CHAPTER 123 Colorectal Cancer
Cancer of the colon and rectum (colorectal cancer [CRC]) is a major cause of cancer-associated morbidity and mortality in North America, Europe, and other regions with similar lifestyles and dietary habits. CRC is the fourth most common newly diagnosed internal cancer overall in the United States, after cancers of the lung, prostate, and breast, and currently constitutes 10% of new cancers in men and women. In 2009, there were an estimated 147,000 new CRC cases in the United States and 50,000 related deaths (a rate second only to that of lung cancer).1 In the United States, CRC incidence rates in men and women are similar, although there appears to be a slight male predominance worldwide. Approximately 6% of the American population eventually will develop invasive colon or rectal cancer, and more than 6 million Americans who are alive today will die of the disease; the lifetime risk of dying from CRC in the United States is 2.5%. Globally, CRC is the fourth most common cancer in men and the third most common in women, with mortality paralleling incidence. Despite evidence that five-year survival is 90% when CRC is diagnosed at an early stage, less than 40% of cases are diagnosed when the cancer is still localized.2
EPIDEMIOLOGY
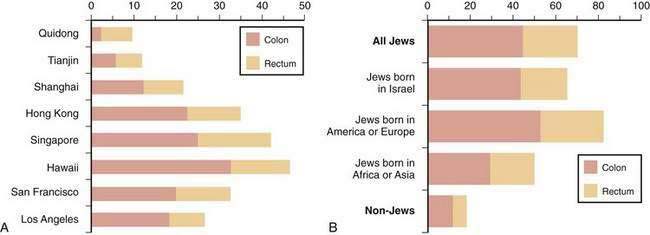
The frequency of CRC varies remarkably among different populations (Fig. 123-1).3 Incidence rates are highest in the developed countries in North America and in Australia and New Zealand; intermediate in Europe; and low in Asia, South America, and especially sub-Saharan Africa. Internationally, the incidence of colon cancer in men differs by a factor of almost 90 between areas with the extreme lowest and highest rates; the incidence of rectal cancer (cancer within 11 cm of the anus) differs by a factor of 13. CRC incidence also differs within countries, depending on region and population (Fig. 123-2). These differences most likely are due to differences in environmental factors, including dietary patterns (discussed later).
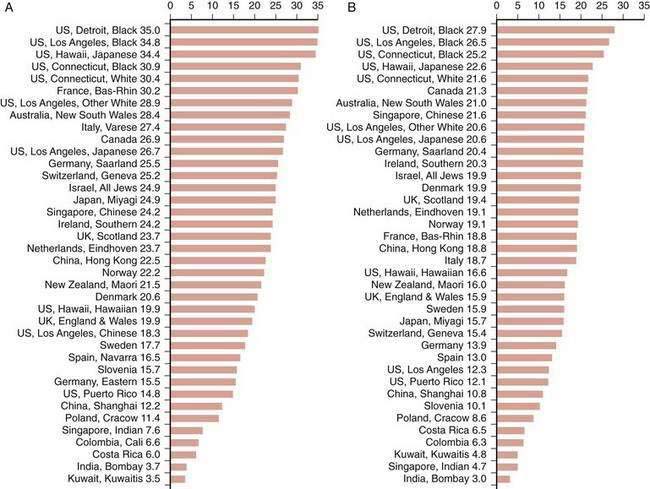
Figure 123-1. Age-standardized incidence of colon cancer per 100,000 population in various populations for men (A) and women (B).
(Data from Parkin DM, Whelen SL, Ferlay J, et al. Cancer incidence in five continents. [IARC Sci. Publ. No. 143]. Lyon: International Agency for Research on Cancer; 1997; and Curado MP, Edwards SB, Shin HR, et al. Cancer incidence in five continents, vol. IX [IARC Sci. Publ. No. 160]. Lyon: International Agency for Research on Cancer; 2007.)
Between 1950 and the mid-1980s, the incidence of colon cancer in the United States rose in the white population, whereas that of rectal cancer remained fairly stable. Mortality rates from CRC were stable among white men but decreased in white women. Both incidence and mortality rates for CRC increased substantially among the nonwhite population during this period. CRC incidence and mortality have declined since 1985 in American adults at an average annual rate of 1.6% and 1.8%, respectively2; these trends are more evident in whites than blacks. Overall death rates from CRC declined between 1990 and 2004 by almost 30% in men and 25% in women.1 Currently, incidence and mortality rates for CRC are higher in the African American population compared with the white population, and these rates in the U.S. Latin American population are slightly lower.1
The risk of CRC rises rapidly in populations that migrate from areas of low risk to areas of high risk. This pattern was demonstrated clearly in Japanese immigrants to Hawaii and to the continental United States during the 1950s and 1960s. Cancer rates for Issei (the migrating generation) rose over a short period to exceed those of native Japanese living in Japan, and the incidence rates for Nissei (their U.S.-born offspring) rose progressively to approximate those of the native white population. A similar upward displacement of CRC risk was noted in Europeans who migrated to Australia after World War II and in Jews who migrated to Israel from low-risk areas in Yemen and North Africa. Longitudinal studies reveal that in many countries where CRC mortality rates were low before 1950, rates have increased sharply, whereas in countries where rates were high or moderate, they have decreased, stabilized, or increased slightly. Japan is a good example of this change: Once a low-risk region for colon cancer, incidence rates have risen to equal or exceed those in North America and Europe.3
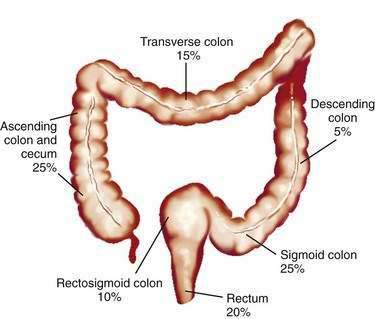
Studies of temporal trends by subsite location of large bowel cancer demonstrate that for both sexes, incidence rates have increased for cancers of the right colon (cecum, ascending colon) and sigmoid colon and have decreased for cancers of the rectum; this change might reflect differing susceptibilities to neoplastic transformation in the proximal and distal colon. Currently in the United States, the prevalence of CRCs in whites is higher in the cecum and ascending colon (22% in men, 27% in women) and in the sigmoid colon (25% in men, 23% in women) than elsewhere in the large bowel (Fig. 123-3).
ETIOLOGY
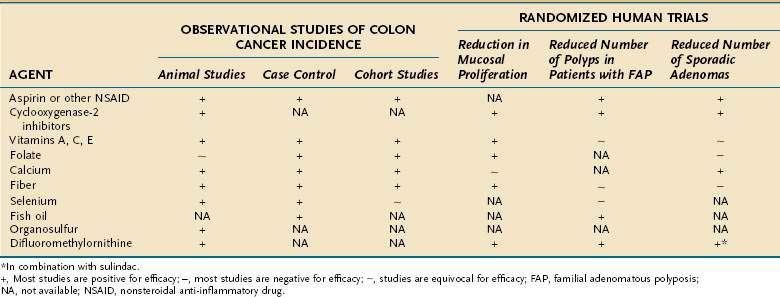
Inter-regional differences in the incidence of CRC, including differences among population groups living in geographic proximity but with different lifestyles, strongly suggest that environment plays a role in the development of this disease. Migrant studies and rapid changes in incidence in countries assimilating Western practices support this concept. Strong circumstantial evidence exists for a link between diet and CRC. Population studies and animal studies have attempted to delineate the effects of various fats and proteins, carbohydrates, vegetable and fiber components, and micronutrients on the genesis of cancer of the large bowel (Table 123-1).
Table 123-1 Factors That May Influence Carcinogenesis in the Colon and Rectum
| Probably Causative |
NSAIDs, nonsteroidal anti-inflammatory drugs.
* Dietary fats and fiber are heterogeneous in composition, and not all fats or fiber components play a role in cause or protection.
