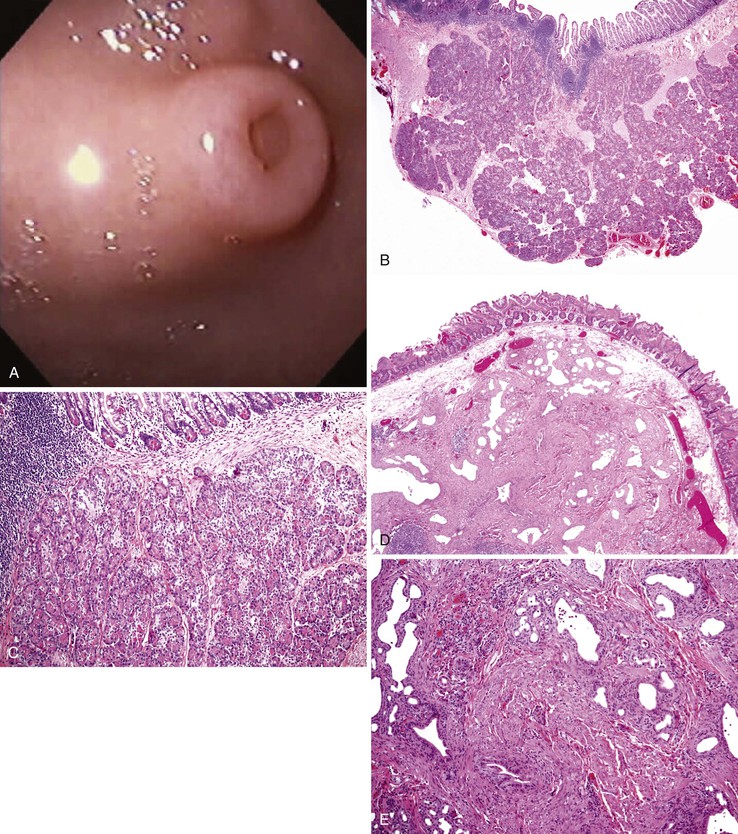
The omphalomesenteric (vitelline) duct is the last point to close after separation of the intestine from the yolk sac; it usually does so by the 10th week of embryonic life.105 The entire duct may remain patent, and it may drain intestinal contents at the umbilicus. The more common Meckel diverticulum results from persistence of the vitelline duct immediately adjacent to the bowel wall; it occurs in approximately 2% of individuals and accounts for approximately 90% of all omphalomesenteric remnants. The other variations are rare and are listed in Table 8.13. The remnants are located 15 to 167 cm proximal to the ileocecal valve, but most are 40 cm from the valve.137 Seventy-five percent of diverticula are between 1 and 5 cm long, but some are up to 26 cm long.138 Meckel diverticula are composed of all layers of the normal intestinal wall, as are most cysts and fistulas. The mucosa is ileal. Heterotopias are common. Gastric mucosa is seen in half of all Meckel diverticula (Fig. 8.27), in 62% of those with diverticulitis, in one third of fistulas, and occasionally in umbilical polyps. In diverticula, 94% of the gastric heterotopias are of fundal type; in fistulas and umbilical polyps, 70% are fundal.139 Pancreatic heterotopias are found in 5% of Meckel diverticula, where they can often be identified at the tip by gross examination, and occasionally in umbilical polyps.44 Duodenal, colonic, and, rarely, biliary mucosa can be found. Omphalomesenteric remnants can be distinguished from urachal remnants by the presence of columnar or intestinal epithelium in the former, whereas the latter are lined by transitional epithelium.
Twenty-five percent of omphalomesenteric remnants are symptomatic.140 Fundal-type gastric mucosa causes ulcers that manifest with pain, hemorrhage, and perforation leading to peritonitis. Meckel diverticulitis mimics appendicitis. Intussusception, volvulus around a solid cord or vascular remnant to the umbilicus, incarceration by a fibrous cord or vascular remnant, a knot formed by a long diverticulum around a loop of intestine—all lead to obstruction. Fistulas may drain intestinal content and mucus at the umbilicus. Omphalomesenteric remnants are common in patients with multiple congenital anomalies, but other anomalies are infrequent in otherwise normal patients with a Meckel diverticulum.
Heterotopias
Heterotopic gastric mucosa can occur in duplications and diverticula of the alimentary tract, and more rarely as isolated lesions. They have been documented from the oropharynx to the rectum and can manifest grossly as nodules, polyps, or erosions.141 The heterotopias can secrete gastric acid, leading to inflammation, bleeding, perforation, and intussusceptions.142 They can be detected by technetium 99m (99mTc pertechnetate scintigraphy. Surgical excision is the treatment of choice.
