[level-membership-for-critical-care-medicine-category]61
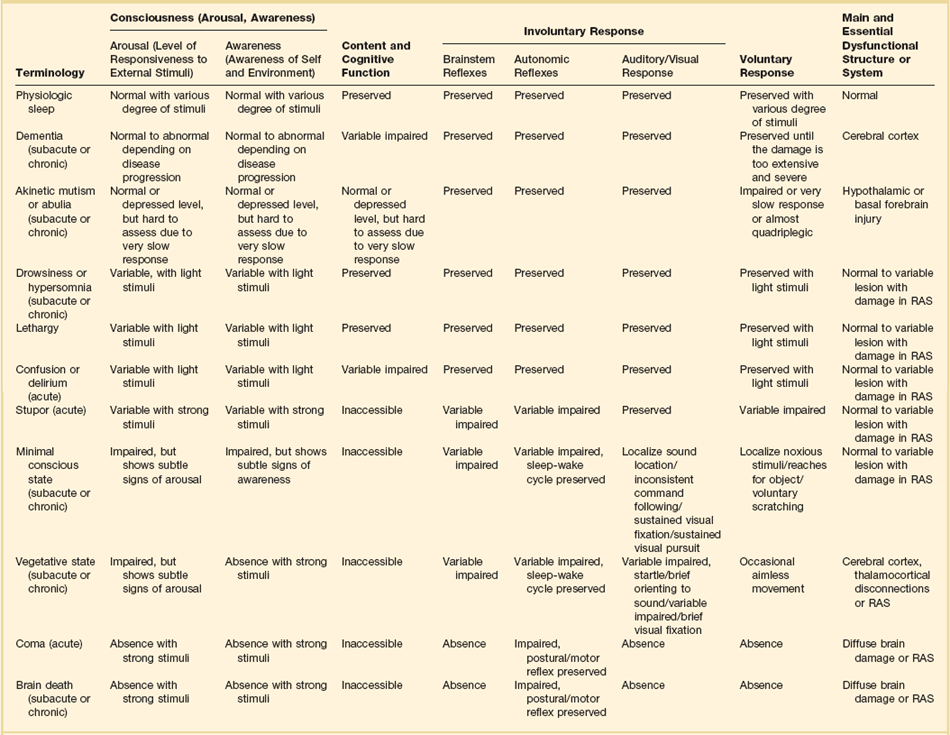
Coma
Concept and Terminology of Impaired Consciousness
Alteration of consciousness is a frequent admission diagnosis to critical care services. Most patients require immediate and often extensive diagnostic workup as both time to diagnosis and treatment initiation are decisive factors for brain recovery. To the public, the portrayal of coma is overly optimistic as 89% of “comatose” patients in a meta-analysis of 64 characters in American soap dramas regained consciousness and around 92% “survived” coma.1 In contrast, medical experience does not compare as favorably, as only about 50% of patients survive in an unselected coma population.2–4 Therefore, defining coma in a succinct and operational manner will provide a deeper understanding of the depth of brain injury and avoid miscommunications and unrealistic prognostications.
When evaluating a patient’s mental status, the state of consciousness should be viewed as a continuum ranging from the patient who has full alertness and cognitive lucidity to the deeply comatose patient; it is not an all-or-nothing phenomenon.5 An all-or-nothing approach limits the interpretation of remaining brain function and, hence, diagnostic certainty. Furthermore, the duration and time development of coma are helpful diagnostic features and should complement the quantitative and qualitative assessment of consciousness. Standard classification systems categorize consciousness based on systematic testing of arousal, awareness, and content. Table 61.1 classifies various abnormalities of consciousness, and the following section delineates clinically important syndromes.
Coma (from Greek κ µα, or “deep sleep”) is a continuous state of unresponsiveness identified by an inability to arouse to vigorous (noxious) external or internal stimuli. The degree of coma can differ; lighter stages (sometimes denoted as semicoma) can be identified by brief moaning to strong stimulation and associated observed changes in autonomic function; whereas the deepest coma examination shows an absence of any response, including brainstem reflex responses (i.e., lack of oculo- and pupillomotoric responses). Some cyclic autonomic activity such as the sleep-wake cycle and changes in motor tone may coexist. A detailed discussion of coma is provided in the classic textbook by Plum and Posner.6
µα, or “deep sleep”) is a continuous state of unresponsiveness identified by an inability to arouse to vigorous (noxious) external or internal stimuli. The degree of coma can differ; lighter stages (sometimes denoted as semicoma) can be identified by brief moaning to strong stimulation and associated observed changes in autonomic function; whereas the deepest coma examination shows an absence of any response, including brainstem reflex responses (i.e., lack of oculo- and pupillomotoric responses). Some cyclic autonomic activity such as the sleep-wake cycle and changes in motor tone may coexist. A detailed discussion of coma is provided in the classic textbook by Plum and Posner.6
The vegetative state is characterized by the complete absence of behavioral evidence for self- or environmental awareness. This state can follow coma and identifies a state in which brainstem and diencephalic (thalamic) activity is present to a degree that clinical signs of spontaneous or stimulus-induced arousal and sleep-wake cycles are observed. Patients often show blink responses to light; intermittent eye movements (sometimes erroneously interpreted as following objects or looking at family members); stimulus-sensitive automatisms such as swallowing, bruxism, and moaning; or primitive motor responses. If this state lasts longer than 30 days, it is referred to as persistent vegetative state (PVS) and is used as a descriptive clinical syndrome rather than a disease-specific entity. The most common causes include cardiac arrest, head trauma, severe brain infections, and various causes of thalamic injury. Vegetative states can also be seen in the terminal phase of degenerative illnesses such as Alzheimer’s disease. Ambiguous terms for PVS such as apallic syndrome and neocortical death should be avoided.2,7
Minimal conscious state can be diagnosed in patients displaying some but often inconsistent behavioral evidence of awareness of the environment, but they cannot communicate their content and are unable to follow instructions reliably.8 It describes a large group of patients who are different from vegetative patients in that they demonstrate some signs of awareness of themselves and their surroundings, albeit inconsistently.
Neuroanatomy, Neurotransmitter, and Pathology
Consciousness, which consists of three main parts, largely depends on the integrity of brain structure for arousal (Fig. 61.1). The ascending reticular activating system (ARAS) is the lowest order arousal system, consisting of a set of brainstem nuclei located within the brainstem and interconnected by neuronal circuits. The ARAS relays arousal signals to the more rostral thalami, which, in turn, act as hemispheric gatekeepers of arousal and regulate consciousness and sleep-wake transitions. In turn, thalamic activated cerebral cortices allow cognitive processing, which then determines the overall content of consciousness.6 Structural damage to or metabolic-chemical derangements of any or all of these elements will affect consciousness. Important components of the ARAS9–11 include the midbrain reticular formation, mesencephalic nuclei (dorsal raphe nucleus, pedunculopontine tegmental nucleus, locus coeruleus), and ventral tegmental areas. Those areas have rich connections to thalamic intralaminar nuclei and further frontally reaching projections to the tuberomammillary nucleus, lateral hypothalamus, and basal forebrain.
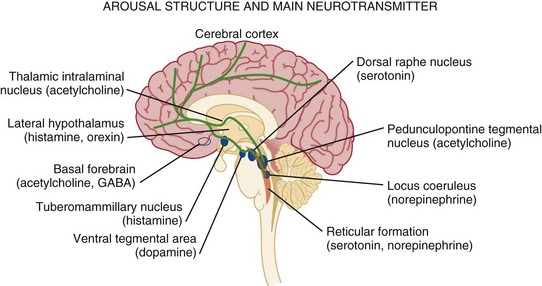
The ascending arousal system of the brainstem contains different neurotransmitter systems. Main cholinergic projections include the ascending mesopontine tegmental and basal forebrain pathways projecting to the thalami and to virtually all subcortical and cortical structures. These projections usually function as synaptic facilitators but at times can act as depressors of transmission.12 Glutamate is the main transmitter influencing the firing patterns of tegmental cholinergic neurons.13 The adrenergic component of the ARAS is closely associated with the noradrenergic neurons of the midbrain locus coeruleus. It runs in parallel with cholinergic projections rostrally to the cortical areas but also descends caudally within the spinal cord.14 Hypocretin/orexin neurons within the hypothalamus activate both adrenergic and cholinergic pathways and coordinate activity of the entire ARAS system, enhancing complementary and synergistic control of arousal and locomotion.15 Histamine-releasing cells from the hypothalamus and tuberomammillary neurons are active during the qualitative activation needed for cognition and EEG activation, but they remain silent during sleep.16–18
The main inhibitor of the arousal system is γ-aminobutyric acid (GABA), without which normal sleep does not occur. The GABAergic circuitry is located in the midbrain and pons, but its activity is regulated and sustained by forebrain structures.19 It is thought that the GABAergic system’s main function is to contain and channel the spread of arousal, both spontaneously and in response to a stimulus. GABAergic processes are responsible for the occurrence of paradoxical sleep (deep sleep characterized by a brain wave pattern similar to that of wakefulness, rapid eye movements, and heavier breathing but without motor responses). The name “waking neurons” has been given to the serotonergic dorsal raphe nucleus in the midbrain,20 which receives convergent excitatory input from the noradrenaline, histamine, and hypocretin/orexin arousal systems21,22 as the main activator of the ARAS. Cortical signals that pass through the striatum (caudate and putamen) are refined by the action of dopamine. Dopaminergic neurons are tonically active and stimulate or inhibit cortical neurons depending on various environmental influences (pain, hunger, etc.).23,24 It is thought that many sensory signals that reach conscious awareness pass through this basal ganglia loop with dopamine as the modulating neurotransmitter. Koch and colleagues suggested that coalitions of neurons compete to dominate conscious thoughts and activities at any given time point and, hence, dopaminergic innervation may facilitate the transient domination of conscious awareness by certain sets of neuron coalitions.25 Therefore, it seems that arousal is an event with a defined time distribution, which is tightly orchestrated by a few key neurotransmitters. As we know, there is no singular “arousal neurotransmitter.”10,26
Pathology Seen in Patients with Impaired Consciousness
There are four major pathologies that can cause severe, acute, and global reductions of consciousness.6,27 (1) One of these pathologies is the presence of diffuse, global, or extensive multifocal bilateral dysfunction of the cerebral cortex. In this injury mechanism, the cortical gray matter is diffusely impaired and so are cortical-subcortical excitatory feedback loops. The clinical examination will reveal disinhibited autonomic brainstem reflexes, which have also been described as “reticular shock.” (2) Another is injury to the paramedian gray matter from the level of the nucleus parabrachialis of the pons (tegmentum) and reaching to the midbrain pretectal areas and ventral posterior hypothalamus. An injury of this type damages the ascending arousal system and normal cortical activation. The affected structures are predominantly the paramedian gray matter, extending rostrally from the level of the nucleus parabrachialis of the pons (tegmentum) and reaching rostrally as far as the adjacent pretectal area and ventral posterior hypothalamus. (3) The widespread disconnection of the cortex from subcortical activating mechanisms acts pathophysiologically to produce effects similar to both of the previously mentioned conditions. (4) Finally, cortical and subcortical arousal mechanisms can be affected by a variety of diffuse disorders and to various degrees—for example, metabolic encephalopathy in a patient with acute liver failure.
Approach to Coma
Emergency Management
Oxygenation and Intubation
A brief neurologic examination is mandatory before any sedative or paralytic required for intubation is administered. The key points of a rapid neurologic examination are the following:28
• Assessment of level of arousal and level of consciousness (i.e., coma, stupor, lethargy)
• Pupillary size and response to light
• Abnormalities of eye movements (i.e., disconjugate eyes; unilateral gaze paralysis; lack of voluntary movements)
The preferred route of emergency intubation is orotracheal, which can be performed rapidly, safely, and reliably with inline stabilization of the neck in patients with suspected cervical spine injury. Intubation causes intense reflexive cardiovascular stimulation that may lead to a deleterious elevation of intracranial pressure (ICP). Therefore, the patient should be adequately sedated. Etomidate is the preferred sedative in patients with suspected ICP elevation as it reliably facilitates induction in less than 1 minute with a duration of action of 4 to 6 minutes. Propofol is another anesthetic agent that does not increase ICP; however, its hypotensive effects, which can ultimately decrease cerebral perfusion pressure, often limit its use. Both medications result in a dose-dependent decrease of cerebral metabolic rate that reduces cerebral blood flow and ICP. Ketamine has a fast onset, but it may elevate ICP and should generally be avoided. Midazolam can alter the patient’s mental status and may prohibit a postintubation examination.