FAT, BILE ACIDS, AND BACTERIA
Activation of protein kinase C (PKC) by bile acids in colonic mucosa also might represent an important intracellular event by which bile acids provoke a proliferative response. Mucin alterations are a common feature of colonic neoplasia, and alterations in MUC2 mucin have been associated with tumor progression in the colon. Bile acids induce mucin expression in human colon carcinoma cells by increasing MUC2 transcription through a process involving mitogen-activated protein (MAP) kinase-independent, PKC-dependent activation of the transcription factor AP-1.4 Bile acids can, in addition, induce release of arachidonate and conversion of arachidonic acid to prostaglandins in the mucosa, which can enhance cell proliferation.5 Preclinical and clinical evidence indicate that nonsteroidal anti-inflammatory drugs (NSAIDs), which reduce prostaglandin synthesis, reduce the incidence of large bowel cancer (discussed later); inhibition of the inducible enzyme cyclooxygenase (COX)-2 may be particularly important in this regard.6
An inverse relationship has been reported between physical activity and risk for CRC in men; obesity is associated with elevated risk of CRC. Serum cholesterol and β-lipoprotein levels have been positively correlated with the development of colorectal adenomas and CRC, but this association has not been demonstrated consistently, and serum cholesterol levels can decline before the development of colon cancer. Adiponectin is a hormone secreted by adipose tissue, serum levels of which are inversely correlated with obesity and hyperinsulinemia. Variants of adiponectin and adiponectin receptor genes have been correlated with differences in CRC risk.7
FIBER
Epidemiologic, case-control, and animal studies suggest that dietary fiber protects against the development of colon cancer. Dietary fiber is plant material that resists digestion and that is composed of a heterogeneous mix of carbohydrates (e.g., cellulose, hemicellulose, pectin) and noncarbohydrates (e.g., lignin). Although the protective role of fiber is not completely clear, epidemiologic studies correlate high-fiber intake with a lower incidence of colorectal neoplasia.8,9 The majority of observational-epidemiologic and case-control studies support the protective effect of fiber-rich diets; however, these data do not define the relationship between fiber-rich food and the importance of nonfiber vegetable components, nutrients, and micronutrients in fruits and vegetables. The effect of fiber components on different portions of the large bowel also can vary. This might explain, in part, the inability to demonstrate a protective effect of fiber in several randomized, controlled trials that have examined the ability of fiber supplementation to prevent adenoma recurrence.10,11
Two large prospective observational studies, including one from the Prostate, Lung, Colon and Ovarian Cancer Trial (PLCO) found that increased fiber intake is significantly associated with reduced risk of colorectal neoplasia.8 Another analysis of 13 prospective cohort studies included in the Pooling Project of Prospective Studies of Diet and Cancer demonstrated that dietary fiber intake was inversely associated with risk of CRC in age-adjusted analyses, but that after accounting for other dietary factors, high intake of dietary fiber was not associated with a reduced risk of CRC.12
CALCIUM AND VITAMIN D
Epidemiologic, clinical, and laboratory evidence suggest that calcium intake might protect against carcinogenesis in the colon. Calcium has numerous biological effects that might reduce colon carcinogenesis, including actions on the cell cycle, cyclic adenosine monophosphate (cAMP), calmodulin, tyrosine kinases, ornithine decarboxylase, and e-cadherin. The calcium-sensing receptor expressed in the intestine senses extracellular calcium with effects on differentiation and proliferation. The potential chemopreventive activity of calcium was suggested originally by epidemiologic studies reporting an inverse relationship between CRC and intake of vitamin D and calcium. Dietary calcium supplementation in the form of low-fat dairy foods can affect a variety of intermediate biomarkers thought to be associated with tumor progression in the colon, and supplemental calcium plus vitamin D alters preneoplastic features of colorectal adenomas.13 Although the relationship between calcium intake and a lower incidence of colonic adenomas and carcinomas has not been uniformly demonstrated, observational studies overall suggest a protective effect. A pooling project of 10 cohort studies strongly suggested that calcium intake is inversely related to the risk of CRC.14
Vitamin D3 metabolites and analogs have been shown to play an important role in the regulation of a number of important cellular processes, including proliferation, differentiation, and apoptosis, in addition to their established role in mineral homeostasis. These steroid compounds have rapid effects that do not involve gene transcription or protein synthesis, as well as genomic effects involving the vitamin D receptor and other transcription factors.15,16 Vitamin D can modulate more than 200 genes involved in cell cycle regulation, growth factor signaling, protection against oxidative stress, bile acid and xenobiotic metabolism, cell adhesion, DNA repair, angiogenesis, inflammation, and immune function. Effects of vitamin D and its metabolites have been demonstrated in normal and malignant colonocytes, and several potential mechanisms have been suggested by which these compounds might prevent carcinogenesis in the colon. Dietary supplementation with calcium and vitamin D in rodents fed colon tumor-inducing Western diets significantly reduced tumor incidence and multiplicity in addition to altering expression of a variety of genes linked to initiating formation of colon tumors.17
ARACHIDONIC ACID, EICOSANOIDS, AND CYCLOOXYGENASE-2
This enzyme exists in two isoforms: COX-1 and COX-2. COX-1, the constitutive form of the enzyme, is present in most tissues and is involved in the physiologic production of prostaglandins to maintain normal homeostasis. COX-2 is induced by cytokines, mitogens, and growth factors, and its level has been shown to be elevated in both murine and human CRCs.18–22 Expression of COX-2 is increased markedly in 85% to 95% of CRCs and in experimental models of CRC. COX-2 inhibition prevents cancer from developing during the initiation and the promotion and progression stages of carcinogenesis.21 Knockout of COX-2 results in suppression of intestinal polyposis in animal models of familial adenomatous polyposis (FAP).19 15-Hydroxyprostaglandin dehydrogenase (15-PGDH) is a prostaglandin-degrading enzyme that is lost in human colon cancers and has been shown to be a physiologic antagonist of the prostaglandin-synthesizing activity of COX-2.22 It has been speculated that NSAIDs might reduce formation of colon tumors by inhibiting prostaglandin-mediated proliferation, but other evidence suggests that part of their effect might result from inducing apoptosis. Overexpression of COX-2 has been demonstrated to decrease apoptosis, whereas inhibition of COX-2 leads to an increase in apoptosis.
Other potential mechanisms by which COX-2 inhibition might affect tumor formation include alterations of cell adhesion to extracellular matrix proteins, inhibition of tumor neovascularization (angiogenesis), and reduction in carcinogen activation. A study using the ApcΔ716 mouse, an animal model of FAP, demonstrated that treatment with the COX-2 specific inhibitor rofecoxib (Vioxx) was associated with a significant dose-dependent reduction in size and number of polyps as well as alterations in polyp morphology. COX-2 inhibition was associated with decreased levels of vascular endothelial growth factor (VEGF) and with lower rates of DNA replication.20
CHEMOPREVENTION
Chemoprevention refers to the use of natural or synthetic agents to reverse, suppress, or prevent progression or recurrence of cancer23,24; this is a cornerstone of primary prevention. Data on chemoprevention of CRC come from studies in laboratory animals (see earlier), observational epidemiologic studies, case-control studies, and randomized controlled trials (RCTs). Because the natural history of CRC is protracted, clinical RCTs often have concentrated on preventing colorectal adenomas, which represent a form of intraepithelial neoplasia and are the precursors to carcinoma. The duration of the studies required, sample sizes necessary, cost, and ethical considerations make the use of cancer as an end point impractical. This difficulty has led to an increasing use of surrogate biomarkers to study chemoprevention of CRCs,25 with the hope that use of such markers will lead to shorter, smaller, and less expensive trials.26 To be valid, however, such biomarkers need to accurately represent the events involved in the process of carcinogenesis. When an intervention such as a chemopreventive agent is tested, there should be a clear relationship among the agent, modulation of the biomarker, and the development of cancer. Surrogate end points for cancer ideally should be validated in the context of clinical studies that use cancer as the ultimate end point. This is a difficult task because these are the very trials that such markers are designed to complement or replace.
There has been interest in the use of magnifying endoscopy to study aberrant crypt foci of the colon as possible markers in chemoprevention trials.24 These foci consist of large, thick crypts that can be detected by chromoendoscopy using agents such as methylene blue or indigo carmine (Fig. 123-4). Aberrant crypt foci, particularly large crypts with dysplastic features, are thought to be precursors of adenomas in the colon. Standardization of techniques to identify and quantify these lesions is crucial to the successful interpretation of data from these trials, because it is unclear whether these lesions, which appear to be precursors to neoplasia in animal models, play a similar role in the human colon.27 Studies suggest that the majority of aberrant crypt foci in the human colon may be hyperplastic rather than dysplastic (see Chapter 122).
The potential benefit of low-fat, high-fiber diets based on descriptive epidemiology and case-control studies already has been discussed, but current data from prospective human trials are thus far equivocal or negative. Two large RCTs examined the effects of fiber supplementation on recurrence of adenomas. The Polyp Prevention Trial11 randomized 2079 subjects with a history of colorectal adenomas to receive counseling together with a low-fat, high-fiber diet rich in fruits and vegetables or to receive their usual diet alone. The incidence of recurrent adenomas at one and four years, as determined by colonoscopy, was similar in both groups. In a study conducted by the Phoenix Colon Cancer Prevention Physicians’ Network,10 1429 patients with a history of colorectal adenoma were randomized to receive 2.0 g or 13.5 g of supplemental wheat bran per day. Colonoscopy failed to show a difference in the incidence of recurrent adenomas at a median follow-up of 34 to 36 months.
A large body of observational and laboratory studies suggests a role for dietary calcium supplementation in chemoprevention. A prospective double-blind placebo-controlled trial showed that supplemental calcium (3000 mg of calcium carbonate per day, equivalent to 1200 mg of elemental calcium) reduced the incidence and number of recurrent adenomas in subjects chosen for a recent history of such lesions.26 The effect of calcium was modest: 19% reduction in recurrence of adenomas and 24% reduction in the number of adenomas over three years, independent of age, sex, or dietary intake of calcium, fat, or fiber. The protective effect of calcium supplementation on the risk of colorectal adenoma recurrence extended up to five years after cessation of active treatment, even in the absence of continued supplementation.28 Analysis of subjects’ serum vitamin D status suggested that calcium supplementation and vitamin D status appear to act together to reduce the risk of adenoma recurrence.29 The results of a Japanese study, the Fukuoka Colorectal Cancer Study,30 support the joint action of calcium and vitamin D in preventing CRC. Human trials using antioxidant vitamins A, C, and E have provided equivocal results, and current data do not support their routine use for colon cancer prevention in average-risk persons.
Folic acid and its metabolites play an important role in DNA synthesis, strand integrity, and methylation. Epidemiologic studies have found a lower incidence of CRC among those with high compared with low dietary intake of folate.31 This protective effect also was suggested by the Nurses’ Health Study, in which high doses of folate (as part of multivitamin supplementation) given over several years were protective against CRC. A large prospective RCT32,33 failed, however, to demonstrate a protective effect of 1 mg/day of folate supplementation on recurrence of adenoma compared with placebo and suggested that folate supplementation in persons with prior adenomas actually might increase adenoma risk. Analysis of baseline dietary and serum folate levels in these subjects supports the idea that although moderate doses of folate may be protective compared with deficiency, at some point of sufficiency, supplementation provides no additional benefit.34 The lack of efficacy of folate (0.5 mg/day) supplementation to prevent adenoma recurrence also was found in another RCT.35
Epidemiologic, case-control, and prospective cohort trials suggest a protective effect against the development of CRC in women taking hormone (estrogen) replacement therapy. It has been postulated that estrogen might protect against colon cancer by decreasing production of secondary bile acids, by decreasing levels of insulin-like growth factor-1, or through as yet undetermined direct effects on colonic mucosal epithelial cells. Data from the Women’s Health Initiative demonstrated that short-term use of estrogen plus progestin was associated with a decreased risk of CRC; however, CRCs in women who took estrogen plus progestin were diagnosed at a more advanced stage than those in women who took placebo.36
Cigarette smoking has been associated with incidence of and mortality from CRC in observational studies,37 but the long-term effects of smoking cessation on CRC have not been studied.