Heterotopic pancreatic tissue is most commonly observed in the stomach and proximal small bowel143,144 (Fig. 8.28), although it may be seen in the liver, spleen, umbilicus, and other sites. Heterotopic pancreatic tissue also affects small bowel stenoses, duplications, and diverticula. Most patients are asymptomatic.144 Rare cases of adenocarcinoma arising in pancreatic heterotopias have been documented.145 Histologically, they consist mostly of ducts and acini. A closely related lesion, consisting of ducts in association with smooth muscle hyperplasia and sometimes referred to as an adenomyoma, is also considered to be a pancreatic heterotopia.146
Anorectal Malformations
Anorectal anomalies constitute an important group of malformations that occur in approximately 1 of every 4000 live births, with one third being isolated and the rest associated with other anomalies.147 Approximately 60% occur in males and 40% in females. They have been associated with mutations in almost every chromosome, although trisomy 21 and microdeletion of chromosome 22q11.2 appear to be the most frequent.147 The pathogenesis and embryologic events leading to these malformations are poorly understood. The early hindgut is the cloaca, a cavity formed at approximately 21 days’ gestation into which the hindgut, tailgut, allantois, and mesonepric ducts empty. It is believed that, during the sixth to seventh weeks of gestation, by a process of differential growth and apoptosis, the urorectal septum (which divides the hindgut and the allantois) proliferates caudally toward the cloacal membrane (which forms the junction between endoderm and ectoderm) and fuses with it, resulting in separate urogenital and anorectal sinuses. Perforation of the cloacal membrane occurs at approximately day 50. Interruptions of any of these steps can result in anorectal anomalies.148 Some studies suggest that, contrary to the traditional concepts, urorectal separation is not accomplished by the active caudal growth of the urorectal septum and that, in fact, the urorectal septum is only passively involved in the process.149 There is increasing evidence that the Hedgehog family of genes, particularly sonic hedgehog (SHH), and the homeobox family play a role in these anomalies.150
Several classifications of the many variations within the spectrum of anorectal atresias and fistulas have been proposed.151–153 The classification presented in Table 8.14 is adapted from the Wingspread classification153 and that of Kiely and Pena.154 Presumably, the higher the lesion, the earlier the time of onset and the more severe the abnormality of development.155 The list is arranged in descending order of level of the lesion (highest level at the top, lowest at the bottom). The interposition of the uterus and vagina between the anorectum and the urinary tract during the 9th and 10th weeks of gestation explains the difference in the fistulas seen in males and females. In males, two thirds are high lesions, and in females, two thirds are low lesions.151
Table 8.14
Types of Anorectal Atresias and Fistulas
| Atresia | Fistula | |
| Male | Female | |
| Anorectal agenesis | Rectocloacal | Rectocloacal |
| Rectovesicle | Rectovesicle | |
| Rectoprostatic urethra | Rectovaginal, high | |
| Without fistula | Without fistula | |
| Rectal atresia | Without fistula | Without fistula |
| Anal agenesis | Rectobulbar urethra | Rectovaginal, low rectovestibular |
| Without fistula | Without fistula | |
| Imperforated anus | Anoraphe, perineal or scrotal | Anovestibular |
| Anocutaneous | Anocutaneous | |
| Anal stenosis | Anocutaneous | Anocutaneous |
| None | Anterior anus | Anterior anus |

Data from Huff DS. Developmental anatomy and anomalies of the gastrointestinal tract, with involvement in major malformative syndromes. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. New York: Springer; 2004:3-37.
Rectal atresia is a high, blind-ending rectum with a normal-appearing perineum, an intact anus, and a blind-ending anal canal. Anorectal agenesis is a high, blind-ending rectum and an abnormal perineum with absence of the anus and anal canal (Fig. 8.29). Anal agenesis is a low, blind-ending rectum and an abnormal-appearing perineum with absence of the anus and anal canal. The most severe form of anorectal atresia is persistent cloaca with or without a perineal opening. It is rare, more common in females, and there are many variations. The colon, genital tract, and urinary tract join together to form a common structure (Fig. 8.30). If the perineal outlet is atretic or severely stenotic, the cloaca may be massively cystic.