Reversal of Drug Overdose
Drug overdose is the largest single cause (30%) of coma in the emergency department. Most drug overdoses are treated by supportive measures alone. Certain antagonists, however, specifically reverse the effects of coma-producing drugs. Intravenous naloxone (0.4 to 2 mg) is used as “test” antidote for opiate-induced coma, and it acts as a µ-receptor antagonist. Caution is needed as the reversal of narcotic effect may precipitate acute withdrawal in an opiate addict. In suspected opiate coma, the minimum naloxone dose (not completely reversal) should be given to establish the diagnosis by pupillary dilation and to reverse depressed breathing and consciousness. Due to its short half-life, patients who respond to naloxone reversal may require additional doses or a continuous infusion to avoid rebound sedation and respiratory depression. Intravenous flumazenil antagonizes benzodiazepine-induced coma and can be administered in 0.2-mg doses every minute for a maximum of 1 mg. In patients who initially respond to flumazenil reversal but experience recurrent sedation, flumazenil 1 mg may be redosed every 20 minutes with a max of 3 mg/hour.29 Careful consideration should be given prior to administration of flumazenil as patients may experience benzodiazepine-withdrawal seizures. Thus, whereas naloxone is commonly used with minimal serious adverse effects, the use of flumazenil should be restricted to select, low-risk cases. The sedative effects of drugs with anticholinergic properties, particularly tricyclic antidepressants, can be reversed with 1 to 2 mg physostigmine intravenously (duration of action about 45 to 60 minutes). Pretreatment with 0.5 mg atropine will reduce the risk for symptomatic bradycardia. Of note, only full awakening is characteristic of an anticholinergic drug overdose because physostigmine has nonspecific arousal properties.
Body Temperature
Hyperthermia is detrimental in brain injury as it increases brain metabolic demands and facilitates secondary brain injury.30 Elevated temperature greater than 104° F (40° C) requires immediate, lifesaving cooling measures, even before the underlying cause is determined and treated. In 2002, two research groups independently published that lowering the body temperature to 33° C for 12 or 24 hours in comatose survivors of cardiac arrest resulted in nearly doubling the number of patients being discharged home or to rehabilitation. Generally speaking, no patient with acute brain injury should be allowed to be hyperthermic, and modern technology provides a variety types of noninvasive and invasive cooling equipment suitable to maintain any target core temperature desired. Core temperatures of less than 93° F (34° C) on admission should be slowly elevated to above 35° C to reduce the risks of unwanted side effects from uncontrolled, persistent hypothermia.
Physical Examination
A systematic, detailed examination is necessary when approaching a comatose patient (Box 61.1, Table 61.2). Important findings include evidence of trauma, acute or chronic medical illnesses, ingestion of drugs (needle marks, alcohol breath), and the presence of nuchal rigidity.

Table 61.2
Correlation Between Levels of Brain Function and Clinical Signs
| Structure | Function | Clinical Sign |
| Cerebral cortex | Conscious behavior | Speech (including any sounds) Purposeful movement Spontaneous To command To pain |
| Brainstem activating and sensory pathways (reticular activating system) | Sleep-wake cycle | Eye opening Spontaneous To command To path |
| Brainstem motor pathways | Reflex limb movements | Flexor posturing (decorticate) Extensor posturing (decerebrate) |
| Midbrain CN III | Innervation of ciliary muscle and certain extraocular muscles | Pupillary reactivity |
| Pontomesencephalic MLF | Connects pontine gaze center with CN III nucleus | Internuclear ophthalmoplegia |
| Upper pons | ||
| CN V | Facial and corneal | Corneal reflex-sensory |
| CN VIII | Facial muscle innervation | Corneal reflex-motor response Blink Grimace |
| Lower pons | ||
| CN VIII (vestibular portion) connects by brainstem pathways with CN III, IV, VI | Reflex eye movements | Doll’s eyes Caloric responses |
| Pontomedullary junction pressure | Spontaneous breathing Maintained blood pressure |
Breathing and blood pressure do not require mechanical or chemical support |
| Spinal cord | Primitive protective responses | Deep tendon reflexes Babinski response |
As outlined earlier, an in-depth physical and neurologic examination followed by serial neurologic evaluations (i.e., hourly) is the most important, indispensable, and readily available method of assessing a comatose patient.31 However, in coma patients the examination remains limited in detecting changes in brain function. Nevertheless, the astute clinician will carefully evaluate for changes in findings (i.e., the appearance of new asymmetry in examination or focal deficits, progressive loss of brainstem reflexes, or loss of reflex motor responses). Finding progression of impairment is of immense clinical value as it indicates a new or worsening “intracranial emergency” demanding immediate clarification and stabilization. Skilled neurologic examination is challenging and provides a limited surveillance window of brain tissue at risk. Intracranial monitoring and, if needed, repeated head imaging have become the standard of care in neurocritical care units.
Assessment of Coma: General Aspects
Correctly characterizing disorders of consciousness continues to pose interesting clinical questions and diagnostic challenges with important ethical consequences (Box 61.2). Not only may an individual patient acutely fluctuate in his or her examination findings, but recovery may take place over prolonged time periods, necessitating constant reassessments. To address this uncertainty, standardized neurobehavioral assessments have become the best tool for categorizing coma and its transitional stages. Predicting long-term outcome has significantly improved when utilizing standardized assessments. Generally speaking, clinical and electrophysiologic markers of coma and its transitional states remain unsatisfactory. The reader is reminded that larger observational studies identified a high rate of misdiagnoses (>40%),32 especially in patients in vegetative state, when assessment is made on clinical grounds only.
Standardized Neurobehavioral Assessment
The behavioral diagnosis of the level of consciousness in severely brain-damaged patients remains challenging. To improve differential diagnostic evaluations, evidence-based recommendations endorse neurobehavioral characteristics using standardized tools.33 The Coma Recovery Scale-Revised (CRS-R) has excellent content validity and is a solid tool addressing all established differential diagnostic criteria. To differentiate vegetative, minimally conscious state (MCS) or emerging MCS, several other tools have demonstrated good validity, including the Sensory Modality Assessment Technique (SMART), Sensory Stimulation Assessment Measure (SSAM), Wessex Head Injury Matrix (WHIM), and Stimulation Profile (WNSSP). The FOUR and CRS-R tools showed substantial evidence of good interpreter reliability. Our recommendations are to use the Coma Recovery Scale-Revised (CRS-R)32 as a valid and reliable tool in most situations for differential diagnostic assessments. With reservations, the SMART, WNSSP, SSAM, WHIM, and DOCS can be used. The lack of evidence with respect to content validity, standardization, and reliability make the FOUR, INNS, Glasgow Coma Scale, Swedish Reaction Level Scale-1985, Loewenstein Communication Scale, and CLOCS undesirable.
Neuroimaging
Novel neuroimaging modalities have not only provided additional insight into residual brain function in patients with disorders of consciousness but have also raised ethical discussions concerning the clinical management of these patients. Neuroimaging studies have provided considerable evidence for the existence of major functional differences in patients with vegetative state (VS) and minimally conscious state (MCS).34–37 For example, positron emission tomography (PET) and functional MRI (fMRI) studies delineated more extended cerebral processing in response to various external stimuli of modalities in patients with MCS.35,36,38 Resting state fMRI studies identified that MCS patients have preservations of functional connectivity to higher-order associative cortices, such as default networks.37,39 Complementary to these results, brain tissue integrity, as assessed by diffusion weighted imaging and gray matter volumetry, is more preserved in MCS as compared to VS.34,40 These findings correlate to the relative improved long-term prognosis in MCS.41
Conventional Magnetic Resonance Imaging (MRI)
FLAIR and T2-SE sequences are commonly used to delineate, measure, and monitor the extent and impact of acute brain injury. Common pathologies seen include brain edema, contusions, intracerebral, dural or subarachnoid blood, herniation syndromes, and hydrocephalus. T2* (also called GRE) and susceptibility-weighted imaging (SWI) are helpful for delineating blood product and detecting hemorrhagic diffuse axonal injuries (DAI). In traumatic Brain Injury (TBI) patients, the total number of lesions detected on FLAIR and T2* has been shown to inversely correlate with admission GCS score.42,43 Three-dimensional rendering allows evaluating and quantifying brain atrophy after injury.44 Conventional MRI is useful in TBI patients. Lesions in the pons, midbrain, and basal ganglia, especially when bilateral, correlate with poor outcome. However, the contrary may not be true as MRI may fail to explain why some vegetative state or marked cognitively impaired patients have minimal or no lesions. Further advanced imaging studies would be especially helpful for those patients (discussed later). Diffusion tensor imaging (DTI) can be viewed as an extension of diffusion-weighted imaging (DWI) where diffusion restricted water molecule movement (i.e., water trapped in ischemic cells or, in DTI, confined to axonal spaces) is preferentially visualized. As with most conventional MRI techniques, obtaining results with DTI is independent of the coma or sedation status of a patient.45 Serial changes in white matter DTI metrics can be used as surrogate markers to evaluate treatment responses even when clinical scores cannot be obtained.46 Whole brain white matter and regional DTI measures have become sensitive markers of TBI impact correlating with acute and discharge injury severity, especially when adjusted for age, gender, and admission GCS score.47,48
Proton Magnetic Resonance Spectroscopy (MRS)
MRS reveals noninvasively regional, metabolic information of targeted brain regions correlating with the functional neuronal state of the examined sample region. To assess brain function, comprehensive MRS protocols looking for changes in axial chemical shift imaging at the level of the cortical white matter,49,50 splenium,51 basal ganglia (thalamus),52 insular cortex, and pons42 have been used. Using this approach, it is possible to detect posttraumatic neurochemical damage and neuronal dysfunction in brain areas where conventional MR imaging techniques show no abnormalities.53 In a TBI case-control study, the N-acetylaspartate (NAA)/creatinine (Cr) ratio correlated as a good neuronal marker with the overall recovery (and therefore lack as reliable index of unfavorable outcome after TBI), whereas less consistency was found using the choline (Cho)/Cr54 or NAA/Cho ratios.55 Moreover, early posttraumatic spectroscopic assessment of axonal damage is clinically relevant for prognostication, and significant increases in brain lactate levels seem to be a dependable index of injury severity and outcome in TBI.56
Positron Emission Tomography (PET)
18-fluorodeoxyglucose (FDG)-PET delineates glucose utilization deficiencies and, hence, it provides insights to neuronal energy metabolism correlating to regional, functional performance. For example, in acute vegetative state, overall glucose utilization was significantly reduced to 50% to 70% in comparison with age-matched, healthy controls.57 As expected, patients with locked-in syndrome (i.e., pontine lesions) showed normal cortical metabolism. TBI patients with diffuse axonal injury had neuronal hyperglycolysis and metabolic depression.58 1C-Flumazenil (FMZ)-PET measures the density of benzodiazepine receptors (BZRs) providing an estimate of neuronal integrity.15 O-H2O PET imaging delineates activation-induced changes in regional cerebral blood flow in response to passive external (auditory, somatosensory or visual) stimulation. The combination of preserved activation of primary cortices (i.e., visual) but lack of co-activation of secondary processing areas provides evidence that “higher-order” associative cortical networks are disconnected from the primary sensory cortex.38,59–63 Thalamocortical disconnections have also been reported using this technique.64
Functional MRI (fMRI)
Functional MR imaging provides activity measures of brain regions at rest and after both passive (internal, though-induced) and active (external, command-induced) stimulation.65 Such acquisitions can obtain estimates of average metabolic activity in relatively short time periods (i.e., 10 minutes) compared to PET (~30 minutes).66 fMRI has been shown to be a good predictor of outcome in patients with impaired consciousness.67–72 The previously discussed PET evidence of preserved but isolated (disconnected) cortical activation areas in a patients in vegetative state was corroborated by fMRI.39,73–76
Electrophysiology
EEG
In patients with alpha coma the underlying brain wave activity is predominantly in the (normal) alpha range, from 8 to 12 Hz. In contrast to the normal EEG in a healthy, awake person, the EEG pattern found in comatose patients lacks alpha reactivity, implying a significant disturbance of thalamocortical pathways. In 36 patients in alpha coma,77 one third showed anterior predominance and two thirds (66%) showed uniform distribution of alpha activity. Three major brain-injury categories have been observed in alpha coma:78,79 status postcardiorespiratory arrest (anoxic encephalopathy), toxic encephalopathies, and de-efferentiated (locked-in) state (i.e., pontine hemorrhage).
