[level-membership-for-pediatrics-category]Chapter 43
Congenital Abnormalities of the Spine
Embryology and Developmental Anatomy
Concurrent with the neural tube folding during primary neurulation, spinal cord development below the caudal neuropore commences within the pluripotent tissue at the caudal eminence in the process of secondary neurulation. The initially solid cell mass canalizes and becomes contiguous with the rostral neural tube that was formed by primary neurulation. By day 48, a transient ventriculus terminalis appears in the future conus. If this persists after birth, it is noted incidentally as a normal variant ventriculus terminalis (“fifth ventricle”), usually of no clinical significance (see Chapter 40). Failure of proper secondary neurulation leads to caudal spine anomalies in the caudal regression, tethered cord, or sacrococcygeal teratoma (SCT) spectra in addition to terminal myelocystocele and anterior sacral meningocele (ASM).
Abnormalities of Primary Neurulation
Premature Dysjunction
Premature dysjunction of the neural tube from overlying ectoderm permits perineural mesenchyme to access the neural groove and ependymal lining. This mesenchyme differentiates into fat and prevents complete neural tube closure, resulting in skin-covered lipomatous malformations with or without posterior spinal dysraphism. The most commonly observed anomalies are lipomyelocele (LMC), LMMC (Fig. 43-1), and intradural spinal lipomas (Fig. 43-2).
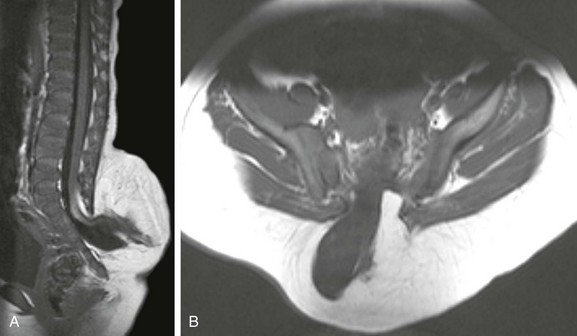
Figure 43-1 Lipomyelomeningocele.
Sagittal (A) and axial (B) T1-weighted magnetic resonance (MR) images demonstrate a typical lipomyelomeningocele, with a low-lying cord tethered into a large lipomatous malformation contiguous with the subcutaneous fat through a posterior dysraphic defect.
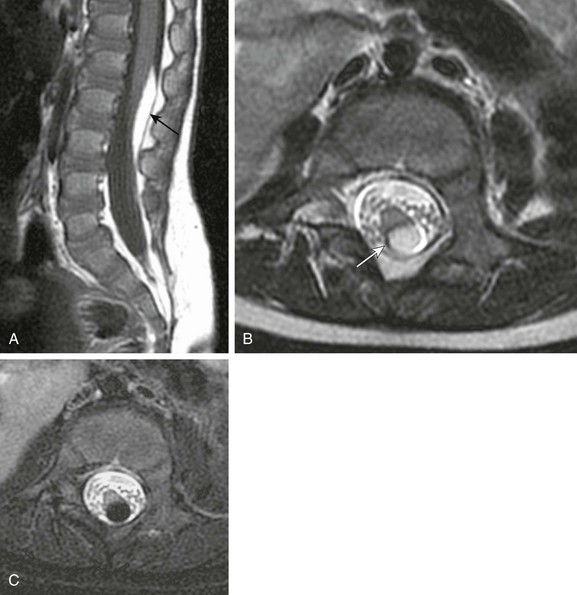
Figure 43-2 Juxtamedullary (subpial) spinal lipoma.
A, Sagittal T1-weighted magnetic resonance (MR) image shows a small subpial intradural lipoma (arrow) adherent to the dorsal conus surface. B, Axial T2-weighted MR image confirms direct contiguity of the neural placode with the lipoma. Note chemical shift artifact (arrow) in the frequency encoding direction, indicating fat. C, Axial fat-saturated T2-weighted MR image confirms fat content by homogeneous signal loss within the lipoma.
Nondysjunction
In contrast to lipomatous malformations, anomalies that result from nondysjunction occur when the neural tube fails to dissociate from adjacent cutaneous tissue. The simplest and least extensive variation is the dorsal dermal sinus, which occurs when a single connection persists and forms a fibrous cord from a skin dimple to the dural sac, conus, or central spinal cord canal. It is important to distinguish dermal sinus from its clinically asymptomatic mimic, simple coccygeal dimple (Fig. 43-3). In this mimic, the low sacral or coccygeal sinus originates from a low skin dimple and attaches to the coccyx via a short fibrous tract. These dimples are nearly always found within the intergluteal cleft, never communicate with the spinal canal, and require no treatment. Simple coccygeal dimples are the most common reason for newborn spinal ultrasound imaging.
