[level-membership-for-allergy-and-immunology-category]
Chapter 4 Complement
• Complement is central to the development of inflammatory reactions and forms one of the major immune defense systems of the body. The complement system serves as one of the links between the innate and adaptive arms of the immune system.
• Complement activation pathways have evolved to label pathogens for elimination. The classical pathway links to the adaptive immune system through antibody. The alternative and lectin pathways provide antibody-independent ‘innate’ immunity, and the alternative pathway is linked to and amplifies the classical pathway.
• The complement system is carefully controlled to protect the body from excessive or inappropriate inflammatory responses. C1 inhibitor controls the classical and lectin pathways. C3 and C5 convertase activity are controlled by decay and enzymatic degradation. Membrane attack is inhibited on host cells by CD59.
• The membrane attack pathway results in the formation of a lytic transmembrane pore. Regulation of the membrane attack pathway by CD59 reduces the risk of ‘bystander’ damage to adjacent host cells.
• Many cells express one or more membrane receptors for complement products. Receptors for fragments of C3 are widely distributed on different leukocyte populations. Receptors for C1q are present on phagocytes, mast cells, and platelets. C5 fragment receptors are present on many cell types. The plasma complement regulator fH binds leukocyte surfaces.
• Complement has a variety of functions. Its principal functions include opsonization, chemotaxis and cell activation, lysis of target cells, and priming of the adaptive immune response.
• Complement deficiencies illustrate the homeostatic roles of complement. Classical pathway deficiencies result in tissue inflammation. Deficiencies of mannan-binding lectin (MBL) are associated with infections in infants and the immunosuppressed. Alternative pathway and C3 deficiencies are associated with bacterial infections. Terminal pathway deficiencies predispose to Gram-negative bacterial infections. C1 inhibitor deficiency leads to hereditary angioedema. Deficiencies in alternative pathway regulators produce a secondary loss of C3.
Complement and inflammation
Complement is now known to comprise some 16 plasma proteins, together constituting nearly 10% of the total serum proteins, and forming one of the major immune defense systems of the body (Fig. 4.1). In addition to serving as a key component of the innate immune system, complement also interfaces with and enhances adaptive immune responses. More than a dozen regulatory proteins are present in plasma and on cells to control complement. The functions of the complement system include:
• triggering and amplification of inflammatory reactions;
• attraction of phagocytes by chemotaxis;
• clearance of immune complexes and apoptotic cells;
• cellular activation for microbial killing;
• direct microbial killing; and
• an important role in the efficient development of antibody responses.
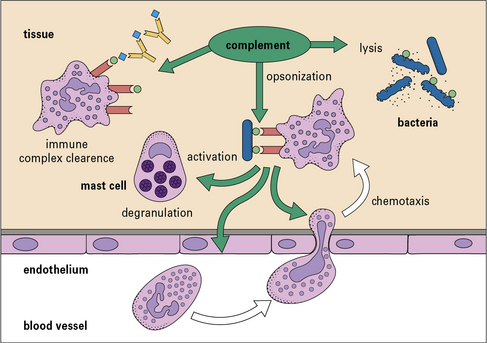
Complement activation pathways
Moreover, there are several different ways to activate the complement system, so providing a large degree of flexibility in response (Fig. 4.2).
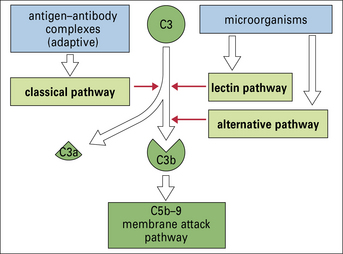
All three pathways – classical, alternative, and lectin pathways:
• involve the activation of C3, which is the most abundant and most important of the complement proteins;
• comprise a proteolytic cascade in which complexes of complement proteins create enzymes that cleave other complement proteins in an ordered manner to create new enzymes, thereby amplifying and perpetuating the activation cascade.
Thus a small initial stimulus can rapidly generate a large effect. Figure 4.3 summarizes how each of the pathways is activated.
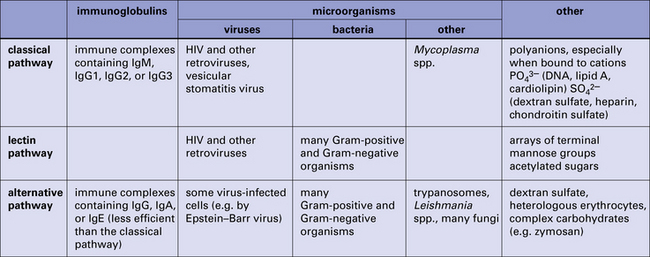
Fig. 4.3 Summary of the activators of the classical, lectin, and alternative pathways
Summary of the activators of the classical, lectin, and alternative complement activation pathways.
• the small chemotactic and proinflammatory fragments C3a and C5a;
The details of complement activation, the nomenclature, and the ways in which the pathways are controlled are shown in Figure 4.4.