Theta coma and alpha-theta coma patients show slower brain wave activities in the theta 4- to 7-Hz range (theta coma), which in some is superseded by a predominance of alpha activity (alpha-theta coma).80 In many coma patients, the alpha and theta activities will change after about 5 days to a more permanent pattern at which time the EEG may be uniformly slow and depressed. EEG reactivity may emerge in subsequent studies as a relatively favorable sign; however, resolution into a burst-suppression pattern without reactivity signifies unfavorable outcome after anoxic encephalopathy.80
Beta coma consists of fast activities in the 12- to 16-Hz range, usually with frontal predominance. Interspersed alpha, theta, delta, and spindle (sleep)-like activities can be found. The most frequent cause for beta coma is sedative-hypnotic overdose from substances such as benzodiazepines and barbiturates.81
Intermittent rhythmic delta activity (IRDA) consists of low amplitude, slow 2- to 3-Hz sinusoidal waves. Those waves can appear intermittently during the early stages of impairment of consciousness and soon after loss of alpha rhythm.78 This nonspecific pattern can be seen in many metabolic and toxic encephalopathies, hemispheric lesions, acute brain injuries, and other conditions affecting cortical and subcortical structures diffusely. The prognosis of IRDA depends on the underlying etiology.
Spindle coma consists of an EEG pattern predominated by 11- to 14-Hz spindle discharges, which are generally symmetric and synchronous, appearing as sleeplike oscillations on a background rhythm dominated by theta and delta activities. It resembles the EEG of a sleeping person and additional findings include anterior, low-voltage rhythmic bursts, K complexes, vertex waves, and delta slowing. However, in these patients, wakefulness is not regained after stimulation and depth of consciousness does not fluctuate in spindle coma. These patterns indicate dysfunction at the brainstem level. Traumatic brain injury, anoxic encephalopathy, infectious encephalitis, drug-induced and metabolic encephalopathy, as well as postictal states can lead to this EEG finding.82–84 The prognosis of spindle coma is uncertain, as both positive85–87 and negative outcomes84 have been reported.
Periodic lateralized epileptiform discharges (PLEDs) occur asymmetrically (lateralized) and are episodic in nature. Morphologically, variations include sharp waves, spikes, slow waves, or a combination of these followed by a slow wave with a periodicity ranging from 0.3 to several seconds.88 PLEDs are nonspecific and can be seen in a wide variety of brain injuries such as after cortical ischemia and with brain tumors. They are also associated with recent seizures, alcohol withdrawal, and toxic-metabolic encephalopathy among many others.89 In contrast, generalized periodic epileptiform discharges (GPEDs) are seen bilaterally and typically occur at a frequency of <1 Hz. Subclassifications define the interval between discharges and include periodic short-interval diffuse discharges (PSIDDs), periodic long-interval diffuse discharges (PLIDDs), and suppression burst patterns.90 GPEDs are seen after global brain injury—for example, after hypoxic or hepatic encephalopathy, drug intoxication, but also with various degenerative disorders.90
Bilateral independent periodic lateralized epileptiform discharges (BIPLEDs) are slow (<1 Hz), bilateral independent epileptiform activity91 seen in coma due to bihemispheric lesions. For example, after anoxic encephalopathy and infections, they are associated with a poorer prognosis91 and about two times higher overall mortality than patients with PLEDS.91,92
The burst-suppression pattern identifies an EEG demonstrating generalized, synchronous bursting of slow but mixed-frequency waves of high-voltage alternating periodically interspersing within longer periods of isoelectric (flat) EEG. It identifies abnormal cortical excitability,93 and with deepening coma, the isoelectric periods become longer. Main etiologies include anoxic encephalopathy, severe intoxication, or sedative/anesthesia-induced coma states. The overall prognosis is determined by the underlying etiology.94
Triphasic waves are distinct, blunt delta waves (~2 to 3 Hz) consisting of a high-voltage positive wave preceded and followed by lower amplitude negative waves. This nonspecific EEG can be seen in various brain injuries, among them serotoninergic syndrome, Creutzfeldt-Jacob disease, lithium and baclofen toxicity, hepatic and renal insufficiency, and neuroleptic malignant syndrome.95
Studies have identified that different EEG patterns seen in patients with hypoxic (postarrest) encephalopathy include suppression, burst suppression, alpha and theta coma, complete generalized suppression (<10 µV), and generalized periodic complexes in combination with burst suppression. Unfortunately, common to all of them is the poor correlation to long-term outcome.94
Event-Related Potentials (ERPs)
These EEG potentials are identifiable after signal-averaging EEG-derived potentials evoked by a specific sensory, cognitive, or motor stimulus. Under normal circumstances, the brain generates event-related, neuronal (electrical) responses to stimulation. This neuronal activation can be detected by surface EEG; however, it requires specific mathematical processing of the EEG signal to identify brain oscillations that are time and phase locked to particular events (stimuli). Detection of such event-related potentials in patients with impaired consciousness has been interpreted as the brain’s ability to process stimulus information and for cortical movement preparation. Advantages of this approach include detected signals that have an excellent time-stimulus correlation within the millisecond range helpful to relate a particular stimulus to certain cortical processing. The method is low cost and noninvasive, and it can be used to monitor the brain function. Fast processors have allowed the development of brain-computer interfaces based on event-related signal responses that assist communication with brain-injured patients. Several ERP paradigms have been developed allowing identification of specific response signals. The N1-P2 complex has been identified in visual ERPs and its presence interpreted as a negative correlation to repressiveness.96 The P300 (or P3) is an average, positive potential with a latency of about 300 milliseconds. It is generated when a patient mentally reacts to a defined cognitive stimulus and is related to positive outcomes in coma.97 Cognitive responses to command have been verified in patients with complete locked-in syndrome after basilar artery thrombosis.98
Evoked Potentials
Brainstem auditory evoked responses (BAER) are a series of seven potentials recorded at specific time intervals at the skull after defined auditory stimulation employing headphones. Based on the redundancy and interconnections of the auditory pathways, BAER are robust and can be obtained in many patients with depressed level of consciousness, including patients under anesthesia or deep sedation. This implies that the absence of BAER, or its lemniscal complexes III-IV reflecting the integrity of upper brainstem, uniformly infers a poorer prognosis in coma (i.e., 3 to 6 days after head injury).99 In addition, BAER have been employed as a “brainstem monitoring” tool to identify progressive downward brain herniation. Frequently, in clinical settings, BAER monitoring is combined with somatosensory evoked potential (SEP) and EEG monitoring. Somatosensory evoked potentials (SSEP) are a series of peaks recorded over the neck and scalp in response to electrical stimulation of a peripheral nerve, usually the median nerve. The N13 wave is a negative peak at 13 milliseconds reflecting activity at the cervical cord, whereas the N20 is a negative peak at 20 milliseconds reflecting the earliest cortical activity. Longer latency responses are also seen at 25, 35, 45, 70, and 95 milliseconds, reflecting higher cortical processing. Multimodality evoked potentials (MEP) combine simultaneous brainstem auditory, somatosensory, and visual evoked potential monitoring100 in order to enhance the predictive value of evoked potential monitoring—for example, in TBI patients101 or during induced barbiturate coma where examinations cannot be obtained.102
Cerebral Blood Flow Monitoring
Transcranial Doppler (TCD) records the velocity and pulsatility of cerebral blood flow, allowing hemodynamic analysis, despite the use of sedatives or moderate hypothermia. TCD has been utilized for serial examinations in comatose patients after initial recovery from cardiac arrest in order to detect and treat early cerebral hemodynamic changes. If a patient has an appropriate temporal ultrasound window, TCD can determine cerebral perfusion arrest and, hence, can be used as an ancillary study to determine brain death.103 Further, velocity and pulsatility indices correlate as a noninvasive measure with ICP elevations and also identify the delayed presence of a hyperemic flow pattern—that is, high velocities with low pulsatility as seen in patients with intracranial hypertension from hyperemia.104
Xenon-Enhanced CT Scanning
Xenon CT utilizes the diffusion of xenon into the CNS to determine cerebral blood flow (CBF)105 in order to assess perfusion deficits after a traumatic brain injury.105 Using hyperventilation, xenon CT can be used to estimate cerebral vasoreactivity, a marker correlating with outcome in comatose TBI patients.106
General Treatment Approach
If known, initial coma evaluation must take the injury mechanism into account. For example, in trauma victims, empiric cervical spine stabilization and early detection of an unstable c-spine injury by reconstructed cervical CT will be needed (i.e., before evaluating oculocephalic responses). Endotracheal intubation is frequently required due to the risk of aspiration and the need to maintain an open upper airway in an unconscious patient. Prehospital endotracheal intubation was associated with a significant decrease in mortality from 36% to 26% in patients with blunt injury and a GCS ≤ 8, especially those with severe head injury by anatomic criteria.107 Furthermore, mechanical ventilation is required to avoid oxygen desaturations (of the brain) or to induce therapeutic hypocapnia to acutely lower uncontrolled ICP. Early tracheotomy is often indicated in moderate to severe brain injuries. For example, a GCS of ≤4 on day 3 of injury or a Simplified Acute Physiology Score (SAPS)108 of >15 on day 4 can indicate tracheotomy with 84% positive predictive value.109 There are many benefits of early tracheotomy including increased patient comfort and subsequent reduction in sedatives and analgesics and ease of oral and pulmonary care, and its association with shorter duration of mechanical ventilation and ICU length of stay.110 At our institution we commonly perform bedside tracheotomy, which is a safe bedside procedure that circumvents the need to transport patients with brain injury and reduces overall costs.111
Any correctable coma etiology such as abnormal glucose and electrolyte levels, hypotension, hypoxemia, or abnormal core temperatures should be managed immediately. Intravenous access is immediately established for the administration of resuscitation fluids, administration of contrast, and therapeutic care. Infusion of hypotonic solutions should be avoided to prevent deterioration of intracranial swelling, and there is a definite role for immediate ICP monitoring (i.e., using an external ventricular drain, which also allows the release of CSF) in any acute brain injury with the potential for ICP. The management of raised ICP is discussed in Chapter 16.
Traumatic and Nontraumatic Coma
Coma is a descriptive term based on examination findings and can result from a range of heterogeneous etiologies that are classically grouped into traumatic versus nontraumatic coma. As outlined earlier, other stages of impaired consciousness, including vegetative state, typically follow nonremitting acute coma. Usually after 1 month of coma, the term persistent vegetative state is used for both nontraumatic and traumatic etiologies; however, some authors prefer the term permanent vegetative state 3 months after nontraumatic and 1 year after traumatic injury. Notably, these terms do not imply irreversibility and do not exclude the potential for further recovery.112
Coma after Severe Traumatic Brain Injury
Noncontrast CT is the most common imaging evaluation tool in TBI patients. A cervical spine CT with reconstructed views and head and neck CT angiogram (CTA) are often added during initial evaluation, if injury to the neck and arteries, respectively, is suspected. Bone and brain windows should be reviewed separately and in experienced hands. Head imaging has a good sensitivity for the presence of pathologic air, skull fractures, hemorrhagic lesions, abnormalities of the brain parenchyma (i.e., shift), and obstruction of CSF spaces (hydrocephalus). Injury to the neuroanatomic structures of consciousness or the presence of additional etiologies for coma (i.e., alcohol) should be considered when disparities between CT findings and examination exist. In this setting, appropriate additional screening and further imaging diagnostics (i.e., MRI) must be initiated.113 Compared to CT, MRI has a higher sensitivity to detect diffuse axonal injury (DAI); however, this advantage has not translated into improved clinical outcomes as there is currently no treatment for this condition. However, both the detection of DAI and delineating injury of major motor (i.e., corticospinal) tracts on diffusion tensor imaging (DTI) can be utilized together with the clinical course to prognosticate TBI.114 MR spectroscopy (MRS) provides additional “chemical” information, especially of the white matter injury process.115
The presence and severity of secondary brain injury should constantly be assessed and managed throughout the acute phase of TBI. Its time course can be variable but is somewhat predictable: for example, ranging from hours (i.e., development of new hematoma) to 1 to 3 days for brain edema development to several days as seen in late brain ischemia from vasospasm due to traumatic subarachnoid blood. There is only limited evidence to support the use of ICP monitoring. ICP monitoring has evolved as an essential tool and also one of the earliest monitoring tools in clinical practice; thus many believe clinical trials cannot ethically be justified. Current TBI management guidelines recommend its use in severe TBI. Of note, head imaging cannot predict ICP. Furthermore, treating for elevated ICP (other than empirically in the emergency setting) and not monitoring ICP can be deleterious and result in a poor outcome.116 In addition to ICP, other brain monitoring techniques utilized in modern TBI management include jugular venous and transcranial oxygen saturations (SjvO2; NIRS), brain tissue oxygen tension (btO2), brain tissue metabolism (microdialysis) and temperature, direct brain perfusion, transcranial Doppler sonography, and electrophysiologic studies such as EEG and evoked potentials. A discussion of these techniques is beyond the scope of this chapter and the reader is referred to recent reviews.