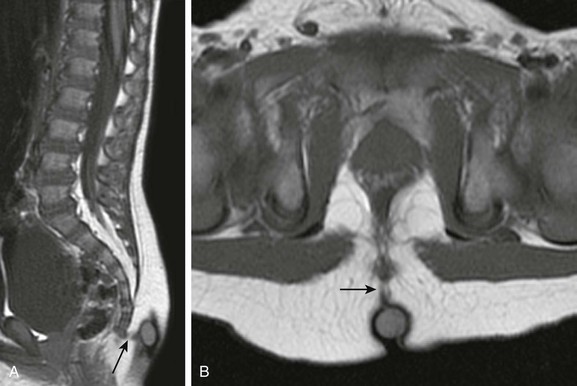
Figure 43-3 Simple sacrococcygeal dimple.
Sagittal (A) and axial (B) T1-weighted magnetic resonance images demonstrate a low sacral dimple within the intergluteal cleft, marked by a hyperintense vitamin E capsule. The short fibrous tract (arrow) that extends from the dimple to the coccyx tip confirms the diagnosis.
Conversely, the true congenital dorsal dermal sinus tract (DST; Fig. 43-4) usually has an atypical dimple at the ostium that is larger (> 5 mm), often asymmetric, and remote (> 2.5 cm) from the anus. It may also be found in combination with other cutaneous anomalies, such as a hair patch or vascular lesion. The most common DST location is in the lumbosacral spine, followed by the occiput. In all dermal sinus cases, there is some degree of focal dysraphism, which may be as subtle as a bifid spinous process. The sinus tract cord is epithelial-cell lined and may or may not be canalized. When patent, it exposes the patient to an elevated risk of meningitis. It is critical to look for this anomaly in all patients with atypical skin dimples, cutaneous back lesions, or lipomas. Additionally, 30% to 50% of DSTs may have an associated dermoid or epidermoid cyst. These patients should be imaged with MRI. The best MRI sequence is usually a sagittal T1-weighted MR image, windowed widely so that the hypointense tract is visualized as a gray cord immersed in bright fat that passes inferiorly and ventrally to the lumbodorsal fascia; it then turns upward to ascend within the spinal canal, often tenting the dorsal dura at the point of entry. Dermal sinuses are surgically excised to prevent meningitis and to untether the spinal cord.
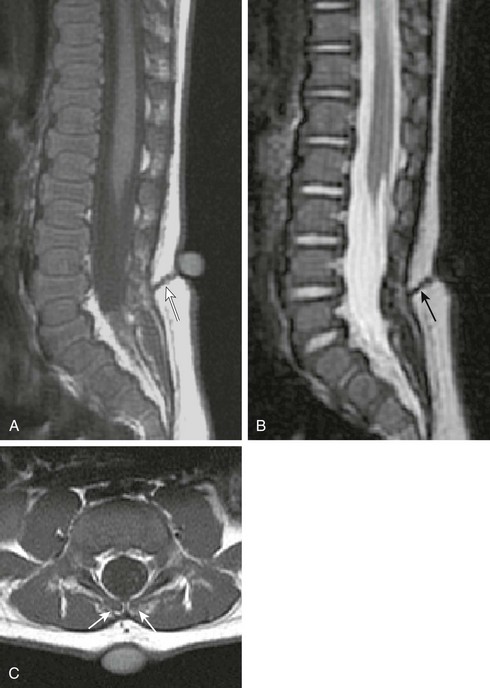
Figure 43-4 Dorsal dermal sinus.
Sagittal T1- (A) and T2-weighted magnetic resonance (MR) images (B) demonstrate a low-lying spinal cord tethered by a dorsal dermal sinus tract (arrows). The skin opening is marked by a vitamin E capsule. Axial T1-weighted MR image (C) confirms the tract accessing the dural sac through a bifid spinous process (arrows).
More extensive nondysjunction produces the MMC lesion (Fig. 43-5) associated with maternal folate deficiency. Infants come to medical attention with an open, red, weeping skin defect on the back that features protruding neural elements. Most MMC lesions are either lumbosacral or thoracolumbar, but cervical and thoracic MMCs occur. Lesion level and severity of associated hydrocephalus determine the patient’s prognosis. MMCs are linked to methylenetetrahydrofolate-reductase mutations with abnormal folate metabolism. PAX3 paired box gene derangements and trisomy 13 or 18 (14% of fetuses with neural tube defect) are also described. These gene derangements and folate metabolism abnormalities are postulated to interfere with carbohydrate molecule expression on the neuroectodermal surface, which causes neural tube closure to fail. Prevalence in the United States is 2 in 10,000 live births, and it is more common in females by a ratio of 3 : 1. The prevalence of MMC decreased 23% between 1995 to 2004, which was attributed to more widespread folate fortification of food.
