[level-membership-for-cardiovascular-category]
CHAPTER 49 Complex Congenital Heart Disease
Complex congenital heart disease is obviously one of the most challenging issues faced by the health care provider who takes care of the pediatric patient with cardiac lesions. Taken as a group, this set of lesions represents only a small portion of congenital heart disease (CHD) and is rare in the population; nevertheless, these diseases take up a considerable amount of the physician’s time. Hoffman and Kaplan1 have reported the results of a meta-analysis of the literature and found the incidence of moderate to severe forms of congenital heart disease to be 6/1,000 live births, rising to 19/1,000 live births if potentially serious bicuspid aortic valves are included. Putting this in perspective, all forms of CHD represent 75/1,000 live births, including such lesions as tiny muscular ventricular septal defects. In addition, the New England Infant Cardiac Program2 has reported that 3/1,000 live births need cardiac catheterization or surgery, or will die with CHD in early infancy (excluding premature infants with patent ductus arteriosus). This number rises to 5/1,000 live births who will need some type of specialized care during their lifetime. All these issues are a measure of the severity of the lesion. With improvements in diagnosis and treatment of CHD, along with a greater understanding of the anatomy and physiology, patients are living longer3 and represent a growing patient base seen by adult cardiologists and internists. In 1980, there were an estimated 300,000 adults with CHD, whereas this rose to approximately 1 million in 2000. In 2020, the number is anticipated to be 1.4 million.
TRANSPOSITION OF THE GREAT ARTERIES
Definition
The classic transposition of the great arteries (TGA) consists of isolated ventriculoarterial discordance, with the aorta arising from the right ventricle and the main pulmonary artery arising from the left ventricle.1,2,3 This results in separation of the systemic and pulmonary circulations, with limited mixing of oxygenated and deoxygenated blood. It usually refers to dextro-TGA (d-TGA; segmental anatomy {S,D,D}), in which the aortic valve is positioned anterior and to the right of the pulmonary valve. However, some refer to any segmental anatomy that results in this physiology as transposition. Occasionally, the conotruncus is further rotated so that the aorta is anterior and to the left of the pulmonary valve (segments {S,D,L}).
Prevalence and Epidemiology
The incidence of transposition of the great arteries is approximately 3 in 10,000 live births. It represents approximately 5% of all newborns with congenital heart disease.1 It is rarely associated with syndromes or extracardiac malformations, but is more common in infants of diabetic mothers.
Manifestations of Disease
Imaging Techniques and Findings
Radiography
The classic radiographic findings of transposition of the great arteries include a narrow superior mediastinum (so-called egg on a string) caused by the anteroposterior orientation of the great vessels and inapparent thymus, increased pulmonary vascular markings, and cardiomegaly. However, these findings are rarely diagnostic and, in fact, were shown to be absent in the vast majority of newborns with d-TGA (Fig. 49-1).4
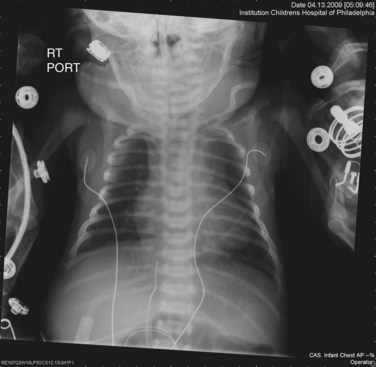
 FIGURE 49-1
FIGURE 49-1Ultrasound
Echocardiography can generally readily identify all the pertinent anatomic findings to manage patients with transposition of the great arteries. Subcostal imaging can readily identify the pulmonary artery arising from the left ventricle and the aorta arising from the right ventricle. The aorta can usually be identified by the coronaries and by its long length before branching (Fig. 49-2). The pulmonary artery branches early and the ductal arch should not be mistaken for the aorta (Fig. 49-3).
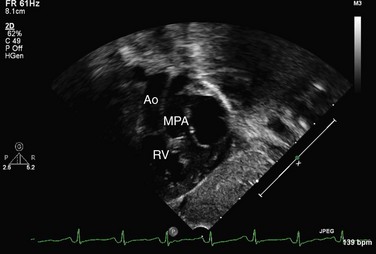
 FIGURE 49-2
FIGURE 49-2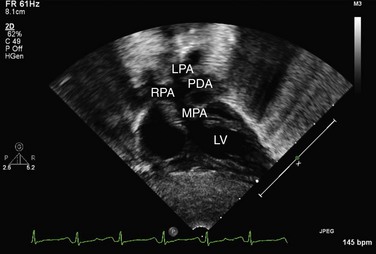
 FIGURE 49-3
FIGURE 49-3It is important to assess the atrial septum to determine whether it is restrictive (Fig. 49-4). In addition, the ventricular septum should be assessed to determine whether there are ventricular septal defects (Fig. 49-5). This is important for both preoperative management and surgical planning.
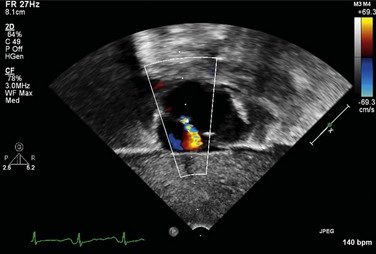
 FIGURE 49-4
FIGURE 49-4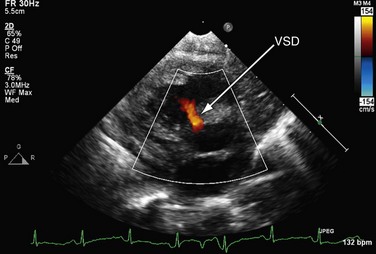
 FIGURE 49-5
FIGURE 49-5Magnetic Resonance Imaging
MRI plays an important role in the postoperative management of TGA. The standard for the treatment of TGA is the arterial switch (Jatene procedure), in which the aorta and main pulmonary artery are transected and sewn to the pulmonary and aortic root, respectively. The pulmonary arteries are generally brought anteriorly and draped over the aorta. The coronary arteries are transferred separately with buttons of tissue from the aorta to avoid ostial stenosis. Several problems can result, for which cardiac MRI is well suited to investigate. Supravalvular stenosis can occur at either anastomosis site (Fig. 49-6). Cine and velocity mapping can be used effectively to define regions of stenosis and quantify the degree of acceleration. Three-dimensional gadolinium sequences can also be useful to define stenoses and their relationships to other structures (Fig. 49-7). Unilateral or occasionally bilateral branch pulmonary artery stenosis may occur from stretching the branch pulmonary arteries after the Lecompte maneuver. Insufficiency of either valve can result from distortion during surgery. Through-plane velocity mapping can be used to quantify the degree of insufficiency of the valves precisely. Furthermore, short-axis cine volume sets can quantify the ventricular size, ejection, and wall motion abnormalities to screen for the effects of valve regurgitation or coronary abnormalities. Whole heart sequences can be used to evaluate for ostial stenosis of the transferred coronaries (Fig. 49-8). Perhaps more importantly, MRI can be used to evaluate for perfusion defects secondary to coronary stenosis. Gadolinium perfusion imaging is generally performed at rest and during adenosine administration. Adenosine administration causes coronary vasodilation and accentuates perfusion abnormalities by “stealing” flow from regions of marginal coronary perfusion.
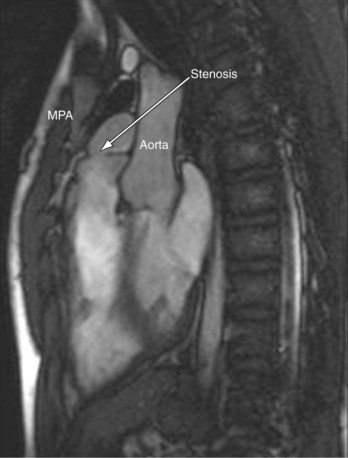
 FIGURE 49-6
FIGURE 49-6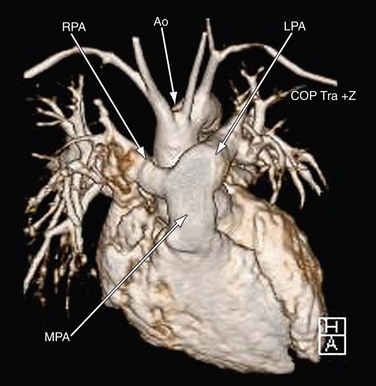
 FIGURE 49-7
FIGURE 49-7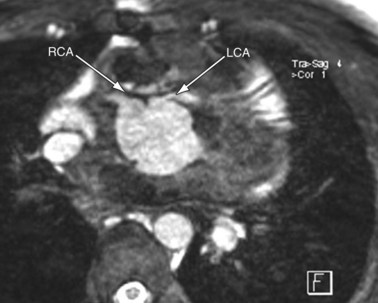
 FIGURE 49-8
FIGURE 49-8See later, “Single Ventricle,” for a more detailed description of the routine cardiac MRI examination.
TETRALOGY OF FALLOT
Prevalence and Epidemiology
The incidence of tetralogy of Fallot is approximately 4 in 10,000 live births, accounting for approximately 7% to 10% of cases of congenital heart disease.5 It occurs equally in males and females, and represents one of the most common lesions requiring intervention in the first year of life. It occurs commonly in association with genetic defects, including Down syndrome, DiGeorge syndrome (22q11 microdeletion), and Alagille syndrome (Jag1 mutation).6,7
Etiology and Pathophysiology
While originally described by Fallot as a constellation of four findings,8 it is now understood that the pathogenesis appears to be related to a single abnormality. The anterior malalignment VSD with normally related great vessels is responsible for the associated aortic override and right ventricular outflow tract obstruction, which classically results in right ventricular hypertrophy.