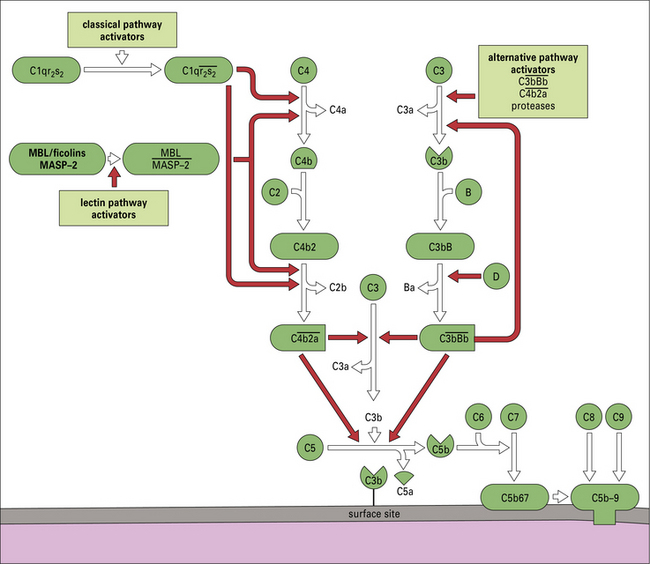
Fig. 4.4 Overview of the complement activation pathways
The proteins of the classical and alternative pathways are assigned numbers (e.g. C1, C2). Many of these are zymogens (i.e. pro-enzymes that require proteolytic cleavage to become active). The cleavage products of complement proteins are distinguished from parent molecules by suffix letters (e.g. C3a, C3b). The proteins of the alternative pathway are called ‘factors’ and are identified by single letters (e.g. factor B, which may be abbreviated to fB or just ‘B’). Components are shown in green, conversion steps as white arrows, and activation/cleavage steps as red arrows. The classical pathway is activated by the cleavage of C1r and C1s following association of C1qr2s2 with classical pathway activators (see Fig. 4.3), including immune complexes. Activated C1s cleaves C4 and C2 to form the classical pathway C3 convertase 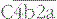 . Cleavage of C4 and C2 can also be effected via MASP-2 of the lectin pathway, which is associated with mannan-binding lectin (MBL) or ficolin. The alternative pathway is activated by the cleavage of C3 to C3b, which associates with factor B and is cleaved by factor D to generate the alternative pathway C3 convertase
. Cleavage of C4 and C2 can also be effected via MASP-2 of the lectin pathway, which is associated with mannan-binding lectin (MBL) or ficolin. The alternative pathway is activated by the cleavage of C3 to C3b, which associates with factor B and is cleaved by factor D to generate the alternative pathway C3 convertase 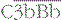 . The initial activation of C3 happens to some extent spontaneously, but this step can also be affected by classical or alternative pathway C3 convertases or a number of other serum or microbial proteases. Note that C3b generated in the alternative pathway can bind more factor B and generate a positive feedback loop to amplify activation on the surface. Note also that the activation pathways are functionally analogous, and the diagram emphasizes these similarities. For example, C3 and C4 are homologous, as are C2 and factor B. MASP-2 is homologous to C1r and C1s. Either the classical or alternative pathway C3 convertases may associate with C3b bound on a cell surface to form C5 convertases,
. The initial activation of C3 happens to some extent spontaneously, but this step can also be affected by classical or alternative pathway C3 convertases or a number of other serum or microbial proteases. Note that C3b generated in the alternative pathway can bind more factor B and generate a positive feedback loop to amplify activation on the surface. Note also that the activation pathways are functionally analogous, and the diagram emphasizes these similarities. For example, C3 and C4 are homologous, as are C2 and factor B. MASP-2 is homologous to C1r and C1s. Either the classical or alternative pathway C3 convertases may associate with C3b bound on a cell surface to form C5 convertases, 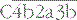 , or
, or 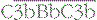 , which split C5. The larger fragment C5b associates with C6 and C7, which can then bind to plasma membranes. The complex of C5b67 assembles C8 and a number of molecules of C9 to form a membrane attack complex (MAC), C5b–9.
, which split C5. The larger fragment C5b associates with C6 and C7, which can then bind to plasma membranes. The complex of C5b67 assembles C8 and a number of molecules of C9 to form a membrane attack complex (MAC), C5b–9.
The classical pathway links to the adaptive immune system
The classical pathway is activated by antibody bound to antigen and requires Ca2+
• IgM is the most efficient activator, but unbound IgM in plasma does not activate complement;
• among IgG subclasses, IgG1 and IgG3 are strong complement activators, whereas IgG4 does not activate because it is unable to bind the first component of the classical pathway.
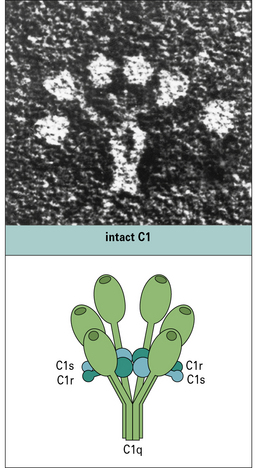
The first component of the pathway, C1, is a complex molecule comprising a large, 6-headed recognition unit termed C1q and two molecules each of C1r and C1s, the enzymatic units of the complex (Fig. 4.5). Assembly of the C1 complex is Ca2+-dependent, and the classical pathway is therefore inactive if Ca2+ ions are absent.
C1s enzyme cleaves C4 and C2
• releasing a small fragment, C4a; and
• exposing a labile thioester group in the large fragment C4b.
Through the highly reactive thioester, C4b becomes covalently linked to the activating surface (Fig. 4.6).
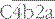 is the classical pathway C3 convertase
is the classical pathway C3 convertase
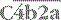 – the classical pathway C3 convertase) is the next activation enzyme. C2a in the
– the classical pathway C3 convertase) is the next activation enzyme. C2a in the 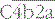 complex cleaves C3, the most abundant of the complement proteins:
complex cleaves C3, the most abundant of the complement proteins:• releasing a small fragment, C3a; and
• exposing a labile thioester group in the large fragment C3b.