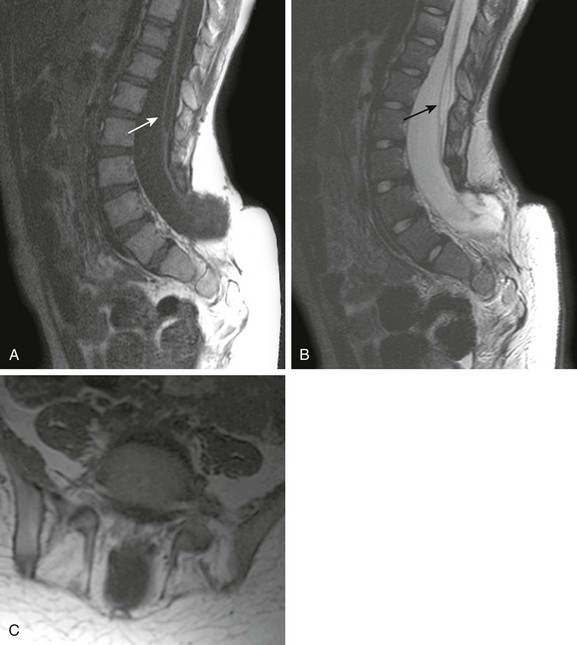
Figure 43-5 Myelomeningocele.
Sagittal T1- (A) and T2-weighted (B) magnetic resonance (MR) images show the postoperative appearance of the spine after myelomeningocele closure. The large dysraphic defect is now covered with skin, and the attenuated distal spinal cord is scarred into the closure. A small syrinx is present (arrow). Axial T1-weighted MR image (C) reveals the protrusion of the distal sac through parallel dysraphic posterior elements.
Anomalies of the Caudal Cell Mass
Hypogenesis or agenesis of the caudal cell mass produces caudal regression syndrome (CRS; Fig. 43-6). Two types are described. The more severe CRS, type 1, features a foreshortened terminal vertebral column, high-lying wedge-shaped conus termination, and more severe associated visceral and orthopedic anomalies. The less severe CRS, type 2, has a low-lying tethered spinal cord with milder associated malformations. In general, the higher the cord termination, the more severe the sacral anomalies. The most severe CRS presentations are lumbosacral agenesis, in which the spine terminates at the lower thoracic level, and severe sacral dysgenesis, with fused lower extremities in a “mermaid” configuration (sirenomyelia). In contrast, the mildest CRS cases may manifest merely as a missing terminal sacral segment identified on imaging that is clinically asymptomatic. CRS is associated with myriad other visceral abnormalities, including renal or pulmonary hypoplasia and anorectal malformations. Other commonly associated spinal malformations include open dysraphism, vertebral SFAs, and split-cord malformations.
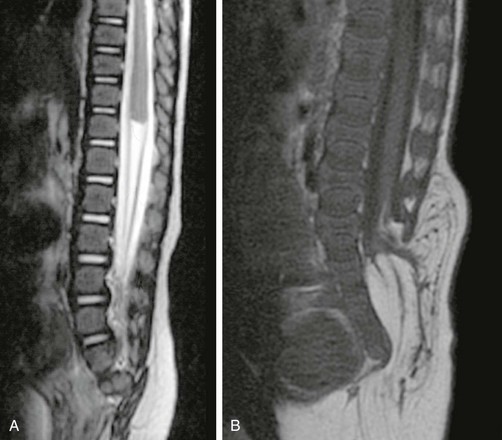
Figure 43-6 Caudal regression syndrome (CRS).
Sagittal T2-weighted image (A) shows the typical appearance of type 1 CRS, with severe sacral dysplasia and the characteristic wedge-shaped high conus termination. Sagittal T1-weighted magnetic resonance image (B) contrasts the appearance of type 2 CRS (with a less severe sacral dysgenesis) with an elongated spinal cord that inserts distally into a terminal lipoma.
Perhaps the most common entity within the caudal cell mass dysplasia spectrum is tethered cord syndrome (TCS), which manifests clinically as gait spasticity, low back and leg pain that is worse in the morning, lower extremity sensory abnormalities, and/or bladder difficulties. TCS patients are most likely to come in during periods of rapid somatic growth. TCS refers strictly to patients with a low-lying cord and thickened filum, not those with other spine and cord abnormalities, although in fact those patients may be similar in clinical presentation and may be considered “tethered.” It is important to consider TCS a distinct clinical diagnosis, with imaging relegated to the role of preoperative planning rather than establishing the primary diagnosis. On imaging, TCS manifests either as a taut spinal cord without definitive conus or a low-lying conus (Fig. 43-7). The filum is frequently thickened and shortened, and an associated terminal lipoma may be present. Symptomatic patients may benefit from surgery, but it is crucial to exclude other associated anomalies before surgery.