Certain associated conditions may be important to the management of tetralogy of Fallot. A right aortic arch occurs in approximately 25% of patients. Coronary anomalies are common (approximately 9%), including the left anterior descending (LAD) artery from the right coronary and single coronary.9 Pulmonary atresia may occur, with or without the presence of major aortopulmonary collaterals. Patients may often have additional muscular VSDs, and it may occur in association with a complete common atrioventricular canal, especially in association with Down syndrome. Stenosis of the left pulmonary artery is common and, rarely, isolation of the pulmonary artery contralateral to the aortic arch may occur. A patent ductus arteriosus is common; when there is significant outflow tract obstruction, the duct may be tortuous.
Manifestations of Disease
Imaging Techniques and Findings
Radiography
The classic radiographic finding is a boot-shaped heart, with an upturned apex (Fig. 49-9). Other findings may range from a normal heart size and decreased pulmonary blood flow to an increased heart size and increased pulmonary blood flow, depending on the degree of obstruction. A right aortic arch can be noted on the radiograph.
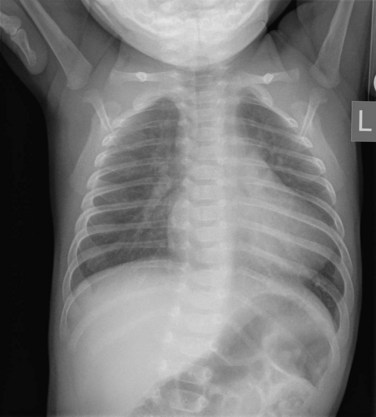
 FIGURE 49-9
FIGURE 49-9Ultrasound
The mainstay of the preoperative evaluation of tetralogy of Fallot is echocardiography. In addition to making the diagnosis (Fig. 49-10), the echocardiogram should focus on the degree and location (subpulmonary, valvular or supravalvular) of right ventricular outflow obstruction (Fig. 49-11), the presence of additional ventricular septal defects, the size and origin of the branch pulmonary arteries, the presence of aortopulmonary collaterals, the coronary origins and courses (with particular attention to whether the LAD or other major branch crosses the right ventricular outflow tract [RVOT]), the arch sidedness and branching pattern, and the patency and course of the ductus arteriosus. With rare exception, these can be evaluated by routine transthoracic imaging.
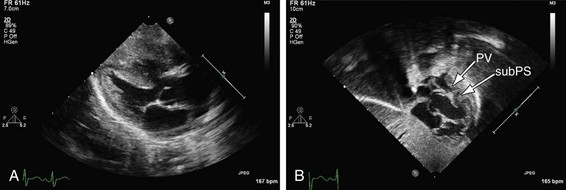
 FIGURE 49-10
FIGURE 49-10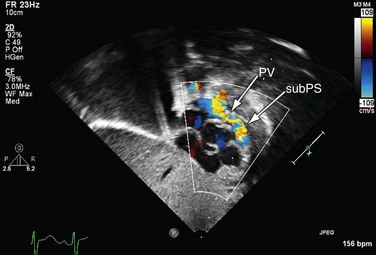
 FIGURE 49-11
FIGURE 49-11Magnetic Resonance Imaging
The preoperative use of cardiac MRI is limited to specific situations. In some cases, it may be useful to use respiratory and cardiac-gated, T2-prepared whole heart imaging to define the origins of the coronary arteries if they are not well seen by echocardiography. Gadolinium angiography is also useful to define aortopulmonary collaterals, and has been shown to be as effective as traditional angiography.10
MRI has become an important part of the postoperative management of tetralogy of Fallot. The repair for tetralogy of Fallot generally involves patch closure of the VSD and relief of the RVOT obstruction. Some patients may have an adequate pulmonary valve annulus and require only resection of an RV muscle bundle and/or pulmonary valvotomy. However, many will require a transannular patch. Patients in which a major coronary branch crosses the RVOT may require an RV to PA conduit, creating a double-barreled outflow. In patients who have had a transannular patch as part of the repair, pulmonary insufficiency may cause progressive RV dilation and decreased RV performance. In addition, left ventricular performance may decline, likely secondary to interactions with the left ventricle. Cardiac MRI can quantify the pulmonary regurgitation and right ventricular size, and is important in the monitoring of patients who have evidence of significant RV dilation by echocardiography or have poor echocardiographic windows (Figs. 49-12 and 49-13). MRI can also effectively evaluate residual RVOT obstruction or conduit stenosis, branch pulmonary artery stenosis, and pulmonary flow distribution to each lung (Fig. 49-14).
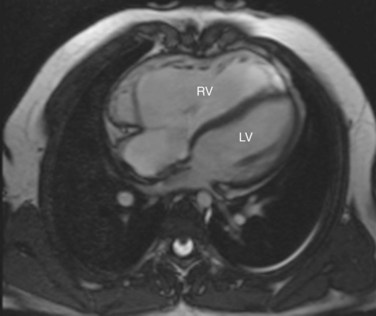
 FIGURE 49-12
FIGURE 49-12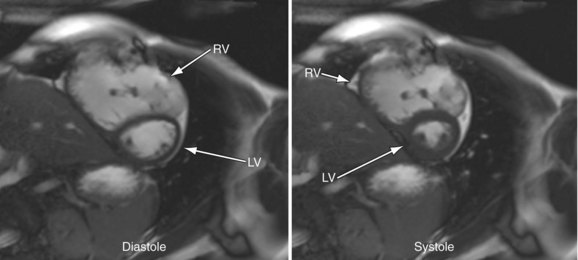
 FIGURE 49-13
FIGURE 49-13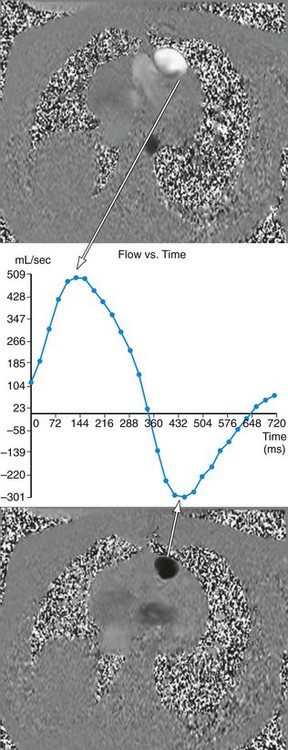
 FIGURE 49-14
FIGURE 49-14See later, “Single Ventricle,” for a more detailed description of the routine cardiac MRI examination.
Nuclear Medicine: Positron Emission Tomography
Nuclear scintigraphy perfusion imaging has been used postoperatively in patients with TOF in the setting of branch pulmonary artery stenosis to quantify pulmonary blood flow distribution.11 However, this has largely been supplanted by cardiac MRI velocity mapping.12,13 It still may be useful for patients in whom a stent or coil artifact precludes assessment by MRI.
TRUNCUS ARTERIOSUS
Definition
There are two major classification schemes in use—those of Collett and Edwards14 and Van Praagh.15 These are invariably based on the position of the main pulmonary artery segment and branch pulmonary arteries, with the Van Praagh classification using types A and B to delineate whether a VSD is present (almost all have VSDs). Of the different forms, 92% of all patients fall into type 1A or 2A. The following are the definitions used in the Van Praagh classification with its differences and similarities with the Collett and Edwards classification noted; the Van Praagh classification takes into account aortic arch anomalies.
Prevalence and Epidemiology
According to Hoffman and Kaplan,1 the mean incidence of truncus arteriosus is 107 per million live births; it is thought to occur in 1% to 2% of patients with CHD at necropsy and represents approximately 0.7% of all congenital heart disease. The DiGeorge syndrome and patients with microdeletion of chromosome 22 have a high incidence of having truncus arteriosus. There is no race or gender predilection.
Etiology and Pathophysiology
Etiology
This rare lesion is caused by a failure in conotruncal septation of the embryonic truncus arteriosus and conus. The truncus is normally divided into the aorta and pulmonary artery by two ridges that appear in the 4- to 5-week-old embryo and that grow toward the midline. Cells from the neural crest directly contribute to this septation; one of the leading theories is that this malformation is a disruption in neural crest migration. Van Praagh and Van Praagh, however, believed this to be a form of tetralogy of Fallot.15
Pathophysiology
There are associated cardiovascular malformations, such as abnormalities of the coronary arteries, a right aortic arch, persistent left superior vena cava, aberrant origin of the left subclavian, patent foramen ovale, partial and complete atrioventricular canal defects, mitral and tricuspid malformations, double-inlet or hypoplastic left ventricle, left pulmonary artery sling, and anomalous pulmonary venous connections.16
Manifestations of Disease
Imaging Techniques and Findings
Ultrasound
Postoperatively, narrowing of the reconstructed right ventricular outflow tract and pulmonary arteries needs to be assessed. Stenosis can be evaluated by color flow mapping and Doppler echocardiography can determine the gradient; this is best performed in the subcostal sagittal or parasternal short-axis views and, occasionally, in the apical view angled extremely anteriorly. Residual ventricular septal defects can be seen by short-axis sweeps. Assessment of the truncal valve, as in the preoperative assessment, must be made routinely (Figs 49-15 and 49-16).
