14 Coarctation of the Aorta
I. CASE
A. Fetal echocardiography findings
1. The fetal echo reveals situs solitus of the atria, levocardia, left aortic arch, heart rate 145 bpm, and a normal cardiothoracic ratio (0.3).
2. The RV is enlarged. The LV has relative hypoplasia but normal length.
3. The aortic valve is bicuspid, with mildly hypoplastic annulus (3.5 mm) but without significant aortic stenosis (velocity 1.5 m/s). There is neither mitral nor aortic valve regurgitation.
4. There is no significant poststenotic dilation of the ascending aorta, but there is mild isthmal hypoplasia (2.5 mm).
5. The RV is moderately dilated, with normal tricuspid and pulmonary artery flow.
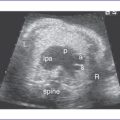
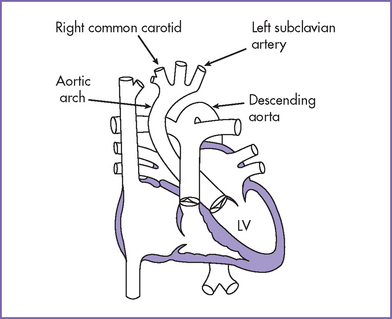
6. Flow in both the aortic and ductal arches is normal and antegrade. The aortic arch, however, is hypoplastic distally between the left common carotid artery and the descending aorta. There appears to be a posterior shelf consistent with coarctation of the aorta (CoA) (Fig. 14-1).
7. There is an unrestricted right-to-left shunt across the foramen ovale.
8. The RV Tei index (myocardial performance index) is mildly increased at 0.50. The LV Tei index is normal.
D. Fetal management and counseling
1. Ultrasound plus amniocentesis with karyotyping for chromosomal anomalies.
c. Fluorescent in situ hybridization (FISH) screen for chromosome 22q11 microdeletion.
2. Follow-up includes serial antenatal fetal echocardiography studies at 4-week intervals.
a. Size and function of the LV (apex forming).
b. Diameters of the mitral and aortic valves and the presence of subaortic stenosis. Assessment of valve morphology might become easier later in gestation.
c. Size of the transverse aortic arch compared to the ductus.
d. Size and direction of flow across the foramen ovale. Right-to-left flow is normal in the fetus.
e. Size and direction of flow through the distal aortic arch.
f. The patency of the ductus arteriosus. Any constriction of the ductus in late gestation could lead to heart failure and fetal death.
g. The flow pattern in the pulmonary veins, especially diastolic a wave reversal as an indirect measure of increased pressure in the LA.
h. Missed ventricular septal defect (VSD), which is common in the context of aortic CoA.
F. Neonatal management
a. Following delivery of a baby with a prenatal diagnosis of probable CoA, prostaglandin E1 (PGE1) may be initiated if the coarctation is clearly more severe, with significant arch hypoplasia, or if there is a concern that the left heart size may be borderline, particularly if there is retrograde distal arch flow.
b. Otherwise, many cases of prenatally diagnosed CoA might not be ductus arteriosus dependent and may not require surgical correction in the neonatal period.
c. Lack of blood pressure differences while the ductus arteriosus is still patent is the norm because the ductus arteriosus may be providing critical blood flow to the lower body beyond the level of the coarctation.
d. The hyperoxia test should be performed in either case because it can provide useful physiologic data that can help in appropriately managing the infant.
e. In critical neonatal CoA, the ductus arteriosus often supplies blood flow to the distal arch. The hyperoxia test would in such a state reveal a normal Pao2 (>250 mm Hg) in the preductal area, whereas the postductal area will fail the hyperoxia test as a result of right-to-left shunting through the ductus arteriosus. Occasionally, where there is a discrete coarctation, the baby passes preductal and postductal hyperoxia tests.
f. The two-dimensional echocardiogram shows the site and extent of coarctation. The Doppler examination reveals a disturbed flow distal to the coarctation and signs of delayed emptying in the proximal descending aorta, with continuous flow through the distal arch beginning at the most proximal point of significant arch hypoplasia or obstruction. In the presence of a patent ductus, there is usually no gradient across the coarctation.
g. In borderline situations, or when the infant passes the hyperoxia test and there is still suspicion of coarctation, the ductus could be allowed to close while the vital signs and blood pressures are monitored in the intensive care unit, with follow-up echocardiography to confirm the diagnosis. Ductal constriction can result in a discrete shelf’s evolving at the site of the distal arch where the ductus joins the aorta. This type of coarctation can continue to evolve even weeks to months beyond the neonatal period.
a. The surgical procedure of choice varies from institution to institution.
b. The appropriate surgical procedure also is determined by the type of coarctation, whether there is significant proximal arch hypoplasia or only a discrete contraductal shelf.
c. Transcatheter intervention.
d. Balloon angioplasty or stenting is the treatment of choice for recurrent coarctation, and in some institutions it is used more routinely for discrete native coarctation identified in older infants and children.