As described above for C4b, C3b covalently binds the activating surface.
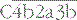 is the classical pathway C5 convertase
is the classical pathway C5 convertase
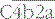 , and the trimolecular complex formed,
, and the trimolecular complex formed, 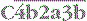 (the classical pathway C5 convertase), can bind C5 and present it for cleavage by C2a:
(the classical pathway C5 convertase), can bind C5 and present it for cleavage by C2a:• a small fragment, C5a, is released; and
• the large fragment, C5b, remains associated with the 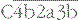 complex.
complex.
Cleavage of C5 is the final enzymatic step in the classical pathway.
The ability of C4b and C3b to bind surfaces is fundamental to complement function
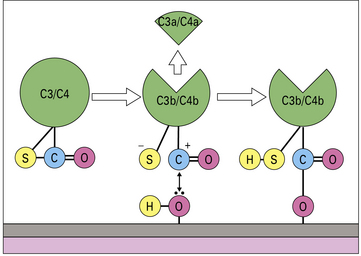
When either C3 or C4 is cleaved by the convertase enzyme, a conformational change takes place that exposes the internal thioester bond in C3b and C4b, making it very unstable and highly susceptible to attack by nucleophiles such as hydroxyl groups (-OH) and amine groups (-NH2) in membrane proteins and carbohydrates. This reaction creates a covalent bond between the complement fragment and the membrane ligand, locking C3b and C4b onto the surface (see Fig. 4.6).
The alternative and lectin pathways provide antibody-independent ‘innate’ immunity
The lectin pathway is activated by microbial carbohydrates
C1q and MBL are members of the collectin family of proteins characterized by globular head regions with binding activities and long collagenous tail regions with diverse roles (see Fig. 6.w3 ). Ficolins are structurally similar but the head regions comprise fibrinogen-like domains.
). Ficolins are structurally similar but the head regions comprise fibrinogen-like domains.
The 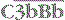 complex is the C3 convertase of the alternative pathway
complex is the C3 convertase of the alternative pathway
The 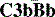 complex is the C3 cleaving enzyme (C3 convertase) of the alternative pathway. C3b generated by this convertase can be fed back into the pathway to create more C3 convertases, thus forming a positive feedback amplification loop (Fig. 4.7). Activation may occur in plasma or, more efficiently, on surfaces.
complex is the C3 cleaving enzyme (C3 convertase) of the alternative pathway. C3b generated by this convertase can be fed back into the pathway to create more C3 convertases, thus forming a positive feedback amplification loop (Fig. 4.7). Activation may occur in plasma or, more efficiently, on surfaces.
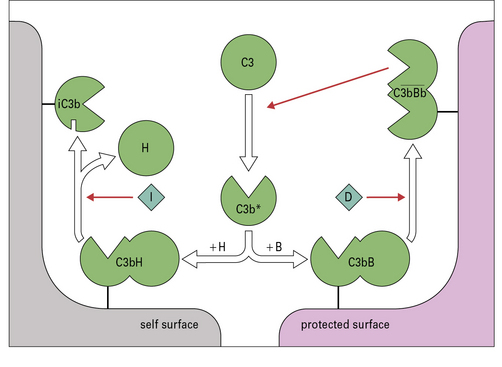
Fig. 4.7 Regulation of the amplification loop
Alternative pathway activation depends on the presence of activator surfaces. C3b bound to an activator surface recruits factor B, which is cleaved by factor D to produce the alternative pathway C3 convertase 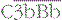 , which drives the amplification loop, by cleaving more C3. However, on self surfaces the binding of factor H is favored and C3b is inactivated by factor I. Thus the binding of factor B or factor H controls the development of the alternative pathway reactions. In addition, proteins such as membrane cofactor protein (MCP) and decay accelerating factor (DAF) also limit complement activation on self cell membranes (see Fig. 4.9).
, which drives the amplification loop, by cleaving more C3. However, on self surfaces the binding of factor H is favored and C3b is inactivated by factor I. Thus the binding of factor B or factor H controls the development of the alternative pathway reactions. In addition, proteins such as membrane cofactor protein (MCP) and decay accelerating factor (DAF) also limit complement activation on self cell membranes (see Fig. 4.9).
On an activating surface such as a bacterial membrane, amplification will occur unimpeded and the surface will rapidly become coated with C3b (see Fig. 4.7). In a manner analogous to that seen in the classical pathway, C3b molecules binding to the C3 convertase will change the substrate specificity of the complex, creating a C5 cleaving enzyme, 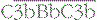 .
.
Complement protection systems
C1 inhibitor controls the classical and lectin pathways
In the activation pathways, the regulators target the enzymes that drive amplification:
• activated C1 is controlled by a plasma serine protease inhibitor (serpin) termed C1 inhibitor (C1inh), which removes C1r and C1s from the complex, switching off classical pathway activation;
• C1inh also regulates the lectin pathway in a similar manner, removing the MASP-2 enzyme from the MBL or ficolin complex to switch off activation.
C3 and C5 convertase activity are controlled by decay and enzymatic degradation
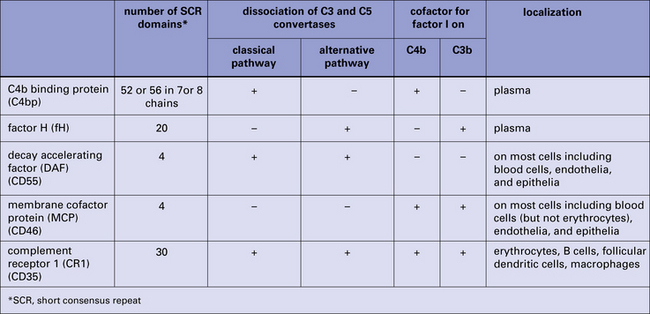
• factor H (fH) and a related protein factor H-like 1 (fHL-1) destroy the convertase enzymes of the alternative pathway;
• C4 binding protein (C4bp) performs the same task in the classical pathway.