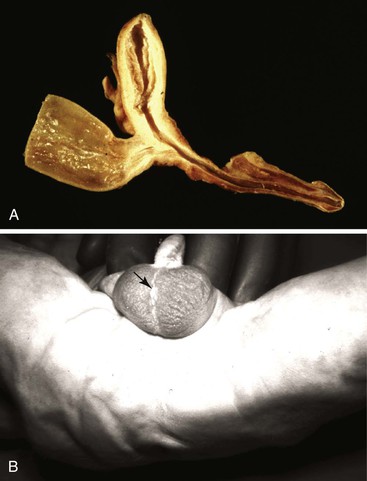
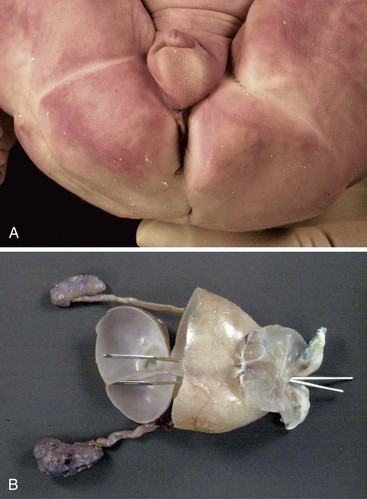
The patients are seen with failure to pass meconium. In most, the anus is absent, severely abnormal, or displaced anteriorly. Patients with high lesions have a higher incidence of associated malformations than those with low lesions; the incidence is highest in patients with cloacal anomalies. Congenital heart disease, usually tetralogy of Fallot or ventricular septal defect, is seen in approximately 20%. GI anomalies are seen in 10% to 15%. Tracheoesophageal malformations predominate, and duodenal atresia and stenosis, malrotation, small and large intestinal atresia, and Hirschsprung disease are also found. Urinary tract anomalies, often vesicoureteral reflux or renal agenesis or dysplasia, are among the most common associated anomalies and have been reported in as many as 60% in some series. Genital anomalies are as frequent as urinary anomalies and include cryptorchidism and hypospadius in males and bicornuate or septate uterus and septate vagina in females. Patterns of associated anomalies include multiple intestinal atresias, the CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital abnormality, and ear abnormality) and VACTERL associations, and many others, as listed by Huff126 and by Roberts and colleagues.105
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Congenital and Developmental Disorders of the Gastrointestinal Tract
Dale Huff
Pierre Russo
Molecular Mechanisms of Gastrointestinal Development
Recent advances in understanding of the molecular controls of gut development have flowed from studies of a number of vertebrate and invertebrate models, including Caenorhabditis elegans, Drosophila, sea urchins, zebrafish, and mice. These studies have provided insights into the genetic mechanisms that direct formation and modeling of the gastrointestinal (GI) tract, highlighted the importance of endodermal-mesenchymal interactions, and demonstrated the high degree of phylogenetic conservation of these mechanisms.
Gut development is controlled by a number of intercellular signaling pathways, such as the bone morphogenetic protein (BMP), Notch, Homeobox, Hedgehog, and Wnt pathways. A discussion of these studies is beyond the scope of this chapter, and several excellent reviews are available.1–4 Early development of the endoderm depends on molecular signaling pathways such as that of Wnt pathway, which acts by stabilizing β-catenin, allowing it to translocate to the nucleus to activate transcription genes. Ablation of β-catenin in the notochord and primitive streak abrogates endoderm formation.5 In Drosophila and in the mouse, there are regional differences in the specific expression of Homeobox (Hox) genes along the gut axis.2,4 For example, hindgut defects in mice can be linked to defective expression of Hoxd-13.4
Another family of signaling genes critical in cellular cross-talk is the Hedgehog (Hh) family, which appears to be essential to anterior-posterior, dorsal-ventral, and radial patterning. Knockout and transgenic mouse models of various hedgehog components result in a variety of malformed phenotypes, ranging from esophageal atresia (EA) to persistent cloaca.6 Vertebrate homologs of Hh exist in three forms: sonic (Shh), Indian (Ihh) and Desert (Dhh), which have different but overlapping expression patterns. For example, Shh−/− mutant embryos die in utero and have overgrown duodenal villi resulting in occlusion, analogous to duodenal stenosis in humans. Selective postnatal blocking of Hh signaling resulted in a wasting and runted phenotype characterized by diarrhea with disorganized intestinal villi, hyperplastic crypts, and enterocyte vacuolization.6
Epigenetic factors may also contribute to phenotypic development as well as disease susceptibility in genetically identical individuals. For example, different degrees of methylation of CpG groups in the agouti mouse, which can vary according to maternal intake of B group vitamins, may result in variation in coat colors.7 Epigenetic factors such as diet and the development of the microbiome appear to play a major role, especially in postnatal development of the GI tract.4
Embryology and Anatomic Development of the GI tract
The development of the GI tract proceeds through three major overlapping steps: formation of the gut tube during blastogenesis, differentiation of the specific segments of the digestive tract and its accessory organs during organogenesis, and histogenesis of the individual organs with their specialized cell types.2 Major developmental milestones are outlined in Table 8.1. The first two steps take place during the embryonic period, which begins on the day of fertilization and ends on the 56th postconceptual day (week 8). The developing human is more susceptible to teratogenetic agents during the embryonic period than at any other period of development. The fetal period, which begins on postconceptual day 57 and ends at birth, is characterized by the final stages of rotation and fixation and by continued elongation and histogenesis of the GI tract. By 15 to 20 weeks’ gestation, the fetal gut essentially resembles that of the newborn.1 An overview of these basic processes, especially pertaining to the GI tract, is presented in Table 8.2. The pattern of congenital anomalies of the GI tract varies depending on the developmental period from which they arise (Table 8.3).
Table 8.1
Developmental Milestones
| Event | Time of First Expression (Weeks after Conception) |
| Gastrulation | 3 |
| Gut tube largely closed | 4 |
| Liver and pancreas buds Growth of intestines into cord |
4 7 |
| Intestinal villus formation | 8 |
| Retraction of intestines into abdominal cavity | 10 |
| Organ formation complete | 12 |
| Parietal cells detectable, pancreatic islets appear, bile secretion, intestinal enzymes detectable | 12 |
| Swallowing detectable | 16 and 17 |
| Mature motility | 36 |
Data from Montgomery RK, Mulberg AE, Grand RJ. Development of the human gastrointestinal tract: twenty years of progress. Gastroenterology. 1999;116:702-731.
