Chapter 60 Coagulopathies
General considerations
Disseminated intravascular coagulation
Disseminated intravascular coagulation (DIC) is often described as a secondary disease process typically associated with other disease entities.1 The presence of DIC increases the risk of mortality beyond that associated with the primary disease.2 A cascading activation of both procoagulants and fibrinolysis leads to simultaneous, uncontrolled hemorrhages and diffuse thrombosis of small and large vessels affecting virtually all mucocutaneous tissues and various organs, ultimately resulting in end-organ failure (see Box 60.1). As such, DIC is regarded as one of the most common causes of death. But not all presentations of DIC are lethal; it may also manifest as low-grade disease with a chronic or compensated course.
The currently established theory of pathogenesis is an initial activation of the coagulation cascade following inflammation or damage to tissue and vascular endothelial cells.3 Fibrin formation leads to microvascular or macrovascular thrombosis. Soon thereafter, fibrinolysis is upregulated and the consumption of platelets in the obstructed microcirculation causes a systemic thrombocytopenia (cell count <100 000/mm3), thereby generating an environment of simultaneous clotting and extrusion of hemorrhages. Despite a common pathophysiologic pathway, clinical manifestations of the disease may vary widely (see Box 60.1), complicating diagnosis and treatment management.
The onset of DIC is usually marked by fever, shock, acidosis and, more specifically, widespread hemorrhaging, acral cyanosis, gangrene and end-organ failure. Bleeding from more than three unrelated sites at once and a recent medical history compatible with known causes of DIC can direct the clinician towards the diagnosis. Definite diagnosis is heavily based on positive laboratory test results for platelet count, D-dimer (fibrin degradation product or FDP), antithrombin-III, protein C, prothrombin time, partial thromboplastin time, and fibrinogen.4
Treatment of DIC is difficult and should primarily address the underlying cause, however, removal of its cause does not necessarily alleviate the process in all cases and prognosis of a fulminant DIC is very poor. Replacement treatment such as frozen plasma transfusion or infusion of antithrombin-III, fibrinogen, and platelet-concentrates can help to limit hemorrhages. In some cases, anticoagulant treatment may be required. Further, activated protein C has been suggested to treat this condition,5 however, the efficacy of this approach for clinically relevant outcome parameters remains unclear.6
Idiopathic thrombocytopenic purpura and thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura (TTP) is a severe thrombotic microangiopathy caused by platelet adhesion and aggregation mediated by endothelial cell-attached ultra-large von Willebrand factor multimers (ULVWF). The underlying cause is a congenital or acquired (autoimmune) deficiency of von Willebrand factor-cleaving proteases such as ADAMTS-13.7 TTP is more frequent in young female adults than in males or older individuals. Most presentations of the disease are idiopathic, although familial and secondary TTP have been described. Pregnancy, treatment with certain drugs (clopidogrel, cyclosporine, tacrolimus), and malignancies are associated with a higher incidence of TTP. TTP is accompanied by profound thrombocytopenia, erythrocyte fragmentation, and increased serum levels of lactatdehydrogenase (LDH).8 Clinically it manifests with fever, renal failure, neurological symptoms, purpura, and signs of hemolytic anemia. There is often a prodromal phase that may consist of headache, dizziness, nausea, and abdominal pain, presumably caused by microinfarction of the viscera. Though there are chronic smoldering patterns of the disease, an acute and fulminant course is not uncommon and, if left untreated, can be lethal. In some cases, ocular manifestations as described below may present as the first signs of the disease; thus, prompt referral to a hematologist may be life-saving.9 Plasmapheresis is the principal treatment for this condition and can be supplemented by immunosuppressive therapy (cyclosporine A, rituximab) in refractory cases, although relapses are generally the rule.10 Of note, systemic treatment with bevacizumab, a monoclonal antibody to VEGF which is increasingly used via intravitreal delivery for ocular neovascular disorders, can induce a TTP-like microangiopathy.11
HELLP syndrome
The incidence of the HELLP syndrome (toxemia of pregnancy) is reported as being between 0.2 and 0.6% of all pregnancies.12 Major symptoms are hemolytic anemia (H), elevated liver enzymes (EL), and low platelet count (LP). While being regarded as a microangiopathy much like TTP, it can also convert into a fulminant DIC in 20% of cases. The mortality rate is around 1.1% with adequate management, yet other serious complications such as abruptio placentae (16%), acute renal failure (7.7%), or pulmonary edema (6%) can occur more frequently.13
