[level-membership-for-pediatrics-category]
Chapter 570 Congenital Adrenal Hyperplasia and Related Disorders
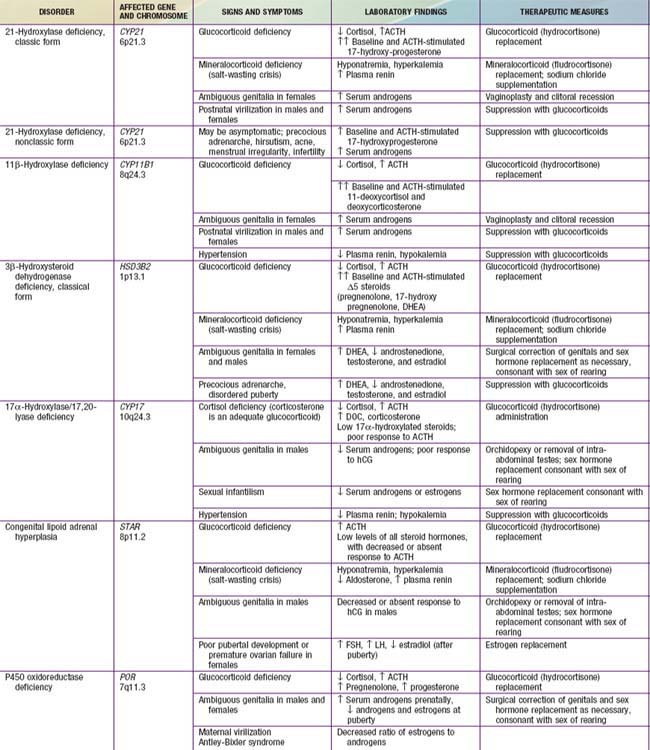
Congenital adrenal hyperplasia (CAH) is a family of autosomal recessive disorders of cortisol biosynthesis (normal adrenal steroidogenesis is discussed in Chapter 568). Cortisol deficiency increases secretion of corticotropin (ACTH), which in turn leads to adrenocortical hyperplasia and overproduction of intermediate metabolites. Depending on the enzymatic step that is deficient, there may be signs, symptoms, and laboratory findings of mineralocorticoid deficiency or excess; incomplete virilization or premature puberty in affected males; and virilization or sexual infantilism in affected females (Figs. 570-1 and 570-2 and Table 570-1).
570.1 Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency
Etiology
More than 90% of congenital adrenal hyperplasia (CAH) cases are caused by 21-hydroxylase deficiency. This P450 enzyme (CYP21, P450c21) hydroxylates progesterone and 17-hydroxyprogesterone (17-OHP) to yield 11-deoxycorticosterone (DOC) and 11-deoxycortisol, respectively (see Fig. 568-1). These conversions are required for synthesis of aldosterone and cortisol, respectively. Both hormones are deficient in the most severe, “salt-wasting” form of the disease. Slightly less severely affected patients are able to synthesize adequate amounts of aldosterone but have elevated levels of androgens of adrenal origin; this is termed simple virilizing disease. These 2 forms are collectively termed classical 21-hydroxylase deficiency. Patients with nonclassical disease have relatively mildly elevated levels of androgens and may have signs of androgen excess after birth.
Pathogenesis and Clinical Manifestations
Aldosterone and Cortisol Deficiency
Because both cortisol and aldosterone require 21-hydroxylation for their synthesis, both hormones are deficient in the most severe, salt-wasting form of the disease. This form constitutes about 70% of cases of classical 21-hydroxylase deficiency. The signs and symptoms of cortisol and aldosterone deficiency, and the pathophysiology underlying them, are essentially those described in Chapter 569. These include progressive weight loss, anorexia, vomiting, dehydration, weakness, hypotension, hypoglycemia, hyponatremia, and hyperkalemia. These problems typically 1st develop in affected infants at approximately 10-14 days of age. Without treatment, shock, cardiac arrhythmias, and death may occur within days or weeks.