On membranes, two proteins, membrane cofactor protein (MCP) and decay accelerating factor (DAF), collaborate to destroy the convertases of both pathways (Fig. 4.8).
Control of the convertases is mediated in two complementary ways
Decay acceleration –
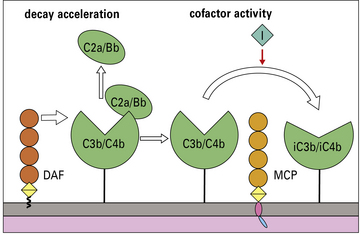
Cofactor activity –
Factor I (fI) is a fluid-phase enzyme that, in the presence of an appropriate cofactor, can cleave and irreversibly inactivate C4b and C3b (see Fig. 4.9). MCP is a cofactor for fI cleavage of both C4b and C3b, whereas:
Efficient regulation of the convertases on membranes therefore requires the concerted action of:
The membrane attack pathway
Activation of the pathway results in the formation of a transmembrane pore
The MAC is a transmembrane pore (Fig. 4.10). Cleavage of C5 creates the nidus for MAC assembly to begin. While still attached to the convertase enzyme, C5b binds first C6 then C7 from the plasma. Conformational changes occurring during assembly of this trimolecular C5b67 complex:
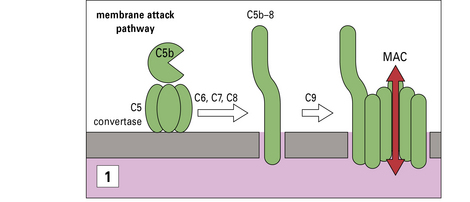
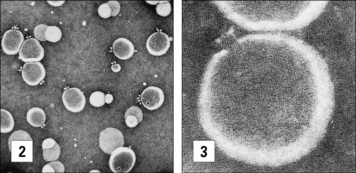
The MAC is clearly visible in electron microscopic images of complement lysed cells as doughnut-shaped protein-lined pores, first observed by Humphrey and Dourmashkin 40 years ago (see Fig. 4.10). The pore has an inner diameter approaching 10 nm:
Regulation of the membrane attack pathway reduces the risk of ‘bystander’ damage to adjacent cells
• binding of one of the fluid-phase regulators of the terminal pathway – S protein (also termed vitronectin) or clusterin – both of which are multifunctional plasma proteins with diverse roles in homeostasis.
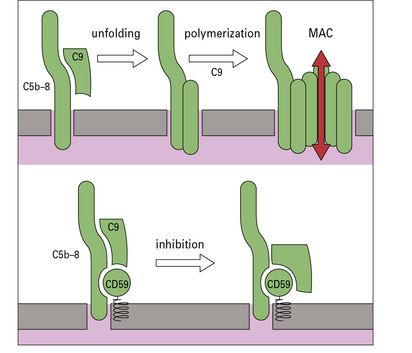
CD59 protects host cells from complement-mediated damage
Complexes that do bind are further regulated on host cells by CD59, a membrane protein that locks into the MAC as it assembles and inhibits binding of C9, thereby preventing pore formation (Fig. 4.11). CD59 is a broadly expressed, glycolipid-anchored protein that is structurally unrelated to the complement regulators described above.
Membrane receptors for complement products
Receptors for fragments of C3 are widely distributed on different leukocyte populations
Many cells express one or more membrane receptors for complement products (Fig. 4.12). An understanding of the receptors is essential because the majority of the effects of complement are mediated through these molecules. The best characterized of the complement receptors are those binding fragments of C3.
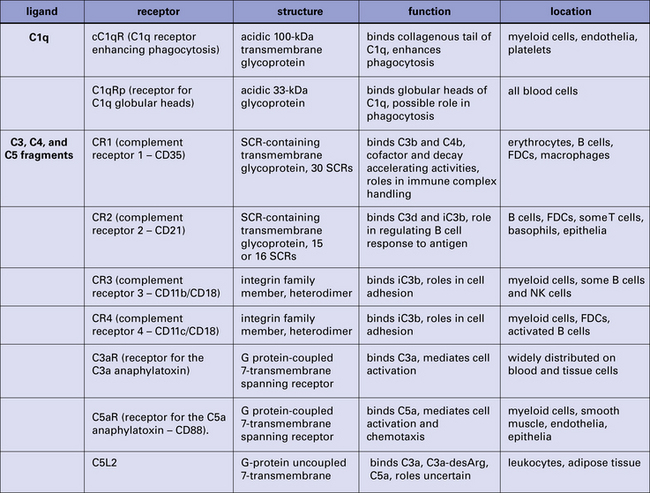
CR1, CR2, CR3, and CR4 bind fragments of C3 attached to activating surfaces
• CR1, expressed on erythrocytes and leukocytes, binds the largest fragment C3b (and also C4b), an interaction that is crucial to the processing of immune complexes (see below);
• CR2, expressed mainly on B cells and follicular dendritic cells (FDCs), binds fragments derived from fI-mediated proteolysis of C3b – iC3b and C3d.
These interactions aid the B cell response to complement-coated particles.