G. Follow-up
1. Most patients are hospitalized for 5 to 10 days after surgery for coarctation.
2. Reexamination is necessary every 6 to 12 months because residual obstruction and/or recoarctation occur in 20% to 25% of the patients who had the repair as neonates or young infants.
3. Subacute bacterial endocarditis (SBE) prophylaxis should be continued on indications because of frequently associated bicuspid aortic valve and residual coarctation.
4. Yearly surveillance by clinical examination, echocardiography, and cardiac magnetic resonance imaging (MRI) is necessary. This surveillance monitors persistent systemic hypertension due to residual narrowing proximal to the arch repair and development of an aneurysm of the aorta near the site of repair.
5. Reassessment for subvalvar and valvar aortic stenosis or mitral stenosis should be performed at follow-up, given the common association and their progressive nature, particularly in the first few years of life.
6. Late repair of coarctation is associated with sudden intracranial bleeding from berry aneurysm in the brain.
I. Outcome of this case
1. The baby was delivered at term weighing 2.3 kg. Apgar scores were 8 at 1 minute and 9 at 5 minutes.
2. The baby passed the preductal hyperoxia test with a Pao2 of 275 mm Hg, but the postductal Pao2 was 80 mm Hg, suggesting significant right-to-left shunting through the ductus arteriosus and a critical coarctation.
3. PGE1 infusion was started at 0.0125 μg/kg per minute.
a. A low dose should be used here, given the prenatal diagnosis and because closure, even with a dose as low as 0.005 μg/kg per minute, is extremely unusual in a newborn just delivered.
b. The PDA responded well to PGE1.
4. The aortic valve annulus (4 mm) with antegrade flow and LV size (LV:RV length ratio was 0.8) was considered borderline but apex forming.
5. There were two surgical options: a univentricular repair (Norwood stage 1 with arch reconstruction) or biventricular repair only addressing the transverse arch hypoplasia.
a. The baby had arch reconstruction with pulmonary homograft and division of the patent ductus arteriosus at 1 week of age.
b. He was discharged on PO digoxin and furosemide at 2 weeks of age.
c. At 6 months of age he had cardiac catheterization for possible recoarctation and a right upper and lower limb cuff blood pressure difference of 50 mm Hg with clear evidence of systemic hypertension in the upper extremities. Balloon dilation of recoarctation was successful.
d. The patient had a follow-up MRI of the aortic arch, and there was no evidence of aneurysm at the site of dilation.
e. The patient is currently doing well and has a yearly follow-up in the outpatient clinic and two yearly follow-up visits with cardiac MRI of the aortic arch.
II. YOUR HANDY REFERENCE
A. Infantile coarctation of the aorta
a. CoA accounts for 8% to 10% of congenital heart disease, with a male preponderance (2:1).
b. Aortic arch anomalies accounted for 6.1% of all cardiovascular lesions detected antenatally in the combined series of Allan and colleagues (1994).
a. Arch obstruction when diagnosed antenatally represents a more severe spectrum of disease than that diagnosed postnatally, because the lesion is more likely to be associated with significant intracardiac pathology. Spontaneous intrauterine death can occur in a small fraction of cases, especially in coarctation with Turner’s syndrome or aortic stenosis where hydrops develops.
b. Counseling should be individualized depending on the natural history of the specific lesion, arch hypoplasia with or without a potential for single-ventricle palliation. Long-term prognosis should be included (Box 14-1).
c. Prenatal diagnosis of nearly all forms of critical left heart disease results in improved neonatal hemodynamics after delivery and improved outcome with delivery in a tertiary care program and early initiation of prostaglandin therapy.
d. Balloon aortoplasty of native coarctation is associated with 91% to 100% primary success (gradient <20 mm Hg) and 25% restenosis over the next 2 to 17 months.
e. Dilation of recurrent coarctation is associated with an 89% to 91% primary success rate, with relatively low morbidity. In one study, with long-term follow-up between 7 months and 12 years, the restenosis rate was between 16% and 28%.
f. Long-term morbidity associated with repair of isolated CoA includes the potential risk of systemic hypertension and potential for recurrent distal arch obstruction requiring either balloon angioplasty or surgical intervention.
g. Long-term survival following repair of isolated CoA is 86% to 93% at 12 to 15 years following repair.