Prenatal Androgen Excess
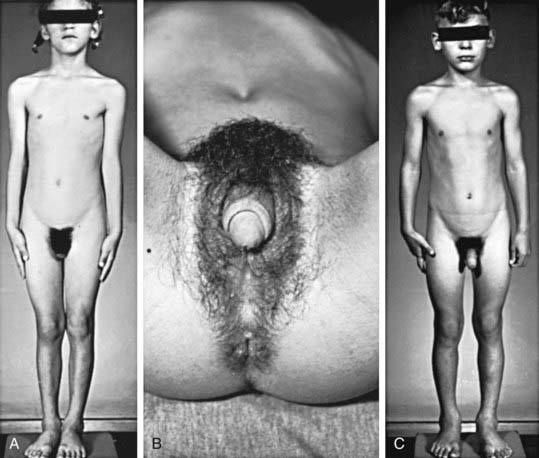
The most important problem caused by accumulation of steroid precursors is that 17-hydroxyprogesterone is shunted into the pathway for androgen biosynthesis, leading to high levels of androstenedione that are converted outside the adrenal gland to testosterone. This problem begins in affected fetuses by 8-10 wk of gestation and leads to abnormal genital development in females (see Figs. 570-1 and 570-2).
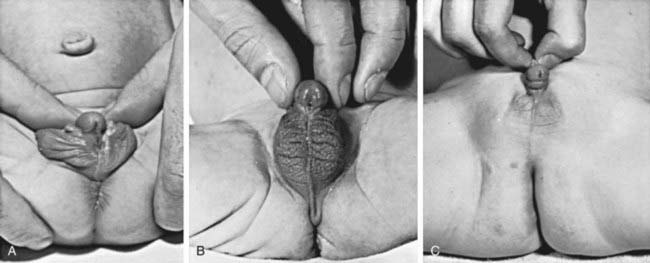
The external genitals of males and females normally appear identical early in gestation (Chapter 576). Affected females, who are exposed in utero to high levels of androgens of adrenal origin, have masculinized external genitalia (see Figs. 570-1 and 570-2). This is manifested by enlargement of the clitoris and by partial or complete labial fusion. The vagina usually has a common opening with the urethra (urogenital sinus). The clitoris may be so enlarged that it resembles a penis; because the urethra opens below this organ, some affected females may be mistakenly presumed to be males with hypospadias and cryptorchidism. The severity of virilization is usually greatest in females with the salt-losing form of 21-hydroxylase deficiency. The internal genital organs are normal, because affected females have normal ovaries and not testes and thus do not secrete antimüllerian hormone.
Male infants appear normal at birth. Thus, the diagnosis may not be made in boys until signs of adrenal insufficiency develop. Because patients with this condition can deteriorate quickly, infant boys are more likely to die than infant girls. For this reason, many states and countries have instituted newborn screening for this condition (see Newborn Screening, later).
Postnatal Androgen Excess
Signs of androgen excess include rapid somatic growth and accelerated skeletal maturation. Thus, affected patients are tall in childhood but premature closure of the epiphyses causes growth to stop relatively early, and adult stature is stunted (see Fig. 570-1). Muscular development may be excessive. Pubic and axillary hair may appear; and acne and a deep voice may develop. The penis, scrotum, and prostate may become enlarged in affected boys; however, the testes are usually prepubertal in size so that they appear relatively small in contrast to the enlarged penis. Occasionally, ectopic adrenocortical cells in the testes of patients become hyperplastic similarly to the adrenal glands, producing testicular adrenal rest tumors (Chapter 578). The clitoris may become further enlarged in affected females (see Fig. 570-1). Although the internal genital structures are female, breast development and menstruation may not occur unless the excessive production of androgens is suppressed by adequate treatment.