Receptors for C1q are present on phagocytes, mast cells, and platelets
Receptors for the collagen tails (cC1qR):
• can recognize C1q attached through its globular head regions to complement-coated particles;
• are present on leukocytes, platelets, and some other cell types; and
• likely play roles in enhancing phagocytosis of C1q-labeled particles.
Complement functions
The principal functions of complement are:
C5a is chemotactic for macrophages and polymorphs
Polymorphs and macrophages express receptors for C3a and C5a. These small (~10 kDa) fragments (i.e. C3a and C5a) diffuse away from the site of activation, creating a chemical gradient along which the motile cells migrate to congregate at the site of activation (Fig. 4.13).
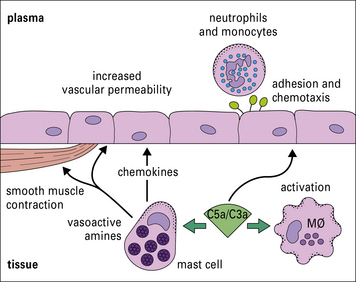
Binding of C3a and C5a to their receptors also causes cell activation:
C3b and iC3b are important opsonins
Complement activation and amplification cause complement fragments to efficiently coat activator surfaces of targets such as bacteria or immune complexes, enhancing their recognition by phagocytes (Fig. 4.14). Phagocytes and other cells carrying receptors for these complement fragments are then able to bind the target, triggering ingestion and cell activation. The key players here are the surface-bound fragments of C3 and the family of C3 fragment receptors described above.
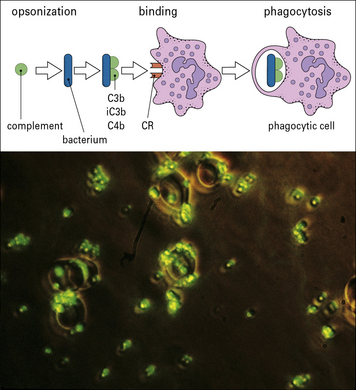
C3b disaggregates immune complexes and promotes their clearance
• first, coating with C3b masks the foreign antigens in the core of the immune complex, blocking further growth;
• second, coating with C3b disaggregates large immune complexes by disrupting interactions between antigen and antibody;
• third, C3b (and C4b) on immune complexes interacts with CR1 on erythrocytes, taking the immune complex out of the plasma – the immune adherence phenomenon.
The MAC damages some bacteria and enveloped viruses
Assembly of the MAC creates a pore that inserts into and through the lipid bilayer, breaching the membrane barrier (see Fig. 4.10). The consequences of MAC attack vary from target to target:
• for most pathogens, opsonization is the most important antibacterial action of complement;
• for Gram-negative bacteria, particularly organisms of the genus Neisseria, MAC attack is a major component of host defense, and individuals deficient in components of the MAC (e.g. patients with C6 deficiency, which is the second most common deficiency of complement) are susceptible to neisserial infection.
• may also play roles in the efficient dispatching of other pathogens, including some viruses;
• can also damage or destroy host cells – in some instances, such as autoimmunity, the host cell is itself the target and complement is directly activated on the cell, leading to MAC attack.
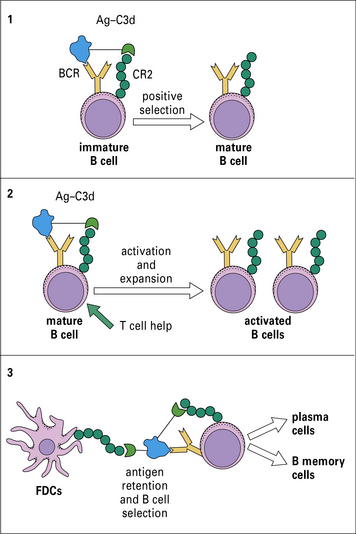
Immune complexes with bound C3b are very efficient in priming B cells
Complement is a key component of the innate immune response. However, it has recently become apparent that complement also plays important roles in adaptive immunity. This realization arose from studies in complement-depleted and complement-deficient mice in which antibody responses to foreign particles were markedly reduced. At least three linked mechanisms contribute to this effect (Fig. 4.15):
• first, immature B cells directly bind foreign particles through the B cell receptor (BCR) recognizing specific antigen in the particle, and through CR2 recognizing attached C3d – this co-ligation triggers B cell maturation, with the mature cells migrating to the lymphoid organs;
• second, while in the lymph nodes, mature B cells encounter opsonized antigen and, in the presence of B cell help, are induced to become activated and to proliferate;
• finally, in the lymphoid organs FDCs capture antigen through attached C3 fragments and use this bait to select the correct activated B cells and switch them on to further maturation and proliferation to form plasma cells and B memory cells (see Chapters 9 and 11).
Complement deficiencies
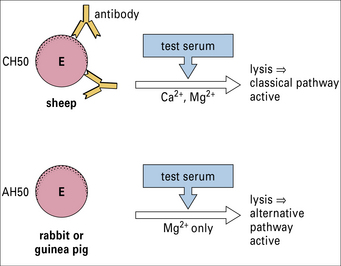
A variety of assays (Method box 4.1) are available for detecting:
are available for detecting:
• the activity of different complement pathways;
• the functional activity of individual components;
• the total amount of individual components (functional or non-functional).
A variety of assays are available for detecting:
• the activity of different complement pathways;
• the functional activity of individual components;
• the total amount of individual components (functional or non-functional).
The consequences of a deficiency in part of the complement system depend upon the pathway(s) affected (Fig. 4.16).