Table 8.2
Overview of Gastrointestinal Development in the First 10 Weeks after Conception
| Feature | Embryo | Fetus | ||||||||
| Blastogenesis | Organogenesis | |||||||||
| week | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Bilaminar disc; endoderm | X | |||||||||
| Yolk sac; connecting stalk | X | |||||||||
| Trilaminar disc | X | |||||||||
| Early: Midline developmental field; induction of gastrointestinal patterning | X | |||||||||
| Late: Primitive foregut, midgut, hindgut; respiratory and hepatic primordia | X | |||||||||
| Cylindrical embryo; definitive tubular gastrointestinal tract; umbilical cord | X | |||||||||
| Beginning intestinal rotation | X | |||||||||
| Remodeling, growth, histogenesis; anus completed | X | X | X | X | ||||||
| Return of bowel to abdomen; final rotation and fixation | X | |||||||||
| Sexual differentiation of perineum | X | |||||||||
Table 8.3
Patterns of Congenital Anomalies Arising during Various Developmental Periods
| Developmental Period | Age (Weeks) | Developmental Events | Congenital Anomalies |
| Embryo | |||
| Early blastogenesis | 1-2 | Basic patterning of body: dorsoventrality, rostrocaudal axis, laterality | Lethal to embryo: empty chorionic sac and global embryonic growth disorganization |
| Late blastogenesis | 3-4 | Midline developmental field Right/left sidedness (visceral situs) Induction of endodermal primordia Basic patterning of the gastrointestinal tract |
Lethal to embryo: empty chorionic sac and global embryonic growth disorganization Severe gastrointestinal anomalies as part of extensive, sometimes monstrous maldevelopment of embryo, not necessarily lethal |
| Organogenesis | 5-8 | Differentiation of endodermal primordia into specific segments of gastrointestinal tract and accessory digestive organs Histogenesis |
Isolated anomalies of gastrointestinal tract Disruptions and deformations |
| Fetus | |||
| 9-10 | Return and final rotation and fixation of intestinal loop Sexual differentiation of perineum |
Isolated abnormalities of rotation and fixation Disruptions and deformations |
|
| 11-34 | Histogenesis, remodeling, growth |
From Huff DS. Developmental anatomy and anomalies of the gastrointestinal tract, with involvement in major malformative syndromes. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. New York: Springer; 2004:3-37.
Blastogenesis extends from fertilization to day 28. During the first half of blastogenesis, the bilaminar disc and the basic body plan of dorsoventrality, rostrocaudal axis, and laterality are established. During the second half of blastogenesis, the midline developmental field directs the process of gastrulation, which establishes all three germ layers (endoderm, mesoderm, and ectoderm). The mammalian digestive system is derived from each of these layers—the epithelial lining from the endoderm, the muscle layers and supportive elements from the mesoderm, and the neurons of the enteric nervous system from the ectoderm. It is during this period that the basic plan of the GI tract is established through the inductive influences of the notochord, primitive streak, emerging mesoderm, and other anatomic components of the midline developmental field on the primitive endoderm. These inductive influences predetermine the sites of the specific segments of the GI tract and the primordia of its accessory organs of digestion. For example, at the end of gastrulation in mice, further patterning of the endoderm is determined by regional expression of factors such as Sox2 and Hhex in the anterior endoderm and Cdx2 in the posterior endoderm.3
Simultaneously, during the third and fourth weeks of development, cephalocaudad and lateral folding of the embryo converts the trilaminar germ disc into an elongated cylinder. The primitive gut is at that point somewhat arbitrarily divided into three major segments: a cranial foregut, a midgut open to the yolk sac via the vitelline duct, and a hindgut (Fig. 8.1). Each of these segments will give rise to specialized regions of the gut and, as in the case of the foregut, to other organs such as the thyroid, lungs, liver, and pancreas. The blood supply to the primitive gut is derived from the vitelline arteries of the yolk sac. The celiac artery, superior mesenteric artery (SMA), and inferior mesenteric artery vascularize the abdominal foregut, midgut, and hindgut, respectively, and by convention determine the boundaries of each (Fig. 8.2).
Organogenesis extends from day 29 to day 56 (weeks 5 through 8). Suddenly, during the fifth week, the entire tubular GI tract, its major divisions, and its accessory organs of digestion, having been predetermined during blastogenesis, emerge from the imprinted primordium of the primitive endodermal tube. The abdominal portion of the foregut is divided into the esophagus, stomach, and proximal duodenum. The common origin of the trachea and esophagus from the foregut results in various types of fistulas if separation is incomplete. The hepatic diverticulum arises from the proximal duodenum, its cephalic portion budding into the transverse septum (precursor of the diaphragm) to become the liver, and its caudal portion giving rise to the gallbladder and the extrahepatic biliary tree. Dorsal and ventral pancreatic buds also emerge from the proximal duodenum.