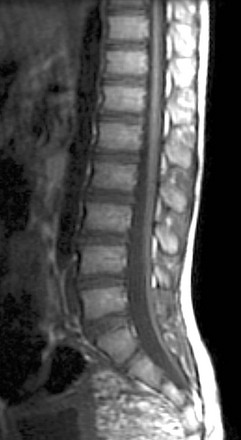
Figure 43-7 Tethered cord syndrome.
Sagittal T1-weighted magnetic resonance image reveals an elongated spinal cord, indistinguishable from the filum, inserting into the terminal thecal sac.
Two other important rare presentations of caudal cell mass dysplasia are the ASM and the terminal myelocystocele. ASM features a large anterior meningocele outpouching that traverses an enlarged sacral foramen and produces a presacral cystic mass. This may be clinically mistaken for a SCT and may prompt neuroimaging. Most ASMs are sporadic, but a minority show an inherited predisposition within the Currarino triad or in syndromes that feature dural dysplasia, such as neurofibromatosis type 1 (NF1) and Marfan syndrome. As with other caudal cell mass dysplasias, additional congenital abnormalities may be found, such as anorectal malformations, caudal dysgenesis, and dermoid or epidermoid cysts. Fortunately, imaging is highly characteristic in the more common simple form, with a presacral cystic mass that connects to the thecal sac through an enlarged sacral neural foramen (Fig. 43-8). Complex ASMs with fat or neural elements are also recognized by their extension through the neural foramen. Terminal myelocystocele is a very rare malformation that manifests as a hydromyelic spinal cord that traverses a meningocele to terminate in a skin-covered myelocystocele (e-Fig. 43-9). These abnormalities are rarely imaged, until the infant survives the commonly associated anorectal and visceral anomalies that drive early management and produce most of the morbidity and mortality. Infants with terminal myelocystocele may be neurologically intact at birth but subsequently lose neurologic function.
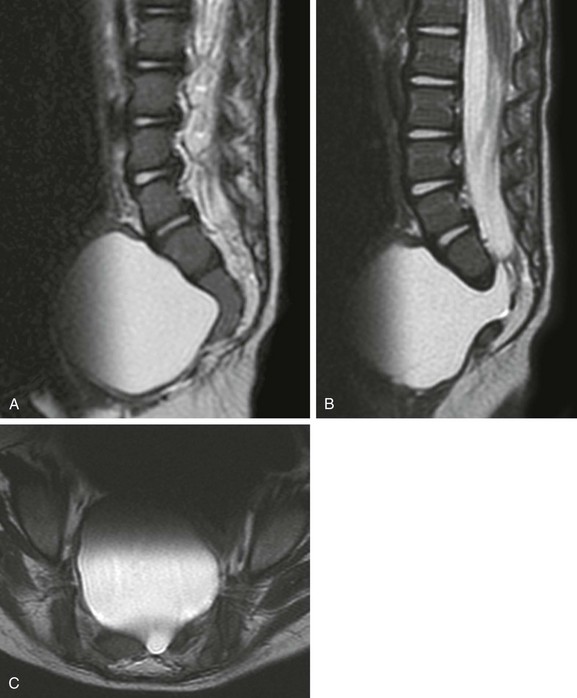
Figure 43-8 Anterior sacral meningocele.
Sagittal T2-weighted magnetic resonance (MR) image (A) in the midline shows the characteristic scimitar shape of the sacrum. Parasagittal (B) and axial (C) T2-weighted MR images confirm a typical, simple, anterior sacral meningocele that appears as a presacral cyst contiguous with the thecal sac through an enlarged left sacral neural foramen.
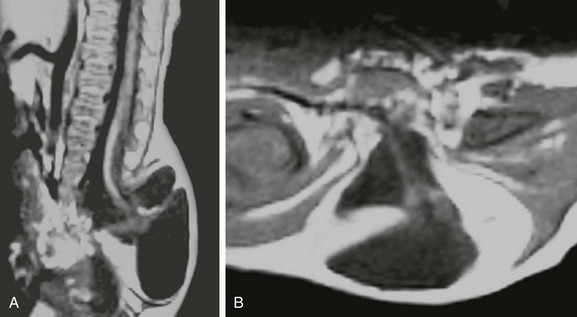
e-Figure 43-9 Terminal myelocystocele.
Sagittal T1-weighted magnetic resonance (MR) image (A) depicts a low-lying hydromyelic cord with terminal cyst projecting through a sacral meningocele. Axial T1-weighted MR image (B) confirms the origin of the terminal cyst as the hydromyelic terminal central canal.