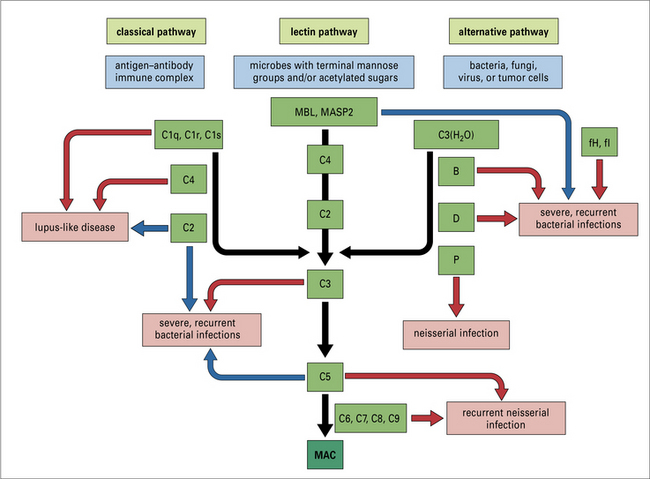
Classical pathway deficiencies result in tissue inflammation
The large majority of cases of SLE are, however, not associated with complement deficiencies, and autoimmune SLE is discussed in Chapter 20. The historical view of immune complex disease in classical pathway deficiency was based upon defective immune complex handling.
Deficiencies of MBL are associated with infection in infants
Terminal pathway deficiencies predispose to Gram-negative bacterial infections
C1 inhibitor deficiency causes hereditary angioedema
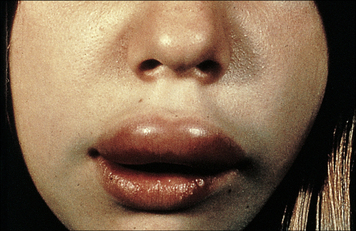
Deficiency of the classical pathway regulator C1inh is responsible for the syndrome hereditary angioedema (HAE). C1inh regulates C1 in the classical pathway and MBL/MASP-2 (or ficolin-MASP-2) in the lectin pathway and also controls activation in the kinin pathway that leads to the generation of bradykinin and other active kinins. The condition and the underlying pathways are outlined in Chapter 16 (see Figs 16.14 & 16.15) and additional detail is given below.
C1inh deficiency is a dominant condition
Episodes of angioedema are often triggered in the skin or mucous membranes by minor trauma – occasionally stress may be sufficient to induce an attack. Swelling, which may be remarkable in severity, rapidly ensues as unregulated activation of the kinin and complement systems occurs in the affected area, inducing vascular leakiness. Swelling of mucous membranes in the mouth and throat may block the airways, leading to asphyxia (Fig. 4.w1). Involvement of gut mucosa can cause intestinal obstruction, presenting with acute abdominal symptoms.
Deficiencies in alternative pathway regulators cause a secondary loss of C3
Complement polymorphisms and disease
Critical thinking: Complement deficiency (see p. 434 for explanations)
1. Why would a deficiency in complement cause the children to be particularly susceptible to bacterial infections? Measurements are made of individual complement components of the classical and alternative pathways to determine which of the components is defective. The results are shown in the table.
| Complement component | Normal concentration (μg/mL) | Levels in affected children (μg/mL) |
|---|---|---|
| C4 | 600 | 480–520 |
| C2 | 20 | 15–22 |
| C3 | 1300 | 10–80 |
| factor B (fB) | 210 | not detectable |
| factor H (fH) | 480 | 200–350 |
| factor I (fI) | 35 | not detectable |
2. Using knowledge of the complement reaction pathways, how can you explain the apparent combined deficiencies in C3, fB, and fI?
3. What is the fundamental deficiency in this family and how would you treat the affected children?
Barrington R., Zhang M., Fischer M., Carroll M.C. The role of complement in inflammation and adaptive immunity. Immunol Rev. 2001;180:5–15.
Botto M., Kirschfink M., Macor P., et al. Complement in human diseases: lessons from complement deficiencies. Mol Immunol. 2009;46:2774–2783.
Cole D.S., Morgan B.P. Beyond lysis: how complement influences cell fate. Clin Sci. 2003;104:455–466.
Colten H.R., Rosen F.S. Complement deficiencies. Annu Rev Immunol. 1992;10:809–834.
Davis A.E., 3rd. The pathogenesis of hereditary angioedema. Transfus Apher Sci. 2003;39:195–203.
De Cordoba S.R., de Jorge E.J. Genetics and disease associations of complement factor H. Clin Exp Immunol. 2008;151:1–13.
Dodds A.W., Ren X.D., Willis A.C., et al. The reaction mechanism of the internal thioester in the human complement component C4. Nature. 1996;379:177–179.
Frank M.M., Fries L.F. The role of complement in inflammation and phagocytosis. Immunol Today. 1991;12:322–326.
Gerard C., Gerard N.P. C5a anaphylatoxin and its seven transmembrane-segment receptor. Annu Rev Immunol. 1994;12:775–808.
Holmskov U., Malhotra R., Sim R.B., et al. Collectins: collagenous C-type lectins of the innate immune defense system. Immunol Today. 1994;14:67–74.
Hourcade D., Holers V.M., Atkinson J.P. The regulators of complement activation (RCA) gene cluster. Adv Immunol. 1989;45:381–416.
Jack D.L., Klein N.J., Turner M.W. Mannose-binding lectin: targeting the microbial world for complement attack and opsonophagocytosis. Immunol Rev. 2001;180:86–99.
Kavanagh D., Richards J., Atkinson J. Complement regulatory genes and hemolytic uremic syndromes. Annu Rev Med. 2008;59:293–309.