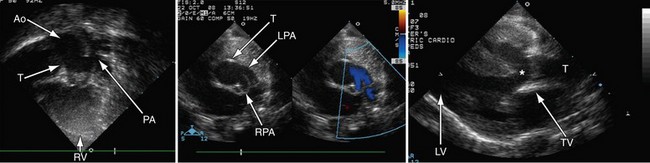
 FIGURE 49-15
FIGURE 49-15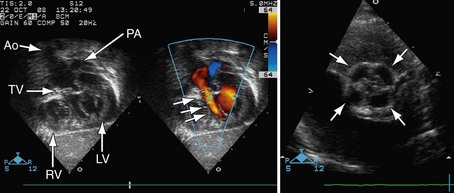
 FIGURE 49-16
FIGURE 49-16Computed Tomography
Because of radiation considerations, CT scanning plays a limited role in the care of the patient with congenital heart disease, and truncus arteriosus is no different. It is done chiefly when there is a contraindication to MRI and, when used, it is generally carried out postoperatively to visualize the right ventricular outflow tract and branch pulmonary arteries along with the aortic reconstruction if the aortic arch was interrupted. Because of its very limited temporal resolution, except if absolutely needed, ventricular function is best determined by MRI or echocardiography (Figs. 49-17 to 49-19).
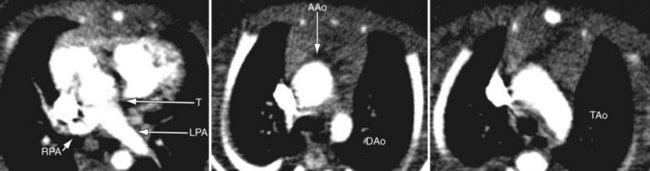
 FIGURE 49-17
FIGURE 49-17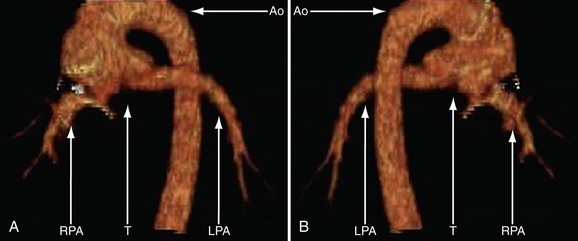
 FIGURE 49-18
FIGURE 49-18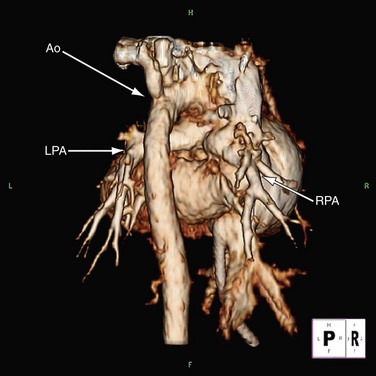
 FIGURE 49-19
FIGURE 49-19Magnetic Resonance Imaging
In the native state, the goal of MRI is to define the type of truncus arteriosus (including origin of pulmonary trunk, branches, and collaterals), functional abnormalities of the truncal valve (regurgitation, stenosis, morphology of the valve, number of cusps), alignment of the truncal valve with respect to the ventricular septum, brachiocephalic vessels, pulmonary veins, and aorta, associated cardiac anomalies, and mediastinal structures (hypoplasia or absence of thymus). This can be performed with the protocol outlined later (see later, “Single Ventricles”). Specifically, static steady-state free precession (SSFP), cine imaging, and gadolinium imaging can visualize the truncus arteriosus and branching pattern of the pulmonary arteries from this vessel. Determining aortic arch interruption or hypoplasia along with the presence of the ductus arteriosus can easily be done. Truncal valve insufficiency and stenosis should be assessed with cine and phase-encoded velocity mapping, which can quantify the regurgitant fraction. The Qp/Qs ratio may be assessed by placing velocity maps on each pulmonary artery and in the aortic arch distal to the takeoff of the main or branch pulmonary arteries. Velocity mapping at the level of the truncal valve not only quantifies truncal insufficiency, but is also used as an internal check on the data—sum of the net flows in the branch pulmonary arteries and aorta distal to the takeoff of the main and branch pulmonary arteries must equal the net flow across the truncal valve—as well as visualizing the number of leaflets. Cine is used to quantify ventricular performance and assess for any additional ventricular septal defects. T2-prepared coronary imaging can be used to identify any coronary artery abnormalities.
Postoperatively, MRI is used more often than in the preoperative state, especially as the patient gets older and the echocardiographic windows become poorer. Similar to echocardiography, imaging of the reconstructed right ventricular outflow tract and pulmonary arteries for stenosis is an important component of the examination and can be done with steady SSFP cine, three-dimensional gadolinium-enhanced MRI, and dark blood imaging. Residual VSDs can be seen by cine imaging. Assessment of the truncal valve, now the neoaorta, as in the preoperative assessment, must be made routinely with cine and velocity mapping in the right ventricular outflow tract, branch pulmonary arteries, and neoaorta. Delayed enhancement imaging is used to determine myocardial scar tissue. Follow-up of ventricular function by quantification by cine is routine (Figs. 49-20 to 49-24).
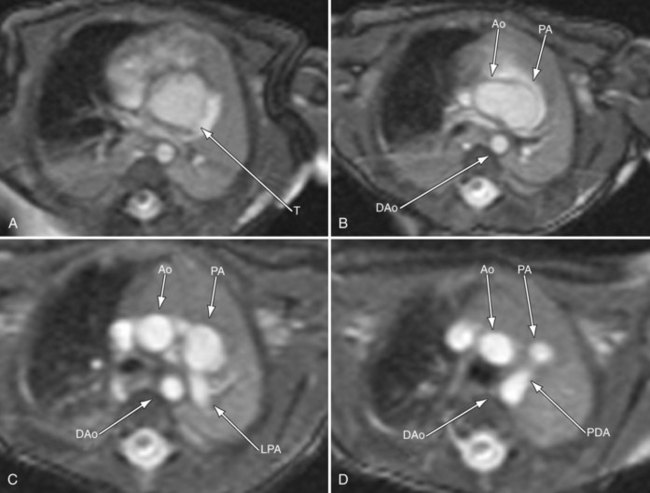
 FIGURE 49-20
FIGURE 49-20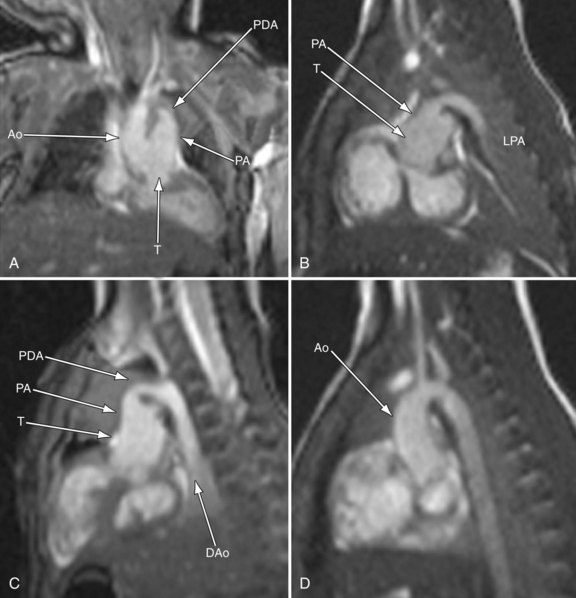
 FIGURE 49-21
FIGURE 49-21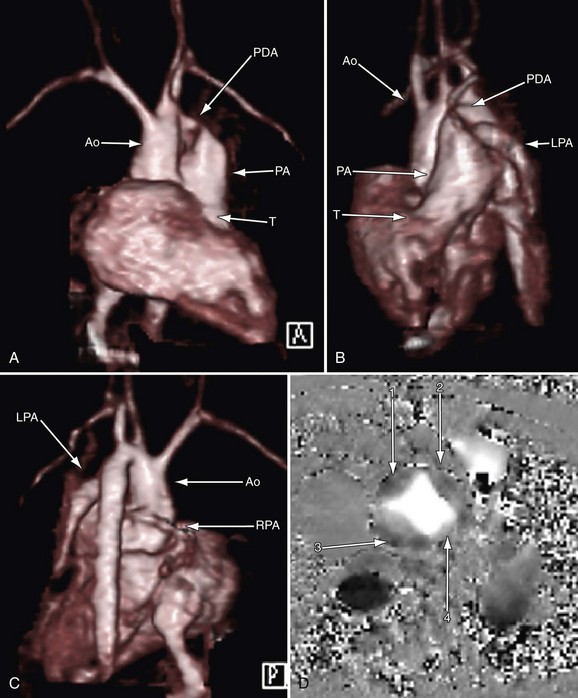
 FIGURE 49-22
FIGURE 49-22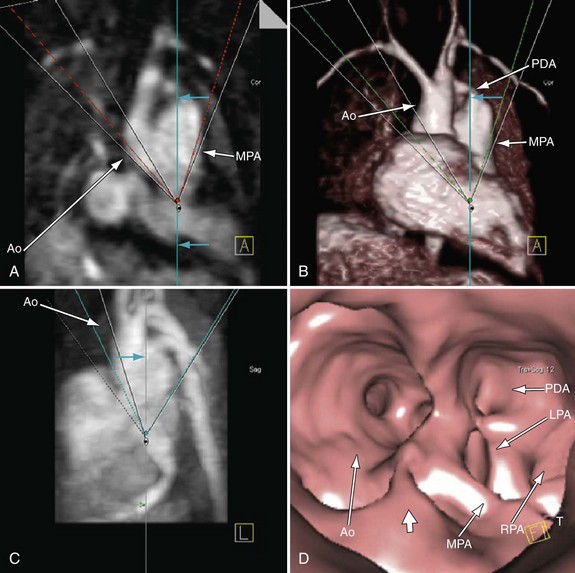
 FIGURE 49-23
FIGURE 49-23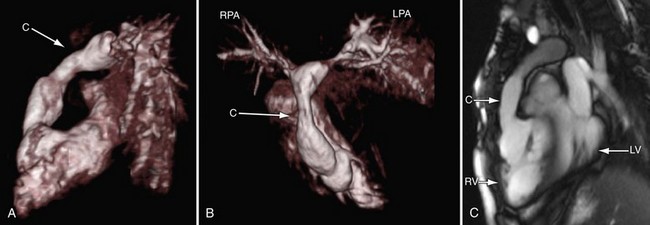
 FIGURE 49-24
FIGURE 49-24SINGLE VENTRICLE
Definition: Functional Single Ventricle
The simple definition of a functional single ventricle, sometimes called the univentricular heart, is a heart that has only one usable pumping chamber in the native state or with surgical correction. The detailed anatomy of functional single ventricles is highly variable; the ventricles can be of the RV or left ventricle (LV) morphologic type, can be D-looped or L-looped,17 or can be a true single ventricle. A true single ventricle is defined as an atrioventricular (AV) valve to ventricle connection in which two AV valves or a common AV valve (excluding atresia) enters into one ventricle only in the presence of only one ventricular sinus. A functional single ventricle can be any type of ventricular arrangement, including a true single ventricle (e.g., two ventricles with multiple large ventricular septal defects, straddling AV valve with hypoplasia of one ventricle), in which the ventricle acts like a single pumping chamber and needs to be treated as such. Examples of single ventricles are hypoplastic left heart syndrome (HLHS; functional single RV), double-inlet left ventricle, and tricuspid atresia (functional single LV).