It is generally agreed and illustrated in most embryology texts that further development and growth of the gut involves a counterclockwise rotation of 270 degrees during development (Fig. 8.3). However, the exact timing of these events is still subject to disagreement.8 The initial event appears to be herniation of the developing gut into the stalk of the yolk sac during the sixth week, likely because elongation of the midgut proceeds much faster than growth of the embryo and because the concomitant rapid growth of the liver displaces the intestines from the abdominal cavity. As the midgut elongates, it rotates 90 degrees counterclockwise (as viewed from the front of the embryo) around the axis of the SMA, so that the cranial limb (“prearterial” in relation to the SMA) moves to the embryo’s right and the caudal limb (“postarterial”) moves to the embryo’s left (see Fig. 8.3, A and B). Continued elongation, especially of the prearterial segment, results in a series of folds called the jejunoileal loops, the identity of which Keibel believed was retained in the adult.9 The postarterial loop, most of which will form the colon, remains relatively straight. At approximately 10 weeks of life, the intestines return to the abdominal cavity. The factors responsible for this step are unclear, but decrease in the relative growth of the liver and rapid expansion of the abdominal cavity have been suggested. As the gut returns, there is a further anticlockwise, 180-degree rotation, which, added to the previous rotation, makes a total of 270 degrees (see Fig. 8.3, C). Alternatively, some authors have suggested that the gut rotates 180 degrees in the cord followed by a 90-degree rotation during return.8
As a net result of this 270-degree rotation, the third portion of the duodenum passes horizontally caudal and dorsal to the artery, and the proximal anchoring point comes to lie near the final position of the ligament of Treitz to the left of the artery. The SMA hangs over the ventral wall of the third portion of the duodenum. As the distal limb then rapidly returns, it swings ventral and rostral to the proximal loop, and the cecum comes to lie in the right abdomen near the liver (see Fig. 8.3, D). Rotation is completed by the 10th week, and fixation continues throughout fetal life as the mesenteries become adherent to the parietal peritoneum. The cecum and liver then separate by unknown mechanisms, and the increasing distance becomes occupied by the lengthening ascending colon, with the final position of the liver being the right upper quadrant and that of the cecum being the right lower quadrant (see Fig. 8.3, E). This separation is referred to, probably incorrectly, as “cecal descent.” See Estrada10 for an extensive review and Kluth and colleagues11 for a recent reevaluation of these events.
Mucosal histogenesis transforms the primitive undifferentiated epithelium of the gut tube into the specific epithelia of the final differentiated segments of the digestive tract. Although histogenesis begins in the late embryonic period, most of the histologic transformation occurs during fetal life. It begins with a transient phase of epithelial proliferation. The proliferating epithelium completely occludes the lumen of the duodenum, significantly narrows the lumen of the esophagus, and may mildly narrow the lumen of the cardia, pylorus, upper jejunum, and distal ileum. These proliferations may be accompanied by the transient formation of multiple antimesenteric diverticula in the duodenum, upper jejunum, and distal ileum. Some instances of congenital atresia, stenosis, or diverticula may be the result of abnormalities in the formation or resolution of the proliferative phase.
Esophagus
Ciliated columnar epithelium covers the epithelial surface of the midesophagus at 10 weeks and spreads to both ends by the 11th week (Fig. 8.4). Stratified squamous epithelium begins to replace the ciliated columnar epithelium in the midesophagus at 16 weeks and spreads proximally and distally to cover the entire esophagus by birth, except for the proximal esophagus, where islands of ciliated columnar epithelium may persist, disappearing shortly after birth.12–14 Intestinal goblet cells in the distal esophagus have been rarely observed in the neonate and fetus, although positive staining with acidic mucins at the squamocolumnar junction in this age group is common.15 Residual embryonic cells that are induced to proliferate after damage to the squamous epithelium have been postulated to be the source of Barrett metaplasia.16
Pancreatic acinar tissue has been observed in young children at the gastroesophageal junction, independent of Barrett esophagus, esophagitis, or gastritis.17 The superficial cardiac glands of the lamina propria appear during the 13th week. The submucosal mucous glands appear in the 27th week. The circular muscle layer is present at 8 weeks and the longitudinal layer at approximately 13 weeks, followed by the muscularis mucosae.
The waves of differentiation begin in the esophagus, propagating caudally, and at the anorectal junction, propagating cranially; the two meet at the ileocecal junction. Ganglion cells become recognizable by 8 weeks. Their density peaks between 16 and 20 weeks, decreases with increasing gestational age until 30 weeks, then becomes constant.18 The timing of development of the muscle layers and myenteric plexuses in the various segments of the GI tract is outlined in Table 8.4.