The management of severe TBI includes emergent decompressive craniectomy in controlling intractable ICP, especially if deterioration is associated with hematoma evacuation and there is some evidence that there will be positive outcomes in surviving patients.117–128 Although timing of decompressive craniectomy remains controversial, early referral seems most intuitive. The Brain Trauma Foundation’s Guidelines for Management of Severe Traumatic Brain Injury regard craniectomy as an option after TBI to manage patients with intracerebral hemorrhage (ICH) and the resultant increased ICP refractory to medical management.116 Intravenous mannitol and hypertonic saline solutions are a main building block in the management of increased ICP.129–141 The superiority of one over the other is uncertain, and both approaches are routinely used in the management of severe TBI.142 Therapeutic hypothermia is postulated to exert neuroprotective effects by several mechanisms, including reducing cerebral metabolic rate/requirement, modulating apoptosis or programmed cell death, reducing intracellular calcium/toxic excitatory neurotransmitters/inflammation, and preserving protein synthesis.116,143–151 However, the reported impact on neurologic outcome in severe TBI remains inconsistent and a meta-analysis of randomized controlled trials suggests its lack of benefit in severe TBI.143,144 We utilize hypothermia as last resort therapy and commonly, together with pentobarbital therapy and ICP-matching, CPP elevation to protect the brain from otherwise intractable ICP elevations. Until well-designed, randomized controlled trials are available, its use must be weighed against potential side effects such as an increased incidence in pneumonia and coagulation abnormalities, among others.143,151
The initial GCS seems to be a reasonable marker to predict mortality from severe TBI. To further improve prediction, models include a combination of clinical data, imaging results, GCS score, and patient demographics.152 In a large, traumatic coma databank study,153 patients with severe injury (GCS 3 to 8) had approximately 36% mortality. In 170 patients, the GCS 5 hours postinjury was compared to the Glasgow Outcome Scale (GOS) at 1 month and positive recovery was seen in 99% of mild TBI (GCS 13 to 15), 71% of moderate TBI(GCS 9 to 12), and in only 35% of severe TBI. In contrast, only 2% to 3% of patients with mild TBI (GCS 13 to 15) died. Generally speaking, clinical outcome after TBI is more variable than in nontraumatic coma (i.e., after hypoxia).154 Furthermore, outcome predictions for patients with intermediate GCS scores identify that a midline shift of less than 4.1 mm on initial CT scanning had a significantly higher favorable outcome rate compared with patients with a larger shift.155
Biomarkers after traumatic brain injury (TBI), such as cellular glial fibrillary acidic protein (GFAP) and S100B released with astrocyte injury, may offer diagnostic and prognostic tools in addition to clinical indices. In 79 patients with TBI (GSC ≤ 12), identified intracranial mass lesion, pupillary reactivity, GFAP, and S100B were the strongest predictors of death and unfavorable outcome, with S100B as the strongest single predictor of unfavorable outcome with 100% discrimination.156 Somatosensory evoked potentials in 58 TBI patients unconscious for >30 days were compared with outcome at 12 months157 and showed an excellent prediction for outcome (persistent coma or MCS, 92% and 86%, respectively) at 12 months when combined with age, sex, and GCS.157 Furthermore, among electrophysiologic testing, SEP and BAEP are more sensitive than EEG (i.e., 45% to 60% sensitive versus 35%) and if EEG and SEP show opposing results, generally speaking, prognosis is linked to the better test result.158 The results of multimodality evoked potentials can be used as a reliable prognostic indicator (91% accuracy) in TBI patients.101
The International Mission for Prognosis and Analysis of Clinical Trials in TBI (IMPACT) is a prognostic model predicting 6-month functional outcome (Glasgow Outcome Scale [GOS]) and mortality in 587 patients with severe TBI (mean age 38 ± 17 years). At the 6th month, the median GOS was 3 (IQR 3) and 6-month mortality 41%, and age, motor score, pupillary reactivity, head CT findings, secondary insults, and laboratory values displayed good prediction ability for unfavorable outcome and mortality.159 In another study of 428 isolated, older adult TBI cases, the in-hospital death rate was 28%, and increasing age, decreasing GCS, and injury type were significant independent predictors of in-hospital mortality. At the 6-month follow-up, age < 75 years and with systolic blood pressure (BP) on arrival at hospital of 131 to 150 mm Hg had higher likelihood of living independently at follow-up. No patients with a GCS < 9 had a favorable 6-month outcome, and most of them died, with the survival rate for brainstem injury at 21%.160
Coma after Cardiac Arrest
The retrospective landmark study by Levy and colleagues,161 which reviewed postcardiac arrest outcome of 210 patients studied in the mid-1970s, is still commonly applied for prognostication. However, several inherent epidemiologic shortcomings of this study exist in addition to the fact that postcardiac arrest management has obviously advanced. The American Academy of Neurology published guidelines in 2006162 based on an in-depth review of the available literature; however, most studies included were performed without the use of hypothermia. The guideline defines poor outcome either as coma or death at 1 month or severe disability at 6 months. Further, the absence of brainstem reflexes or absence of extensor motor responses at 72 hours was shown to be highly specific (or have a false positive rate near zero) and so were absence of somatosensory evoked responses (SSEPs) and a high level of neuron-specific enolase (NSE).
Hypothermia has multiple actions not only within the CNS but also systemically. Among them are depression of neuronal activity, amelioration of intracellular postischemic reactions mitigating secondary cell injury, cardiac arrhythmias, hypocoagulability, decrease in immune defense, and reduction in drug metabolism and clearance. Notably, the latter can lead to misinterpretation of the prolonged effects of sedatives and paralytics. Therefore, interpretation of the neurologic examination and prognostication in patients undergoing hypothermia or rewarming is complex and challenging. Generally speaking and without the use of hypothermia, absence of brainstem reflexes implies that the cortex is severely affected as the brainstem neurons are more resistant to hypoxia-ischemia. Absence of pupillary reflexes at 72 hours predicts poor outcome with high specificity; however, the opposite, the presence of reflexive pupils, only predicts good outcome in about 21%. Studies performed after the introduction of hypothermia163–165 delineate predictive findings for absent papillary responses but delineate that the corneal reflex seems to be more vulnerable to prolonged sedative and hypothermic effects. With respect to motor responses, studies performed prior to hypothermia identify a high predictive value for poor outcome for the absence of motor responses at 72 hours, a clinical finding that outperformed even SSEP prediction.166 On the contrary, studies suggest that the lack of motor responses does not invariably predict poor outcome in patients treated with hypothermia (i.e., positive predictive values 92% and negative predictive value 85%) and exceptions have been reported.163–165,167
Electroencephalography (EEG) is used in postcardiac arrest patients to determine outcome but also to differentiate myoclonus from shivering and detect seizures. Generalized EEG suppression, burst suppression with or without epileptiform activity, and a flat background with intermittent generalized periodic complexes are indicators of poorer prognosis. The opposite is true for finding reactivity and variability of the EEG pattern in patients treated with or without cooling. Larger prospective studies are currently lacking; however, some reports identify some hypothermia-treated patients with postanoxic status epilepticus who can achieve good outcome,168 especially when the status evolves from a continuous EEG pattern and not from burst suppression.169 As seizures and status epilepticus are seen in up to 65% of patients with less severe cortical damage, continuous EEG is indicated to guide management and prognostication in many postarrest patients.
According to current practice parameters from 2006, the absence of the N20 (primary cortical somatosensory) wave on SSEPs in the first 3 days after cardiac arrest identifies a high likelihood for poor outcome. In contrast, only about 41% of patients with present N20 wave will recover. Prognosticating based on SSEP findings in patients treated with hypothermia has only been reported in a few smaller studies with uncertain overall results170,171; however, SSEP likely remains useful as evoked potentials are relatively resistant to metabolic and temperature derangements. Also, the NSE is a frequently used and validated biochemical marker used for prognostication. Levels >33 mcg/L at 72 hours or earlier were found to have zero false predictive rate in patients treated without hypothermia. However, hypothermia decreases NSE levels. Some studies have shown that a similar cutoff also holds true for the hypothermia-treated patient population,172–174 whereas others found unacceptable false-positive rates for the cutoff value 33 mcg/L.175,176 With respect to imaging, certain MRI findings such as severe cortical abnormalities on diffusion-weighted imaging have been shown to correlate to poor outcome,177 but imaging studies often report on smaller patient numbers and are performed unblinded, reducing their value for prediction. Some studies identify that most patients who regained consciousness had normal cortical structures, some of them despite involvement of the deep gray nuclei.178,179
Myoclonic status epilepticus are generalized, multifocal, repetitive muscle jerks involving some or all parts of the body. They are frequently seen within 24 hours after arrest, and EEG often shows burst suppression or bursts of generalized spikes. Whether myoclonic discharges actually represent seizures remains controversial, and as patients may have severe cortical damage and clinical myoclonus at the same time, it is postulated that myoclonus status epilepticus is subcortical in origin. In contrast, postanoxic status epilepticus is cortical in origin (but some overlap to myoclonic status may exist), whereas the posthypoxic myoclonus (Lance-Adams syndrome) is generally seen after primary respiratory failure. It is triggered by startle, often subsides over time, and the patient may be aware and distressed by it. Myoclonic status epilepticus has been associated with poor prognosis and remains poor even in the setting of hypothermia.163,165
Ethical Considerations
A surrogate decision maker is a person who directs care when the patient is unable to provide consent. Legally and ethically, surrogate decisions should follow the expressed wishes of the patient. In the absence of known prior wishes, surrogates should utilize the best standard with respect to what the patient would do when confronted by the circumstance. A surrogate designated by the patient has precedence over other potential decision makers and is delineated by an advanced directive, also called a health care proxy, health care agent, or durable power of attorney for health care. In contrast, a living will expresses patient preferences but does not authorize a surrogate decision maker or spokesperson. If there is no designated surrogate, family members and close friends are selected in order of their relationship to the patient; the highest priority is given to the spouse, then parents, then children, then siblings followed by other relatives and finally friends. In a comatose patient, surrogates or family members may authorize a do no resuscitate (DNR) order or request limitations of care (i.e., no hemodialysis). Approximately two thirds of DNR orders are put into place by surrogate decision makers.180
It is important to strive for the best possible diagnostic and prognostic correctness when discussing a DNR order or withdrawal of care. Understandably, relatives and surrogates place a great deal of decision making on the significance of conscious experience and the level of expected patient interactions with the environment (recovery). Therefore, the diagnostic accuracy of a patient’s present level of consciousness is requested from the treating physician as well as the best estimate of improvement of consciousness.181 Identifying medical futility in a patient with markedly impaired consciousness allows the surrogate and physician to withdraw life-supporting therapy. However, surrogate decision making may change if a patient is in a minimal conscious state, as this disorder implies different prognostication, especially in TBI patients.182 Further, it is important for physicians to reiterate to the family that coma progression into the vegetative state is not considered an improvement. This progression is seen in most coma patients and sleep-wake cycles, roving eye movements, startle, and other witnessed phenomena are merely reflexive and do not indicate consciousness or awareness. Of note, “persistence” is a diagnosis given to a patient who remains in a vegetative state for greater than 1 month. This state is considered permanent when it persists for 3 months after anoxic brain injury and 12 months after TBI.2 Greater prognostic uncertainties exist for patients in a state of minimal consciousness, as these patients have consistent evidence of consciousness, awareness, and, in some, the ability to communicate. Because these patients have an unpredictable recovery, it is necessary to continue standardized assessment and therapy.
References
1. Casarett, D, Fishman, JM, MacMoran, HJ, et al. Epidemiology and prognosis of coma in daytime television dramas. BMJ. 2005; 331:1537–1539.