The most promising results for CRC prevention come from trials using aspirin and NSAIDs. Case-control and cohort studies have suggested that the risk for development of adenoma and carcinoma may be reduced substantially (40% to 50%) among aspirin and NSAID users, compared with controls.6,24,38 A prospective cohort study among male health professionals demonstrated that persons who take aspirin more than twice per week were at lower risk for CRC (relative risk [RR], 0.68) than were controls. An RCT that assessed the effect of low-dose aspirin in an average-risk population demonstrated no significant reduction in the number of CRC cases during the first six years of follow-up; longer follow-up may be necessary to demonstrate a significant effect of aspirin on development of cancer, because the Nurses’ Health Study demonstrated that the benefits of aspirin might not be evident until after at least a decade of regular aspirin consumption.6 Three prospective adenoma prevention trials (see later) now provide compelling evidence that aspirin use reduces the risk of developing colorectal adenoma in persons with a personal history of adenoma or carcinoma.39–41
Given the long natural history of CRC, preventing recurrence of adenoma after endoscopic removal often is used as an intermediate or surrogate end point in chemoprevention trials.24 In FAP, in which hundreds of adenomas occur in the colon and rectum, chemoprevention trials often use reductions of the number and size of adenomas as end points in short-term studies. Such trials have suggested a potential role for NSAIDs as chemopreventive agents in this setting. There is a significant decrease in the mean number and size of polyps in patients treated with the NSAID sulindac. In a small, double-blind RCT of 22 patients with FAP, treatment with sulindac reduced the number and mean diameter of colorectal polyps by 44% and 35% of respective baseline values after nine months; three months after treatment was stopped, however, the number and size of polyps had increased. A subsequent prospective cohort study42 confirmed that long-term use of sulindac is effective in reducing the number of adenomas in patients with FAP. A 76% reduction in the number of polyps was seen at one year and was sustained (74% reduction) through 63 months of follow-up. Sulindac might not be effective, however, for primary prevention of development of adenomas in genetically disposed persons with FAP who have not yet developed macroscopic adenomas.43
A double-blind RCT studied the effects of celecoxib (Celebrex), a selective COX-2 inhibitor, on colorectal polyps in patients with FAP.44 Treatment with high doses of celecoxib for six months was associated with a significant reduction (28%) in the number of colorectal polyps compared with placebo (4.5%). This drug is now approved in the United States as an adjunct to standard therapy in patients with FAP.
Data are now available on the role of nonselective NSAIDs and COX-2 inhibitors as chemopreventive agents for preventing sporadic adenomas. Four published trials have demonstrated a reduction in recurrence of adenoma in chemoprevention trials involving aspirin. An RCT of aspirin versus placebo40 demonstrated that daily use of 81 mg of aspirin was associated with a 19% reduction in occurrence of adenoma and a 41% reduction in occurrence of advanced neoplasms (adenomas measuring at least 1 cm or with tubulovillous or villous features, severe dysplasia, or invasive cancer) compared with results of placebo treatment three years after removal of an index sporadic adenoma. In a similar trial, aspirin (300 mg/day) was associated with a 21% overall risk reduction for all adenomas and a 37% reduction in advanced adenomas at three years.35 A prospective RCT in a higher-risk group for adenoma recurrence, those with a previous history of sporadic CRC, demonstrated a 45% risk reduction in those taking 325 mg of aspirin daily compared with those taking placebo with a mean follow-up of almost three years.39
Given biological plausibility, preclinical in vitro and animal data, and data on regression of adenoma in patients with FAP, three randomized trials were undertaken to examine the effect of COX-2 selective inhibitors on formation of new adenomas in patents with a history of sporadic adenomas. The Adenoma Prevention with Celecoxib (APC) trial45 randomized 2035 subjects to celecoxib at a dose of 200 mg or 400 mg twice daily. Use of celecoxib was associated with a dose-dependent 33% to 45% reduction in the development of new adenomas by three years, with a 57% to 66% reduction in the number of patients developing advanced adenomas. The Prevention of Sporadic Adenomatous Polyps (PreSAP) trial46 randomized 1561 patients at a ratio of two to three to receive 400 mg celecoxib once daily or placebo. Use of celecoxib was associated with a 36% overall reduction in formation of new adenomas and a 51% reduction in advanced adenomas over three years. The Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial47,48 was a double-blind RCT of the efficacy of oral rofecoxib, 25 mg daily, to prevent colorectal adenomas in 2587 subjects. The risk of adenoma recurrence over three years was lower for subjects on rofecoxib than on placebo, with a 24% overall reduction and a 30% reduction in advanced adenomas. The effect of drug was more pronounced in the first year (RR 0.65; 95% confidence interval [CI]: 0.57-0.73) than in the subsequent two years.
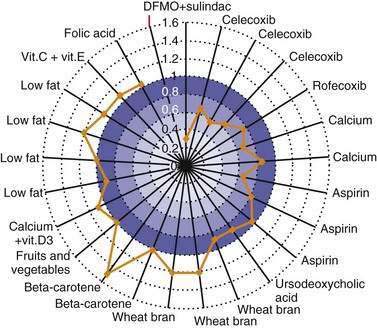
Other agents currently undergoing study for chemoprevention of colorectal neoplasia include the ornithine decarboxylase inhibitor difluoromethylornithine (DFMO); the bile acid ursodiol; 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, such as pravastatin and lovastatin; epidermal growth factor receptor (EGFR) inhibitors; and matrix metalloproteinase (MMP) inhibitors.24 The combination of DFMO and the NSAID sulindac were studied in an RCT to assess their efficacy in preventing sporadic adenoma recurrence in 375 subjects. Use of this regimen was associated with a 70% reduction in new adenomas at three years compared with placebo.49 Larger studies are needed to confirm this result and to fully assess the toxicity of this combination. A population-based case-control study of persons with CRC and matched controls demonstrated a 47% relative reduction of CRC associated with statin use, but further investigation is needed to assess this benefit.50 Table 123-2 and Figure 123-5 summarize the status of current studies that examine the effect of chemopreventive agents on colorectal neoplasia.
BIOLOGY
Current concepts concerning environmental causes for CRC have been discussed in the preceding sections. It has been suggested that carcinogens introduced into the bowel act in concert with other luminal factors (e.g., bile acid and other tumor promoters) to affect epithelial cells in the colonic mucosa; however, carcinogenesis is a multistage process. Cells must be genetically primed (through either hereditary disposition or genotoxic events) and induced to proliferate, after which they must pass through a series of stages en route to immortalization and uncontrolled growth. Our knowledge of this sequence of events in the colon, although fragmentary, is growing rapidly (see Chapter 3).
ABNORMAL CELLULAR PROLIFERATION
Abnormal cellular proliferation is a hallmark of neoplasia (see Chapter 3). Actively proliferating cells are more susceptible to initiators of carcinogenesis (primary carcinogens) and genetic alterations than are resting cells. In the normal colon, DNA synthesis occurs and cells divide and proliferate only in the lower and middle regions of the crypts. As cells normally migrate upward from deeper in the crypt, the number of cells that continue to proliferate decreases, and, on reaching the upper crypt region, cells become terminally differentiated and can no longer divide. A substantial body of literature indicates that this sequence of events is disordered during the evolution of neoplastic lesions in the colon. Increased proliferative activity and characteristic differences in the distribution of tritiated thymidine-labeled cells (i.e., those that actively synthesize DNA) within the colonic crypts have been demonstrated and distinguish at-risk and affected members of kindreds with familial polyposis, as well as nonpolyposis-inherited colon cancer from lower-risk groups. Correlations between rectal mucosal proliferation and the clinical and pathologic features of nonfamilial colorectal neoplasia also have been demonstrated. Conversely, populations at low risk for developing colon cancer, such as Seventh Day Adventist vegetarians, have relatively quiescent proliferative activity in their colonic mucosa.
An explosion of knowledge in the field of molecular genetics has demonstrated how alterations in proto-oncogenes and tumor suppressor genes might lead to disruption of mechanisms that regulate the normal cell cycle and cell proliferation. In some cases, the cell is predisposed to abnormal proliferation by germline mutations, such as FAP, whereas in other cases, somatic mutations occur as the result of complex interactions with environmental factors,51 as detailed earlier.
MOLECULAR BIOLOGY AND BIOCHEMICAL CHANGES
Molecular Genetics
Tumor cells in the colon, as elsewhere, are characterized by heritable phenotypic changes that are the result of quantitative or qualitative alterations in gene expression (see Chapters 3 and 122). A large body of evidence demonstrates that CRCs are associated with an accumulation of such genetic alterations (Table 123-3; Fig. 123-6). Genetic changes that might lead to the development of CRC traditionally have been categorized into three major classes: alterations in proto-oncogenes, loss of tumor suppressor gene activity, and abnormalities in genes involved in DNA mismatch repair (MMR).52–54 Adenomas and carcinomas arise in the context of genomic instability, by which epithelial cells acquire the number of mutations needed to attain a neoplastic state. Destabilization of the genome is a prerequisite to tumor formation and most commonly involves chromosomal instability (CIN), which is found in 80% to 85% of colorectal tumors with subsequent allelic loss; chromosomal amplifications and translocations; or increased rates of intragenic mutation in tandemly repeated DNA sequences known as microsatellites (microsatellite instability [MSI]).54
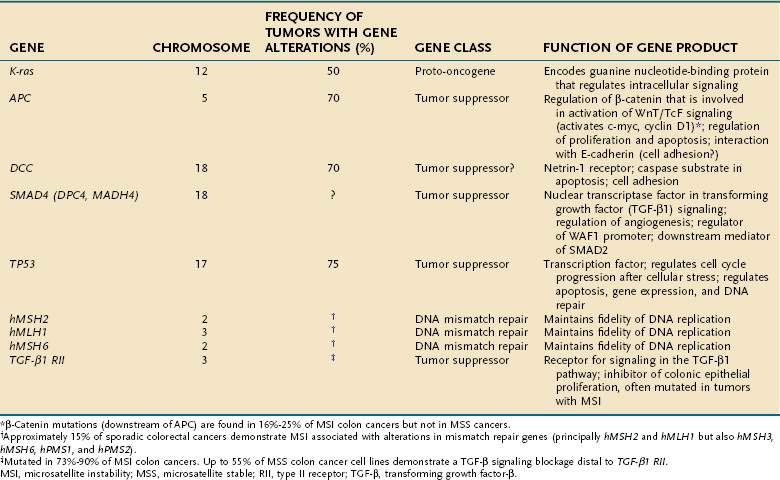
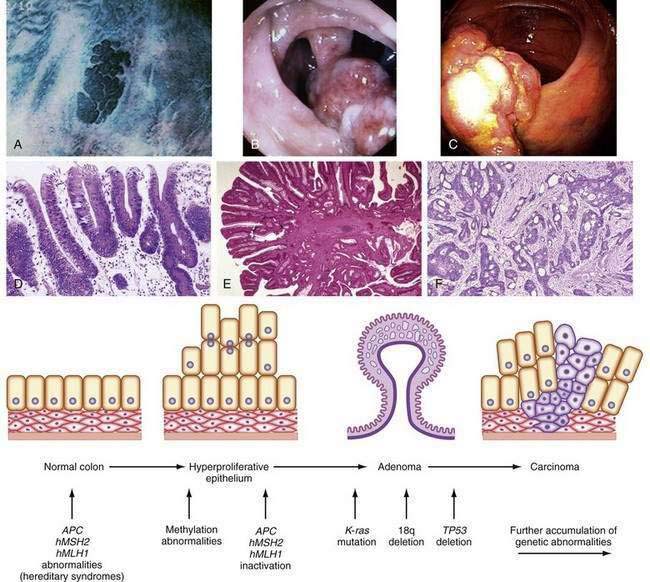
Cellular proto-oncogenes are evolutionarily conserved human genes that contain DNA sequences homologous to those of acute transforming retroviruses. Many of these genes play a role in signal transduction and the normal regulation of cell growth. Inappropriate activation of these genes leads to abnormal transmission of regulatory messages from the cell surface to the nucleus that results in abnormal proliferation and, eventually, tumor formation. Three human ras genes—K-ras, N-ras, and H-ras—encode guanine nucleotide-binding proteins that regulate intracellular signaling pathways. Approximately 65% of sporadic CRCs have activating point mutations in a ras gene, most in K–ras. Most ras mutations appear to occur during intermediate stages of adenoma growth (see Chapter 122). Ras gene mutations occur in 47% of carcinomas, in 58% of adenomas larger than 1 cm, but in only 10% of adenomas smaller than 1 cm, suggesting that earlier events must contribute to formation of neoplasms. Alterations in signal transduction might lead to abnormal cell growth and thus participate in neoplastic transformation, but activation of ras alone is not sufficient for progression to carcinoma. The exact functional relationship between ras mutations and carcinogenesis remains to be established, but understanding the role of ras mutations in stimulating proliferation might lead to development of anti-tumor therapies aimed at interrupting signals that alter tumor cell growth.