Table 8.4
Histogenesis of the Muscular Coats and Myenteric Plexus
| Layer | Esophagus | Stomach | Small Bowel, Distal | Colon | |
| Proximal | Distal | ||||
| Circular | 6 | 7 | 7 | 9 | 8 |
| Longitudinal | 8 | 11 | 10 | 26 | 11 |
| Muscularis mucosae | 12 | 14 | 21 | 26 | 21 |
| Myenteric plexus | 6 | 7 | 7 | 8 | 12 |
| Bidirectional peristalsis | — | — | 12 | 12 | — |
| Unidirectional peristalsis | — | — | 30 | 30 | — |
Stomach
During the fifth week of life, differential growth of the dorsal wall of the stomach results in formation of the greater curvature. Subsequent rotation of the stomach 90 degrees along a craniocaudal axis during the seventh week, followed by fixation of the second part of the duodenum to the dorsal body wall, forms the lesser sac of the peritoneal cavity. Prenatal ultrasound examinations have shown that the stomach continues to grow in a linear fashion from 13 to 39 weeks. Studies of the development of the mouse stomach have established that epithelial stem cells of the gastric pits reside in the neck region, producing different cell populations that move either upward or downward.19,20
Intestinal villi appear in the cardia and pylorus, where they are normally abundant by 30 weeks (Fig. 8.5). They disappear by birth. Intestinal metaplasia of cardiac or pyloric epithelium may represent a dedifferentiation to the normal fetal condition. The cardiac mucosa is thought to arise from undifferentiated gastric mucosa and not from esophageal metaplasia.21 The development of the gastric glands occurs early during fetal life. Glandular pits are formed during the 11th to 12th weeks of fetal life, along with emergence of the first cells of the parietal lineage. By 15 to 17 weeks of gestation, the fetal gastric glands are essentially similar to an adult’s, with compartmentalization into foveolus, isthmus, neck, and base containing the various phenotypically differentiated cell types.1
Further development of the stomach involves thickening of the glandular region with proliferation and maturation of the chief cells, which are relatively fewer in the neonatal stomach than in the adult stomach and which do not produce pepsin in the newborn.22 Gastric pH is relatively high in the neonate; it becomes comparable to that of adults by the age of 2 years. This may result in part from buffering by amniotic fluid but also from a relative lack of gastrin, levels of which increase during the first few postnatal months.22
Small and Large Intestine
Rearrangement of the endodermal epithelium resulting from elongation of the gut tube, rather than epithelial proliferation as previously thought, leads to temporary occlusion of the lumen by the end of the sixth week.23 Defects in subsequent recanalization of the lumen can result in stenoses or duplications of the digestive tract. As the lumen expands, the epithelium undergoes folding, which will eventually lead to the formation of villi. Mesenchymal cells grow toward the lumen to form early villi, and this process is orchestrated by an elaborate endodermal-mesodermal cross-talk under the control of signaling pathways including the BMP, Hedgehog, platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), and Wnt pathways and the mesenchymal transcription factors FoxL1, FoxF1, and FoxF2.3
Villi and crypts appear first in the duodenum in the 8th week, spread to the middle of the small intestine by the 9th week, and reach the distal ileum by the 12th week. The early intestinal mucosa consists of stratified epithelium; columnar epithelium appears gradually, first at the apices and then along the sides of villi. By the 10th week, only intervillus epithelium remains stratified.2 By 9 to 10 weeks, absorptive cells of the proximal intestine display a brush border with an array of microvilli.24 Eosinophilic globules frequently can be observed within the fetal intestinal epithelium (Fig. 8.6); these have been referred to as “thanatosomes,” and seem to reflect apoptotic activity.25
Both Wnt and BMP signaling pathways appear to be involved in the formation of crypts.3 These crypts contain the stem cells that will serve as the source of the epithelial cells, which are renewed every 4 days.4 Cytologic differentiation of the crypts begins with the appearance of goblet cells in the eighth week, followed by Paneth cells and enteroendocrine cells in the ninth week. Clusters of enteroendocrine cells occurring on top of villi in the duodenum and upper jejunum during the 20th week of gestation have been described as Segi’s cap.26
The Notch pathway plays a critical role in epithelial differentiation by regulating the specification of absorptive versus secretory lineages, which is controlled by differences in expression of factors such as Hes1, Atoh1, and Neurog3.3 Atoh1 (also called Math1) is a basic loop-helix-loop transcription factor that appears to be a key regulator of the development of secretory cells (goblet, Paneth, and enteroendocrine cells), whereas absorptive cells appear to be Math1 independent.27 Neonates have been reported with an absence of gut secretory cells.28 Patients with mutations in a gene called neurogenin 3 (i.e., NEUROG3) have presented with malabsorptive diarrhea and a complete absence of enteroendocrine cells.29 The time of appearance and electronmicroscopic features of 13 enteroendocrine cells have been described in human embryos between 9 and 22 weeks’ gestation.30 Brunner glands appear in the proximal duodenum in the 12th week. The histologic appearance of the small intestine resembles that of a newborn by 20 weeks.31
The enteric nervous system results from contributions from the sympathetic system, growing along the arterial supply, and from the parasympathetic system, with branches of the vagal nerve innervating the upper GI tract; the pelvic splanchnic nerves innervate the descending colon and rectum. Postganglionic neurons are derived from neural crest cells, which appear in the fetal gut at approximately 8 weeks’ gestation and migrate in a craniocaudal direction; they may be detected in the rectum by 12 weeks.2 The development of the enteric nervous system is beyond the scope of this chapter, and recent references should be consulted.32–36
Similar to the intestinal epithelium, the colonic mucosa consists of a stratified epithelium beginning at about 8 weeks. At approximately the 10th week, villi with developing crypts cover the surface of the large intestine and persist until the 28th week. Therefore, intestinal villi are normally seen in the embryo not only in the small intestine but also in the cardia, pylorus, and colon (Fig. 8.7). The intervillous surface epithelium differentiates into a single layer containing goblet cells by the 13th to 16th week. After birth, a 100-fold increase in the number of intestinal crypts occur, along with an expansion of crypt cells.22 An outline of the histogenesis of the muscular coats and myenteric plexus is presented in Table 8.4.