Differential Diagnosis
Intersex conditions are discussed more generally in Chapter 582. The initial step in evaluating an infant with ambiguous genitals is a thorough physical examination to define the anatomy of the genitals, locate the urethral meatus, palpate the scrotum or labia and the inguinal regions for testes (palpable gonads almost always indicate the presence of testicular tissue and thus that the infant is a genetic male), and look for any other anatomic abnormalities. Ultrasonography is helpful in demonstrating the presence or absence of a uterus and can often locate the gonads. A rapid karyotype (such as fluorescence in situ hybridization of interphase nuclei for X and Y chromosomes) can quickly determine the genetic sex of the infant. These results are all likely to be available before the results of hormonal testing and together allow the clinical team to advise the parents as to the genetic sex of the infant and the anatomy of internal reproductive structures. Injection of contrast medium into the urogenital sinus of female pseudohermaphrodites demonstrates a vagina and uterus, and most surgeons utilize this information to formulate a plan for surgical management.
Treatment
Berenbaum SA, Korman BK, Duck SC, et al. Psychological adjustment in children and adults with congenital adrenal hyperplasia. J Pediatr. 2004;144:741-746.
Bonfig W, Bechtold S, Schmidt H, et al. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. 2007;92:1635-1639.
Crouch NS, Liao LM, Woodhouse CR, et al. Sexual function and genital sensitivity following feminizing genitoplasty for congenital adrenal hyperplasia. J Urol. 2008;179:634-638.
Green-Golan L, Yates C, Drinkard B, et al. Patients with classic congenital adrenal hyperplasia have decreased epinephrine reserve and defective glycemic control during prolonged moderate-intensity exercise. J Clin Endocrinol Metab. 2007;92:3019-3024.
Hagenfeldt K, Janson PO, Holmdahl G, et al. Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Reprod. 2008;23:1607-1613.
Lajic S, Nordenstrom A, Hirvikoski T. Long-term outcome of prenatal treatment of congenital adrenal hyperplasia. Endocr Dev. 2008;13:82-98.
Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365:2125-2136.
Meyer-Bahlburg HF, Dolezal C, Baker SW, et al. Sexual orientation in women with classical or non-classical congenital adrenal hyperplasia as a function of degree of prenatal androgen excess. Arch Sex Behav. 2008;37:85-99.
Nimkarn S, New MI. Prenatal diagnosis and treatment of congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Nat Clin Pract Endocrinol Metab. 2007;3:405-413.
Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydooxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4133-4160.
Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776-788.
White PC: Newborn screening for congenital adrenal hyperplasia owing to steroid 21-hydroxylase deficiency, Nat Clin Pract Endocrinol Metab in press
570.2 Congenital Adrenal Hyperplasia Due to 11β-Hydroxylase Deficiency
Peter M. Congenital adrenal hyperplasia: 11beta-hydroxylase deficiency. Semin Reprod Med. 2002;20:249-254.
White PC. Steroid 11 beta-hydroxylase deficiency and related disorders. Endocrinol Metab Clin North Am. 2001;30:61-79.
Zhao LQ, Han S, Tian HM. Progress in molecular-genetic studies on congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. World J Pediatr. 2008;4:85-90.
570.3 Congenital Adrenal Hyperplasia Due to 3β-Hydroxysteroid Dehydrogenase Deficiency
Etiology
Deficiency of 3β-hydroxysteroid dehydrogenase (3β-HSD) occurs in fewer than 2% of patients with adrenal hyperplasia. This enzyme is required for conversion of Δ5 steroids (pregnenolone, 17-hydroxypregnenolone, dehydroepiandrosterone [DHEA]) to Δ4 steroids (progesterone, 17-hydroxyprogesterone, and androstenedione). Thus, deficiency of the enzyme results in decreased synthesis of cortisol, aldosterone, and androstenedione but increased secretion of DHEA (see Fig. 568-1). The 3β-HSD enzyme expressed in the adrenal cortex and gonad is encoded by the HSD3B2 gene located on chromosome 1p13.1. Over 30 mutations in the HSD3B2 gene have been described in patients with 3β-HSD deficiency.
Simard J, Moisan AM, Morel Y. Congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase/delta(5)-delta(4) isomerase deficiency. Semin Reprod Med. 2002;20:255-276.
Simard J, Ricketts ML, Gingras S, et al. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr Rev. 2005;26:525-582.