2. Medical aspects of the persistent vegetative state (2). The Multi-Society Task Force on PVS. N Engl J Med. 1994; 330:1572–1579.
3. Hamel, MB, Goldman, L, Teno, J, et al. Identification of comatose patients at high risk for death or severe disability. JAMA. 1995; 273:1842–1848.
4. Sazbon, L, Zagreba, F, Ronen, J, et al. Course and outcome of patients in vegetative state of nontraumatic aetiology. J Neurol Neurosurg Psychiatry. 1993; 56:407–409.
5. Jennett, B, Plum, F. Persistent vegetative state after brain damage: A syndrome in search of a name. Lancet. 1972; 1:734–737.
6. Posner, JB, Saper, CB, Schiff, ND, Plum, F. Diagnosis of Stupor and Coma, 4th ed. New York, NY: Oxford University Press; 2007.
7. Medical aspects of the persistent vegetative state (1). The Multi-Society Task Force on PVS. N Engl J Med. 1994; 330:1499–1508.
8. Giacino, JT, Ashwal, S, Childs, N, et al. The minimally conscious state: Definition and diagnostic criteria. Neurology. 2002; 58:349–353.
9. Rothballer, AB. Studies on the adrenaline-sensitive component of the reticular activating system. Electroencephalogr Clin Neurophysiol. 1956; 8:603–621.
10. Kinomura, S, Larsson, J, Gulyas, B, Roland, PE. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science. 1996; 271:512–515.
11. Brodal A: Neurological anatomy in relation to clinical medicine, Oxford Press, NY 1981.
12. Shute, CC, Lewis, PR. The ascending cholinergic reticular system: Neocortical, olfactory and subcortical projections. Brain. 1967; 90:497–520.
13. Vincent, SR. The ascending reticular activating system—from aminergic neurons to nitric oxide. J Chem Neuroanat. 2000; 18:23–30.
14. Garcia-Rill, E. Disorders of the reticular activating system. Med Hypotheses. 1997; 49:379–387.
15. Burlet, S, Tyler, CJ, Leonard, CS. Direct and indirect excitation of laterodorsal tegmental neurons by hypocretin/orexin peptides: Implications for wakefulness and narcolepsy. J Neurosci. 2002; 22:2862–2872.
16. Lin, JS, Hou, Y, Sakai, K, Jouvet, M. Histaminergic descending inputs to the mesopontine tegmentum and their role in the control of cortical activation and wakefulness in the cat. J Neurosci. 1996; 16:1523–1537.
17. Takahashi, K, Lin, JS, Sakai, K. Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J Neurosci. 2006; 26:10292–10298.
18. Anaclet, C, Parmentier, R, Ouk, K, et al. Orexin/hypocretin and histamine: Distinct roles in the control of wakefulness demonstrated using knock-out mouse models. J Neurosci. 2009; 29:14423–14438.
19. Gottesmann, C. Brain inhibitory mechanisms involved in basic and higher integrated sleep processes. Brain Res Brain Res Rev. 2004; 45:230–249.
20. Levine, ES, Jacobs, BL. Neurochemical afferents controlling the activity of serotonergic neurons in the dorsal raphe nucleus: Microiontophoretic studies in the awake cat. J Neurosci. 1992; 12:4037–4044.
21. Brown, RE, Sergeeva, OA, Eriksson, KS, Haas, HL. Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline). J Neurosci. 2002; 22:8850–8859.
22. Sakai, K, Crochet, S. Serotonergic dorsal raphe neurons cease firing by disfacilitation during paradoxical sleep. Neuroreport. 2000; 11:3237–3241.
23. Schultz, W. Behavioral dopamine signals. Trends Neurosci. 2007; 30:203–210.
24. Brischoux, F, Chakraborty, S, Brierley, DI, Ungless, MA. Phasic excitation of dopamine neurons in ventral VTA by noxious stimuli. Proc Natl Acad Sci U S A. 2009; 106:4894–4899.
25. Koch, C. The quest for consciousness. Englewood, CO: Roberts & Co; 2004.
26. Svorad, D. Reticular activating system of brain stem and animal hypnosis. Science. 1957; 125:156.
27. Plum, F. Encyclopedia of neuroscience. Amsterdam: Elsevier Science; 1999.
28. Goldberg, S, The Four Minute Neurologic Exam Medmaster, Miami, FL. 1992.
29. Winkler, E, Almog, S, Kriger, D, et al. Use of flumazenil in the diagnosis and treatment of patients with coma of unknown etiology. Crit Care Med. 1993; 21:538–542.
30. Hund, EF, Lehman-Horn, F. Life-threatening hyperthermic syndromes. Neurocritical Care. Berlin: Springer-Verlag; 1994.
31. Lemke, DM. Sympathetic storming after severe traumatic brain injury. Crit Care Nurse. 2007; 27:30–37.
32. Schnakers, C, Vanhaudenhuyse, A, Giacino, J, et al. Diagnostic accuracy of the vegetative and minimally conscious state: Clinical consensus versus standardized neurobehavioral assessment. BMC Neurol. 2009; 9:35.
33. Seel, RT, Sherer, M, Whyte, J, et al. Assessment scales for disorders of consciousness: Evidence-based recommendations for clinical practice and research. Arch Phys Med Rehabil. 2010; 91:1795–1813.
34. Fernandez-Espejo, D, Bekinschtein, T, Monti, MM, et al. Diffusion weighted imaging distinguishes the vegetative state from the minimally conscious state. Neuroimage. 2011; 54:103–112.
35. Qin, P, Di, H, Liu, Y, et al. Anterior cingulate activity and the self in disorders of consciousness. Hum Brain Mapp. 2010; 31:1993–2002.
36. Schnakers, C, Chatelle, C, Majerus, S, et al. Assessment and detection of pain in noncommunicative severely brain-injured patients. Expert Rev Neurother. 2010; 10:1725–1731.
37. Soddu, A, Vanhaudenhuyse, A, Bahri, MA, et al. Identifying the default-mode component in spatial IC analyses of patients with disorders of consciousness. Hum Brain Mapp. 2012; 33:778–796.
38. Boly, M, Faymonville, ME, Schnakers, C, et al. Perception of pain in the minimally conscious state with PET activation: An observational study. Lancet Neurol. 2008; 7:1013–1020.
39. Vanhaudenhuyse, A, Noirhomme, Q, Tshibanda, LJ, et al. Default network connectivity reflects the level of consciousness in non-communicative brain-damaged patients. Brain. 2010; 133(Pt 1):161–171.
40. Fernandez-Espejo, D, Junque, C, Bernabeu, M, et al. Reductions of thalamic volume and regional shape changes in the vegetative and the minimally conscious states. J Neurotrauma. 2010; 27:1187–1193.
41. Luaute, J, Maucort-Boulch, D, Tell, L, et al. Long-term outcomes of chronic minimally conscious and vegetative states. Neurology. 2010; 75:246–252.
42. Carpentier, A, Galanaud, D, Puybasset, L, et al. Early morphologic and spectroscopic magnetic resonance in severe traumatic brain injuries can detect “invisible brain stem damage” and predict “vegetative states. ”. J Neurotrauma. 2006; 23:674–685.
43. Yanagawa, Y, Tsushima, Y, Tokumaru, A, et al. A quantitative analysis of head injury using T2*-weighted gradient-echo imaging. J Trauma. 2000; 49:272–277.
44. Trivedi, MA, Ward, MA, Hess, TM, et al. Longitudinal changes in global brain volume between 79 and 409 days after traumatic brain injury: Relationship with duration of coma. J Neurotrauma. 2007; 24:766–771.
45. Zazulia, AR, Videen, TO, Diringer, MN, Powers, WJ. Poor correlation between perihematomal MRI hyperintensity and brain swelling after intracerebral hemorrhage. Neurocrit Care. 2011; 15:436–441.
46. Kumar, R, Husain, M, Gupta, RK, et al. Serial changes in the white matter diffusion tensor imaging metrics in moderate traumatic brain injury and correlation with neuro-cognitive function. J Neurotrauma. 2009; 26:481–495.
47. Betz, J, Zhuo, J, Roy, A, et al. Prognostic value of diffusion tensor imaging parameters in severe traumatic brain injury. J Neurotrauma. 2012; 29:1292–1305.
48. Huisman, TA, Schwamm, LH, Schaefer, PW, et al. Diffusion tensor imaging as potential biomarker of white matter injury in diffuse axonal injury. AJNR Am J Neuroradio. 2004; 25:370–376.
49. Friedman, SD, Brooks, WM, Jung, RE, et al. Quantitative proton MRS predicts outcome after traumatic brain injury. Neurology. 1999; 52:1384–2391.
50. Ross, BD, Ernst, T, Kreis, R, et al. 1H MRS in acute traumatic brain injury. J Magn Reson Imaging. 1998; 8:829–840.
51. Sinson, G, Bagley, LJ, Cecil, KM, et al. Magnetization transfer imaging and proton MR spectroscopy in the evaluation of axonal injury: Correlation with clinical outcome after traumatic brain injury. AJNR Am J Neuroradiol. 2001; 22:143–151.
52. Uzan, M, Albayram, S, Dashti, SG, et al. Thalamic proton magnetic resonance spectroscopy in vegetative state induced by traumatic brain injury. J Neurol Neurosurg Psychiatry. 2003; 74:33–38.
53. Signoretti, S, Marmarou, A, Aygok, GA, et al. Assessment of mitochondrial impairment in traumatic brain injury using high-resolution proton magnetic resonance spectroscopy. J Neurosurg. 2008; 108:42–52.
54. Choe, BY, Suh, TS, Choi, KH, et al. Neuronal dysfunction in patients with closed head injury evaluated by in vivo 1H magnetic resonance spectroscopy. Invest Radiol. 1995; 30:502–506.
55. Cecil, KM, Lenkinski, RE, Meaney, DF, et al. High-field proton magnetic resonance spectroscopy of a swine model for axonal injury. J Neurochem. 1998; 70:2038–2044.
56. Marino, S, Zei, E, Battaglini, M, et al. Acute metabolic brain changes following traumatic brain injury and their relevance to clinical severity and outcome. J Neurol Neurosurg Psychiatry. 2007; 78:501–507.
57. Tommasino, C, Grana, C, Lucignani, G, et al. Regional cerebral metabolism of glucose in comatose and vegetative state patients. J Neurosurg Anesthesiol. 1995; 7:109–116.
58. Bergsneider, M, Hovda, DA, Shalmon, E, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: A positron emission tomography study. J Neurosurg. 1997; 86:241–251.
59. Boly, M, Faymonville, ME, Peigneux, P, et al. Cerebral processing of auditory and noxious stimuli in severely brain injured patients: Differences between VS and MCS. Neuropsychol Rehabil. 2005; 15:283–289.
60. Boly, M, Faymonville, ME, Peigneux, P, et al. Auditory processing in severely brain injured patients: Differences between the minimally conscious state and the persistent vegetative state. Arch Neurol. 2004; 61:233–238.
61. Laureys, S, Faymonville, ME, Degueldre, C, et al. Auditory processing in the vegetative state. Brain. 2000; 123(Pt 8):1589–1601.
62. Meilof, JF. Cortical processing in persistent vegetative state. Lancet. 1998; 352:1148–1149.
63. Owen, AM, Menon, DK, Johnsrude, IS, et al. Detecting residual cognitive function in persistent vegetative state. Neurocase. 2002; 8:394–403.
64. Laureys, S, Faymonville, ME, Luxen, A, et al. Restoration of thalamocortical connectivity after recovery from persistent vegetative state. Lancet. 2000; 35:1790–1791.
65. Coleman, MR, Davis, MH, Rodd, JM, et al. Towards the routine use of brain imaging to aid the clinical diagnosis of disorders of consciousness. Brain. 2009; 132(Pt 9):2541–2552.
66. Gosseries, O, Bruno, MA, Chatelle, C, et al. Disorders of consciousness: What’s in a name? NeuroRehabilitation. 2011; 28:3–14.
67. Bekinschtein, T, Tiberti, C, Niklison, J, et al. Assessing level of consciousness and cognitive changes from vegetative state to full recovery. Neuropsychol Rehabil. 2005; 15:307–322.
68. Di, HB, Yu, SM, Weng, XC, et al. Cerebral response to patient’s own name in the vegetative and minimally conscious states. Neurology. 2007; 68:895–899.
69. Eickhoff, SB, Dafotakis, M, Grefkes, C, et al. fMRI reveals cognitive and emotional processing in a long-term comatose patient. Exp Neurol. 2008; 214:240–246.