Chromosomal abnormalities have been reported in CRCs for more than a decade, and abundant evidence has shown that allelic losses, particularly at chromosome locations 5q, 17p, and 18q, play major roles in the genesis of large bowel tumors.52–55 A deletion within chromosome 5 in patients with FAP led to the identification of the APC gene on the long arm of this chromosome (5q21). Positional cloning identified a single tumor suppressor gene, which is mutated in both the germline of FAP patients and in sporadic colorectal tumors. The protein encoded by APC consists of 2843 amino acids and is located at the basolateral membrane of colorectal epithelial cells; expression is increasingly pronounced as cells migrate up through the colonic crypt.
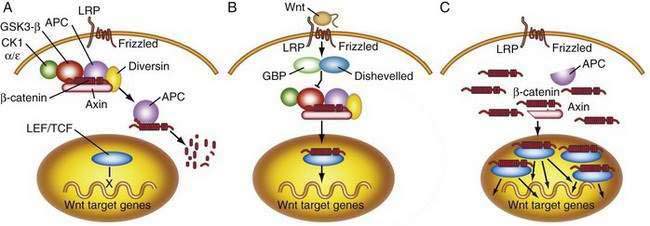
APC functions to modulate extracellular signals that are transmitted to the nucleus through the cytoskeletal protein β-catenin, as part of the Wnt signaling pathway (Fig. 123-7). Nuclear β-catenin binds to transcription factors in the nucleus that are members of the lymphoid enhancer factor/T-cell factor (LEF/TCF) family including Tcf-4, which in turn activate various target genes (e.g., c-myc, cyclin D1)56 that affect cell cycling and growth. APC is a tumor suppressor gene that binds to β-catenin and causes its degradation through phosphorylation. Loss of APC function, therefore, leads to accumulation of β-catenin and unopposed stimulation through the Wnt-Tcf signaling pathway, which in turn leads to increased and unregulated proliferation and decreased programmed cell death (apoptosis). APC gene abnormalities also might lead to disruption of normal cell-cell adhesion through altered association with the cellular adhesion molecule E-cadherin. Disruption of APC-mediated regulation of transcriptional activation is critical for colorectal tumorigenesis and is achieved most commonly through inactivating mutations of both APC alleles; disruption also can occur through dominant mutations of the β-catenin gene that render β-catenin-Tcf-regulated transcription insensitive to the regulatory effects of normal wild-type APC.
Other genetic changes occur later in the adenoma-to-carcinoma sequence. Stepwise tumor progression is associated in more than 75% of cases with loss of the tumor suppressor gene activity located on chromosome 18q. Several candidate genes are present on this chromosome, and loss of chromosome 18 is associated with a poor prognosis.57 One gene, DCC (deleted in CRC), originally was thought to be important because its loss from a stage II (Dukes B) cancer was associated with a worse prognosis in some studies; its role as an important tumor suppressor gene has been questioned.
Loss of TP53 also may be associated with reduced apoptosis of damaged cells. Inactivation of the TP53 gene mediates the conversion from adenoma to carcinoma, a late and important event in colon carcinogenesis. Distant metastases from CRCs are associated with high fractional allelic loss and deletions of 17p and 18q. A distinct set of metastasis-suppressor genes also has been postulated.58
Genomic instability creates a permissive state in which a cell acquires sufficient mutations to be transformed to a cancer cell; this is a common mechanism central to the development of most, if not all, colon cancers.53 Several forms of genomic instability are common in colon cancer, including chromosome instability (CIN) and chromosome translocations, and MSI in which subtle sequence changes, including base substitutions, deletions, or insertions, lead to a hypermutable state (Fig. 123-8). Candidates responsible for CIN include genes responsible for the human mitotic spindle checkpoint (hBUB1 and hBUBR1), genes involved in the DNA damage checkpoint (ATM, BRCA1 and BRCA2, TP53, and hRad17), and genes that control centrosome number. Inactivation of genes involved in base excision repair (BER) resulting from oxidative damage are found in a subset of CRCs. Inactivation of one of the BER genes (MYH) is a cause of an autosomal recessive form of FAP. Although tumors arising as the result of MYH germline mutations demonstrate chromosomal instability, they appear to have a unique pathogenesis compared with sporadic CRC.
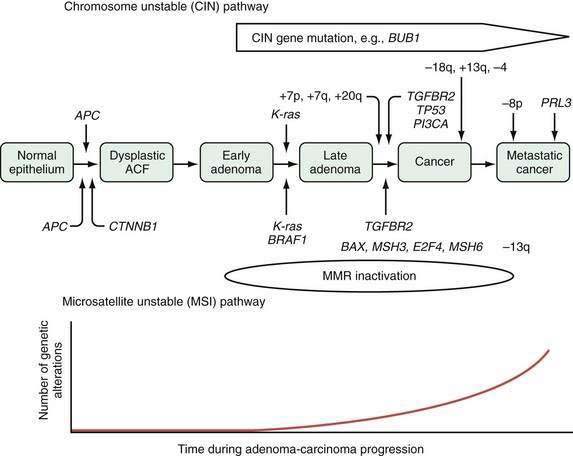
The significance of genomic instability in the pathogenesis of a subset of colon cancers became evident with the discovery of MSI in colon cancers associated with hereditary nonpolyposis colorectal cancer (HNPCC). Alterations in genes that help maintain DNA fidelity during replication are characteristic of patients with HNPCC.53,59 Alterations in MMR genes designated hMLH1, hPMS1, hPMS2, hMSH2, hMSH3, and hMSH6 might lead to the inability to repair base pair mismatches and result in DNA replication errors or MSI. Inactivation of the MMR system causes genomic instability by increasing the rate of polymerase-generated replication errors and degrading the fidelity of DNA replication, particularly at microsatellite repeat sequences.53 MSI involves mutations or instability in short, tandemly repeated DNA sequences such as (A)n, (CA)n, and (GATA)n. Such DNA sequences are found in several key genes that are important for maintaining normal cellular function (Table 123-4). The receptor for TGF-β (TGF-βRII), for example, often is mutated as the result of MSI.
Table 123-4 Frequency of Some Target Gene Mutations in Colon Cancers with Microsatellite Instability (MSI)
| TARGET GENE | FREQUENCY OF MUTATION IN COLON CANCERS WITH MSI (%) | NORMAL FUNCTION OF GENE PRODUCT |
|---|---|---|
| TGF-β R2 | 90 | TGF-β signaling |
| ACVR2 | 86 | Activin signaling |
| IGFIIR | 10 | Insulin-like growth factor and TGF-β signaling |
| BAX | 50 | Apoptosis |
| hMSH 3 | 50 | DNA mismatch repair |
| hMSH 6 | 33 | DNA mismatch repair |
| E2F-4 | 65 | Cell cycle control |
| PTEN | 19-34 | Growth regulation |
| MBD4 (MED1) | 40 | DNA repair and binding to methylated DNA |
| TCF4 | 39 | Growth regulation |
| CHK1 | 10 | G2 cell cycle checkpoint |
| STK11 (LKB1) | <2 | Signal transduction |
| BLM | <18 | Chromosome stability, DNA repair; helicase |
| Caspase 5 (ICErel-III) | 62 | Apoptosis |
| CDX2 | <2 | Homeobox protein |
| TPB | 83 | TATA binding protein |
| RIZ | 26 | Interacts with RB |
| hRAD50 | 31 | DNA repair |
| SEC63 | 49 | ER chaperone protein |
| AIM2 | 48 | Interferon-inducible protein |
Note: Most mutations cause frame shifts that prematurely truncate the protein, leading to inactivation of the affected allele.
ER, endoplasmic reticulum; RB, retinoblastoma protein; TGF, transforming growth factor.
Modified from Grady WM, Carethers J. Genomic and epigenetic instability in colorectal cancer progression. Gastroenterology 2008; 135:1079-99.
Multiple lines of evidence suggest that the TGF-β pathway is an important tumor-suppressing pathway in the colon and that alterations in this pathway lead to tumor development. Less frequently targeted genes include the insulin growth factor 2 receptor; Bax and caspase 5, proteins that regulate apoptosis; E2F4, a transcription factor; and MSH3 and MSH6, DNA MMR proteins. β-Catenin mutations are present in up to 25% of MSI colon cancers. MSI, therefore, leads to accumulation of mutations in vulnerable genes, eventually resulting in the acquisition of the malignant phenotype. Although a high frequency of MSI (instability at more than 40% of microsatellite loci) is characteristic of HNPCC, similar alterations can be found in about 15% of sporadic CRCs and also in premalignant lesions. MSI tumors remain diploid. Patients whose tumors demonstrate MSI might have a better prognosis60 and respond differently to chemotherapy,54,61,62 than those whose tumors are characterized by chromosomal instability. Most patients with MSI colon cancers do not possess mutations in the known MMR genes, and evidence indicates that MSI in these tumors might arise through epigenetic mechanisms. Epigenetics is the study of clonal changes in gene expression without accompanying changes in DNA coding sequences.