Finally, if the primitive streak incompletely regresses and leaves a caudal totipotential cell rest remnant, an SCT (Fig. 43-10) may result. SCTs demonstrate tissue from all three cell layers and contain varying proportions of mature and immature elements. These are surgically graded, according to the American Academy of Pediatrics (AAP) classification, based on proportion of internal (pelvic) and external components; prognosis is determined by the AAP grade and by the presence or absence of mature or malignant components. External tumors, mature elements, and younger patient age predict a better outcome. The surgeon must resect the coccyx to prevent recurrence. Fetal and early infancy morbidity and mortality are related mostly to cardiac failure from intratumoral shunting and associated visceral anomalies, whereas later mortality is related to lesion malignancy.
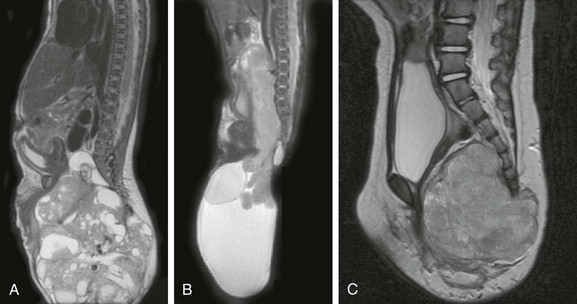
Figure 43-10 Sacrococcygeal teratoma.
Sagittal T2-weighted magnetic resonance images in three different infants show large, mixed cystic and solid pelvic masses confirmed pathologically as sacrococcygeal teratomas. The proportions of internal and external tumor produce diagnoses of grade 1 (A), grade 2 (B), and grade 3 (C) using the American Academy of Pediatrics classification.
Anomalies of Notochord Development
Neurenteric cyst (e-Fig. 43-11) consists of an intraspinal cyst lined by enteric mucosa, and it is most common in the thoracic spine, followed by the cervical spine. Such cysts putatively arise from an abnormal connection between primitive endoderm and ectoderm that persists beyond the third embryonic week. Whereas normally the notochord separates ventral endoderm (foregut) and dorsal ectoderm (skin, spinal cord) during embryogenesis, in a neurenteric cyst, a separation failure “splits” the notochord and hinders the development of mesoderm, which traps a small piece of primitive gut within the developing spinal canal. This gut remnant may become isolated, forming a cyst, or it may maintain connections with gut or skin (or both); this produces the spectrum of fistulas and sinuses that constitute the spectrum of dorsal-enteric spinal anomalies. The most severe malformations remain in communication through the primitive vertebral osseous canal of Kovalevsky, but even mild cases usually show some vertebral segmentation anomalies on close inspection. Multiplanar MRI best demonstrates the cyst and its relationship to the spinal cord, as well as connections to the mediastinal or abdominal viscera. CT, particularly with multiplanar and three-dimensional reformats, optimally demonstrates osseous vertebral anomalies for preoperative planning.
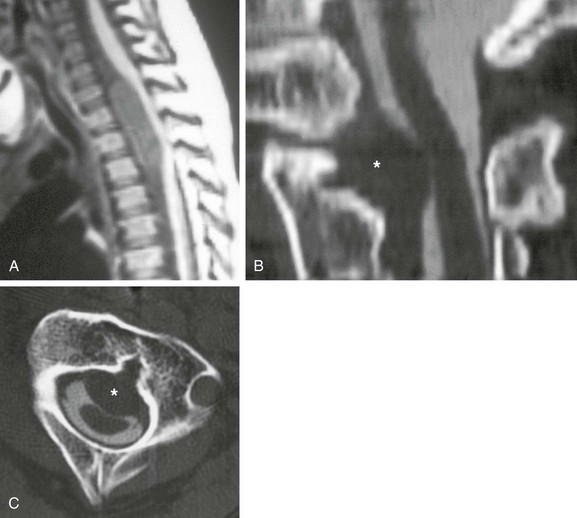
e-Figure 43-11 Neurenteric cyst.
Sagittal T1-weighted magnetic resonance image (A) shows a ventral neurenteric cyst in the spinal canal that compresses the spinal cord. Additionally, a small cyst of similar signal characteristics was found along the anterior spine that on axial imaging (not shown) was contiguous with the neurenteric cyst through a midline sagittal vertebral cleft. Sagittal (B) and axial (C) computed tomographic myelogram images in a different patient with Klippel-Feil syndrome show a ventral extradural neurenteric cyst (asterisk) compressing the ventral thecal sac and spinal cord, contiguous with a sagittal vertebral body cleft.