70. Fernandez-Espejo, D, Junque, C, Vendrell, P, et al. Cerebral response to speech in vegetative and minimally conscious states after traumatic brain injury. Brain Inj Oct. 2008; 22:882–890.
71. Schiff, ND, Rodriguez-Moreno, D, Kamal, A, et al. fMRI reveals large-scale network activation in minimally conscious patients. Neurology. 2005; 64:514–523.
72. Staffen, W, Kronbichler, M, Aichhorn, M, et al. Selective brain activity in response to one’s own name in the persistent vegetative state. J Neurol Neurosurg Psychiatry. 2006; 77:1383–4138.
73. Boly, M, Tshibanda, L, Vanhaudenhuyse, A, et al. Functional connectivity in the default network during resting state is preserved in a vegetative but not in a brain dead patient. Hum Brain Mapp. 2009; 30:2393–2400.
74. Cauda, F, Micon, BM, Sacco, K, et al. Disrupted intrinsic functional connectivity in the vegetative state. J Neurol Neurosurg Psychiatry. 2009; 80:429–431.
75. Heelmann, V, Lippert-Gruner, M, Rommel, T, et al. Abnormal functional MRI BOLD contrast in the vegetative state after severe traumatic brain injury. Int J Rehabil Res. 2010; 33:151–157.
76. Rousseau, MC, Confort-Gouny, S, Catala, A, et al. A MRS-MRI-fMRI exploration of the brain: Impact of long-lasting persistent vegetative state. Brain Inj. 2008; 22:123–134.
77. Kaplan, PW, Genoud, D, Ho, TW, Jallon, P. Etiology, neurologic correlations, and prognosis in alpha coma. Clin Neurophysiol. 1999; 110:205–213.
78. Husain, AM. Electroencephalographic assessment of coma. J Clin Neurophysiol. 2006; 23:208–220.
79. Westmoreland, BF, Klass, DW, Sharbrough, FW, Reagan, TJ. Alpha-coma: Electroencephalographic, clinical, pathologic, and etiologic correlations. Arch Neurol. 1975; 32:713–718.
80. Young, GB, Blume, WT, Campbell, VM, et al. Alpha, theta and alpha-theta coma: A clinical outcome study utilizing serial recordings. Electroencephalogr Clin Neurophysiol. 1994; 91:93–99.
81. Herkes, GK, Wszolek, ZK, Westmoreland, BF, Klass, DW. Effects of midazolam on electroencephalograms of seriously ill patients. Mayo Clin Proc. 1992; 67:334–338.
82. Bortone, E, Bettoni, L, Buzio, S, et al. Spindle coma and alternating pattern in the course of measles encephalitis. Clin Electroencephalogr. 1996; 27:210–214.
83. Britt, CW, Jr. Nontraumatic “spindle coma”: Clinical, EEG, and prognostic features. Neurology. 1981; 31:393–397.
84. Hansotia, P, Gottschalk, P, Green, P, Zais, D. Spindle coma: Incidence, clinicopathologic correlates, and prognostic value. Neurology. 1981; 31:83–87.
85. Chatrian, GE, White, LE, Jr., Daly, D. Electroencephalographic patterns resembling those of sleep in certain comatose states after injuries to the head. Electroencephalogr Clin Neurophysiol. 1963; 15:272–380.
86. Rumpl, E. [Electro-neurological correlations in early stages of post-traumatic comatose states. II. The EEG at the transition stage to, and at the full stage of the traumatic apallic syndrome (author’s transl)]. EEG EMG Z Elektroenzephalogr Elektromyogr Verwandte Geb. 1980; 11:43–50.
87. Rumpl, E, Prugger, M, Bauer, G, et al. Incidence and prognostic value of spindles in post-traumatic coma. Electroencephalogr Clin Neurophysiol. 1983; 56:420–429.
88. Chatrian, GE, Shaw, CM, Leffman, H. The significance of periodic lateralized epileptiform discharges in eeg: An electrographic, clinical and pathological study. Electroencephalogr Clin Neurophysiol. 1964; 17:177–193.
89. Garcia-Morales, I, Garcia, MT, Galan-Davila, L, et al. Periodic lateralized epileptiform discharges: Etiology, clinical aspects, seizures, and evolution in 130 patients. J Clin Neurophysiol. 2002; 19:172–177.
90. Brenner, RP, Schaul, N. Periodic EEG patterns: Classification, clinical correlation, and pathophysiology. J Clin Neurophysiol. 1990; 7:249–267.
91. de la Paz, D, Brenner, RP. Bilateral independent periodic lateralized epileptiform discharges: Clinical significance. Arch Neurol. 1981; 38:713–715.
92. Fitzpatrick, W, Lowry, N. PLEDs: Clinical correlates. Can J Neurol Sci. 2007; 34:443–450.
93. Amzica, F, Kroeger, D. Cellular mechanisms underlying EEG waveforms during coma. Epilepsia. 2011; 52(Suppl 8):25–27.
94. Sethi, N. EEG in anoxic coma. J Clin Neurophysiol. 2012; 29:199.
95. Kaplan, PW. The EEG in metabolic encephalopathy and coma. J Clin Neurophysiol. 2004; 21:307–318.
96. Fritzler, DE, Shevrin, H, Smith, WH. Subliminally stimulated brain and verbal responses of twins differing in repressiveness. J Abnorm Psychol. 1970; 76:39–46.
97. Rodeck, H. [Neurosecretion and posterior pituitary gland hormones]. Dtsch Krankenpflegez. 1975; 28:701–707.
98. Schnakers, C, Perrin, F, Schabus, M, et al. Detecting consciousness in a total locked-in syndrome: An active event-related paradigm. Neurocase. 2009; 15:271–277.
99. Rogowski, M, Michalska, BI. [The importance of brain stem evoked potentials in the diagnosis of neurosurgical patients]. Neurol Neurochir Pol. 2001; 35:667–679.
100. Greenberg, RP, Ducker, TB. Evoked potentials in the clinical neurosciences. J Neurosurg. 1982; 56:1–18.
101. Narayan, RK, Greenberg, RP, Miller, JD, et al. Improved confidence of outcome prediction in severe head injury: A comparative analysis of the clinical examination, multimodality evoked potentials, CT scanning, and intracranial pressure. J Neurosurg. 1981; 54:751–762.
102. Sutton, LN, Frewen, T, Marsh, R, et al. The effects of deep barbiturate coma on multimodality evoked potentials. J Neurosurg. 1982; 57:178–185.
103. Marinoni, M, Alari, F, Mastronardi, V, et al. The relevance of early TCD monitoring in the intensive care units for the confirming of brain death diagnosis. Neurol Sci. 2011; 32:73–77.
104. Gura, M, Elmaci, I, Sari, R, Coskun, N. Correlation of pulsatility index with intracranial pressure in traumatic brain injury. Turk Neurosurg. 2011; 21:210–215.
105. Zink, BJ. Traumatic brain injury outcome: Concepts for emergency care. Ann Emerg Med. 2001; 37:318–332.
106. Rebet, O, Leclerc, C, Sillard, B, et al. [Complete heart block and myocardial ischaemia during a severe anaphylactic reaction]. Ann Fr Anesth Reanim. 2008; 27:1026–1029.
107. Winchell, RJ, Hoyt, DB. Endotracheal intubation in the field improves survival in patients with severe head injury. Trauma Research and Education Foundation of San Diego. Arch Surg. 1997; 132:592–597.
108. Le Gall, JR, Loirat, P, Alperovitch, A, et al. A simplified acute physiology score for ICU patients. Crit Care Med. 1984; 12:975–977.
109. Major, KM, Hui, T, Wilson, MT, et al. Objective indications for early tracheostomy after blunt head trauma. Am J Surg. 2003; 186:615–619.
110. Arabi, Y, Haddad, S, Shirawi, N, et al. Early tracheostomy in intensive care trauma patients improves resource utilization: A cohort study and literature review. Crit Care. 2004; 8:R347–R352.
111. Kornblith, LZ, Burlew, CC, Moore, EE, et al. One thousand bedside percutaneous tracheostomies in the surgical intensive care unit: Time to change the gold standard. J Am Coll Surg. 2011; 212:163–170.
112. Howsepian, AA. The 1994 Multi-Society Task Force consensus statement on the Persistent Vegetative State: A critical analysis. Issues Law Med. 1996; 12:3–29.
113. Broder, JS. Head computed tomography interpretation in trauma: A primer. Psychiatr Clin North Am. 2010; 33:821–854.
114. Jang, SH. Diffusion tensor imaging studies on corticospinal tract injury following traumatic brain injury: A review. NeuroRehabilitation. 2011; 29:339–345.
115. Kubal, WS. Updated imaging of traumatic brain injury. Radiol Clin North Am Jan. 2012; 50:15–41.
116. Bratton, SL, Chestnut, RM, Ghajar, J, et al. Guidelines for the management of severe traumatic brain injury. VI. Indications for intracranial pressure monitoring. J Neurotrauma. 2007; 24(Suppl 1):S37–S44.
117. Aarabi, B, Hesdorffer, DC, Ahn, ES, et al. Outcome following decompressive craniectomy for malignant swelling due to severe head injury. J Neurosurg. 2006; 104:469–479.
118. Bao, YH, Liang, YM, Gao, GY, et al. Bilateral decompressive craniectomy for patients with malignant diffuse brain swelling after severe traumatic brain injury: A 37-case study. J Neurotrauma. 2010; 27:341–347.
119. Bell, RS, Mossop, CM, Dirks, MS, et al. Early decompressive craniectomy for severe penetrating and closed head injury during wartime. Neurosurg Focus. 2010; E1:28.
120. Cooper, DJ, Rosenfeld, JV, Murray, L, et al. Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med. 2011; 364:1493–1502.
121. Daboussi, A, Minville, V, Leclerc-Foucras, S, et al. Cerebral hemodynamic changes in severe head injury patients undergoing decompressive craniectomy. J Neurosurg Anesthesiol. 2009; 21:339–345.
122. Ecker, RD, Mulligan, LP, Dirks, M, et al. Outcomes of 33 patients from the wars in Iraq and Afghanistan undergoing bilateral or bicompartmental craniectomy. J Neurosurg. 2011; 115:124–129.
123. Howard, JL, Cipolle, MD, Anderson, M, et al. Outcome after decompressive craniectomy for the treatment of severe traumatic brain injury. J Trauma. 2008; 65:380–385.
124. Olivecrona, M, Rodling-Wahlstrom, M, Naredi, S, Koskinen, LO. Effective ICP reduction by decompressive craniectomy in patients with severe traumatic brain injury treated by an ICP-targeted therapy. J Neurotrauma. 2007; 24:927–935.
125. Qiu, W, Guo, C, Shen, H, et al. Effects of unilateral decompressive craniectomy on patients with unilateral acute post-traumatic brain swelling after severe traumatic brain injury. Crit Care. 2009; 13:R185.
126. Timofeev, I, Kirkpatrick, PJ, Corteen, E, et al. Decompressive craniectomy in traumatic brain injury: Outcome following protocol-driven therapy. Acta Neurochir Suppl. 2006; 96:11–16.
127. Wen, L, Wang, H, Wang, F, et al. A prospective study of early versus late craniectomy after traumatic brain injury. Brain Inj. 2011; 25:1318–1324.
128. Williams, RF, Magnotti, LJ, Croce, MA, et al. Impact of decompressive craniectomy on functional outcome after severe traumatic brain injury. J Trauma. 2009; 66:1570–1574.
129. Francony, G, Fauvage, B, Falcon, D, et al. Equimolar doses of mannitol and hypertonic saline in the treatment of increased intracranial pressure. Crit Care Med. 2008; 36:795–800.
130. Hartl, R, Ghajar, J, Hochleuthner, H, Mauritz, W. Hypertonic/hyperoncotic saline reliably reduces ICP in severely head-injured patients with intracranial hypertension. Acta Neurochir. 1997; 70(Suppl):126–129.
131. Huang, SJ, Chang, L, Han, YY, et al. Efficacy and safety of hypertonic saline solutions in the treatment of severe head injury. Surg Neurol. 2006; 65:539–546.
132. Ichai, C, Armando, G, Orban, JC, et al. Sodium lactate versus mannitol in the treatment of intracranial hypertensive episodes in severe traumatic brain-injured patients. Intensive Care Med. 2009; 35:471–479.