Epigenetic silencing is recognized now as an important mechanism in the evolution of a subgroup of CRCs. DNA methylation within promoters and alterations in histone modifications appear to be primary mediators of epigenetic inheritance in cancer cells, and hypermethylation of the hMLH1 promoter has been reported in up to 70% of sporadic MSI tumors. In the large intestine, aberrant methylation may be an important early event in the age-related field defect observed in sporadic colorectal neoplasia. Aberrant methylation also contributes to tumor progression through a hypermethylator phenotype (CPG island methylator phenotype [CIMP]) responsible for most cases of MSI related to hMLH1 inactivation (associated with about 15% of sporadic CRCs).54,63 The hallmark of CIMP is abnormal methylation of several tumor promoters. In one model, hyperplastic aberrant crypt foci may be the initial lesions in a pathway leading to the development of serrated adenomas (see Chapter 122). Methylated promoters of the MGMT, EVL, HLTF, SFRP2, SLC5A8, and MINT1 genes develop during tumor initiation, and hMLH1 promoter hypermethylation corresponds to the development of a serrated adenoma, with methylated TSP1 and TIMP3 helping to drive tumor progression.54 DNA methylation and histone H3 lysine 9 hypoacetylation and methylation appear to form a mutually reinforcing loop that contributes to tumor suppressor gene inactivation in CRCs.
MicroRNAs are 18- to 25-nucleotide noncoding RNA molecules that regulate translation of many genes. Expression patterns of microRNAs are altered in colon cancers, and they may be associated with survival and therapeutic outcome.64
Biochemical and Other Changes
Chapter 3 deals in depth with the biological and biochemical changes that occur during the development of colorectal neoplasia. Alterations in cell surface and secreted proteins and glycoproteins, including a number of important cell adhesion molecules, are characteristic of CRC.65 Interactions between tumor cells or between tumor cells and their environment may be homotypic (involving like molecules) or heterotypic (involving different adhesion molecules). Homotypic interactions often maintain the integrity of primary tumors by fostering adhesion between neighboring tumor cells, whereas heterotypic interactions might occur among tumor cells and platelets, lymphocytes, vascular endothelial cells, and components of the basement membrane matrix. Most tumor-associated molecules represent quantitatively or qualitatively altered molecules found either on normal tissues or during development, such as oncofetal antigens—for example, carcinoembryonic antigen [CEA]). Many of these molecules appear to play a role in maintaining normal tissue homeostasis or targeting blood-borne cells to specific sites. Altered expression, therefore, might contribute to tumor invasion and metastasis.
MMPs (matrix metalloproteinases) are a family of enzymes that degrade extracellular matrix. Overexpression of MMP-1, -2, -3, -7, -9, and -13 and MT1-MMP has been demonstrated in human CRCs. The degree of overexpression of some MMPs correlates with stage of disease, prognosis, or both.66
Metastasis is a multistage process by which tumor cells escape the primary tumor and establish secondary foci at distant sites (Fig. 123-9). Cells in the primary tumor must become vascularized (angiogenesis via vascular endothelial growth factors). Then they must escape the primary tumor by overcoming adhesive interactions (e.g., loss of E-cadherin) and by disrupting basement membranes (metalloproteinases such as type IV collagenase, matrilysin, loss of tissue inhibitors of collagenase). Finally they must enter lymphatics, the circulation, or both. In the bloodstream, they must survive interactions with blood components and the immune system and be transported to distant organ sites (principally the liver). At distant sites, tumor cells adhere to target endothelia via specific interactions (e.g., tumor-associated sialoglycoproteins and endothelial selectins) (Fig. 123-10), extravasate, interact with the microenvironment (e.g., growth factors), and establish secondary tumor foci.
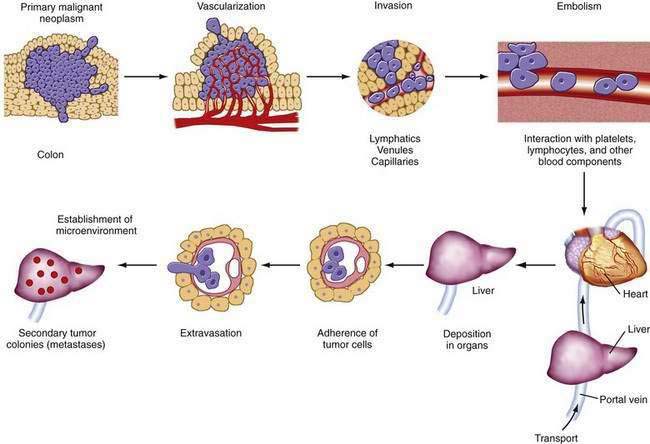
Tumor cell subpopulations with different metastatic potentials exist within the same primary tumor, and metastases result from the selective dissemination of tumor cells that possess the ability to participate in all stages of this complex process. Several carbohydrate antigens have been studied in relation to their potential usefulness as markers of metastatic potential and for their possible role in determining prognosis.65–68
FAMILIAL COLORECTAL CANCER
It has become increasingly clear that genetic predisposition plays a role in a substantial number of CRCs. Although it is convenient to categorize CRCs into hereditary (or familial) and nonhereditary (or sporadic) types, it is more appropriate to assume that all cancers have genetic components that may be inherited or acquired to varying degrees. Accordingly, persons with familial colon cancer are born with an altered genome, and the environment might contribute additional genotoxic events, leading to the malignant phenotype. In the case of sporadic cancers, multiple somatic mutations are contributed by the environment (see Chapter 3).
The role of heredity in the genesis of colon cancer is manifested most obviously in those with the heritable polyposis syndromes (Chapter 122). These syndromes are inherited in an autosomal dominant manner and are characterized by the presence of hundreds to thousands of colonic adenomas, with or without extracolonic tumors. Adenomas develop approximately a decade before the appearance of cancer, and virtually all affected persons eventually develop large bowel cancer if the colon is not removed. Nevertheless, these dramatic syndromes account for less than 1% of all cases of CRC.
HNPCC is an inherited disease in which colon cancers arise in discrete adenomas, but polyposis—hundreds of polyps—does not occur.59,69,70 HNPCC accounts for approximately 6% of colonic adenocarcinoma. It is an autosomal dominant disorder with high penetration; approximately 80% are caused by germline mutations in genes responsible for repair of DNA errors, called mismatches, that occur during DNA replication (discussed earlier). During DNA synthesis, DNA polymerase might create single base-pair mismatches, resulting in structural abnormalities (loop-outs) involving unpaired bases. These errors tend to occur at repetitive DNA sequences termed microsatellites, and they are repaired by enzymes coded for by MMR genes. The majority of reported germline mutations in DNA MMR genes have been associated with the hMSH2 gene on chromosome 2 (40% to 50%) and the hMLH1 gene on chromosome 3 (20% to 30%).
Mutations in hMSH6, hPMS1, and hPMS2 also have been reported in a small number of patients with HNPCC; no locus has been identified, however, for many HNPCC families. The definition of HNPCC was standardized and most strictly defined by the International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer. These Amsterdam criteria (Table 123-5) include at least three relatives with histologically verified CRC, one of them a first-degree relative of the other two (FAP excluded); at least two successive generations affected; and, in one of the individuals, diagnosis of CRC before age 50 years. Because these criteria do not account for the frequent occurrence of extracolonic cancers in such families, or for small kindreds, broader clinical criteria have been developed, including the Bethesda guidelines published by a National Cancer Institute-sponsored workshop on HNPCC (Table 123-6).70
Table 123-5 Amsterdam Criteria for Hereditary Nonpolyposis Colorectal Cancer
Criteria defined by the International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer.
Table 123-6 Bethesda Guidelines for Testing of Colorectal Tumors for Microsatellite Instability71
HNPCC, hereditary nonpolyposis colorectal cancer.
* Endometrial, ovarian, gastric, hepatobiliary, or small intestinal cancer or transitional cell carcinoma of the renal pelvis or ureter.
† Poorly differentiated or undifferentiated carcinoma composed of irregular solid sheets of large eosinophilic cells and containing small gland-like spaces.
‡ Composed of >50% signet ring cells.
HNPCC families include members whose heritable cancer is limited to the colon and rectum (site-specific HNPCC, HNPCC type a, Lynch’s syndrome I) and families whose members also are prone to cancer of the female genital tract and other sites (cancer family syndrome, HNPCC type b, Lynch’s syndrome II) (Fig. 123-11). In HNPCC syndromes, discrete polyps, but not polyposis, might antedate the cancers. Adenomas in the proximal colon sometimes are flat or slightly raised lesions with foci of adenomatous change confined to the upper half of crypts (flat adenomas). HNPCC tumors have a tendency to involve proximal sites of the colon and to be multiple (synchronous and metachronous), with a higher incidence of mucinous carcinomas (Table 123-7). These CRCs usually appear at age 40 to 50 years, two decades earlier than CRC in the general population. In a study of a small, defined population in central Finland, HNPCC accounted for 4% to 6% of CRCs identified. This figure is similar to estimates of Lynch and others for the U.S. population.
Table 123-7 Comparison of HNPCC and Sporadic Colorectal Cancer
| CLINICAL FEATURE | HNPCC | SPORADIC COLORECTAL CANCER |
|---|---|---|
| Mean age at diagnosis (years) | 45 | 67 |
| Multiple colon cancers | 35% | 4-11% |
| Synchronous colon cancers | 18% | 3-6% |
| Metachronous colon cancers | 24% | 1-5% |
| Proximal location* | 72% | 35% |
| Increased risk of malignant tumors at other sites | Yes | No |
| Mucinous and poorly differentiated colon cancers | Common | Infrequent |
| Prognosis | Favorable† | Variable |
HNPCC, hereditary nonpolyposis colorectal cancer.
* Proximal to the splenic flexure; location of the initial cancer.
† Patients whose tumors demonstrate microsatellite instability have a more favorable prognosis than those with microsatellite-stable tumors.