Because the notochord influences vertebral development, vertebral segmentation anomalies are very commonly associated with DSM. Therefore important factors for preoperative planning include the presence (type 1 DSM; Fig. 43-12) or absence (type 2 DSM; Fig. 43-13) of an osseous or fibrous spur and whether the cords reside in separate or single dural tubes (see also Chapter 42). Occasionally in type 2 DSM, a nerve root or roots may become adherent to the dura and may tether the spinal cord, producing the meningocele manqué. DSM may be isolated or found in conjunction with other spinal anomalies, particularly MMC, thus it is critical to search for DSM before any spinal anomaly repair or scoliosis correction. A patient with MMC whose symptoms progress after surgical closure is a relatively common presentation of undiagnosed DSM. Cutaneous abnormalities such as skin dimples or discoloration, vascular lesions, or hair patches are sometimes present and can guide attention to the most likely level of DSM.
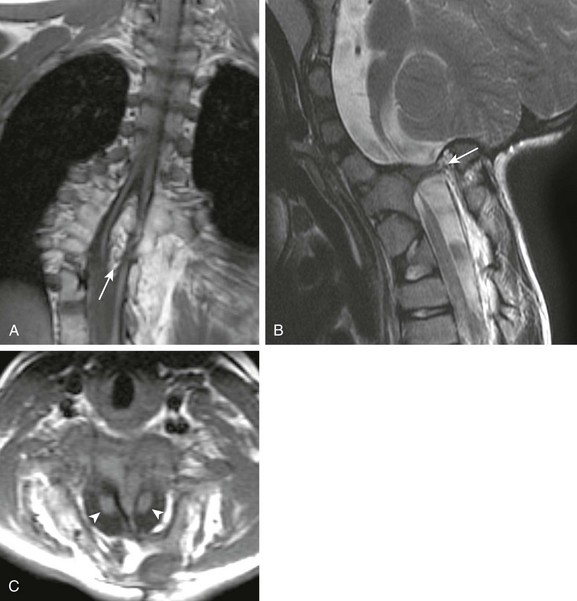
Figure 43-12 Diastematomyelia (DSM) type 1.
Coronal (A) T1-weighted magnetic resonance (MR) images in a patient with type 1 DSM show a large ossified spur (arrow) that splits the thoracic spinal cord. Numerous vertebral segmentation anomalies with posterior rib fusions are present. Sagittal T2- (B) and axial T1-weighted (C) MR images of a different patient show a type 1 cervical DSM with ossified spur (arrow in B) and two hemicords (arrowheads in C).
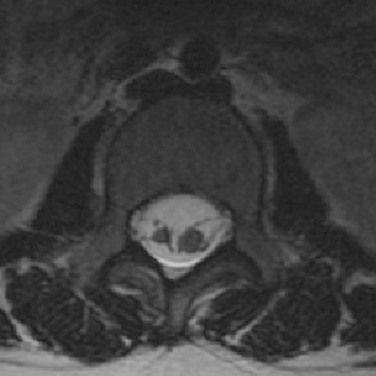
Other Congenital and Developmental Anomalies
Dorsal meningoceles (e-Fig. 43-14) by definition occur dorsally, most often over the lumbosacral spine, and feature a skin-covered meningocele devoid of neural elements that protrudes through a posterior dysraphic defect. In practice, however, it is not uncommon to find a dysplastic nerve root or other neural tissue within a meningocele.
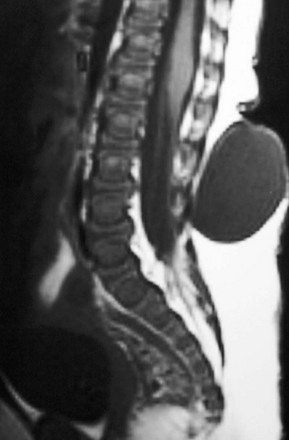
e-Figure 43-14 Dorsal meningocele.
Sagittal T1-weighted magnetic resonance image shows a large, skin-covered cystic back mass connected to the thecal sac by a thin cerebrospinal fluid–filled stalk. The tethered conus terminates at L2–L3. (Image courtesy of Marvin Nelson Jr, MD.)
Lateral meningoceles (e-Fig. 43-15) manifest as paraspinal masses filled with cerebrospinal fluid that are contiguous with the thecal sac and extend through the neural foramen, with adjacent pedicular and foraminal osseous remodeling. They are generally “simple,” but some may contain fat or neural tissue and are then better termed “complex.” Important associations include Marfan syndrome and NF1.
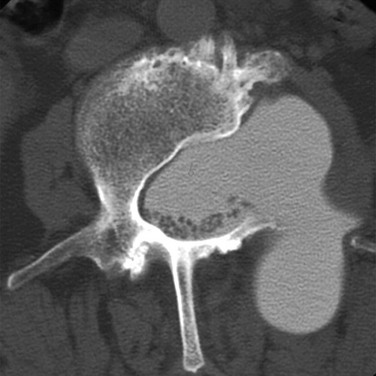
e-Figure 43-15 Lateral meningocele.