Prevalence and Epidemiology
Because this section deals with a series of lesions grouped under the rubric of single ventricle, it is difficult to be precise regarding the epidemiology. According to Hoffman and Kaplan,1 the mean incidence of hypoplastic left heart complexes, hypoplastic right heart complexes, single ventricle, and tricuspid atresia is 266, 222, 106, and 79 per million live births, respectively.
Hypoplastic left heart syndrome, one of the most common cyanotic CHD lesions, has been reported to occur in 0.016% to 0.036% of all live births and in 1.4% to 3.8% of pathologic series,18 with a male predominance (55% to 70%). Recurrence risk in siblings has been reported to be 0.5% and up to 13.5% for other forms of CHD. As a comparison, tricuspid atresia occurs in approximately 1 in 15,000 live births and has a prevalence in clinical series ranging from 0.3% to 3.7%, with no apparent gender predilection. In autopsy series, the rate is 2.9%.
Etiology and Pathophysiology
Etiology
As with prevalence, because functional single ventricle encompasses many disease states, there are a myriad of theories about the etiology of the lesions, which is beyond our scope here. Suffice it to say that from an embryologic standpoint, three theories have been presented, all with evidence to support their viewpoint. One is a flow-related phenomenon, in which blood is directed in an abnormal direction and results in the pathologic state. For example, in HLHS, if septum primum is deviated posteriorly, blood flow is directed away from the developing left-sided valves, LV, and aorta, causing these structures to be hypoplastic. A second theory is that the lesions are genetically determined based on the fact that some of these lesions are familial; as noted for HLHS, relatives of the affected patient have a higher incidence of cardiac disease than the general population. First-degree relatives of patients with HLHS have a 12% prevalence of cardiac abnormalities involving the left ventricular outflow tract. Multiple genetic syndromes have a high incidence of HLHS, such as Noonan, mosaic Turner, and Holt-Oram syndrome.19 Finally, toxic or infectious causes are thought to result in these lesions.
Pathophysiology
Prior to bidirectional superior cavopulmonary connection (BSCC), no surgery may be needed, as in the case of tricuspid atresia with normally related great arteries and a restrictive ventricular septal defect or pulmonary stenosis. In this particular case, adequate but restricted pulmonary blood flow is maintained by the usable LV. The systemic venous return crosses an atrial septal defect, mixes with pulmonary venous return in the left atrium and LV, and is pumped to both circulations. Other patients, such as those with HLHS, need immediate surgery—the Norwood Stage I procedure20—which involves the following:
At approximately 5 to 6 months of age, pulmonary vascular resistance has dropped enough so that the BSCC is performed, which can be done as a hemi-Fontan or bidirectional Glenn procedure. This creates a superior vena cava to pulmonary artery anastomosis and prevents blood from flowing into the atrium from the superior vena cava. Ligation of the systemic to pulmonary artery shunt is done at this time. The ventricle is thus volume-unloaded because it does not have direct access to the pulmonary circulation; instead, part of the systemic circulation’s venous return (blood from the brain and upper body) is shunted into the lungs via the superior vena cava to pulmonary artery anastomosis. It is not clear from cardiac MRI data, however, that it remains volume-unloaded while the patient is in this physiologic state.21,22 Because only part of the systemic venous return enters the lungs, cardiac output is maintained at the expense of cyanosis.
Finally, at about 2 years of age, the systemic and pulmonary circulations (with the possible exception of coronary venous flow) are finally separated by baffling inferior vena cava blood into the lungs via an intra-atrial baffle or extracardiac conduit, the Fontan completion.23 This was formerly done as an atriopulmonary connection but it is now only of historical interest. The entire systemic venous return flows passively into the lungs and the ventricle is volume-unloaded again. To improve outcome, a communication is purposely created between the systemic and pulmonary venous pathways (a fenestration) to allow for right to left shunting when there is increased pulmonary vascular resistance. This allows for maintenance of the cardiac output at the expense of cyanosis, similar in concept to the bidirectional superior cavopulmonary connection. Usually the fenestrations close on their own.
Manifestations of Disease
Imaging Techniques and Findings
Radiography
In the postoperative patient, the chest radiograph is used to assess heart size (e.g., pericardial effusion) and pulmonary vascular markings for assessment of pulmonary flow, pleural effusions, or ascites and to check for device placement (e.g. stent, coils) that may have been placed during cardiac catheterization (Fig. 49-25).
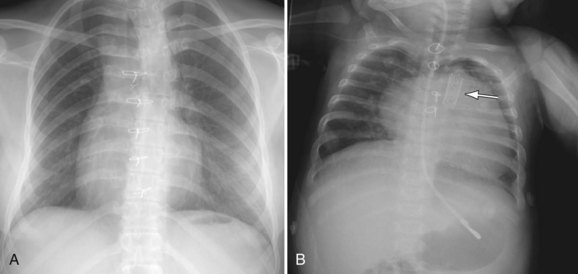
 FIGURE 49-25
FIGURE 49-25Ultrasound
Postoperative assessment is also a key role of echocardiography in the patient’s care; however, at different stages of reconstruction, it is important to focus on different structures. There are parameters such as ventricular function that are significant at all stages and one must endeavor to evaluate these as well. See later, “Magnetic Resonance Imaging,” for important structures to image at each stage of surgical reconstruction (Figs. 49-26 to 49-28).
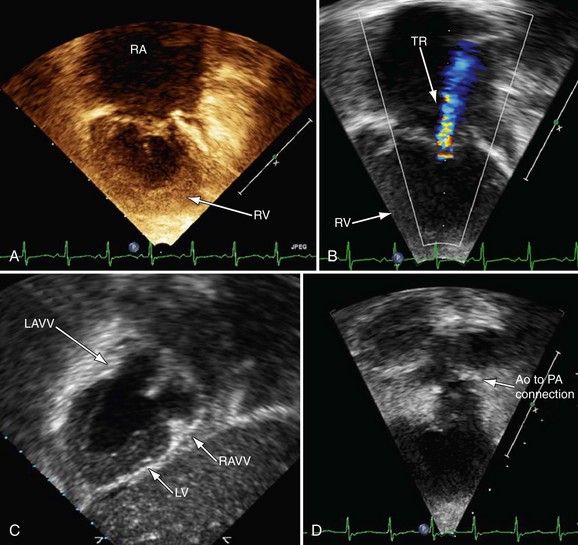
 FIGURE 49-26
FIGURE 49-26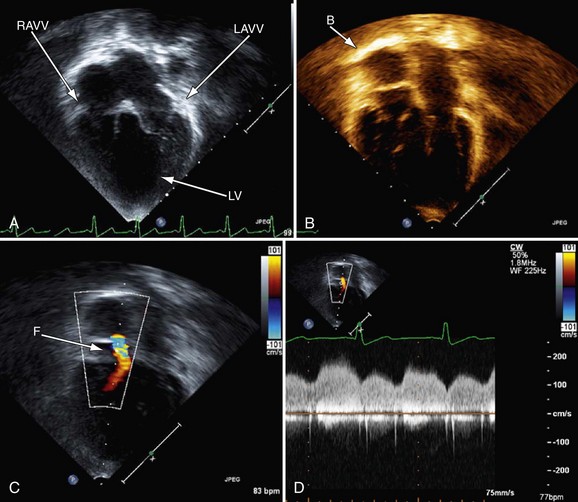
 FIGURE 49-27
FIGURE 49-27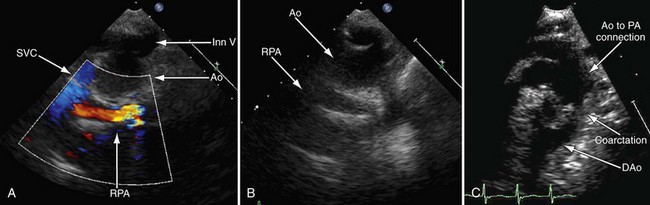
 FIGURE 49-28
FIGURE 49-28Magnetic Resonance Imaging
At each stage, the following are important to image.