133. Kerwin, AJ, Schinco, MA, Tepas, JJ, 3rd., et al. The use of 23. 4% hypertonic saline for the management of elevated intracranial pressure in patients with severe traumatic brain injury: A pilot study. J Trauma. 2009; 67:277–282.
134. Lescot, T, Degos, V, Zouaoui, A, et al. Opposed effects of hypertonic saline on contusions and noncontused brain tissue in patients with severe traumatic brain injury. Crit Care Med. 2006; 34:3029–3033.
135. Munar, F, Ferrer, AM, de Nadal, M, et al. Cerebral hemodynamic effects of 7. 2% hypertonic saline in patients with head injury and raised intracranial pressure. J Neurotrauma. 2000; 17:41–51.
136. Oddo, M, Levine, JM, Frangos, S, et al. Effect of mannitol and hypertonic saline on cerebral oxygenation in patients with severe traumatic brain injury and refractory intracranial hypertension. J Neurol Neurosurg Psychiatry. 2009; 80:916–920.
137. Qureshi, AI, Suarez, JI, Castro, A, Bhardwaj, A. Use of hypertonic saline/acetate infusion in treatment of cerebral edema in patients with head trauma: Experience at a single center. J Trauma. 1999; 47:659–665.
138. Rockswold, GL, Solid, CA, Paredes-Andrade, E, et al. Hypertonic saline and its effect on intracranial pressure, cerebral perfusion pressure, and brain tissue oxygen. Neurosurgery. 2009; 65:1035–1041.
139. Sakellaridis, N, Pavlou, E, Karatzas, S, et al. Comparison of mannitol and hypertonic saline in the treatment of severe brain injuries. J Neurosurg. 2011; 114:545–548.
140. Sakowitz, OW, Stover, JF, Sarrafzadeh, AS, et al. Effects of mannitol bolus administration on intracranial pressure, cerebral extracellular metabolites, and tissue oxygenation in severely head-injured patients. J Trauma. 2007; 62:292–298.
141. Ware, ML, Nemani, VM, Meeker, M, et al. Effects of 23. 4% sodium chloride solution in reducing intracranial pressure in patients with traumatic brain injury: A preliminary study. Neurosurgery. 2005; 57:727–736.
142. Blissitt, PA. Controversies in the management of adults with severe traumatic brain injury. AACN Adv Crit Care. 2012; 23:188–203.
143. Alderson, P, Gadkary, C, Signorini, DF. Therapeutic hypothermia for head injury. Cochrane Database Syst Rev. 2004.
144. Harris, OA, Colford, JM, Jr., Good, MC, Matz, PG. The role of hypothermia in the management of severe brain injury: A meta-analysis. Arch Neurol. 2002; 59:1077–1083.
145. Henderson, WR, Dhingra, VK, Chittock, DR, et al. Hypothermia in the management of traumatic brain injury: A systematic review and meta-analysis. Intensive Care Med. 2003; 29:1637–1644.
146. McIntyre, LA, Fergusson, DA, Hebert, PC, et al. Prolonged therapeutic hypothermia after traumatic brain injury in adults: A systematic review. JAMA. 2003; 289:2992–2999.
147. Meyer, MJ, Megyesi, J, Meythaler, J, et al. Acute management of acquired brain injury part I: An evidence-based review of non-pharmacological interventions. Brain Inj. 2010; 24:694–705.
148. Peterson, K, Carson, S, Carney, N. Hypothermia treatment for traumatic brain injury: A systematic review and meta-analysis. J Neurotrauma. 2008; 25:62–71.
149. Sahuquillo, J, Vilalta, A. Cooling the injured brain: How does moderate hypothermia influence the pathophysiology of traumatic brain injury. Curr Pharm Des. 2007; 13:2310–2322.
150. Sinclair, HL, Andrews, PJ. Bench-to-bedside review: Hypothermia in traumatic brain injury. Crit Care. 2010; 14:204.
151. Sydenham, E, Roberts, I, Alderson, P. Hypothermia for traumatic head injury. Cochrane Database Syst Rev. 2009.
152. Mushkudiani, NA, Hukkelhoven, CW, Hernandez, AV, et al. A systematic review finds methodological improvements necessary for prognostic models in determining traumatic brain injury outcomes. J Clin Epidemiol. 2008; 61:331–343.
153. Rimel, RW, Giordani, B, Barth, JT, Jane, JA. Moderate head injury: Completing the clinical spectrum of brain trauma. Neurosurgery. 1982; 11:344–351.
154. Attia, J, Cook, DJ. Prognosis in anoxic and traumatic coma. Crit Care Clin. 1998; 14:497–511.
155. Young, B, Rapp, RP, Norton, JA, et al. Early prediction of outcome in head-injured patients. J Neurosurg. 1981; 54:300–303.
156. Vos, PE, Jacobs, B, Andriessen, TM, et al. GFAP and S100B are biomarkers of traumatic brain injury: An observational cohort study. Neurology. 2010; 75:1786–1793.
157. Xu, W, Jiang, G, Chen, Y, et al. Prediction of minimally conscious state with somatosensory evoked potentials in long-term unconscious patients after traumatic brain injury. J Trauma Acute Care Surg. 2012; 72:1024–1029.
158. Chen, R, Bolton, CF, Young, B. Prediction of outcome in patients with anoxic coma: A clinical and electrophysiologic study. Crit Care Med. 1996; 24:672–678.
159. Panczykowski, DM, Puccio, AM, Scruggs, BJ, et al. Prospective independent validation of IMPACT modeling as a prognostic tool in severe traumatic brain injury. J Neurotrauma. 2012; 29:47–52.
160. Utomo, WK, Gabbe, BJ, Simpson, PM, Cameron, PA. Predictors of in-hospital mortality and 6-month functional outcomes in older adults after moderate to severe traumatic brain injury. Injury. 2009; 40:973–977.
161. Levy, DE, Caronna, JJ, Singer, BH, et al. Predicting outcome from hypoxic-ischemic coma. JAMA. 1985; 253:1420–1426.
162. Wijdicks, EF, Hijdra, A, Young, GB, et al. Practice parameter: Prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006; 67:203–210.
163. Al Thenayan, E, Savard, M, Sharpe, M, et al. Predictors of poor neurologic outcome after induced mild hypothermia following cardiac arrest. Neurology. 2008; 71:1535–1537.
164. Rossetti, AO, Oddo, M, Logroscino, G, Kaplan, PW. Prognostication after cardiac arrest and hypothermia: A prospective study. Ann Neurol. 2010; 67:301–307.
165. Samaniego, EA, Mlynash, M, Caulfield, AF, et al. Sedation confounds outcome prediction in cardiac arrest survivors treated with hypothermia. Neurocrit Care. 2011; 15:113–119.
166. Lee, YC, Phan, TG, Jolley, DJ, et al. Accuracy of clinical signs, SEP, and EEG in predicting outcome of hypoxic coma: A meta-analysis. Neurology. 2010; 74:572–580.
167. Schefold, JC, Storm, C, Kruger, A, et al. The Glasgow Coma Score is a predictor of good outcome in cardiac arrest patients treated with therapeutic hypothermia. Resuscitation. 2009; 80:658–661.
168. Rossetti, AO, Logroscino, G, Liaudet, L, et al. Status epilepticus: An independent outcome predictor after cerebral anoxia. Neurology. 2007; 69:255–260.
169. Rundgren, M, Westhall, E, Cronberg, T, et al. Continuous amplitude-integrated electroencephalogram predicts outcome in hypothermia-treated cardiac arrest patients. Crit Care Med. 2010; 38:1838–1844.
170. Bouwes, A, Binnekade, JM, Zandstra, DF, et al. Somatosensory evoked potentials during mild hypothermia after cardiopulmonary resuscitation. Neurology. 2009; 73:1457–1461.
171. Leithner, C, Ploner, CJ, Hasper, D, Storm, C. Does hypothermia influence the predictive value of bilateral absent N20 after cardiac arrest? Neurology. 2010; 74:965–969.
172. Cronberg, T, Rundgren, M, Westhall, E, et al. Neuron-specific enolase correlates with other prognostic markers after cardiac arrest. Neurology. 2011; 77:623–630.
173. Oksanen, T, Tiainen, M, Skrifvars, MB, et al. Predictive power of serum NSE and OHCA score regarding 6-month neurologic outcome after out-of-hospital ventricular fibrillation and therapeutic hypothermia. Resuscitation. 2009; 80:165–170.
174. Rundgren, M, Karlsson, T, Nielsen, N, et al. Neuron specific enolase and S-100B as predictors of outcome after cardiac arrest and induced hypothermia. Resuscitation. 2009; 80:784–789.
175. Fugate, JE, Wijdicks, EF, Mandrekar, J, et al. Predictors of neurologic outcome in hypothermia after cardiac arrest. Ann Neurol. 2010; 68:907–914.
176. Steffen, IG, Hasper, D, Ploner, CJ, et al. Mild therapeutic hypothermia alters neuron specific enolase as an outcome predictor after resuscitation: 97 prospective hypothermia patients compared to 133 historical non-hypothermia patients. Crit Care. 2010; 14:R69.
177. Wijdicks, EF, Campeau, NG, Miller, GM. MR imaging in comatose survivors of cardiac resuscitation. AJNR Am J Neuroradiol. 2001; 22:1561–1565.
178. Jarnum, H, Knutsson, L, Rundgren, M, et al. Diffusion and perfusion MRI of the brain in comatose patients treated with mild hypothermia after cardiac arrest: A prospective observational study. Resuscitation. 2009; 80:425–430.
179. Mlynash, M, Campbell, DM, Leproust, EM, et al. Temporal and spatial profile of brain diffusion-weighted MRI after cardiac arrest. Stroke. 2010; 41:1665–1672.
180. Fins, JJ, Miller, FG, Acres, CA, et al. End-of-life decision-making in the hospital: Current practice and future prospects. J Pain Symptom Manage. 1999; 17:6–15.
181. Giacino, JT, Kalmar, K. Diagnostic and prognostic guidelines for the vegetative and minimally conscious states. Neuropsychol Rehabil. 2005; 15:166–174.
182. Lammi, MH, Smith, VH, Tate, RL, Taylor, CM. The minimally conscious state and recovery potential: A follow-up study 2 to 5 years after traumatic brain injury. Arch Phys Med Rehabil. 2005; 86:746–754.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]61
Coma
Concept and Terminology of Impaired Consciousness
Alteration of consciousness is a frequent admission diagnosis to critical care services. Most patients require immediate and often extensive diagnostic workup as both time to diagnosis and treatment initiation are decisive factors for brain recovery. To the public, the portrayal of coma is overly optimistic as 89% of “comatose” patients in a meta-analysis of 64 characters in American soap dramas regained consciousness and around 92% “survived” coma.1 In contrast, medical experience does not compare as favorably, as only about 50% of patients survive in an unselected coma population.2–4 Therefore, defining coma in a succinct and operational manner will provide a deeper understanding of the depth of brain injury and avoid miscommunications and unrealistic prognostications.
When evaluating a patient’s mental status, the state of consciousness should be viewed as a continuum ranging from the patient who has full alertness and cognitive lucidity to the deeply comatose patient; it is not an all-or-nothing phenomenon.5 An all-or-nothing approach limits the interpretation of remaining brain function and, hence, diagnostic certainty. Furthermore, the duration and time development of coma are helpful diagnostic features and should complement the quantitative and qualitative assessment of consciousness. Standard classification systems categorize consciousness based on systematic testing of arousal, awareness, and content. Table 61.1 classifies various abnormalities of consciousness, and the following section delineates clinically important syndromes.
Coma (from Greek κµα, or “deep sleep”) is a continuous state of unresponsiveness identified by an inability to arouse to vigorous (noxious) external or internal stimuli. The degree of coma can differ; lighter stages (sometimes denoted as semicoma) can be identified by brief moaning to strong stimulation and associated observed changes in autonomic function; whereas the deepest coma examination shows an absence of any response, including brainstem reflex responses (i.e., lack of oculo- and pupillomotoric responses). Some cyclic autonomic activity such as the sleep-wake cycle and changes in motor tone may coexist. A detailed discussion of coma is provided in the classic textbook by Plum and Posner.6
The vegetative state is characterized by the complete absence of behavioral evidence for self- or environmental awareness. This state can follow coma and identifies a state in which brainstem and diencephalic (thalamic) activity is present to a degree that clinical signs of spontaneous or stimulus-induced arousal and sleep-wake cycles are observed. Patients often show blink responses to light; intermittent eye movements (sometimes erroneously interpreted as following objects or looking at family members); stimulus-sensitive automatisms such as swallowing, bruxism, and moaning; or primitive motor responses. If this state lasts longer than 30 days, it is referred to as persistent vegetative state (PVS) and is used as a descriptive clinical syndrome rather than a disease-specific entity. The most common causes include cardiac arrest, head trauma, severe brain infections, and various causes of thalamic injury. Vegetative states can also be seen in the terminal phase of degenerative illnesses such as Alzheimer’s disease. Ambiguous terms for PVS such as apallic syndrome and neocortical death should be avoided.2,7
Minimal conscious state can be diagnosed in patients displaying some but often inconsistent behavioral evidence of awareness of the environment, but they cannot communicate their content and are unable to follow instructions reliably.8 It describes a large group of patients who are different from vegetative patients in that they demonstrate some signs of awareness of themselves and their surroundings, albeit inconsistently.