Ophthalmic involvement
Although there are many differences regarding the underlying etiology or management of the various coagulopathies discussed in this chapter, they all may present with similar ophthalmic manifestations (see Fig. 60.1). The precise frequency of ocular complications however is unknown, as many cases may remain unrecognized due to the life-threatening nature of those systemic diseases. Relatively few reports are available regarding ocular complications of DIC as most cases are diagnosed and reported after the patients have died.14 By comparison, many more reports can be found in the literature describing ophthalmic findings in TTP and ITP.15–17 Some groups have reported that ocular signs and symptoms occur in up to 14% of patients with TTP.18
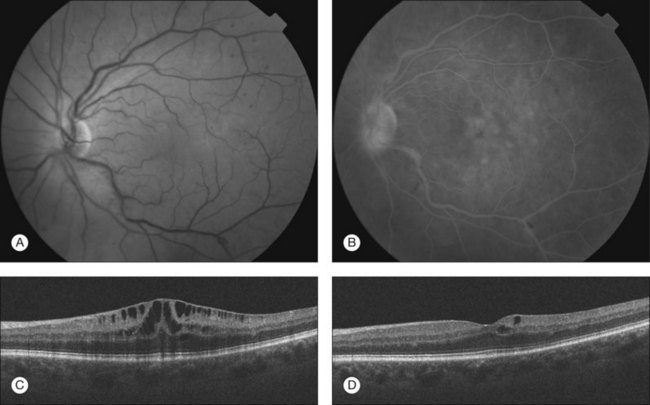
The hypercoagulable state in these individuals typically leads to choriocapillaris occlusion by fibrin-platelet clots, whereas subcutanenous or subconjunctival,19 choroidal, retinal or vitreous hemorrhages15,17,20 are thought to be indicative of thrombocytopenia and anemia. A patchy delay in filling of the choroidal vessels on fluorescein angiography can be an early sign of ophthalmic complications. Pathophysiologically, microthrombi in the choriocapillaris cause localized ischemic injury to the retinal pigment epithelium (RPE), resulting in a dysfunction of the outer blood–retinal barrier as well as a decreased ability of the RPE to transport fluid out of the subretinal space.21 Fluid extravasation from choroidal vessels can then pass through small disruptions of the RPE, extending into the subretinal space and manifesting as serous retinal detachments.22 Both subretinal and intraretinal fluid accumulations have been reported.9 Some groups observed large tears of the RPE leading to acute decrease in vision.23 Hemorrhage in the posterior segment may vary from subclinical small intraretinal dot hemorrhages to widespread sub-RPE, subretinal or intravitreal hemorrhage resulting in dramatic decrease in vision.15,17,20 Choroidal hemorrhage seems to be more specific and has rarely been described in conditions other than the coagulopathic disorders described in this chapter. Cotton-wool spots, typically located around the optic disc, are generally related to a secondary cause such as hypertension or other systemic disease.24 Whereas the decrease in vision from fluid accumulation and hemorrhage may recover following resolution and resorption,16 visual impairment due to choroidal infarction is frequently permanent.25
The preferential localization of fibrin-formation and occlusion in the choroidal bed at the posterior pole has not yet been fully explained. Cogan proposed the hypothesis that a sudden deceleration of the blood flow between the short posterior ciliary arteries and the choriocapillaris sinusoids greatly facilitates clot precipitation in this region.14 Although early case reports did not demonstrate any involvement of the retinal vasculature, more recently, Lewis et al. published a case of severe DIC in meningococcemia with impaired retinal circulation.26 The authors speculated that this may also have been due to septic emboli and not a direct effect of DIC. Schwartz et al. reported a case with bilateral central retinal artery occlusion and central retinal vein occlusion in TTP secondary to adult-onset Still’s disease.27
There are also reports describing ocular involvement of DIC and TTP in neonates.28 Common causes of mechanisms in this setting include placenta previa, sepsis, trauma, hepatocellular failure or infant respiratory distress syndrome (IRDS). The ocular fundi show extensive hemorrhages in the retina or the vitreous, usually with concomitant focal retinal detachments. In selected neonatal cases, anterior segment manifestations were also observed with hyphema, thrombi in the iris and the ciliary body, and subconjunctival hemorrhage.29
Ocular manifestations of toxemia of pregnancy are discussed in detail in Chapter 92 (Pregnancy-related disease). As the HELLP syndrome may be associated with DIC, all of the above-mentioned complications may occur in this condition. Additionally, hypertensive retinal vascular changes are not uncommon. Mere presence of serous retinal detachments should not be used as an indicator of DIC, as these may also occur in pregnancy independent of the presence or absence of DIC and pregnancy-associated hypertension.30,31
Conclusion
DIC and related coagulopathies generally exhibit very similar ocular manifestations, which include widespread retinal or vitreous hemorrhage, subretinal or intraretinal fluid accumulation and choroidal infarction (see Fig. 60.1). The anterior segment is rarely involved in these disease processes. While visual loss related to these conditions is usually not the chief driver in the treatment of the underlying potentially life-threatening systemic diseases, ocular findings may be the initial presentation that enables the diagnosis of these disorders.