570.4 Congenital Adrenal Hyperplasia Due to 17-Hydroxylase Deficiency
Etiology
Less than 1% of CAH cases are caused by 17-hydroxylase deficiency. A single polypeptide, CYP17, catalyzes 2 distinct reactions: 17-hydroxylation of pregnenolone and progesterone to 17-hydroxypregnenolone and 17-hydroxyprogesterone, respectively, and the 17,20-lyase reaction mediating conversion of 17-hydroxypregnenolone to DHEA and, to a lesser extent, 17-hydroxyprogesterone to Δ4-androstenedione. DHEA and androstenedione are steroid precursors of testosterone and estrogen (see Fig. 568-1). The enzyme is expressed in both the adrenal cortex and the gonads and is encoded by a gene on chromosome 10q24.3. Most mutations affect both the hydroxylase and lyase activities, but rare mutations can affect either activity alone.
Clinical Manifestations and Laboratory Findings
Patients with 17-hydroxylase deficiency cannot synthesize cortisol, but their ability to synthesize corticosterone is intact. Because corticosterone is an active glucocorticoid, patients do not develop adrenal insufficiency. Deoxycorticosterone, the immediate precursor of corticosterone, is synthesized in excess. This can cause hypertension, hypokalemia, and suppression of renin and aldosterone secretion, as occurs in 11-hydroxylase deficiency. In contrast to 11-hydroxylase deficiency, patients with 17-hydroxylase deficiency are unable to synthesize sex hormones. Affected males are incompletely virilized and present as phenotypic females (but gonads are usually palpable in the inguinal region or the labia) or with sexual ambiguity (male pseudohermaphroditism). Affected females usually present with failure of sexual development at the expected time of puberty. 17-Hydroxylase deficiency in females must be considered in the differential diagnosis of primary hypogonadism (Chapter 580). In addition to the increased DOC, suppressed renin and aldosterone, and decreased 17-hydroxylated steroids, cortisol, and sex steroids are unresponsive to stimulation with ACTH and human chorionic gonadotropin, respectively.
Treatment
Patients with 17-hydroxylase deficiency require cortisol replacement to suppress secretion of deoxycorticosterone and thus control hypertension. Additional antihypertensive medication may be required. Females require estrogen replacement at puberty. Genetic males may require either estrogen or androgen supplementation depending on the sex of rearing. Because of the possibility of malignant transformation of abdominal testes with androgen insensitivity syndrome (Chapter 577), genetic males with severe 17-hydroxylase deficiency being reared as females require gonadectomy at or before adolescence.
570.5 Lipoid Adrenal Hyperplasia
Hiort O, Holterhus PM, Werner R, et al. Homozygous disruption of P450 side-chain cleavage (CYP11A1) is associated with prematurity, complete 46,XY sex reversal, and severe adrenal failure. J Clin Endocrinol Metab. 2005;90:538-541.
Miller WL. StAR search—what we know about how the steroidogenic acute regulatory protein mediates mitochondrial cholesterol import. Mol Endocrinol. 2007;21:589-601.
Stocco DM. Clinical disorders associated with abnormal cholesterol transport: mutations in the steroidogenic acute regulatory protein. Mol Cell Endocrinol. 2002;191:19-25.
570.6 Deficiency of P450 Oxidoreductase (Antley-Bixler Syndrome)
Etiology, Pathogenesis, and Clinical Manifestations
P450 oxidoreductase (POR, gene located on chromosome 7q11.3) is required for the activity of all microsomal cytochrome P450 enzymes (Chapter 568) including the adrenal enzymes CYP17 and CYP21. Thus, complete POR deficiency abolishes all microsomal P450 activity. This is embryonically lethal in mice and presumably in humans as well. Patients with mutations that decrease but do not abolish POR activity have partial deficiencies of 17-hydroxylase and 21-hydroxylase activities in the adrenals. Deficiency of 17-hydroxylase leads to incomplete masculinization in males; 21-hydroxylase deficiency may lead to virilization in females. Additionally, aromatase (CYP19) activity in the placenta is decreased, leading to unopposed action of androgens produced by the fetal adrenal. This exacerbates virilization of female fetuses and may virilize the mother of an affected fetus as well. Although it is puzzling that affected females could be virilized despite a partial deficiency in CYP17 (which is required for androgen biosynthesis), an alternative biosynthetic pathway may be utilized in which 17-hydroxyprogesterone is converted to 5α-pregnane-3α,17α-diol-20-one, a metabolite that is a much better substrate for the 17,20-lyase activity of CYP17 than the usual substrate, 17-hydroxypregnenolone (Chapter 568). The metabolite is then converted in several enzymatic steps to dihydrotestosterone, a potent androgen.