The development of the mucosal lymphoid system is outlined in Table 8.5. The fetal mucosal lymphoid system has the capacity to respond to an abnormal intrauterine antigenic stimulus with the formation of germinal centers and plasma cells possibly as early as 20 weeks. The neonate responds to the antigenic stimulus of colonization at birth with the formation of germinal centers and plasma cells 2 to 4 weeks after birth.
Table 8.5
Histogenesis of Mucosal Lymphoid Tissue
| Week | Feature |
| Intrauterine | |
| 7 | Intraepithelial lymphocytes. |
| 10 | T cells with surface recognition |
| 12 | PHA-responsive lymphocytes |
| 14 | Lymphocytes with PHA cytotoxicity and ability to mediate graft-vs-host response |
| 17-20 | Mast cells in small intestine |
| 21-24 | Mast cells in large intestine; solitary lymphoid follicle in distal ileum, appendix, and colon |
| 24 | Peyer patches in distal ileum |
| 40 | Solitary lymphoid follicles in duodenum, rectum, and possibly stomach |
| Postnatal | |
| 2-4 | Germinal centers and plasma cells |
PHA, Phytohemagglutinin.
From Huff DS. Developmental anatomy and anomalies of the gastrointestinal tract, with involvement in major malformative syndromes. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. New York: Springer; 2004:3-37.
Congenital Anomalies of the GI Tract
General Aspects
The causes of anomalies of the GI tract include chromosomal abnormalities (numerical and structural); single gene defects; maternal diseases, especially diabetes; and maternal exposure to drugs, especially hydantoin (pyloric stenosis, duodenal and anal atresia). Causes of disruptions include inherited37 and noninherited maternal and fetal thrombophilic diseases, intrauterine hypoxic/ischemic events, intrauterine infection including varicella,38 iatrogenic vascular disruptions,39 and maternal exposure to vasoactive drugs.40 Other diseases of the embryo, such as cystic fibrosis and epidermolysis bullosa, underlie some GI anomalies. Deformations are limited to abnormal shapes of the liver and abnormal rotation and fixation associated with defects of the diaphragm, body wall, and umbilicus. The cause of most anomalies is unknown. Anomalies that arise after completion of organogenesis are often disruptions or deformations; otherwise, the causes are not specific to any developmental period.
Esophagus
Short Esophagus
A congenital short esophagus is a rare anomaly that is associated with intrathoracic development of the stomach. It may be difficult to distinguish from the more common congenital hiatal hernia. Features that favor the diagnosis of a congenital short esophagus include early identification of the intrathoracic stomach during the second trimester and the consistent absence of an abdominal stomach bubble on antenatal ultrasound.41 Furthermore, in the congenital short esophagus, the intrathoracic stomach is supplied by segmental arteries from the descending thoracic aorta, rather than by intrathoracic extensions of the gastric artery, as observed in hiatal hernia.14 Congenital hiatal hernia, by contrast, appears to develop later in gestation and is caused by defective development of the lumbar part of the diaphragm. The differences between these conditions are of more than academic interest, because the outcome of repair of a congenital short esophagus is more guarded than that of hiatal hernia.41
Esophageal Atresia and Tracheoesophageal Fistula
Atresias and stenoses may occur at any site along the tract, but some sites are more commonly involved than others (Table 8.6). Several disorders associated with multiple atresias or stenoses of the GI tract are listed in Table 8.7. Key distinguishing features of the various types of congenital atresias and stenoses of the GI tract are outlined in Table 8.8. EA with or without tracheoesophageal fistula (TEF) occurs in approximately 1 of every 3000 live births. There is a slight male predominance. A history of polyhydramnios is found in a majority of patients with atresia. EA without a fistula is associated with a small stomach42 and absence of a GI gas pattern. A fistula from the distal esophageal segment allows passage of gastric content into the respiratory tract, causing respiratory symptoms. Approximately 50% of patients have associated congenital anomalies, of which the VATER/VACTERL association (consisting of a combination of vertebral anomalies, anal atresia, cardiac defect, tracheoesophageal fistula, renal and limb anomalies) is the most common.43 Conditions associated with EA and TEF are listed in Table 8.9. Familial forms not associated with hereditary syndromes are probably multifactorial.