After the Bidirectional Superior Cavopulmonary Connection
The major extra focus in this examination is the superior vena cava to pulmonary artery anastomosis—either the bidirectional Glenn or hemi-Fontan. This can be done with cine or gadolinium (rarely dark blood) sequences. The Qp/Qs ratio can be calculated by flow in the superior vena cava or in both branch pulmonary arteries. However, recent data suggest that pulmonary vein mapping is much more appropriate because it also captures aortic collateral flow to the lungs,22 much different from catheterization-derived data. If a hemi-Fontan procedure was performed, leaks into the atrium from the superior vena cava to pulmonary artery anastomosis should be assessed.
After the Fontan Procedure
See Figures 49-29 to 49-32.
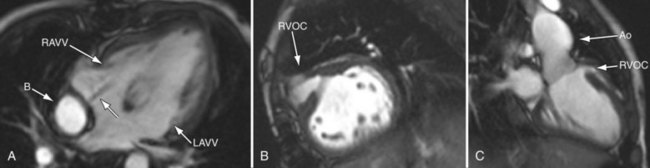
 FIGURE 49-29
FIGURE 49-29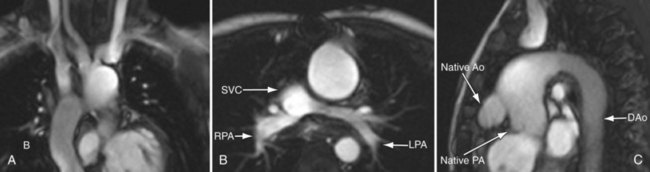
 FIGURE 49-30
FIGURE 49-30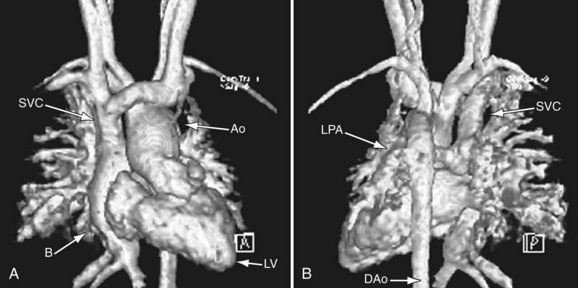
 FIGURE 49-31
FIGURE 49-31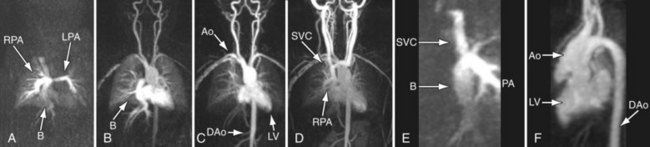
 FIGURE 49-32
FIGURE 49-32Angiography
During catheterization, angiography is used to assess all the structures listed earlier in the cardiac MRI subsection. Generally, in most cases, noninvasive means have been able to assess almost all important structures so angiography at this point is usually used during interventional procedures (e.g., angioplasty of the aorta or pulmonary arteries, device placement; Fig. 49-33).
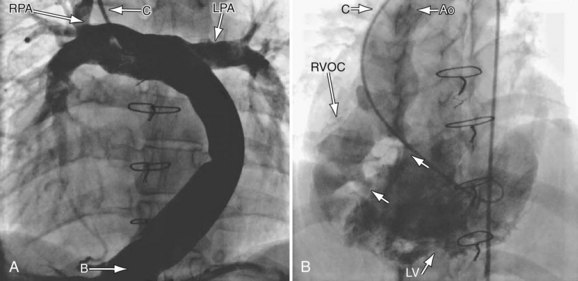
 FIGURE 49-33
FIGURE 49-33LEVO-TRANSPOSITION OF THE GREAT ARTERIES
Definition
Levo-transposition of the great arteries (L-TGA) refers to a wide spectrum of structural heart disease, encompassing two-ventricle and single-ventricle hearts and dextro-, meso-, and levocardia. For the purposes of this section, L-TGA will consist of situs solitus and two good-sized ventricles in the setting of mesocardia or levocardia. The resultant setup gives rise to both AV and ventriculoarterial (VA) discordance (double discordance). The resultant anatomy results in physiologically corrected (atrioarterial concordance) flow so that systemic venous flow is directed to the lungs and pulmonary venous flow is directed to the body. Some have referred to this disorder as congenitally corrected TGA. Segmentation for this anatomy is classically {S,L,L} and includes situs solitus {S} to position the right atrium (RA) to the right, L-looping of the ventricles {L} to position the RV to the left, and L malposition of the great arteries to position the aorta (Ao) anterior and leftward to the PA. By definition, the PA arises from the LV and the Ao arises from the RV.24
Etiology and Pathophysiology
The cause of L-TGA is still under debate. There are ventricular inversion and conotruncal components. One theory has proposed L-looping of the ventricles along with inversion of the infundibulum (subaortic) and normal aortopulmonary septation (involving the cardiac neural crest), but absence of aortopulmonary rotation. Another theory has proposed that L-TGA involves not only L-looping with ventricular inversion but also suggests that the LV structures appear upside-down—there are posterolateral and anteromedial LV papillary muscles. This would help account for the inverted position of the dominant anterior AV node and the course of the AV bundle superior to a VSD. In contrast, normal looping of the ventricles gives rise to a posterior AV node and the course of the AV bundle is inferior to a VSD.25
Manifestations of Disease
Imaging Techniques and Findings
Radiography
Typically, the AP chest radiograph shows situs solitus, levocardia or mesocardia with a prominent leftward and anterior aortic knob, a left aortic arch, a normal-sized cardiac silhouette and normal to increased pulmonary vascular markings, depending on the size of the VSD (Fig. 49-34).
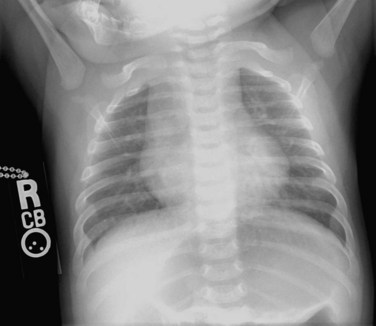
 FIGURE 49-34
FIGURE 49-34Ultrasound
Transthoracic echocardiography is a standard imaging procedure for L-TGA. All associated defects can be delineated with great accuracy, and echocardiography is superior for the evaluation of AV straddle. A careful search for common associated lesions should be undertaken, including delineation of TV anomalies, VSDs, and subpulmonary stenosis (Figs. 49-35 to 49-38).
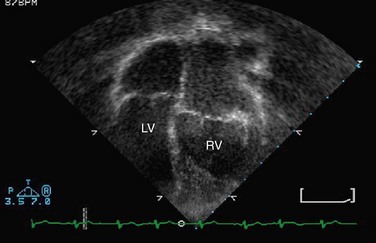
 FIGURE 49-35
FIGURE 49-35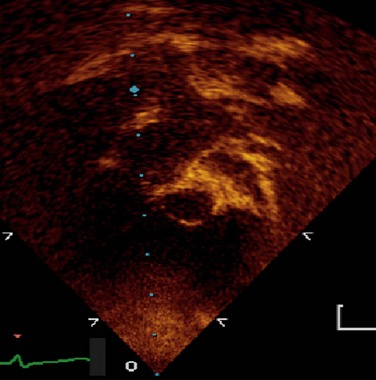
 FIGURE 49-36
FIGURE 49-36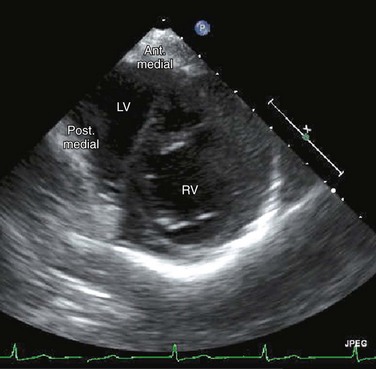
 FIGURE 49-37
FIGURE 49-37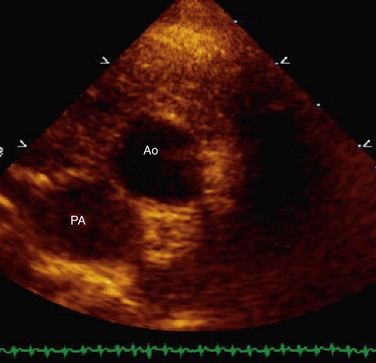
 FIGURE 49-38
FIGURE 49-38Magnetic Resonance Imaging
It also plays an important role in the management of the late double switch, during which a pulmonary artery band is placed to retrain the left ventricle. Several centers have advocated monitoring the LV mass by serial cardiac MRI to ensure that the LV is properly trained before performing the double switch (Figs. 49-39 and 49-40).26
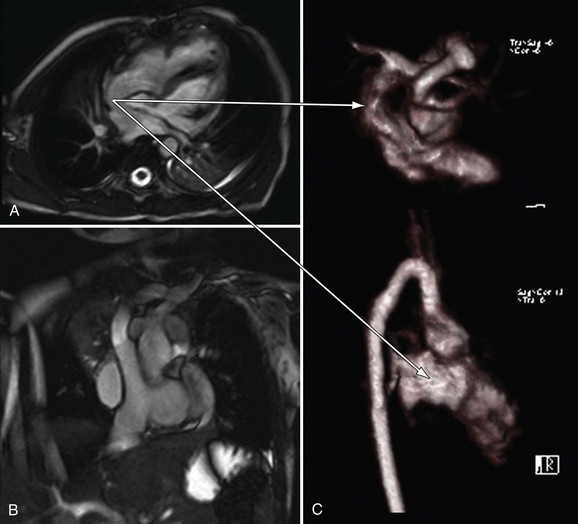
 FIGURE 49-39
FIGURE 49-39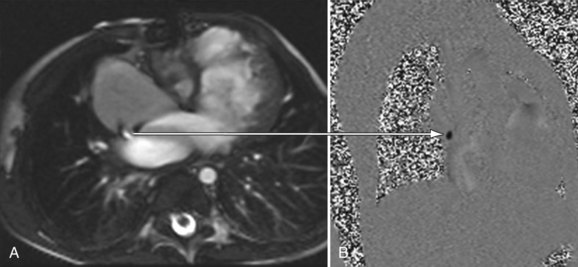
 FIGURE 49-40
FIGURE 49-40DOUBLE-OUTLET RIGHT VENTRICLE
Manifestations of Disease
Imaging Techniques and Findings
Radiography
Typically, the AP chest radiograph shows situs solitus, levocardia with a prominent heart size, a left aortic arch, and normal to increased pulmonary vascular markings, depending on the position of the VSD and conal septum deviation (Fig. 49-41).