3. Associated syndromes and extracardiac anomalies.
a. CoA occurring in isolation or with VSDs or other forms of left heart obstruction may be associated with chromosomal abnormalities including:
b. Chromosome 22q11 microdeletion can be associated with:
c. Chromosomal anomalies were found in nearly 30% of cases of coarctation in one fetal series.
d. Valvar aortic stenosis is not commonly associated with extracardiac malformation.
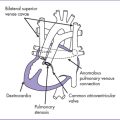
e. Interrupted aortic arch (Fig. 14-2) has a high incidence of microdeletion of chromosome 22q11. About 75% of patients with interrupted aortic arch type B diagnosed prenatally have this microdeletion.
4. Clues to fetal sonographic diagnosis.
b. Pulmonary artery larger than the aorta.
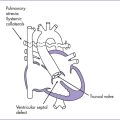
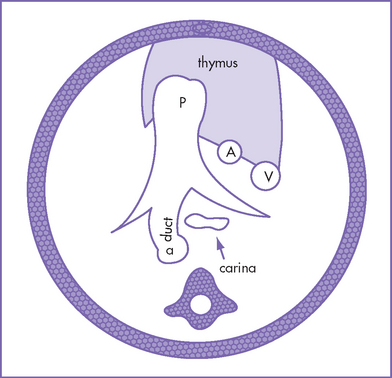
c. Abnormal aorta–to–pulmonary artery diameter ratio (diameter measured at the annulus) for gestation. Here the three-vessel view is extremely useful because the aorta size may be similar to the superior vena cava (SVC) size and significantly smaller than the main pulmonary artery (Fig. 14-3).
d. Hypoplastic transverse arch.
e. Bidirectional or left-to-right shunting at the foramen ovale (usually seen in more severe left heart pathology).
f. If a VSD is present, there will be left-to-right shunt across the VSD instead of the usual bidirectional shunt seen in isolated VSD.
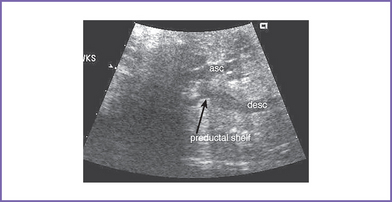
g. Discrete shelf is sometimes seen in the distal arch. The shelf confirms the presence of aortic coarctation; however, its absence does not exclude the diagnosis and its presence does not necessarily indicate a ductus arteriosus–dependent coarctation.
a. Size of the transverse arch, isthmus, and LV (including mitral and aortic valve annuli) can be evaluated sequentially for progression of the disease.
b. Critical aortic stenosis can rarely progress to hypoplastic left heart syndrome (HLHS) by term due to failure of the growth of the LV and the development of complete aortic atresia.
6. Immediate postnatal management for patients without prenatal diagnosis of CoA.
a. After birth, the clinical presentation of CoA is variable. The presentation depends on the severity of CoA and the presence of associated symptoms.
b. In neonates and young infants who present with clinical evidence of significant CoA, PGE1 infusion is initiated to support the lower body circulation (Box 14-2).
c. Surgical repair of CoA without significant intracardiac disease.
d. In the presence of a large hemodynamically significant VSD, the arch repair and VSD closure are usually performed in one stage, but mortality risk is higher.
e. Not all infants with a prenatal diagnosis of CoA require surgery as neonates. Some might not have ductus-dependant circulation and therefore may be clinically well for weeks to months.
7. Pathophysiology (Fig. 14-4).
a. Obstruction at the aortic isthmus has been subdivided into two types.
b. Two theories have tried to explain the pathogenesis of both forms of coarctation.
c. Continuous forward flow through the distal arch in utero and postnatally is a sign of significant coarctation.
d. In coarctation with more severe left heart obstruction (mitral stenosis, aortic stenosis, or LV dysfunction):
e. Diagnosis is made by echocardiography (discrete posterior shelf). This diagnosis is difficult in the presence of a patent ductus or prostaglandin infusion.
f. The infantile type is more severe than the adult type, and the young child can present with metabolic acidosis secondary to severe LV dysfunction and low cardiac output.