Patients with HNPCC and some unaffected family members have biological markers that resemble those in patients with FAP syndromes, including abnormal proliferative activity of colonic crypt cells; increased tetraploidy (twice the normal DNA content) in skin fibroblasts cultured in vitro; decreased degradation of fecal cholesterol; and cell-mediated immune defects in vitro that might interfere with the recognition of killing of incipient tumor cells in vivo. The genetic defect in HNPCC, however, results from loss of the hMSH2, hMLH1, and other genes, leading to increased susceptibility to mutation from failure to repair base-pair mismatches.59,71
Although CRC syndromes with readily apparent patterns of inheritance currently account for only a small portion of total colon cancer cases, hereditary factors may be present in a larger proportion of cases.72 Genetic susceptibility to CRC in the general population is suggested by the two-fold to three-fold increase in CRC in first-degree relatives of patients with sporadic adenomas and CRCs. The relative risk is even stronger when cancer occurs in family members younger than 50 years. The precise role of genetic factors in this group and their interaction with the environment in the evolution of CRC remains to be defined. The identification of susceptibility genes in this group is of great interest and has been aided by genome-wide scanning techniques in sibling pairs with colorectal neoplasia.73
PREDISPOSING FACTORS
The risk of developing CRC depends on a number of demographic factors (Table 123-8). The probable influence of diet and other environmental factors has been discussed (see Tables 123-1 and 123-2). Other factors include age, personal history of adenoma or of carcinoma, existence of predisposing diseases (particularly inflammatory bowel disease [IBD]), and family history (discussed earlier). It has been proposed that risk scores may be developed to aid in identifying and treating susceptible persons.74
Table 123-8 Risk Factors for Colorectal Cancer
* Based on descriptive epidemiology; data from case-control, cohort, and randomized trials are less convincing.
† Especially with high-grade dysplasia or dysplasia-associated mass lesions.
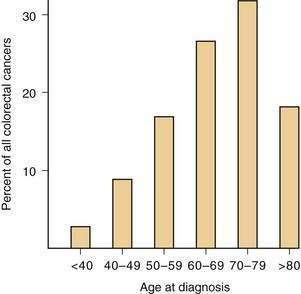
AGE
The risk of developing CRC rises sharply after age 40 years in the general population, 90% of cancers occurring in persons aged 50 years and older (Fig. 123-12). A 50-year-old person has approximately a 5% chance of developing CRC if he or she survives to age 80 years and a 2.5% risk of dying from the disease; this has important implications for screening (see later). Sporadic CRCs arise in other age groups, such as the third and fourth decades, however, and the diagnosis must be considered in younger persons with signs and symptoms characteristic of this disease, especially if they have a family history of colorectal neoplasia.
PRIOR ADENOMA AND CARCINOMA
Adenoma
Present evidence strongly indicates that most CRCs arise from preexisting adenomas (see Chapter 122). The risk of CRC increases with the number of adenomas, the most extreme example being the familial polyposis syndromes. Clinical and morphologic evidence suggest that as adenomas grow larger, they progressively dedifferentiate, become dysplastic, and then become malignant. With increasing size or increasing villous architecture, the incidence of nuclear atypia, dysplasia, and in situ or invasive carcinoma increases. Despite the potential for adenomas to evolve to carcinomas, however, the actual risk of this phenomenon is unknown.
Adenomatous polyps are common, especially after age 50 years, in populations that consume a Western diet, but the prevalence of adenomas is high, compared with the incidence of cancer. It has been estimated that 29% of the living population older than 35 years in Norway have colorectal adenomas, with an annual conversion rate of adenomas to carcinoma (based on cancer incidence from multiple tumor registries in Norway) of 0.25. The malignancy rate is higher in large adenomas, adenomas with villous architecture, and adenomas with cytologic nuclear atypia or dysplasia (advanced adenomas). The estimated annual rate of conversion to invasive cancer in persons with adenomas larger than 1 cm, villous components, or severe dysplasias has been reported to be 3%, 17%, and 37%, respectively. Data from a national colonoscopy data base in Germany suggested that the annual transition rates from advanced adenoma to cancer increase with age.75
FAMILY HISTORY
The risk of CRC in first-degree relatives of those with sporadic CRC is increased two-fold to three-fold (see earlier). The risk is higher when adenoma or carcinoma has occurred in a relative at an early age or when more than one relative has had carcinoma. These factors have been taken into account in screening guidelines that stratify patients according to potential cancer risk.72–83
FAP and its variants are inherited in an autosomal dominant manner, and without colectomy virtually all affected members with polyps eventually develop carcinoma. HNPCC also has an autosomal dominant pattern of inheritance (see Chapter 122).
Turcot’s syndrome is a rare combination of inherited adenomatous polyposis and malignant brain tumors. Inheritance is autosomal dominant, and germline mutations in the APC gene or mutations of hMLH1 and hPMS2 characteristic of HNPCC have been described (see Chapter 122).
Peutz-Jeghers syndrome and the familial form of juvenile polyposis both have an increased risk of small and large bowel cancer (see Chapter 122). Peutz-Jeghers syndrome is an autosomal dominant disease, which, in most families, has been mapped to chromosome 19 p13.3 and the STK11 gene (serine threonine kinase 11). Adenomatous changes have been reported in 3% to 6% of hamartomas from these patients. Extracolonic cancers are common and have been reported to occur in 50% to 90% of patients with Peutz-Jeghers syndrome (relative risk for all cancers was 15.2).84 A significant increase in a variety of cancers (esophagus, stomach, small intestine, pancreas, lung, breast, uterus, and ovary) has been reported.
Familial juvenile polyposis is a rare autosomal dominant disease associated with polyps that may be limited to the colon or the stomach or that occur throughout the gastrointestinal tract (see Chapter 122).85 The genetic basis of this syndrome is not fully understood, but germline mutations in a gene (SMAD4) located on chromosome 18q21.1 that encodes an intracellular mediator in the TGF-β signaling pathway have been identified in some affected patients, as have mutations in the BMPR1A (bone morphogenetic protein receptor type 1A) gene; each accounts for approximately 20% of cases.86 The bone morphogenetic receptors (BMPs) are a family of transmembrane serine-threonine kinases whose ligands are members of the TGF-β superfamily. BMPs repress WNT signaling, and BMPR1A plays a role in apoptosis. The PTEN (phosphatase and tensin homologue deleted on chromosome 10) gene located on chromosome 10 also has been linked to some cases of juvenile polyposis. PTEN encodes a dual phosphatase, loss of function of which leads to protection of cells from various apoptotic stimuli. Overexpression of PTEN leads to cell cycle arrest or induction of apoptosis, or both. In patients with juvenile polyposis, the cumulative lifetime risk for CRC might approach 40%.87
INFLAMMATORY BOWEL DISEASE
Patients with idiopathic IBD (Crohn’s disease, UC) are at increased risk for developing adenocarcinoma of the colon (see Chapters 111 and 112).88 Actuarially derived (life table) cumulative cancer incidences from tertiary referral centers (Fig. 123-13) agree that the risk of cancer in patients with UC begins after a disease duration of seven years and rises about 10% per decade, reaching approximately 30% at 25 years. Nonetheless, difficulties related to sources of referral, sampling, recognition and characterization of disease, differences in follow-up procedures, and methods used to detect neoplastic disease cloud many such studies. The assessed cancer risk for patients with UC followed in one private practice has been reported to be only 6.6% 26 years after disease onset and 11.4% at 32 years. The true risk of developing CRC in UC must lie between predictions from tertiary centers and the primary care setting.
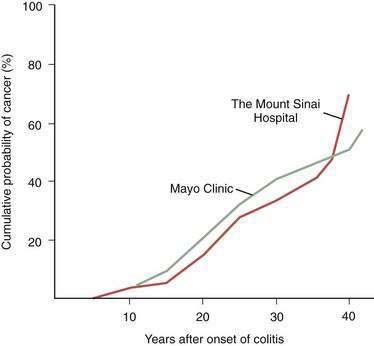
Figure 123-13. Cumulative probability of developing colorectal carcinoma in patients with ulcerative colitis seen at two tertiary referral centers.
Data from primary care settings indicate a similar pattern but lower incidence rates (see text and Chapter 112).
The risk of cancer is not related to severity of the initial attack of colitis or age at onset of colitis, independent of disease duration. The relationship to disease activity per se is unclear, although chronic inflammation is a common theme in epithelial carcinogenesis. CRC in persons with UC is a risk factor for CRC in their relatives without colitis and CRC in relatives without UC is a risk factor for those with colitis. There is an association of backwash ileitis with CRC in patients with UC who undergo proctocolectomy.89 Cancer arising in the setting of UC traditionally has been thought to be a highly malignant lesion with a poor prognosis, but studies using matched controls from colon cancer populations without colitis have failed to show a significant difference in survival between the two groups.
Carcinomas do not develop de novo from normal mucosa but from mucosa that has undergone a sequence of morphologic changes that culminate in invasive carcinoma. As in precancerous adenomas, dysplasia is a precursor to carcinoma in IBD. Dysplasia comprises abnormalities in crypt architecture and cytologic detail (Figs. 123-14 and 123-15). Epithelial crypts are reduced in number, irregularly branched, and crowded together (“back-to-back” glands). Cell nuclei may be enlarged and hyperchromatic, may have increased numbers of mitoses, and may be located at different levels in the cell, producing a picket-fence appearance (pseudostratification). Dysplasia is classified by grade as mild (or low grade) to severe (or high grade).
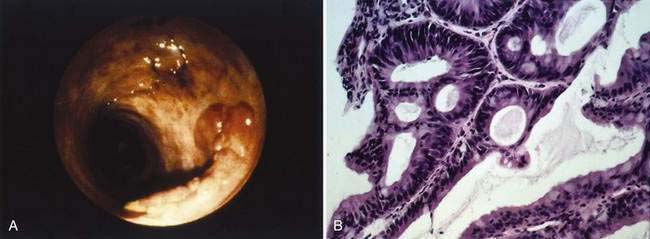
Results suggest that biopsy surveillance programs can be effective in helping control the risk of carcinoma in patients with long-standing UC. The risk of cancer appears highest in those with high-grade dysplasia that arises in visible plaques or masses (dysplasia-associated lesion or mass [DALM], as illustrated in Fig. 123-15). A computer cohort decision analysis suggested that surveillance should increase life expectancy for patients with UC. Most investigators believe that patients who have UC for more than seven to eight years should undergo colonoscopy with multiple mucosal biopsies annually to identify areas of dysplasia; they advocate colectomy for severe dysplasia or DALM. Because the significance of low-grade or moderate dysplasia is less clear, immediate resection for patients with these levels of dysplasia is controversial. Prophylactic colectomy also has been recommended as an option for those with disease of at least 10 years’ duration. Studies employing flow cytometry have detected aneuploid cell populations in colons resected for UC with dysplasia or early cancer. Chromosomal alterations can occur early in UC-related neoplastic progression and appear to precede the histologic development of dysplasia; relative loss of chromosome 18q also may be important in neoplastic progression.