Fluck CE, Pandey AV, Huang N, et al. P450 oxidoreductase deficiency—a new form of congenital adrenal hyperplasia. Endocr Dev. 2008;13:67-81.
Huang N, Pandey AV, Agrawal V, et al. Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet. 2005;76:729-749.
Schmidt K, Hughes C, Chudek JA, et al. Cholesterol metabolism: the main pathway acting downstream of cytochrome P450 oxidoreductase in skeletal development of the limb. Mol Cell Biol. 2009;29:2716-2729.
570.7 Aldosterone Synthase Deficiency
Differential Diagnosis
It is important to distinguish aldosterone synthase deficiency from primary adrenal insufficiency in which both cortisol and aldosterone are affected (including salt-wasting forms of congenital adrenal hyperplasia) because the latter condition is usually associated with a much greater risk of shock and hyponatremia. This becomes apparent after the appropriate laboratory studies. Pseudohypoaldosteronism (Chapter 52) may have similar electrolyte abnormalities and hyperreninemia, but aldosterone levels are high, and this condition usually does not respond to fludrocortisone treatment.
570.8 Glucocorticoid-Suppressible Hyperaldosteronism
Differential Diagnosis
This condition should be distinguished from primary aldosteronism due to bilateral hyperplasia or an aldosterone-producing adenoma (Chapter 572). Most cases of primary aldosteronism are sporadic although rare affected kindreds have been reported. Patients with primary aldosteronism may also have elevated levels of 18-hydroxycortisol and 18-oxocortisol, and these biochemical tests should be used cautiously to distinguish primary and glucocorticoid-suppressible aldosteronism. A therapeutic trial of dexamethasone may be helpful if aldosterone secretion is suppressed, and genetic testing should identify the hybrid gene of glucocorticoid-suppressible hyperaldosteronism if it is present.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 570 Congenital Adrenal Hyperplasia and Related Disorders
Congenital adrenal hyperplasia (CAH) is a family of autosomal recessive disorders of cortisol biosynthesis (normal adrenal steroidogenesis is discussed in Chapter 568). Cortisol deficiency increases secretion of corticotropin (ACTH), which in turn leads to adrenocortical hyperplasia and overproduction of intermediate metabolites. Depending on the enzymatic step that is deficient, there may be signs, symptoms, and laboratory findings of mineralocorticoid deficiency or excess; incomplete virilization or premature puberty in affected males; and virilization or sexual infantilism in affected females (Figs. 570-1 and 570-2 and Table 570-1).
570.1 Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency
Etiology
More than 90% of congenital adrenal hyperplasia (CAH) cases are caused by 21-hydroxylase deficiency. This P450 enzyme (CYP21, P450c21) hydroxylates progesterone and 17-hydroxyprogesterone (17-OHP) to yield 11-deoxycorticosterone (DOC) and 11-deoxycortisol, respectively (see Fig. 568-1). These conversions are required for synthesis of aldosterone and cortisol, respectively. Both hormones are deficient in the most severe, “salt-wasting” form of the disease. Slightly less severely affected patients are able to synthesize adequate amounts of aldosterone but have elevated levels of androgens of adrenal origin; this is termed simple virilizing disease. These 2 forms are collectively termed classical 21-hydroxylase deficiency. Patients with nonclassical disease have relatively mildly elevated levels of androgens and may have signs of androgen excess after birth.