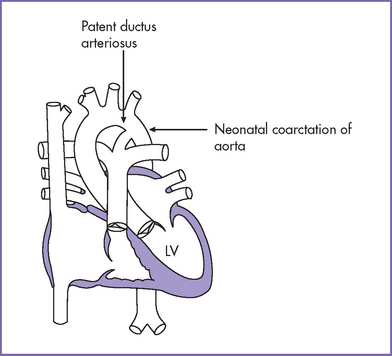
g. Neonatal coarctation involves narrowing of the aorta, most commonly the upper thoracic aorta. Up to 85% of patients with CoA have a bicuspid aortic valve.
h. In neonatal critical coarctation, the narrowing is so severe that the affected neonate might depend on the arterial duct to supply sufficient blood flow to the lower extremities.
i. In symptomatic infants, RV hypertrophy (rather than LV hypertrophy) is usually present on the electrocardiogram (ECG).
j. In asymptomatic children with CoA, the descending aorta is supplied by the LV via the aortic isthmus. Associated anomalies are uncommon, except for bicuspid aortic valve.
k. Other cardiac defects include:
l. Collateral circulation between the upper and lower body is poorly developed in the neonate.
a. The spectrum of LVOT obstruction consists of hypoplastic left heart or LV, aortic valve stenosis and bicuspid aortic valve, hypoplastic aortic arch, and CoA.
b. Wessels and colleagues (2005) described four families with presumed autosomal dominant inheritance of LVOT obstruction. In these families, LVOT obstruction showed a wide clinical spectrum, with some members having severe anomalies such as hypoplastic left heart and others having only minor anomalies such as mild aortic valve stenosis.
c. Wessels and colleagues concluded that their findings supported the suggestion that all anomalies of the LVOT obstruction spectrum are developmentally related and sometimes are caused by a single gene defect.
BOX 14-1 FACTORS AFFECTING PRENATAL COUNSELING IN ARCH ABNORMALITIES
The presence of significant extracardiac abnormalities that can affect both the prenatal and postnatal outcome
Long-term morbidity and mortality associated with repair of coarctation or interruption, such as recoarctation or systemic hypertension
Possibility of the development and progression of mitral and aortic valve obstruction
Additional intracardiac lesions that would affect the prognosis, such as single ventricle physiology
Associated syndromes or chromosomal abnormalities, such as chromosome 22 microdeletion
B. Adult coarctation of the aorta
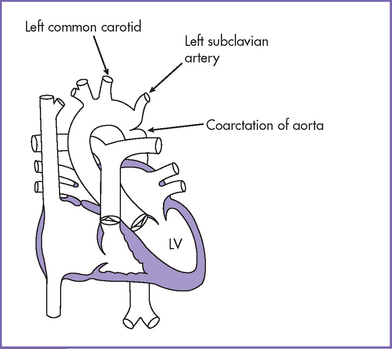
1. In the adult form of CoA (Fig 14-5), the distal arch narrowing is due to a discrete shelf in the area of the ligamentum arteriosum or the closed arterial duct. There is typically no transverse arch or isthmic hypoplasia.
2. Coarctation occurs most commonly in the region of the thoracic aorta just distal to the left subclavian artery.
3. Coarctation symptoms range widely from none to cardiac collapse. Hypertension usually occurs in the upper body. As the condition progresses, post-stenotic aneurysms often develop in the distal portion of the coarctate aorta but rarely in the proximal portion.
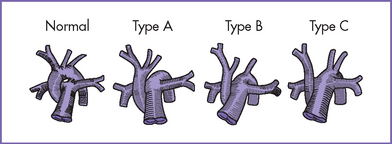
1. Interrupted aortic arch is a significantly less common form of aortic arch obstruction (Fig. 14-6).
2. Incidence is 0.019 per 1000 live births.
a. In type A (30–40% of patients) the interruption is between the left subclavian artery and descending aorta.
b. In type B (50% of patients) the interruption is between the left subclavian artery and the left common carotid (left aortic arch) or between the right subclavian artery and the right carotid artery (right aortic arch).
c. In type C (3% of patients), the interruption is between the right brachiocephalic artery and the left common carotid artery.
4. Prenatal diagnosis is confirmed by demonstrating the pathology at the level of the arch in addition to ventricular and great artery size discrepancy.
5. There are no current data on the natural history of all types of aortic interruption. However, as expected in fetal life, there is progressive ascending aortic hypoplasia. The interruption might also be associated with progressive LV outflow tract obstruction.
6. Allan and colleagues’ (1994) fetal series included six cases of aortic arch interruption (all type B). Three pregnancies were terminated and the other three babies died in the neonatal period.