Kemper C., Atkinson J.P. T cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18.
Lambris J.D., Reid K.B.M., Volanakis J.E. The evolution, structure, biology and pathophysiology of complement. Immunol Today. 1999;20:207–211.
Lambris J.D., Ricklin D., Geisbrecht B.V. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142.
Liszewski M.K., Farries T.C., Lublin D.M., et al. Control of the complement system. Adv Immunol. 1996;61:201–283.
Manderson A.P., Botto M., Walport M.J. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:432–456.
Moffitt M.C., Frank M.M. Complement resistance in microbes. Springer Semin Immunopathol. 1994;15:327–344.
Morgan B.P. Complement regulatory molecules: application to therapy and transplantation. Immunol Today. 1995;16:257–259.
Morgan B.P. Complement in inflammation. In: Rey K., ed. Physiology of inflammation. Oxford: Oxford University Press; 2001:131–145.
Morgan B.P. Hereditary angioedema: therapies old and new. N Engl J Med. 2010;363:581–583.
Morgan B.P., Harris C.L. Complement therapeutics; history and current progress. Mol Immunol. 2003;40:159–170.
Morgan B.P., Meri S. Membrane proteins that protect against complement lysis. Springer Semin Immunopathol. 1994;15:369–396.
Morgan B.P., Walport M.J. Complement deficiency and disease. Immunol Today. 1991;12:301–306.
Muller-Eberhard H.J. The membrane attack complex of complement. Annu Rev Immunol. 1986;4:503–528.
Nonaka M., Yoshizaki F. Evolution of the complement system. Mol Immunol. 2004;40:879–902.
Norris M., Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–1687.
Ricklin D., Hajishengallis G., Yang J., Lambris J.D. Complement: a key system for immunosurveillance and homeostasis. Nat Immunol. 2010;11:785–797.
Walport M.J. Complement: first of two parts. N Engl J Med. 2001;344:1058–1066.
Walport M.J. Complement: second of two parts. N Engl J Med. 2001;344:1140–1144.
Zipfel P.F., Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740.
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Chapter 4 Complement
• Complement is central to the development of inflammatory reactions and forms one of the major immune defense systems of the body. The complement system serves as one of the links between the innate and adaptive arms of the immune system.
• Complement activation pathways have evolved to label pathogens for elimination. The classical pathway links to the adaptive immune system through antibody. The alternative and lectin pathways provide antibody-independent ‘innate’ immunity, and the alternative pathway is linked to and amplifies the classical pathway.
• The complement system is carefully controlled to protect the body from excessive or inappropriate inflammatory responses. C1 inhibitor controls the classical and lectin pathways. C3 and C5 convertase activity are controlled by decay and enzymatic degradation. Membrane attack is inhibited on host cells by CD59.
• The membrane attack pathway results in the formation of a lytic transmembrane pore. Regulation of the membrane attack pathway by CD59 reduces the risk of ‘bystander’ damage to adjacent host cells.
• Many cells express one or more membrane receptors for complement products. Receptors for fragments of C3 are widely distributed on different leukocyte populations. Receptors for C1q are present on phagocytes, mast cells, and platelets. C5 fragment receptors are present on many cell types. The plasma complement regulator fH binds leukocyte surfaces.
• Complement has a variety of functions. Its principal functions include opsonization, chemotaxis and cell activation, lysis of target cells, and priming of the adaptive immune response.
• Complement deficiencies illustrate the homeostatic roles of complement. Classical pathway deficiencies result in tissue inflammation. Deficiencies of mannan-binding lectin (MBL) are associated with infections in infants and the immunosuppressed. Alternative pathway and C3 deficiencies are associated with bacterial infections. Terminal pathway deficiencies predispose to Gram-negative bacterial infections. C1 inhibitor deficiency leads to hereditary angioedema. Deficiencies in alternative pathway regulators produce a secondary loss of C3.
Complement and inflammation
Complement is now known to comprise some 16 plasma proteins, together constituting nearly 10% of the total serum proteins, and forming one of the major immune defense systems of the body (Fig. 4.1). In addition to serving as a key component of the innate immune system, complement also interfaces with and enhances adaptive immune responses. More than a dozen regulatory proteins are present in plasma and on cells to control complement. The functions of the complement system include:
• triggering and amplification of inflammatory reactions;
• attraction of phagocytes by chemotaxis;
• clearance of immune complexes and apoptotic cells;
• cellular activation for microbial killing;
• direct microbial killing; and
• an important role in the efficient development of antibody responses.
Complement activation pathways
Moreover, there are several different ways to activate the complement system, so providing a large degree of flexibility in response (Fig. 4.2).
All three pathways – classical, alternative, and lectin pathways:
• involve the activation of C3, which is the most abundant and most important of the complement proteins;
• comprise a proteolytic cascade in which complexes of complement proteins create enzymes that cleave other complement proteins in an ordered manner to create new enzymes, thereby amplifying and perpetuating the activation cascade.
Thus a small initial stimulus can rapidly generate a large effect. Figure 4.3 summarizes how each of the pathways is activated.
Fig. 4.3 Summary of the activators of the classical, lectin, and alternative pathways
Summary of the activators of the classical, lectin, and alternative complement activation pathways.
• the small chemotactic and proinflammatory fragments C3a and C5a;
The details of complement activation, the nomenclature, and the ways in which the pathways are controlled are shown in Figure 4.4.