Pathogenesis and Clinical Manifestations
Aldosterone and Cortisol Deficiency
Because both cortisol and aldosterone require 21-hydroxylation for their synthesis, both hormones are deficient in the most severe, salt-wasting form of the disease. This form constitutes about 70% of cases of classical 21-hydroxylase deficiency. The signs and symptoms of cortisol and aldosterone deficiency, and the pathophysiology underlying them, are essentially those described in Chapter 569. These include progressive weight loss, anorexia, vomiting, dehydration, weakness, hypotension, hypoglycemia, hyponatremia, and hyperkalemia. These problems typically 1st develop in affected infants at approximately 10-14 days of age. Without treatment, shock, cardiac arrhythmias, and death may occur within days or weeks.
Prenatal Androgen Excess
The most important problem caused by accumulation of steroid precursors is that 17-hydroxyprogesterone is shunted into the pathway for androgen biosynthesis, leading to high levels of androstenedione that are converted outside the adrenal gland to testosterone. This problem begins in affected fetuses by 8-10 wk of gestation and leads to abnormal genital development in females (see Figs. 570-1 and 570-2).
The external genitals of males and females normally appear identical early in gestation (Chapter 576). Affected females, who are exposed in utero to high levels of androgens of adrenal origin, have masculinized external genitalia (see Figs. 570-1 and 570-2). This is manifested by enlargement of the clitoris and by partial or complete labial fusion. The vagina usually has a common opening with the urethra (urogenital sinus). The clitoris may be so enlarged that it resembles a penis; because the urethra opens below this organ, some affected females may be mistakenly presumed to be males with hypospadias and cryptorchidism. The severity of virilization is usually greatest in females with the salt-losing form of 21-hydroxylase deficiency. The internal genital organs are normal, because affected females have normal ovaries and not testes and thus do not secrete antimüllerian hormone.
Male infants appear normal at birth. Thus, the diagnosis may not be made in boys until signs of adrenal insufficiency develop. Because patients with this condition can deteriorate quickly, infant boys are more likely to die than infant girls. For this reason, many states and countries have instituted newborn screening for this condition (see Newborn Screening, later).
Postnatal Androgen Excess
Signs of androgen excess include rapid somatic growth and accelerated skeletal maturation. Thus, affected patients are tall in childhood but premature closure of the epiphyses causes growth to stop relatively early, and adult stature is stunted (see Fig. 570-1). Muscular development may be excessive. Pubic and axillary hair may appear; and acne and a deep voice may develop. The penis, scrotum, and prostate may become enlarged in affected boys; however, the testes are usually prepubertal in size so that they appear relatively small in contrast to the enlarged penis. Occasionally, ectopic adrenocortical cells in the testes of patients become hyperplastic similarly to the adrenal glands, producing testicular adrenal rest tumors (Chapter 578). The clitoris may become further enlarged in affected females (see Fig. 570-1). Although the internal genital structures are female, breast development and menstruation may not occur unless the excessive production of androgens is suppressed by adequate treatment.
Differential Diagnosis
Intersex conditions are discussed more generally in Chapter 582. The initial step in evaluating an infant with ambiguous genitals is a thorough physical examination to define the anatomy of the genitals, locate the urethral meatus, palpate the scrotum or labia and the inguinal regions for testes (palpable gonads almost always indicate the presence of testicular tissue and thus that the infant is a genetic male), and look for any other anatomic abnormalities. Ultrasonography is helpful in demonstrating the presence or absence of a uterus and can often locate the gonads. A rapid karyotype (such as fluorescence in situ hybridization of interphase nuclei for X and Y chromosomes) can quickly determine the genetic sex of the infant. These results are all likely to be available before the results of hormonal testing and together allow the clinical team to advise the parents as to the genetic sex of the infant and the anatomy of internal reproductive structures. Injection of contrast medium into the urogenital sinus of female pseudohermaphrodites demonstrates a vagina and uterus, and most surgeons utilize this information to formulate a plan for surgical management.