III. TAKE-HOME MESSAGE
A. Diagnosis
1. Discrepancy in both ventricular and great artery size (right heart structures larger than left heart structures) before 34 weeks’ gestation is a useful marker in the diagnosis of isolated fetal coarctation provided there is no large VSD.
2. The most sensitive diagnostic feature of fetal coarctation is the narrowing of the transverse aortic arch and isthmus.
3. Pulsed Doppler is not useful in the majority of isolated CoA, because there is usually antegrade flow across the distal arch. However, color flow mapping can help to delineate the hypoplastic lumen of the distal arch for comparison with the large ductal arch.
4. When a fetus is seen for the first time late in gestation with borderline findings such as mild ventricular discrepancy, mildly abnormal aorta–to–pulmonary artery diameter ratio, and aortic arch mildly smaller than the ductal arch, it is safer to assume the diagnosis of a possible coarctation, recognizing that some may be false-positive predictions.
5. In aortic coarctation with VSD, the flow in the VSD is exclusively left to right, in contrast to bidirectional shunting in an isolated VSD.
6. The hyperoxia test might not be conclusive in the diagnosis of aortic coarctation in the presence of large arterial duct or large VSD, which provide left-to-right intracardiac shunt, thus raising the oxygen content in the lower extremity (postductal).
7. Pulse oximeter saturation in the hyperoxia test could be misleading because some babies with HLHS can have oxygen saturation in room air in the 90s. Documentation of PaO2 in an arterial blood gas is mandatory.
8. In the infantile (neonatal) type of coarctation, an arterial duct is present in about 50% of cases at the time of diagnosis.
9. Interrupted aortic arch has a high incidence of microdeletion of chromosome 22.
10. In interrupted aortic arch the aorta proximal to the site of interruption has an unusual configuration. The aorta bluntly ends at the interruption and the brachiocephalic innominate vessels are directed straight toward the upper body.
11. If there is narrowing of the LV outflow tract just under the aortic valve created by posterior malalignment of the infundibular septum, interrupted aortic arch type B could be suspected.
Allan LD, Sharland GK, Milburn A, et al. Prospective diagnosis of 1,006 consecutive cases of congenital heart disease in the fetus. J Am Coll Cardiol. 1994;23(6):1452-1458.
Clark EB. Mechanisms in the pathogenesis of congenital cardiac malformations. In: Pierpont ME, Moller JH, editors. Genetics of Cardiovascular Disease. Boston: Nijhoff; 1986:3-11.
Hanley FL. The various therapeutic approaches to aortic coarctation: Is it fair to compare? J Am Coll Cardiol. 1996;27(2):471-472.
Hornberger LK, Sahn DJ, Kleinman CS, et al. Antenatal diagnosis of coarctation of the aorta: A multicenter experience. J Am Coll Cardiol. 1994;23(2):417-423.
Hornberger LK, Weintraub RG, Pesonen E, et al. Echocardiographic study of the morphology and growth of the aortic arch in the human fetus. Observations related to the prenatal diagnosis of coarctation. Circulation. 1992;86(3):741-747.
Scott WA, Rocchini AP, Bove EL, et al. Repair of interrupted aortic arch in infancy. J Thorac Cardiovasc Surg. 1988;96(4):564-568.
Shinebourne EA, Tam AS, Elseed AM, et al. Coarctation of the aorta in infancy and childhood. Br Heart J. 1976;38(4):375-380.
Skoda J. Demonstration eines Falles Ven Obliteration de Aorta. Wochenblatt Zeischrift de kaiserlichen-Konighiche Gesellschaft der Aerttze Zur Wien. 1995;1:710-720.
Trines J, Hornberger LK. Evolution of heart disease in utero. Pediatr Cardiol. 2004;25(3):287-298.
Van Son JA, Falk V, Schneider P, et al. Repair of coarctation of the aorta in neonates and young infants. J Card Surg. 1997;12(3):139-146.
Wessels MW, Berger RM, Frohn-Mulder IM, et al. Autosomal dominant inheritance of left ventricular outflow tract obstruction. Am J Med Genet A. 2005;134(2):171-179.
Yetman AT, Nykanen D, McCrindle BW, et al. Balloon angioplasty of recurrent coarctation: A 12-year review. J Am Coll Cardiol. 1997;30(3):811-816.
Ziemer G, Jonas RA, Perry SB, et al. Surgery for coarctation of the aorta in the neonate. Circulation. 1986;74(3 Pt 2):I25-I31.