 FIGURE 49-41
FIGURE 49-41Ultrasound
Transthoracic echocardiography remains one of the standard imaging techniques for DORV. Associated defects can be delineated and echocardiography is superior for the evaluation of AV straddle. A careful search for common associated lesions should be undertaken, including delineation of inlet and outlet anomalies, VSD location, subpulmonary or subaortic stenosis, and great artery caliber and narrowing. One important echo measurement for consideration of surgical management is the distance from the medial portion of the TV to the pulmonary valve annulus. Depending on the relation of the aorta to the PA, this distance can be accurately measured from subcostal views in varying planes. It represents the potential baffle pathway diameter to the aorta in a two-ventricle repair (Figs. 49-42 to 49-46).
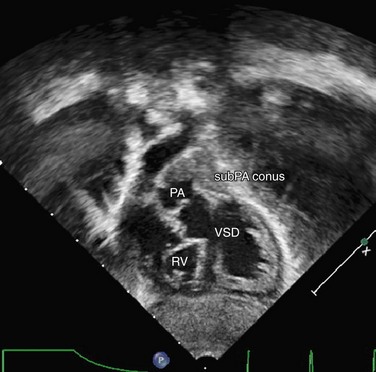
 FIGURE 49-42
FIGURE 49-42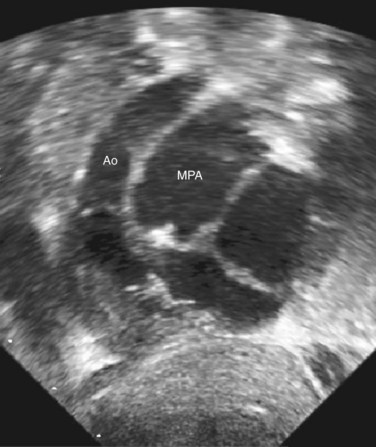
 FIGURE 49-43 Subcostal LAO view in the same patient with DORV as shown in Figure 49-42. The aorta is significantly smaller than the PA, despite the fact that the conal septum appears slightly deviated toward the PA.
FIGURE 49-43 Subcostal LAO view in the same patient with DORV as shown in Figure 49-42. The aorta is significantly smaller than the PA, despite the fact that the conal septum appears slightly deviated toward the PA.
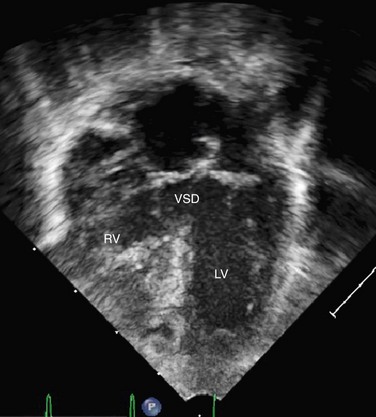
 FIGURE 49-44 Apical four-chamber in the same patient with DORV as shown in Figure 49-42. The VSD is large and nonrestrictive.
FIGURE 49-44 Apical four-chamber in the same patient with DORV as shown in Figure 49-42. The VSD is large and nonrestrictive.
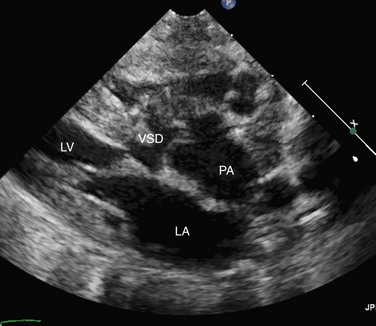
 FIGURE 49-45
FIGURE 49-45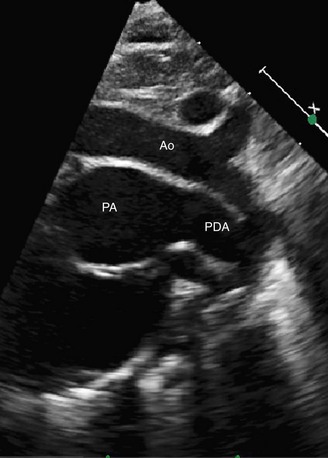
 FIGURE 49-46
FIGURE 49-46Magnetic Resonance Imaging
MRI is complementary for imaging complex anomalies of the systemic and pulmonary venous connections, aortic arch and pulmonary artery anomalies, and calculation of cardiac output, regurgitant valve fractions, and systolic and diastolic volumes of the RV and LV (Fig. 49-47). Because DORV can take on many different types of physiology and anatomic variants, see the discussions of individual lesions for the use of MRI. It can also be useful for surgical planning.
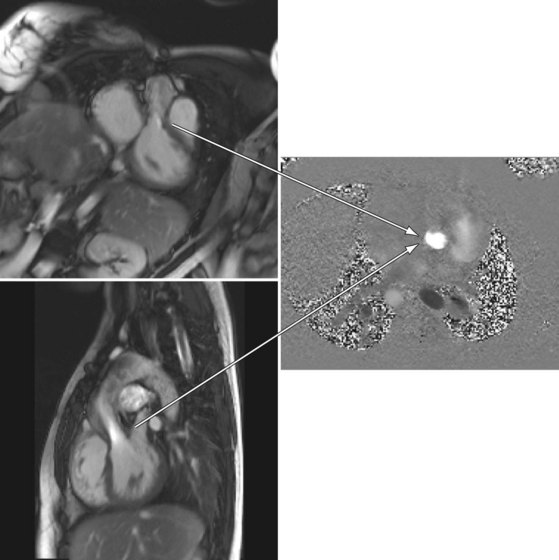
 FIGURE 49-47
FIGURE 49-47SYNOPSIS OF TREATMENT OPTIONS
Surgical and Interventional Treatment
If the patient has L-TGA with an intact ventricular septum, no significant subpulmonary stenosis, and good TV and RV function, most centers advocate diligent observation. Surveillance intervals vary with age but generally would be twice yearly. Medium- and long-term RV function will determine eventual therapy. Natural history studies have shown that most patients begin to show symptoms of significant heart failure by the fourth or fifth decade of life, with worsening systemic right ventricular failure and tricuspid regurgitation.27 This has prompted some centers to adopt a more aggressive approach, primarily performing a so-called double switch, consisting of an atrial switch and arterial switch operation with the goal of preventing right ventricular failure. Patients who are diagnosed at a later age, generally those without associated lesions, must first undergo pulmonary artery banding to retrain the left ventricle and prepare it for the increased workload of supporting the systemic circulation. This is often done in two stages, with a loose band first applied and then tightened at a later date once the left ventricle has acclimated. The pulmonary artery band itself can often improve symptoms by shifting the ventricular septum and reducing RV-LV interactions and tricuspid regurgitation, leading some to propose this as a palliative procedure. However, most advocate proceeding to a double switch as a long-term strategy, assuming the that LV tolerates the banding.26
For patients with L-TGA, large VSD, no evidence of subpulmonary stenosis, and good TV, surgery would be recommended, usually consisting of VSD patch closure along with an atrial switch and an arterial switch (a double switch). Postoperative surveillance intervals vary with age but generally would be twice yearly. For patients with L-TGA, large VSD, and significant subpulmonary stenosis, surgery would be recommended, usually consisting of a Rastelli operation (VSD baffle closure to the aorta) along with an atrial switch and an RV-PA valved homograft conduit.28 Postoperative surveillance intervals vary with age but generally would be twice yearly. The RV-PA conduit will require future replacement. For patients with L-TGA and hemodynamically significant left-sided TV insufficiency, surgical intervention would be recommended. In general, direct surgical evaluation of the TV must be performed to determine if valvuloplasty or replacement benefits the patient the most. For some L-TGA patients, failed supraventricular tachycardia medical therapy will lead to an invasive electrophysiology study along with radiofrequency ablation of the accessory left-sided pathway. For L-TGA patients with symptomatic complete AV block, insertion of a dual-chamber pacemaker will be required.