Axial computed tomography myelogram image shows a large left lateral meningocele with pedicular erosion in a patient with neurofibromatosis type 1. The image confirms vertebral remodeling and contiguity of the meningocele with the thecal sac. In addition to lateral extension, the meningocele also extends posteriorly into the paraspinal muscles.
Vertebral Formation and Segmentation Anomalies
Anomalies of vertebral formation and segmentation arise from aberrancies in vertebral column formation. These are generally divided into anomalies that result from either partial or total failure of vertebral formation (Fig. 43-16) and failure to correctly segment after vertebral formation (vertebral segmentation failure; e-Fig. 43-17). The abnormal vertebra may be supernumerary, or it may replace a normal vertebral body. Abnormal PAX1 expression is a postulated etiology in the development of segmentation anomalies, and other visceral and neuraxis anomalies are also commonly identified. More severe SFAs tend to have a higher incidence of concurrent visceral organ or other neuraxis anomalies. The degree and location of vertebral formation failure predicts morphology; unilateral chondral center deficiency and failure of ossification produces a hemivertebra, whereas central failure of ossification centers to unite produces a butterfly vertebra. Conversely, vertebral segmentation failure presents with composite or “block” vertebra and posterior element fusions (Fig. 43-18). Not surprisingly, block vertebrae frequently coexist with hemivertebrae and butterfly vertebrae (Fig. 43-19), leading many to lump these various vertebral anomalies together into the working term SFAs. Many clinical syndromes prominently feature SFAs, including Klippel-Feil and Jarcho-Levin (spondylothoracic dysplasia) syndromes. Therefore SFAs are not findings that confirm a specific disorder, but rather they are imaging markers that prompt further consideration of a possible syndromic process.
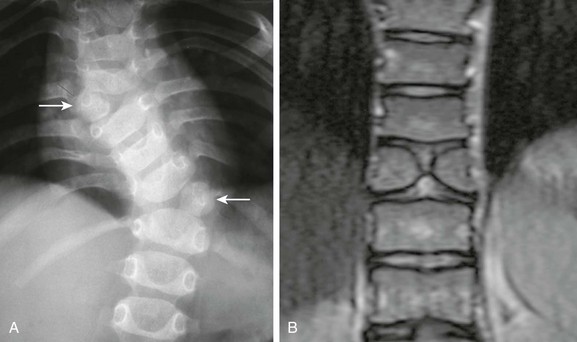
Figure 43-16 Failure of vertebral formation.
A, Frontal radiography demonstrates balanced thoracic hemivertebrae (arrows) detected during evaluation of congenital scoliosis. B, Coronal T2-weighted magnetic resonance image shows a classic butterfly vertebra. Both of these may coexist with each other and with malformations of vertebral segmentation failure.
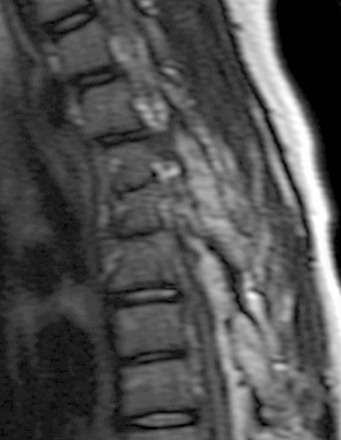
Figure 43-18 Posterior element segmentation failure with fusion.
Sagittal T2-weighted image shows a multilevel pedicular and facet bar associated with vertebral segmentation failure at the same levels.
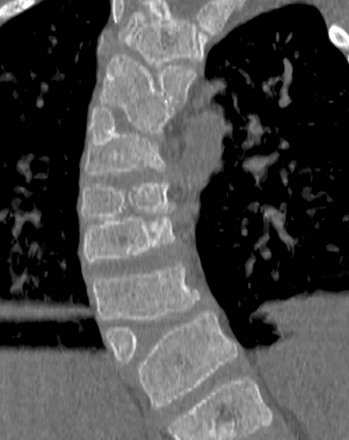
Figure 43-19 Coexistent vertebral segmentation and vertebral formation failure anomalies.
Coronal computed tomography image in a patient with VACTERL syndrome (vertebral, anal atresia, cardiac, tracheal, esophageal, renal, limb), shows multiple combinations of both segmentation and formation failure with resultant congenital dextroscoliosis.
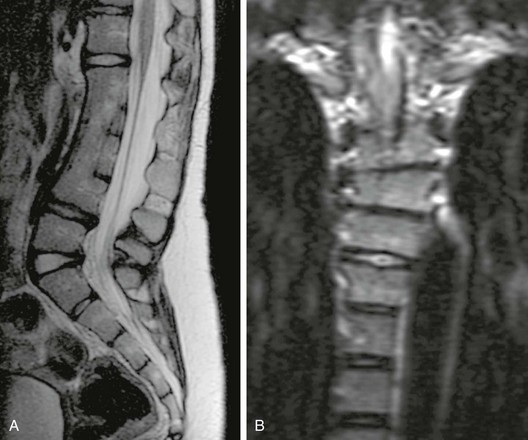
e-Figure 43-17 Vertebral segmentation failure.