Both dysplasia and increased risk of colon carcinoma have been reported in patients with Crohn’s disease. As in UC, dysplasia appears in diseased colon segments, and its presence correlates with duration of disease. In one screening trial,90 the finding of definite dysplasia was associated with age older than 45 years and increased symptoms. By life-table analysis, the probability of detecting dysplasia or cancer after a negative screening colonoscopy was 22% by the fourth surveillance examination.
OTHER ASSOCIATIONS
A retrospective population-based cohort study that used the Swedish Inpatient Register evaluated almost 23,000 persons who had had cholecystectomy up to 31 years previously.91 Patients having had cholecystectomy had an increased risk of proximal intestinal adenocarcinoma, which declined with increasing distance from the common bile duct. The risk was significantly increased for adenocarcinoma of the small bowel and ascending colon but not the remaining colon or rectum.
PATHOLOGY, NATURAL HISTORY, AND STAGING
GROSS PATHOLOGY
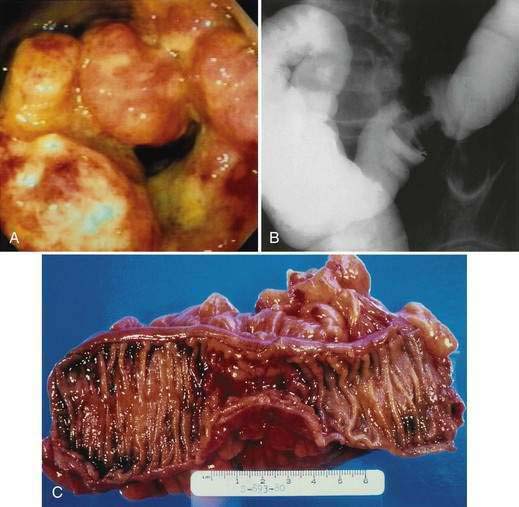
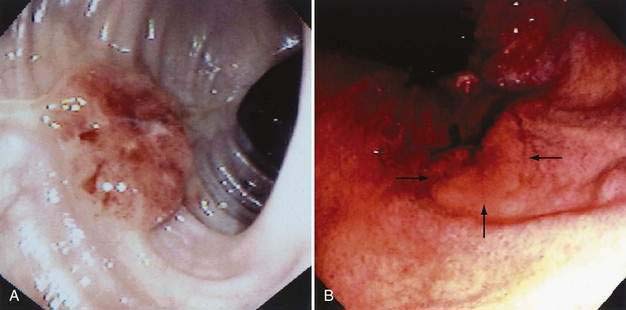
The gross morphologic features of CRC depend on the tumor’s site. Carcinomas of the proximal colon, particularly those of the cecum and ascending colon, tend to be large and bulky, often outgrowing their blood supply and undergoing necrosis (Fig. 123-16); this polypoid configuration, however, also may be found elsewhere in the colon and rectum. In the more distal colon and rectum, tumors more commonly involve a greater circumference of the bowel, producing an annular constriction or napkin-ring appearance (Fig. 123-17). The fibrous stroma of these tumors accounts for the constriction and narrowing of the bowel lumen, whereas the circular arrangement of colonic lymphatics is responsible for their annular growth. These tumors also can ulcerate, and occasionally they have a flatter appearance with predominantly intramural spread (Fig. 123-18); the latter are seen most often in the setting of IBD. Morphologic features of CRCs have clinical, diagnostic, and prognostic implications.
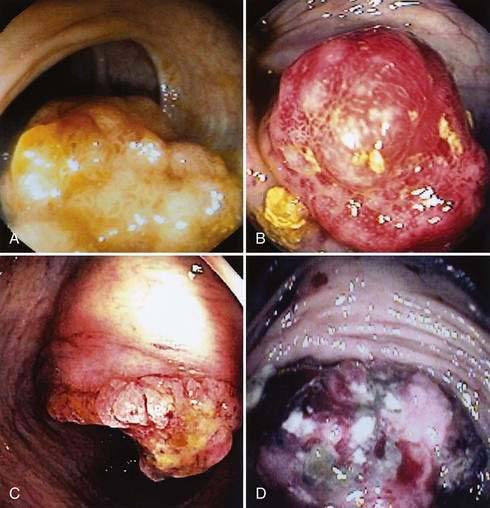
HISTOLOGY
Carcinomas of the large bowel characteristically are adenocarcinomas that form moderately differentiated to well-differentiated glands and secrete variable amounts of mucin (Fig. 123-19). Mucin, a high-molecular-weight glycoprotein, is the major product secreted by both normal and neoplastic glands of the colon, and may be seen best with histochemical stains such as periodic acid–Schiff (PAS). In poorly differentiated tumors (see Fig. 123-19), gland formation and mucin production are present but less prominent. Signet ring cells, in which a large vacuole of mucin displaces the nucleus to one side, are a feature of some tumors (Fig. 123-20A). In approximately 15% of tumors, large lakes of mucin contain scattered collections of tumor cells (see Fig. 123-20B). Such mucinous or colloid carcinomas are most common in patients with HNPCC or UC and in patients whose carcinomas occur at an early age. Scirrhous carcinomas are uncommon and are characterized by sparse gland formation, with marked desmoplasia and fibrous tissue surrounding glandular structures. Sometimes tumors demonstrate a mixed histologic picture, with glands of varying degrees of differentiation.
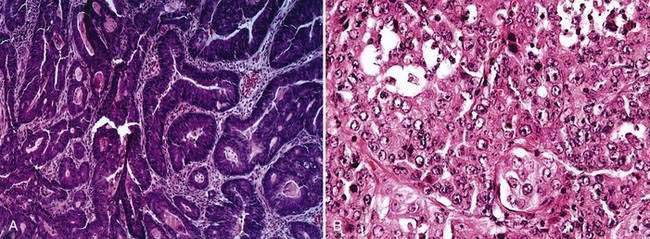
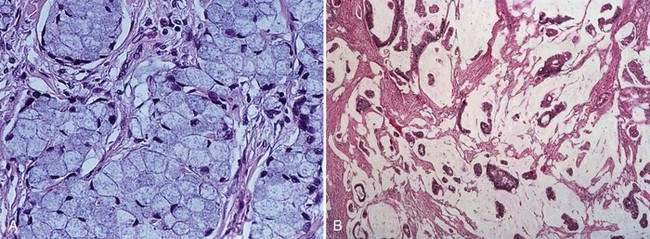
Cancers other than adenocarcinomas account for less than 5% of malignant tumors of the large bowel. Tumors arising at the anorectal junction include squamous cell carcinomas, cloacogenic or transitional cell carcinomas, and melanomas (see Chapter 125). Primary lymphomas and carcinoid tumors of the large bowel account for less than 0.1% of all large bowel neoplasms (see Chapters 29 and 31).
NATURAL HISTORY AND STAGING
CRCs begin as intramucosal epithelial lesions, usually arising in adenomatous polyps or glands. As cancers grow, they become invasive, penetrating the muscularis mucosae of the bowel and invading lymphatic and vascular channels to involve regional lymph nodes, adjacent structures, and distant sites. Although CRCs grow at varying rates, they most often have long periods of silent growth before producing bowel symptoms. The mean doubling time of colon cancers determined radiologically in one study was 620 days. Comparative lesion sequencing using modern molecular techniques combined with clinical observation suggests that it can take approximately 17 years for a large benign tumor to evolve to advanced cancer but less than two years for cells within that cancer to acquire the ability to metastasize.92 Patterns of spread depend on the anatomy of the individual bowel segment as well as its lymph and blood supplies.
On the basis of observations of what he believed to be an orderly progression of locoregional invasion by rectal cancers, Cuthbert Dukes proposed a staging classification in 1929, which has since been modified many times in attempts to increase its prognostic value for cancers of both the rectum and the colon. The most commonly employed modification of the Dukes system is that of Astler and Coller (Table 123-9). This classification uses the following designations:

Stage C tumors are divided further into primary tumors limited to the bowel wall (C1) and those that penetrate the bowel wall (C2). By contrast, in the system proposed by the Gastrointestinal Tumor Study Group (GITSG), C1 lesions are those in which one to four regional lymph nodes contain tumor, and C2 lesions are those in which more than four lymph nodes contain tumor (see Table 123-9). Another modification, by Turnbull and associates, added a D category referring to distant metastases.
In an attempt to provide a uniform and orderly classification for CRCs, the American Joint Committee on Cancer (AJCC) has introduced the tumor-node-metastases (TNM) classification for CRC.93 This system classifies the extent of the primary tumor (T), the status of regional lymph nodes (N), and the presence or absence of distant metastases (M). Cases are assigned the highest value of TNM that describes the full extent of disease and are grouped into five stages (0 through IV). The five stages have become important in uniformly randomizing patients for therapeutic trials (Table 123-10) and have, in most cases, replaced the Dukes classification for CRC.
| STAGE* | CRITERIA† |
|---|---|
| 0 | Carcinoma in situ: intraepithelial or invasion of lamina propria‡ (Tis N0 M0) |
| I | Tumor invades submucosa (T1 N0 M0) [Dukes A] |
| Tumor invades muscularis propria (T2 N0 M0) | |
| II | Tumor invades through the muscularis propria into subserosa or into nonperitonealized pericolic or perirectal tissues (T3 N0 M0) [Dukes B] |
| Tumor perforates the visceral peritoneum or directly invades other organs or structures and/or perforates visceral peritoneum§ (T4 N0 M0) | |
| III | Any degree of bowel wall perforation with regional lymph node metastasis |
| N1: metastasis in 1-3 regional lymph nodes | |
| N2: metastasis in ≥4 regional lymph nodes | |
| Any T N1 M0 [Dukes C] | |
| Any T N2 M0 | |
| IV | Any invasion of bowel wall with or without lymph node metastasis, but with evidence of distant metastasis |
| Any T Any N M1 |
AJCC, American Joint Committee on Cancer; N0, no regional lymph node metastasis; M0, no distant metastasis; M1, distant metastasis.