Almost all patients with DORV require palliative or corrective surgery for optimal long-term survival. Exceptions to this would include concomitant lethal anomalies. For patients with DORV, subaortic VSD, and no significant pulmonary stenosis, surgery is usually performed in the neonatal period, usually consisting of VSD baffle closure to the aorta. Postoperative surveillance intervals vary with age but generally would be twice yearly. For patients with DORV, subaortic VSD, and significant pulmonary stenosis, surgery is usually performed in the infant period, usually consisting of VSD baffle closure to the aorta and relief of the subpulmonary stenosis. Postoperative surveillance intervals vary with age but generally would be twice yearly. For patients with DORV and subpulmonary VSD, surgery would be recommended early, usually consisting of VSD patch closure to align the PA with the LV, along with an arterial switch procedure. Aortic arch problems require attention at the same procedure. Postoperative surveillance intervals vary with age but generally would be twice yearly. For patients with DORV and a doubly committed VSD, surgery would be recommended early, usually consisting of VSD baffle closure to the aorta. Postoperative surveillance intervals vary with age but generally would be twice yearly. For patients with DORV and noncommitted VSD, the surgical pathway may involve complex palliation while waiting for possible corrective repair. For patients with complex DORV, including inlet abnormalities, AV valve straddle, unbalanced ventricles, and/or complex outlet anatomy, the surgical pathway often involves offering staged Fontan palliation or transplantion.29














1 Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890-1900.
2 Fyler DC, Buckley LP, Hellenbrand WE. Report of the New England Regional Infant Cardiac Program. Pediatrics. 1980;65:377-461.
3 Perloff JK. Congenital heart disease in adults. A new cardiovascular subspecialty. Circulation. 1991;84:1881-1890.
4 Donnelly LF, Hurst DR, Strife JL, Shapiro R. Plain-film assessment of the neonate with D-transposition of the great vessels. Pediatric Radiology. 1995;25:195-197.
5 Centers for Disease Control and Prevention (CDC). Improved national prevalence estimates for 18 selected major birth defects—United States, 1999-2001. MMWR Morb Mortal Wkly Rep. 2006;54:1301-1305.
6 Goldmuntz E, Clark BJ, Mitchell LE, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998;32:492-498.
7 Krantz ID, Smith R, Colliton RP, et al. Jagged1 mutations in patients ascertained with isolated congenital heart defects. Am J Med Genet. 1999;84:56-60.
8 Fallot E-LA. Contribution a l’anatomie pathologique de la maladie bleue (cyanose cardiaque). Marseille Médical. 1888;25:77-93.
9 Dabizzi RP, Caprioli G, Aiazzi L, et al. Distribution and anomalies of coronary arteries in tetralogy of Fallot. Circulation. 1980;61:95-102.
10 Roche KJ, Rivera R, Argilla M, et al. Assessment of vasculature using combined MRI and MR angiography. Am J Roentgenol. 2004;182:861-866.
11 Ming-Ting W, Yi-Luan H, Kai-Sheng H, et al. Influence of pulmonary regurgitation inequality on differential perfusion of the lungs in tetralogy of Fallot after repair: a phase-contrast magnetic resonance imaging and perfusion scintigraphy study [abstract]. J Am Coll Cardiol. 2007;49:1880-1886.
12 Harris MA, Weinberg PM, Whitehead KK, Fogel MA. Usefulness of branch pulmonary artery regurgitant fraction to estimate the relative right and left pulmonary vascular resistances in congenital heart disease. Am J Cardiol. 2005;95:1514-1517.
13 Kang IS, Redington AN, Benson LN, et al. Differential regurgitation in branch pulmonary arteries after repair of tetralogy of Fallot: a phase-contrast cine magnetic resonance study. Circulation. 2003;107:2938-2943.
14 Collett RW, Edwards JE. Persistent truncus arteriosus: a classification according to anatomic types. Surg Clin North Am. 1949;29:1245-1270.
15 Van Praagh R, Van Praagh S. The anatomy of common aorticopulmonary trunk (truncus arteriosus communis) and its embryologic implications. A study of 57 necropsy cases. Am J Cardiol. 1965;16:406-425.
16 Jacobs ML. Congenital Heart Surgery Nomenclature and Database Project: truncus arteriosus. Ann Thorac Surg. 2000;69:S50-S55.
17 Van Praagh R. Terminology of congenital heart disease. Glossary and commentary. Circulation. 1977;56:139-143.
18 Edwards JE. Congenital malformations of the heart and great vessels. In: Gould SE, editor. Pathology of the Heart. Springfield, Ill: Charles C Thomas; 1953:406-407.
19 Natowicz M, Kelley RI. Association of Turner syndrome with hypoplastic left-heart syndrome. Am J Dis Child. 1987;141:218-220.
20 Norwood WI, Lang P, Hansen DD. Physiologic repair of aortic atresia–hypoplastic left heart syndrome. N Engl J Med. 1983;308:23-26.
21 Fogel MA, Weinberg PM, Chin AJ, et al. Late ventricular geometry and performance changes of functional single ventricle throughout staged fontan reconstruction assessed by magnetic resonance imaging. J Am Coll Cardiol. 1996;28:212-221.
22 Whitehead KK, Gillespie MJ, Harris MA, et al. Non-invasive quantification of systemic to pulmonary collateral flow: a major source of inefficiency in patients with superior cavopulmonary connections. Circ Cardiovasc Imaging. 2009;2:405-411.
23 Fontan F, Baudet E. Surgical repair of tricuspid atresia. Thorax. 1971;26:240-248.
24 Allen HD, Gutgesell HP, Clark EB, Driscoll DJ. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 7th ed. Lippincott Williams & Wilkins; 2008. pp 1087-1227
25 Kuehl KS, Loffredo CA. Genetic and environmental influences on malformations of the cardiac outflow tract. Expert Rev Cardiovasc Ther. 2005;3:1125-1130.
26 Duncan BW, Mee RB, Mesia CI, et al. Results of the double switch operation for congenitally corrected transposition of the great arteries. Eur J Cardiothorac Surg. 2003;24:11-19.
27 Graham TPJr., Bernard YD, Mellen BG, et al. Long-term outcome in congenitally corrected transposition of the great arteries: a multi-institutional study. J Am Coll Cardiol. 2000;36:255-261.
28 Konstantinov IE, Williams WG. Atrial switch and Rastelli operation for congenitally corrected transposition with ventricular septal defect and pulmonary stenosis. Oper Tech Thorac Cardiovasc Surg. 2003;8:160-166.
29 Lecompte Y, Batisse A, DiCarlo D. Double-outlet right ventricle: a surgical synthesis. Adv Card Surg. 1993;4:109-136.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
CHAPTER 49 Complex Congenital Heart Disease
Complex congenital heart disease is obviously one of the most challenging issues faced by the health care provider who takes care of the pediatric patient with cardiac lesions. Taken as a group, this set of lesions represents only a small portion of congenital heart disease (CHD) and is rare in the population; nevertheless, these diseases take up a considerable amount of the physician’s time. Hoffman and Kaplan1 have reported the results of a meta-analysis of the literature and found the incidence of moderate to severe forms of congenital heart disease to be 6/1,000 live births, rising to 19/1,000 live births if potentially serious bicuspid aortic valves are included. Putting this in perspective, all forms of CHD represent 75/1,000 live births, including such lesions as tiny muscular ventricular septal defects. In addition, the New England Infant Cardiac Program2 has reported that 3/1,000 live births need cardiac catheterization or surgery, or will die with CHD in early infancy (excluding premature infants with patent ductus arteriosus). This number rises to 5/1,000 live births who will need some type of specialized care during their lifetime. All these issues are a measure of the severity of the lesion. With improvements in diagnosis and treatment of CHD, along with a greater understanding of the anatomy and physiology, patients are living longer3 and represent a growing patient base seen by adult cardiologists and internists. In 1980, there were an estimated 300,000 adults with CHD, whereas this rose to approximately 1 million in 2000. In 2020, the number is anticipated to be 1.4 million.
TRANSPOSITION OF THE GREAT ARTERIES
Definition
The classic transposition of the great arteries (TGA) consists of isolated ventriculoarterial discordance, with the aorta arising from the right ventricle and the main pulmonary artery arising from the left ventricle.1,2,3 This results in separation of the systemic and pulmonary circulations, with limited mixing of oxygenated and deoxygenated blood. It usually refers to dextro-TGA (d-TGA; segmental anatomy {S,D,D}), in which the aortic valve is positioned anterior and to the right of the pulmonary valve. However, some refer to any segmental anatomy that results in this physiology as transposition. Occasionally, the conotruncus is further rotated so that the aorta is anterior and to the left of the pulmonary valve (segments {S,D,L}).
Prevalence and Epidemiology
The incidence of transposition of the great arteries is approximately 3 in 10,000 live births. It represents approximately 5% of all newborns with congenital heart disease.1 It is rarely associated with syndromes or extracardiac malformations, but is more common in infants of diabetic mothers.
Manifestations of Disease
Imaging Techniques and Findings
Radiography
The classic radiographic findings of transposition of the great arteries include a narrow superior mediastinum (so-called egg on a string) caused by the anteroposterior orientation of the great vessels and inapparent thymus, increased pulmonary vascular markings, and cardiomegaly. However, these findings are rarely diagnostic and, in fact, were shown to be absent in the vast majority of newborns with d-TGA (Fig. 49-1).4
Ultrasound
Echocardiography can generally readily identify all the pertinent anatomic findings to manage patients with transposition of the great arteries. Subcostal imaging can readily identify the pulmonary artery arising from the left ventricle and the aorta arising from the right ventricle. The aorta can usually be identified by the coronaries and by its long length before branching (Fig. 49-2). The pulmonary artery branches early and the ductal arch should not be mistaken for the aorta (Fig. 49-3).