A, Sagittal T2 weighted magnetic resonance image demonstrates multilevel vertebral segmentation failure producing a large block vertebra and kyphotic lumbar curve. Incomplete segmentation of the posterior elements is also evident. B, Coronal T2-weighted imaging in a different patient shows a three-level thoracic block vertebra and mild scoliotic curve related to a slightly better attempt at segmentation on the left.
Giampietro, PF, Dunwoodie, SL, Kusumi, K, et al. Progress in the understanding of the genetic etiology of vertebral segmentation disorders in humans. Ann NY Acad Sci. 2009;1151:38–67.
Pang, D, Dias, MS, Ahab-Barmada, M. Split cord malformation: part I. A unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451–480.
Pang, D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755–778. discussion 778-779
Rufener, SL, Ibrahim, M, Raybaud, CA, et al. Congenital spine and spinal cord malformations—pictorial review. AJR Am J Roentgenol. 2010;194(suppl 3):S26–S37.
Tortori-Donati, P, Rossi, A, Biancheri, R, et al. Magnetic resonance imaging of spinal dysraphism. Top Magn Reson Imaging. 2001;12:375–409.
Tortori-Donati, P, Rossi, A, Cama, A. Spinal dysraphism: a review of neuroradiological features with embryological correlations and proposal for a new classification. Neuroradiology. 2000;42:471–491.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 43
Congenital Abnormalities of the Spine
Embryology and Developmental Anatomy
Concurrent with the neural tube folding during primary neurulation, spinal cord development below the caudal neuropore commences within the pluripotent tissue at the caudal eminence in the process of secondary neurulation. The initially solid cell mass canalizes and becomes contiguous with the rostral neural tube that was formed by primary neurulation. By day 48, a transient ventriculus terminalis appears in the future conus. If this persists after birth, it is noted incidentally as a normal variant ventriculus terminalis (“fifth ventricle”), usually of no clinical significance (see Chapter 40). Failure of proper secondary neurulation leads to caudal spine anomalies in the caudal regression, tethered cord, or sacrococcygeal teratoma (SCT) spectra in addition to terminal myelocystocele and anterior sacral meningocele (ASM).
Abnormalities of Primary Neurulation
Premature Dysjunction
Premature dysjunction of the neural tube from overlying ectoderm permits perineural mesenchyme to access the neural groove and ependymal lining. This mesenchyme differentiates into fat and prevents complete neural tube closure, resulting in skin-covered lipomatous malformations with or without posterior spinal dysraphism. The most commonly observed anomalies are lipomyelocele (LMC), LMMC (Fig. 43-1), and intradural spinal lipomas (Fig. 43-2).
Figure 43-1 Lipomyelomeningocele.
Sagittal (A) and axial (B) T1-weighted magnetic resonance (MR) images demonstrate a typical lipomyelomeningocele, with a low-lying cord tethered into a large lipomatous malformation contiguous with the subcutaneous fat through a posterior dysraphic defect.
Figure 43-2 Juxtamedullary (subpial) spinal lipoma.
A, Sagittal T1-weighted magnetic resonance (MR) image shows a small subpial intradural lipoma (arrow) adherent to the dorsal conus surface. B, Axial T2-weighted MR image confirms direct contiguity of the neural placode with the lipoma. Note chemical shift artifact (arrow) in the frequency encoding direction, indicating fat. C, Axial fat-saturated T2-weighted MR image confirms fat content by homogeneous signal loss within the lipoma.
Nondysjunction
In contrast to lipomatous malformations, anomalies that result from nondysjunction occur when the neural tube fails to dissociate from adjacent cutaneous tissue. The simplest and least extensive variation is the dorsal dermal sinus, which occurs when a single connection persists and forms a fibrous cord from a skin dimple to the dural sac, conus, or central spinal cord canal. It is important to distinguish dermal sinus from its clinically asymptomatic mimic, simple coccygeal dimple (Fig. 43-3). In this mimic, the low sacral or coccygeal sinus originates from a low skin dimple and attaches to the coccyx via a short fibrous tract. These dimples are nearly always found within the intergluteal cleft, never communicate with the spinal canal, and require no treatment. Simple coccygeal dimples are the most common reason for newborn spinal ultrasound imaging.
Figure 43-3 Simple sacrococcygeal dimple.
Sagittal (A) and axial (B
[/not-level-membership-for-pediatrics-category]