Fig. 4.4 Overview of the complement activation pathways
The proteins of the classical and alternative pathways are assigned numbers (e.g. C1, C2). Many of these are zymogens (i.e. pro-enzymes that require proteolytic cleavage to become active). The cleavage products of complement proteins are distinguished from parent molecules by suffix letters (e.g. C3a, C3b). The proteins of the alternative pathway are called ‘factors’ and are identified by single letters (e.g. factor B, which may be abbreviated to fB or just ‘B’). Components are shown in green, conversion steps as white arrows, and activation/cleavage steps as red arrows. The classical pathway is activated by the cleavage of C1r and C1s following association of C1qr2s2 with classical pathway activators (see Fig. 4.3), including immune complexes. Activated C1s cleaves C4 and C2 to form the classical pathway C3 convertase . Cleavage of C4 and C2 can also be effected via MASP-2 of the lectin pathway, which is associated with mannan-binding lectin (MBL) or ficolin. The alternative pathway is activated by the cleavage of C3 to C3b, which associates with factor B and is cleaved by factor D to generate the alternative pathway C3 convertase . The initial activation of C3 happens to some extent spontaneously, but this step can also be affected by classical or alternative pathway C3 convertases or a number of other serum or microbial proteases. Note that C3b generated in the alternative pathway can bind more factor B and generate a positive feedback loop to amplify activation on the surface. Note also that the activation pathways are functionally analogous, and the diagram emphasizes these similarities. For example, C3 and C4 are homologous, as are C2 and factor B. MASP-2 is homologous to C1r and C1s. Either the classical or alternative pathway C3 convertases may associate with C3b bound on a cell surface to form C5 convertases, , or , which split C5. The larger fragment C5b associates with C6 and C7, which can then bind to plasma membranes. The complex of C5b67 assembles C8 and a number of molecules of C9 to form a membrane attack complex (MAC), C5b–9.
The classical pathway links to the adaptive immune system
The classical pathway is activated by antibody bound to antigen and requires Ca2+
• IgM is the most efficient activator, but unbound IgM in plasma does not activate complement;
• among IgG subclasses, IgG1 and IgG3 are strong complement activators, whereas IgG4 does not activate because it is unable to bind the first component of the classical pathway.
The first component of the pathway, C1, is a complex molecule comprising a large, 6-headed recognition unit termed C1q and two molecules each of C1r and C1s, the enzymatic units of the complex (Fig. 4.5). Assembly of the C1 complex is Ca2+-dependent, and the classical pathway is therefore inactive if Ca2+ ions are absent.
C1s enzyme cleaves C4 and C2
• releasing a small fragment, C4a; and
• exposing a labile thioester group in the large fragment C4b.
Through the highly reactive thioester, C4b becomes covalently linked to the activating surface (Fig. 4.6).
is the classical pathway C3 convertase
• releasing a small fragment, C3a; and
• exposing a labile thioester group in the large fragment C3b.
As described above for C4b, C3b covalently binds the activating surface.
is the classical pathway C5 convertase
• a small fragment, C5a, is released; and
• the large fragment, C5b, remains associated with the complex.
Cleavage of C5 is the final enzymatic step in the classical pathway.
The ability of C4b and C3b to bind surfaces is fundamental to complement function
When either C3 or C4 is cleaved by the convertase enzyme, a conformational change takes place that exposes the internal thioester bond in C3b and C4b, making it very unstable and highly susceptible to attack by nucleophiles such as hydroxyl groups (-OH) and amine groups (-NH2) in membrane proteins and carbohydrates. This reaction creates a covalent bond between the complement fragment and the membrane ligand, locking C3b and C4b onto the surface (see Fig. 4.6).
The alternative and lectin pathways provide antibody-independent ‘innate’ immunity
The lectin pathway is activated by microbial carbohydrates
C1q and MBL are members of the collectin family of proteins characterized by globular head regions with binding activities and long collagenous tail regions with diverse roles (see Fig. 6.w3). Ficolins are structurally similar but the head regions comprise fibrinogen-like domains.
The complex is the C3 convertase of the alternative pathway
The complex is the C3 cleaving enzyme (C3 convertase) of the alternative pathway. C3b generated by this convertase can be fed back into the pathway to create more C3 convertases, thus forming a positive feedback amplification loop (Fig. 4.7). Activation may occur in plasma or, more efficiently, on surfaces.
Fig. 4.7 Regulation of the amplification loop
Alternative pathway activation depends on the presence of activator surfaces. C3b bound to an activator surface recruits factor B, which is cleaved by factor D to produce the alternative pathway C3 convertase , which drives the amplification loop, by cleaving more C3. However, on self surfaces the binding of factor H is favored and C3b is inactivated by factor I. Thus the binding of factor B or factor H controls the development of the alternative pathway reactions. In addition, proteins such as membrane cofactor protein (MCP) and decay accelerating factor (DAF) also limit complement activation on self cell membranes (see Fig. 4.9).
On an activating surface such as a bacterial membrane, amplification will occur unimpeded and the surface will rapidly become coated with C3b (see Fig. 4.7). In a manner analogous to that seen in the classical pathway, C3b molecules binding to the C3 convertase will change the substrate specificity of the complex, creating a C5 cleaving enzyme, .
Complement protection systems
C1 inhibitor controls the classical and lectin pathways
In the activation pathways, the regulators target the enzymes that drive amplification:
• activated C1 is controlled by a plasma serine protease inhibitor (serpin) termed C1 inhibitor (C1inh), which removes C1r and C1s from the complex, switching off classical pathway activation;
[/not-level-membership-for-allergy-and-immunology-category]