Neuroanatomy, Neurotransmitter, and Pathology
Consciousness, which consists of three main parts, largely depends on the integrity of brain structure for arousal (Fig. 61.1). The ascending reticular activating system (ARAS) is the lowest order arousal system, consisting of a set of brainstem nuclei located within the brainstem and interconnected by neuronal circuits. The ARAS relays arousal signals to the more rostral thalami, which, in turn, act as hemispheric gatekeepers of arousal and regulate consciousness and sleep-wake transitions. In turn, thalamic activated cerebral cortices allow cognitive processing, which then determines the overall content of consciousness.6 Structural damage to or metabolic-chemical derangements of any or all of these elements will affect consciousness. Important components of the ARAS9–11 include the midbrain reticular formation, mesencephalic nuclei (dorsal raphe nucleus, pedunculopontine tegmental nucleus, locus coeruleus), and ventral tegmental areas. Those areas have rich connections to thalamic intralaminar nuclei and further frontally reaching projections to the tuberomammillary nucleus, lateral hypothalamus, and basal forebrain.
The ascending arousal system of the brainstem contains different neurotransmitter systems. Main cholinergic projections include the ascending mesopontine tegmental and basal forebrain pathways projecting to the thalami and to virtually all subcortical and cortical structures. These projections usually function as synaptic facilitators but at times can act as depressors of transmission.12 Glutamate is the main transmitter influencing the firing patterns of tegmental cholinergic neurons.13 The adrenergic component of the ARAS is closely associated with the noradrenergic neurons of the midbrain locus coeruleus. It runs in parallel with cholinergic projections rostrally to the cortical areas but also descends caudally within the spinal cord.14 Hypocretin/orexin neurons within the hypothalamus activate both adrenergic and cholinergic pathways and coordinate activity of the entire ARAS system, enhancing complementary and synergistic control of arousal and locomotion.15 Histamine-releasing cells from the hypothalamus and tuberomammillary neurons are active during the qualitative activation needed for cognition and EEG activation, but they remain silent during sleep.16–18
The main inhibitor of the arousal system is γ-aminobutyric acid (GABA), without which normal sleep does not occur. The GABAergic circuitry is located in the midbrain and pons, but its activity is regulated and sustained by forebrain structures.19 It is thought that the GABAergic system’s main function is to contain and channel the spread of arousal, both spontaneously and in response to a stimulus. GABAergic processes are responsible for the occurrence of paradoxical sleep (deep sleep characterized by a brain wave pattern similar to that of wakefulness, rapid eye movements, and heavier breathing but without motor responses). The name “waking neurons” has been given to the serotonergic dorsal raphe nucleus in the midbrain,20 which receives convergent excitatory input from the noradrenaline, histamine, and hypocretin/orexin arousal systems21,22 as the main activator of the ARAS. Cortical signals that pass through the striatum (caudate and putamen) are refined by the action of dopamine. Dopaminergic neurons are tonically active and stimulate or inhibit cortical neurons depending on various environmental influences (pain, hunger, etc.).23,24 It is thought that many sensory signals that reach conscious awareness pass through this basal ganglia loop with dopamine as the modulating neurotransmitter. Koch and colleagues suggested that coalitions of neurons compete to dominate conscious thoughts and activities at any given time point and, hence, dopaminergic innervation may facilitate the transient domination of conscious awareness by certain sets of neuron coalitions.25 Therefore, it seems that arousal is an event with a defined time distribution, which is tightly orchestrated by a few key neurotransmitters. As we know, there is no singular “arousal neurotransmitter.”10,26
Pathology Seen in Patients with Impaired Consciousness
There are four major pathologies that can cause severe, acute, and global reductions of consciousness.6,27 (1) One of these pathologies is the presence of diffuse, global, or extensive multifocal bilateral dysfunction of the cerebral cortex. In this injury mechanism, the cortical gray matter is diffusely impaired and so are cortical-subcortical excitatory feedback loops. The clinical examination will reveal disinhibited autonomic brainstem reflexes, which have also been described as “reticular shock.” (2) Another is injury to the paramedian gray matter from the level of the nucleus parabrachialis of the pons (tegmentum) and reaching to the midbrain pretectal areas and ventral posterior hypothalamus. An injury of this type damages the ascending arousal system and normal cortical activation. The affected structures are predominantly the paramedian gray matter, extending rostrally from the level of the nucleus parabrachialis of the pons (tegmentum) and reaching rostrally as far as the adjacent pretectal area and ventral posterior hypothalamus. (3) The widespread disconnection of the cortex from subcortical activating mechanisms acts pathophysiologically to produce effects similar to both of the previously mentioned conditions. (4) Finally, cortical and subcortical arousal mechanisms can be affected by a variety of diffuse disorders and to various degrees—for example, metabolic encephalopathy in a patient with acute liver failure.
Approach to Coma
Emergency Management
Oxygenation and Intubation
A brief neurologic examination is mandatory before any sedative or paralytic required for intubation is administered. The key points of a rapid neurologic examination are the following:28
• Assessment of level of arousal and level of consciousness (i.e., coma, stupor, lethargy)
• Pupillary size and response to light
• Abnormalities of eye movements (i.e., disconjugate eyes; unilateral gaze paralysis; lack of voluntary movements)
The preferred route of emergency intubation is orotracheal, which can be performed rapidly, safely, and reliably with inline stabilization of the neck in patients with suspected cervical spine injury. Intubation causes intense reflexive cardiovascular stimulation that may lead to a deleterious elevation of intracranial pressure (ICP). Therefore, the patient should be adequately sedated. Etomidate is the preferred sedative in patients with suspected ICP elevation as it reliably facilitates induction in less than 1 minute with a duration of action of 4 to 6 minutes. Propofol is another anesthetic agent that does not increase ICP; however, its hypotensive effects, which can ultimately decrease cerebral perfusion pressure, often limit its use. Both medications result in a dose-dependent decrease of cerebral metabolic rate that reduces cerebral blood flow and ICP. Ketamine has a fast onset, but it may elevate ICP and should generally be avoided. Midazolam can alter the patient’s mental status and may prohibit a postintubation examination.
Reversal of Drug Overdose
Drug overdose is the largest single cause (30%) of coma in the emergency department. Most drug overdoses are treated by supportive measures alone. Certain antagonists, however, specifically reverse the effects of coma-producing drugs. Intravenous naloxone (0.4 to 2 mg) is used as “test” antidote for opiate-induced coma, and it acts as a µ-receptor antagonist. Caution is needed as the reversal of narcotic effect may precipitate acute withdrawal in an opiate addict. In suspected opiate coma, the minimum naloxone dose (not completely reversal) should be given to establish the diagnosis by pupillary dilation and to reverse depressed breathing and consciousness. Due to its short half-life, patients who respond to naloxone reversal may require additional doses or a continuous infusion to avoid rebound sedation and respiratory depression. Intravenous flumazenil antagonizes benzodiazepine-induced coma and can be administered in 0.2-mg doses every minute for a maximum of 1 mg. In patients who initially respond to flumazenil reversal but experience recurrent sedation, flumazenil 1 mg may be redosed every 20 minutes with a max of 3 mg/hour.29 Careful consideration should be given prior to administration of flumazenil as patients may experience benzodiazepine-withdrawal seizures. Thus, whereas naloxone is commonly used with minimal serious adverse effects, the use of flumazenil should be restricted to select, low-risk cases. The sedative effects of drugs with anticholinergic properties, particularly tricyclic antidepressants, can be reversed with 1 to 2 mg physostigmine intravenously (duration of action about 45 to 60 minutes). Pretreatment with 0.5 mg atropine will reduce the risk for symptomatic bradycardia. Of note, only full awakening is characteristic of an anticholinergic drug overdose because physostigmine has nonspecific arousal properties.
Body Temperature
Hyperthermia is detrimental in brain injury as it increases brain metabolic demands and facilitates secondary brain injury.30 Elevated temperature greater than 104° F (40° C) requires immediate, lifesaving cooling measures, even before the underlying cause is determined and treated. In 2002, two research groups independently published that lowering the body temperature to 33° C for 12 or 24 hours in comatose survivors of cardiac arrest resulted in nearly doubling the number of patients being discharged home or to rehabilitation. Generally speaking, no patient with acute brain injury should be allowed to be hyperthermic, and modern technology provides a variety types of noninvasive and invasive cooling equipment suitable to maintain any target core temperature desired. Core temperatures of less than 93° F (34° C) on admission should be slowly elevated to above 35° C to reduce the risks of unwanted side effects from uncontrolled, persistent hypothermia.
Physical Examination
A systematic, detailed examination is necessary when approaching a comatose patient (Box 61.1, Table 61.2). Important findings include evidence of trauma, acute or chronic medical illnesses, ingestion of drugs (needle marks, alcohol breath), and the presence of nuchal rigidity.
Table 61.2
Correlation Between Levels of Brain Function and Clinical Signs
| Structure | Function | Clinical Sign |
| Cerebral cortex | Conscious behavior | Speech (including any sounds) Purposeful movement Spontaneous To command To pain |
| Brainstem activating and sensory pathways (reticular activating system) | Sleep-wake cycle | Eye opening Spontaneous To command To path |
| Brainstem motor pathways | Reflex limb movements | Flexor posturing (decorticate) Extensor posturing (decerebrate) |
| Midbrain CN III | Innervation of ciliary muscle and certain extraocular muscles | Pupillary reactivity |
| Pontomesencephalic MLF | Connects pontine gaze center with CN III nucleus | Internuclear ophthalmoplegia |
| Upper pons | ||
| CN V | Facial and corneal | Corneal reflex-sensory |
| CN VIII | Facial muscle innervation | Corneal reflex-motor response Blink Grimace |
| Lower pons | ||
| CN VIII (vestibular portion) connects by brainstem pathways with CN III, IV, VI | Reflex eye movements | Doll’s eyes Caloric responses |
| Pontomedullary junction pressure | Spontaneous breathing Maintained blood pressure |
Breathing and blood pressure do not require mechanical or chemical support |
| Spinal cord | Primitive protective responses | Deep tendon reflexes Babinski response |
As outlined earlier, an in-depth physical and neurologic examination followed by serial neurologic evaluations (i.e., hourly) is the most important, indispensable, and readily available method of assessing a comatose patient.31 However, in coma patients the examination remains limited in detecting changes in brain function. Nevertheless, the astute clinician will carefully evaluate for changes in findings (i.e., the appearance of new asymmetry in examination or focal deficits, progressive loss of brainstem reflexes, or loss of reflex motor responses). Finding progression of impairment is of immense clinical value as it indicates a new or worsening “intracranial emergency” demanding immediate clarification and stabilization. Skilled neurologic examination is challenging and provides a limited surveillance window of brain tissue at risk. Intracranial monitoring and, if needed, repeated head imaging have become the standard of care in neurocritical care units.
Assessment of Coma: General Aspects
Correctly characterizing disorders of consciousness continues to pose interesting clinical questions and diagnostic challenges with important ethical consequences (Box 61.2). Not only may an individual patient acutely fluctuate in his or her examination findings, but recovery may take place over prolonged time periods, necessitating constant reassessments. To address this uncertainty, standardized neurobehavioral assessments have become the best tool for categorizing coma and its transitional stages. Predicting long-term outcome has significantly improved when utilizing standardized assessments. Generally speaking, clinical and electrophysiologic markers of coma and its transitional states remain unsatisfactory. The reader is reminded that larger observational studies identified a high rate of misdiagnoses (>40%),32 especially in patients in vegetative state, when assessment is made on clinical grounds only.