Table 8.6
Occurrence of Atresia and Stenosis of the Gastrointestinal Tract
| No. Per Live Births* | % of All Intestinal Atresia | |
| Esophagus | 1 : 3000 | |
| Stomach | Rare | |
| Duodenum | 1 : 1500 | 50 |
| Jejunum | 1 : 2000 | 20 |
| Ileum | 1 : 2000 | 25 |
| Colon | Rare | 5 |
| Rectum anus | 1 : 5000 | |
| Multiple | 15 |
* Number per live births vary widely from series to series.
Data from Huff DS. Developmental anatomy and anomalies of the gastrointestinal tract, with involvement in major malformative syndromes. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. New York: Springer; 2004:3-37.
Table 8.7
Disorders with Multiple Gastrointestinal Atresias or Stenoses
Table 8.8
Congenital Atresias and Stenoses of the Gastrointestinal Tract
| Location | Incidence | Key Clinical Features | Key Pathologic Features |
| Esophagus | Atresia 1 : 3000 Stenosis 1 : 30,000 |
>50% associated anomalies; VACTERL association most common Choking and respiratory distress first day of life Absence of normal gastrointestinal gas pattern Stenoses may manifest later in life |
Atresias associated with tracheoesophageal fistula in 85% of cases Stenosis in middle third usually membranous Stenosis in distal third usually due to tracheobronchial remnant |
| Stomach | 1 : 100,000 | Variable; birth to childhood Prenatal history of polyhydramnios Nonbilious vomiting; single large gastric bubble “Single bubble” sign on radiographs in cases of complete obstruction |
Some cases may be associated with epidermolysis bullosa, aplasia cutis, or due to pancreatic heterotopias or adenomyoma |
| Idiopathic hypertrophic pyloric stenosis | 1 : 500 | Projectile vomiting in first weeks of life Palpable epigastric mass Associated with several multiple congenital anomalies syndromes |
Concentric hyperplasia and hypertrophy of pyloric muscularis propria |
| Duodenum | 1 : 1500 | Most common site of intestinal stenoses Nonbilious vomiting if proximal to ampulla; bilious if distal “Double-bubble” gas pattern |
Atresias are usually membranous 50% of stenoses are associated with other anomalies, including annular pancreas, or malrotation |
| Jejunum/ileum | 1 : 1500 | 85% single; 15% multiple Rare familial forms Abdominal distention; gas-filled loops Meconium ileus is presenting feature in 20% of cases of cystic fibrosis |
95% are atresias; 5% are stenoses 50% of atresias are type III with associated mesenteric defect |
| Anorectal | 1 : 4000 | 60% associated with other malformations Imperforate anus with abnormal perineum Failure to pass meconium with cutaneous, vesicular, urethral, or vaginal (females) fistulas |
High atresias more common in males; low atresias more common in females Most severe form is persistent cloaca |
Table 8.9
Disorders with Esophageal Atresia and Tracheoesophageal Fistula
| Single gene disorders |
CHARGE, Coloboma, heart defect, atresia choanae, retarded growth and development, genital abnormality, and ear abnormality; VACTERL, vertebral anomalies, anal atresia, cardiac defect, tracheoesophageal fistula, renal and limb anomalies)
Data from de Jong EM, Felix JF, de Klein A, Tibboel D. Etiology of esophageal atresia and tracheoesophageal fistula: “mind the gap.” Curr Gastroenterol Rep. 2010;12:215-222.
Gross44 and Swenson and colleagues45 proposed the most commonly used classifications of EA and TEF. Figure 8.8 schematically depicts five common variations found in many current publications. The most common form, comprising approximately 85% of cases, consists of a blind-ending proximal pouch with a fistula from the trachea to the distal portion (Fig. 8.9). The tracheal ostium of a fistula to the distal esophageal segment is often at the carina but may be higher in the trachea. Likewise, the ostium of a fistula to the proximal esophageal pouch is often in the upper trachea but is sometimes lower. The length of the distal segment varies. In some cases, it is a short stump barely visible above the diaphragm, and in some it is subdiaphragmatic.46 The distance between the upper esophageal pouch and the distal esophageal segment may be short or long, depending on these variations. Short gap lesions are more easily and successfully repaired than long gap lesions. A fistula from the upper esophageal pouch or the distal esophageal segment may arise from one or both bronchi forming a bronchoesophageal fistula (or fistulas). The trachea may be absent, in which case the distal trachea or both main bronchi arise from the esophagus. An esophageal stenosis may be present at the site of a TEF without EA.
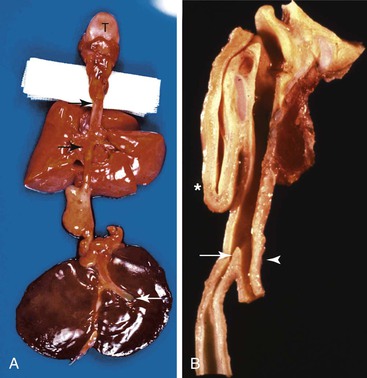
[/not-level-membership-for-pathology-category]