It is important to assess the atrial septum to determine whether it is restrictive (Fig. 49-4). In addition, the ventricular septum should be assessed to determine whether there are ventricular septal defects (Fig. 49-5). This is important for both preoperative management and surgical planning.
Magnetic Resonance Imaging
MRI plays an important role in the postoperative management of TGA. The standard for the treatment of TGA is the arterial switch (Jatene procedure), in which the aorta and main pulmonary artery are transected and sewn to the pulmonary and aortic root, respectively. The pulmonary arteries are generally brought anteriorly and draped over the aorta. The coronary arteries are transferred separately with buttons of tissue from the aorta to avoid ostial stenosis. Several problems can result, for which cardiac MRI is well suited to investigate. Supravalvular stenosis can occur at either anastomosis site (Fig. 49-6). Cine and velocity mapping can be used effectively to define regions of stenosis and quantify the degree of acceleration. Three-dimensional gadolinium sequences can also be useful to define stenoses and their relationships to other structures (Fig. 49-7). Unilateral or occasionally bilateral branch pulmonary artery stenosis may occur from stretching the branch pulmonary arteries after the Lecompte maneuver. Insufficiency of either valve can result from distortion during surgery. Through-plane velocity mapping can be used to quantify the degree of insufficiency of the valves precisely. Furthermore, short-axis cine volume sets can quantify the ventricular size, ejection, and wall motion abnormalities to screen for the effects of valve regurgitation or coronary abnormalities. Whole heart sequences can be used to evaluate for ostial stenosis of the transferred coronaries (Fig. 49-8). Perhaps more importantly, MRI can be used to evaluate for perfusion defects secondary to coronary stenosis. Gadolinium perfusion imaging is generally performed at rest and during adenosine administration. Adenosine administration causes coronary vasodilation and accentuates perfusion abnormalities by “stealing” flow from regions of marginal coronary perfusion.
See later, “Single Ventricle,” for a more detailed description of the routine cardiac MRI examination.
TETRALOGY OF FALLOT
Prevalence and Epidemiology
The incidence of tetralogy of Fallot is approximately 4 in 10,000 live births, accounting for approximately 7% to 10% of cases of congenital heart disease.5 It occurs equally in males and females, and represents one of the most common lesions requiring intervention in the first year of life. It occurs commonly in association with genetic defects, including Down syndrome, DiGeorge syndrome (22q11 microdeletion), and Alagille syndrome (Jag1 mutation).6,7
Etiology and Pathophysiology
While originally described by Fallot as a constellation of four findings,8 it is now understood that the pathogenesis appears to be related to a single abnormality. The anterior malalignment VSD with normally related great vessels is responsible for the associated aortic override and right ventricular outflow tract obstruction, which classically results in right ventricular hypertrophy.
Certain associated conditions may be important to the management of tetralogy of Fallot. A right aortic arch occurs in approximately 25% of patients. Coronary anomalies are common (approximately 9%), including the left anterior descending (LAD) artery from the right coronary and single coronary.9 Pulmonary atresia may occur, with or without the presence of major aortopulmonary collaterals. Patients may often have additional muscular VSDs, and it may occur in association with a complete common atrioventricular canal, especially in association with Down syndrome. Stenosis of the left pulmonary artery is common and, rarely, isolation of the pulmonary artery contralateral to the aortic arch may occur. A patent ductus arteriosus is common; when there is significant outflow tract obstruction, the duct may be tortuous.
Manifestations of Disease
Imaging Techniques and Findings
Radiography
The classic radiographic finding is a boot-shaped heart, with an upturned apex (Fig. 49-9). Other findings may range from a normal heart size and decreased pulmonary blood flow to an increased heart size and increased pulmonary blood flow, depending on the degree of obstruction. A right aortic arch can be noted on the radiograph.
Ultrasound
The mainstay of the preoperative evaluation of tetralogy of Fallot is echocardiography. In addition to making the diagnosis (Fig. 49-10), the echocardiogram should focus on the degree and location (subpulmonary, valvular or supravalvular) of right ventricular outflow obstruction (Fig. 49-11), the presence of additional ventricular septal defects, the size and origin of the branch pulmonary arteries, the presence of aortopulmonary collaterals, the coronary origins and courses (with particular attention to whether the LAD or other major branch crosses the right ventricular outflow tract [RVOT]), the arch sidedness and branching pattern, and the patency and course of the ductus arteriosus. With rare exception, these can be evaluated by routine transthoracic imaging.
Magnetic Resonance Imaging
The preoperative use of cardiac MRI is limited to specific situations. In some cases, it may be useful to use respiratory and cardiac-gated, T2-prepared whole heart imaging to define the origins of the coronary arteries if they are not well seen by echocardiography. Gadolinium angiography is also useful to define aortopulmonary collaterals, and has been shown to be as effective as traditional angiography.10
MRI has become an important part of the postoperative management of tetralogy of Fallot. The repair for tetralogy of Fallot generally involves patch closure of the VSD and relief of the RVOT obstruction. Some patients may have an adequate pulmonary valve annulus and require only resection of an RV muscle bundle and/or pulmonary valvotomy. However, many will require a transannular patch. Patients in which a major coronary branch crosses the RVOT may require an RV to PA conduit, creating a double-barreled outflow. In patients who have had a transannular patch as part of the repair, pulmonary insufficiency may cause progressive RV dilation and decreased RV performance. In addition, left ventricular performance may decline, likely secondary to interactions with the left ventricle. Cardiac MRI can quantify the pulmonary regurgitation and right ventricular size, and is important in the monitoring of patients who have evidence of significant RV dilation by echocardiography or have poor echocardiographic windows (Figs. 49-12 and 49-13). MRI can also effectively evaluate residual RVOT obstruction or conduit stenosis, branch pulmonary artery stenosis, and pulmonary flow distribution to each lung (Fig. 49-14).
See later, “Single Ventricle,” for a more detailed description of the routine cardiac MRI examination.
Nuclear Medicine: Positron Emission Tomography
Nuclear scintigraphy perfusion imaging has been used postoperatively in patients with TOF in the setting of branch pulmonary artery stenosis to quantify pulmonary blood flow distribution.11 However, this has largely been supplanted by cardiac MRI velocity mapping.12,13 It still may be useful for patients in whom a stent or coil artifact precludes assessment by MRI.
TRUNCUS ARTERIOSUS
Definition
There are two major classification schemes in use—those of Collett and Edwards14 and Van Praagh.15 These are invariably based on the position of the main pulmonary artery segment and branch pulmonary arteries, with the Van Praagh classification using types A and B to delineate whether a VSD is present (almost all have VSDs). Of the different forms, 92% of all patients fall into type 1A or 2A. The following are the definitions used in the Van Praagh classification with its differences and similarities with the Collett and Edwards classification noted; the Van Praagh classification takes into account aortic arch anomalies.
Prevalence and Epidemiology
According to Hoffman and Kaplan,1 the mean incidence of truncus arteriosus is 107 per million live births; it is thought to occur in 1% to 2% of patients with CHD at necropsy and represents approximately 0.7% of all congenital heart disease. The DiGeorge syndrome and patients with microdeletion of chromosome 22 have a high incidence of having truncus arteriosus. There is no race or gender predilection.
Etiology and Pathophysiology
Etiology
This rare lesion is caused by a failure in conotruncal septation of the embryonic truncus arteriosus and conus. The truncus is normally divided into the aorta and pulmonary artery by two ridges that appear in the 4- to 5-week-old embryo and that grow toward the midline. Cells from the neural crest directly contribute to this septation; one of the leading theories is that this malformation is a disruption in neural crest migration. Van Praagh and Van Praagh, however, believed this to be a form of tetralogy of Fallot.15
Pathophysiology
There are associated cardiovascular malformations, such as abnormalities of the coronary arteries, a right aortic arch, persistent left superior vena cava, aberrant origin of the left subclavian, patent foramen ovale, partial and complete atrioventricular canal defects, mitral and tricuspid malformations, double-inlet or hypoplastic left ventricle, left pulmonary artery sling, and anomalous pulmonary venous connections.16
Manifestations of Disease
Imaging Techniques and Findings
Ultrasound
Postoperatively, narrowing of the reconstructed right ventricular outflow tract and pulmonary arteries needs to be assessed. Stenosis can be evaluated by color flow mapping and Doppler echocardiography can determine the gradient; this is best performed in the subcostal sagittal or parasternal short-axis views and, occasionally, in the apical view angled extremely anteriorly. Residual ventricular septal defects can be seen by short-axis sweeps. Assessment of the truncal valve, as in the preoperative assessment, must be made routinely (Figs 49-15 and 49-16).
Computed Tomography
Because of radiation considerations, CT scanning plays a limited role in the care of the patient with congenital heart disease, and truncus arteriosus is no different. It is done chiefly when there is a contraindication to MRI and, when used, it is generally carried out postoperatively to visualize the right ventricular outflow tract and branch pulmonary arteries along with the aortic reconstruction if the aortic arch was interrupted. Because of its very limited temporal resolution, except if absolutely needed, ventricular function is best determined by MRI or echocardiography (Figs. 49-17 to 49-19).
[/not-level-membership-for-cardiovascular-category]