* Stage II is subdivided into IIA (for T3 tumors) and IIB (for T4 tumors). Stage III is subdivided into IIIA (T1-T2 N1 M0), IIB (T3-T4 N1 M0), and IIIC (any T N2 M0). N1 lesions have 1-3 positive nodes; N2 tumors have ≥4 positive nodes. Smooth metastatic nodules in the pericolic or perirectal fat are considered lymph node metastases. Irregularly contoured metastatic nodules in the peritumoral fat are considered vascular invasion.
† Dukes B, which corresponds to stage II, is a composite of better (T3 N0 M0) and worse (T4 N0 M0) prognostic groups, as is Dukes C, which corresponds to stage III (any T N1 M0 and any T N2 M0).
‡ Tis includes cancer cells confined within the glandular basement membrane (intraepithelial) or lamina propria (intramucosal) with no extension through the muscularis mucosae into the submucosa.
§ Direct invasion in T4 includes invasion of other segments of the colorectum by way of serosa; e.g., invasion of the sigmoid colon by a carcinoma of the cecum.
From the American Joint Committee on Cancer. AJCC Cancer Staging Manual: Colon and Rectum, 6th ed. New York: Springer-Verlag; 2002.
PROGNOSIS
Clinical and pathologic variables that can affect the prognosis of patients with CRC are outlined in Table 123-11. These variables are important in predicting clinical outcome and in designing optimal strategies for treatment and follow-up. Their identification has led to a progressive modification of the staging classifications for CRC. The roles of histologic differentiation; tumor size, location, configuration, and degree of invasion; and lymph node status must be evaluated on the basis of prospective analyses of patients who undergo curative resections for CRC.
Table 123-11 Pathologic, Molecular, and Clinical Features That May Affect Prognosis in Patients with Colorectal Cancer
| FEATURE OR MARKER | EFFECT ON PROGNOSIS |
|---|---|
| Pathologic | |
| Surgical-pathologic Stage | |
| Depth of colon wall penetration | Increased penetration diminishes prognosis |
| Number of regional nodes involved by tumor | 1-4 nodes is better than >4 nodes |
| Tumor Morphology and Histology | |
| Degree of differentiation | Well-differentiated is better than poorly differentiated |
| Mucinous (colloid) or signet ring cell histology | Diminished prognosis |
| Scirrhous histology | Diminished prognosis |
| Invasion | |
| Venous | Diminished prognosis |
| Lymphatic | Diminished prognosis |
| Perineural | Diminished prognosis |
| Other Features | |
| Local inflammation and immunologic reaction | Improved prognosis |
| Tumor morphology | Polypoid or exophytic is better than ulcerating or infiltrating |
| Tumor DNA content | Increased DNA content (aneuploidy) diminishes prognosis |
| Tumor size | No effect in most studies |
| Molecular | |
| Loss of heterozygosity at chromosome 18q (DCC, DPC4) | Diminished prognosis |
| Loss of heterozygosity at chromosome 17p (TP53) | Diminished prognosis |
| Loss of heterozygosity at chromosome 8p | Diminished prognosis |
| Increased labeling index for p21WAF/CIP1 protein | Improved prognosis |
| Microsatellite instability | Improved prognosis |
| Mutation in BAX gene | Diminished prognosis |
| Clinical | |
| Diagnosis in asymptomatic patients | Possibly improved prognosis |
| Duration of symptoms | No demonstrated effect |
| Rectal bleeding as a presenting symptom | Improved prognosis |
| Colon obstruction | Diminished prognosis |
| Colon perforation | Diminished prognosis |
| Tumor location | May be better for colonic than for rectal tumors May be better for left colonic than right colonic tumors |
| Age <30 yr | Diminished prognosis |
| Preoperative CEA level | Diminished prognosis with a high CEA level |
| Distant metastases | Markedly diminished prognosis |
CEA, carcinoembryonic antigen.
SURGICAL-PATHOLOGIC STAGING
The depth of transmural tumor penetration and the extent of regional lymph node spread are the most important determinants of CRC prognosis (Fig. 123-21). The degree of bowel wall penetration also affects prognosis, independently of lymph node status, and correlates with the number of involved nodes as well as with the incidence of local recurrence after surgical resection. The number of involved regional lymph nodes also correlates independently with outcome (see Fig. 123-21B).
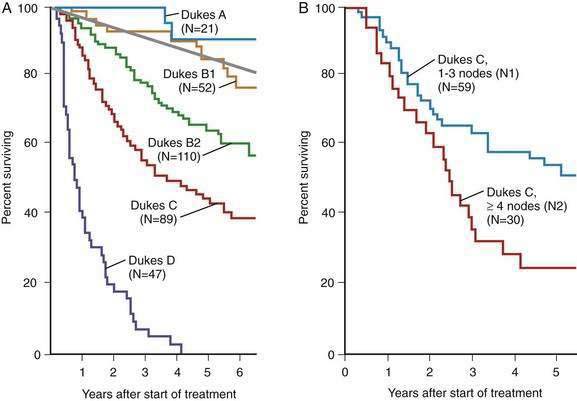
Figure 123-21. A, Survival probabilities according to the Dukes stage as modified by Astler-Coller (see Table 123-9) in patients undergoing potentially curative surgery for colorectal cancer. Expected survival among age- and sex-matched general populations is indicated by the straight line. B, Survival probabilities according to the number of nodes involved in patients with stage C colorectal carcinoma. Data using TNM staging confirm a substantial difference in five-year survival among patients with lesions having one to three positive nodes (N1) and four or more positive nodes (N2).
(From Moertel CG, O’Fallon JR, Go VL, et al. The preoperative carcinoembryonic antigen test in the diagnosis, staging, and prognosis of colorectal cancer. Cancer 1986; 58:603.)
TUMOR MORPHOLOGY AND HISTOLOGY
The TNM classification (see Table 123-10) is based in part on the observation that for most cancers, tumor size correlates with local and distant spread and, thus, indirectly with prognosis. Numerous studies suggest that CRC is an exception, however, and that the size of the primary tumor per se does not correlate with prognosis. In fact, patients with exophytic or polypoid tumors appear to have a better prognosis than those with ulcerating or infiltrating tumors.
Tumor prognosis correlates with histologic grade: poor differentiation confers a worse prognosis than does a high degree of differentiation. Mucinous65 and scirrhous carcinomas appear to be biologically more aggressive, and patients with these tumors do not survive as long as those who have other types of adenocarcinoma. Mucin-associated antigens might play a role in tumor progression and metastasis of colon cancer cells. Signet ring carcinomas have been reported to present at an advanced stage and to be highly invasive tumors.
Venous invasion by CRC (Fig. 123-22A) usually correlates with local recurrence after resection, visceral metastases, and decreased survival, but it might not have independent prognostic value for tumors confined to the bowel wall. Although lymphatic invasion is associated with decreased survival, it is not clear whether this variable is independent of depth of tumor invasion and regional nodal metastasis. Perineural invasion (see Fig. 123-22B) also is linked to increased local recurrence and decreased survival, but such data are limited. The degree of inflammatory response and lymphocytic infiltration in and around a cancer may be related to outcome; increased inflammation and immune reaction appear to confer a better prognosis, but data are limited.
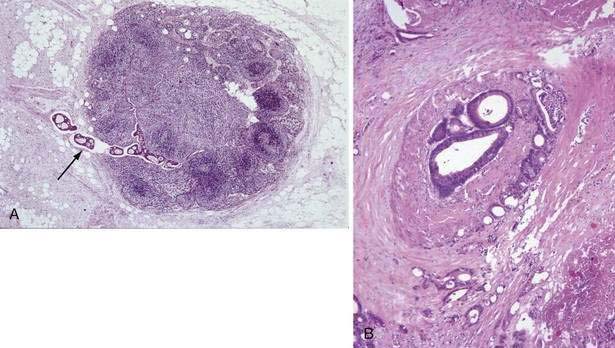
The prognosis of patients with CRC may be related to the DNA content of the primary tumor, because survival is shorter for patients with nondiploid or aneuploid tumors than for those whose tumor cells have a normal or diploid DNA content. Although the DNA content of the primary tumor might correlate with the potential for local or distant recurrence after primary resection, the value of routine flow cytometric measurements or image analysis of the DNA content of tumor cells for assessing clinical prognosis and planning treatment remains to be determined. Deletions in chromosomes 18q and 17p (TP53) may be important indicators of prognosis, independent of stage.55,57 As indicated in Table 123-11, a growing number of other molecular markers also might predict prognosis or response to therapy.57
CLINICAL PREDICTORS OF PROGNOSIS
Whereas screening programs for CRC suggest that tumors diagnosed in asymptomatic patients are less advanced, assessment of the impact of early diagnosis on survival of asymptomatic persons awaits the results of prospective randomized, controlled studies such as the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial.94 Duration of symptoms might not correlate directly with prognosis, and some presenting symptoms, such as rectal bleeding, may be associated with better rates of survival.
As many as 3% of CRCs develop before 30 years of age, and only 11% of such persons have a predisposing condition such as FAP or UC. The prognosis is worse than for older patients and is particularly poor in the pediatric age group. Poor prognosis may be related to a higher percentage of more-advanced cancers and mucinous adenocarcinomas in these young patients. Alternatively, patients with tumors demonstrating MSI appear to have a better prognosis irrespective of age.60 Thus, whereas CRCs occur at a younger age in those with HNPCC, these patients have a better prognosis than those with microsatellite-stable cancers.
Outcome also is related to preoperative serum CEA levels. Tumor recurrence is higher, and the estimated mean time to recurrence is shorter, in patients with Dukes B and C cancers who have high preoperative CEA levels. The preoperative CEA level may be of prognostic value only in patients with Dukes C CRCs who also have four or more involved lymph nodes (stage C2), but not in patients with Dukes A and B lesions or Dukes C lesions with fewer than four nodes involved. Expression of mucin-associated carbohydrate antigens other than CEA, such as sialyl Lewisx, also can correlate with prognosis.65 Expression of the carbohydrate-binding protein galectin-3 correlates with tumor progression in the colon and can confer a poor prognosis.65,67