1 Bick RL. Disseminated intravascular coagulation: current concepts of etiology, pathophysiology, diagnosis, and treatment. Hematol/Oncol Clin North Am. 2003;17:149–176.
2 Gando S, Kameue T, Nanzaki S, et al. Disseminated intravascular coagulation is a frequent complication of systemic inflammatory response syndrom. Thromb Haemost. 1996;75:224–228.
3 Levi M, Nieuwdorp M, vander Poll T, et al. Metabolic modulation of inflammation-induced activation of coagulation. Semin Thromb Hemost. 2008;34:26–32.
4 Favaloro EJ. Laboratory testing in disseminated intravascular coagulation. Semin Thromb Hemost. 2010;36:458–468.
5 Bernard GR, Vincent AL, Laterre PF, et al. Efficiacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709.
6 Levi M, Schultz M. Coagulopathy and platelet disorders in critically ill patinets. Minerva Anestesiol. 2010;76:851–859.
7 Tsai HM. Platelet activation and the formation of the platelet plug: deficiency of ADAMTS13 causes thrombotic thrombocytopenic purpura. Arterioscler Thromb Vasc Biol. 2003;23:388–396.
8 Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600.
9 Hay-Smith G, Sagoo MS, Raina J. Fatal thrombotic thrombocytopenic pupura presentng with choroidal vasculopathy and serous retinal detachment. Eye. 2006;20:982–984.
10 Tsai H. Current concepts in thrombotic thrombocytopenic purpura. Ann Rev Med. 2006;57:419–436.
11 Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136.
12 Stella C, Malik K, Sibai B. HELLP syndrome: an atypical presentation. Am J Obstet Gynecol. 2008;198:e6–e8.
13 Sibai BM, Ramadan MK, Usta I, et al. Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome). Am J Obstet Gynecol. 1993;169:1000–1006.
14 Cogan DG. Ocular involvement in disseminated intravascular coagulopathy. Arch Ophthalmol. 1975;93:1–8.
15 Majji A, Bhatia K, Mathai A. Spontaneous bilateral peripapillay, subhyalid and vitreous hemorrhage with severe anemia secondary to idiopathic thrombocytopenic purpura. Ind J Ophthalmol. 2010;58:234–236.
16 Meyer CH, Callizo J, Mennel S, et al. Complete resorption of retinal hemorrhages in idiopathic thrombocytopenic purpura. Eur J Ophthalmol. 2007;17:128–129.
17 Karagiannis D, Gregor Z. Valsalva retinopathy associated with idiopathic thrombocytopenic purpura and positive antiphospholipid antibodies. Eye. 2006;20:1447–1449.
18 Wyszynski RE, Fran KE, Grossniklaus HE. Bilateral retinal detachments in thrombotic thrombocytopenic purpura. Grafes. Arch Clin Exp Ophalmol. 1988;226:501–504.
19 Sodhi PK, Jose R. Subconjunctival hemorrhage: the first presenting clinical feature of idiopathic thrombocytopenic purpura. Jpn J Ophthalmol. 2003;47:316–318.
20 Okuda A, Inoue M, Shinoda K, et al. Massive bilateral vitreoretinal hemorrhage in patient with chronic refractory idiopathic thrombocytopenic purpura. Graefes Arch Clin Exp Ophthalmol. 2005;243:1190–1193.
21 Nanayakkara P, Gans RO, Reichert-Thoen J, et al. Serous retinal detachment as an early presentation of thrombotic thrombocytopenic purpura. Eur J Intern Med. 2000;11:286–288.
22 Jellie HG, Gonder JR, Canny CL, et al. Ocular involvement in thrombotic thrombocytopenic purpura: the angiographic and histopathological features. Can J Ophthalmol. 1984;19:279–283.
23 Hartley KL, Benz MS. Retinal pigment epithelium tear associated with a serous detachment in a patient with thrombotic thrombocytopenic purpura and hypertension. Retina. 2004;24:806–808.
24 Matsuo T, Matsuura S, Nakagawa H. Retinopathy in a patient with thrombotic thrombocytopenic purpura complicated by polymyositis. Jpn J Ophthalmol. 2000;44:161–164.
25 Patel N, Riordan-Eva P, Chong V. Persistent visual loss after retinochoroidal infarction in pregnancy-induced hypertension and disseminated intravascular coagulation. J Neuroophthalmol. 2005;25:128–130.
26 Lewis K, Herbert EN, Williamson TH. Severe ocular involvement in disseminated intravascular coagulation complicating meningococcaemia. Graefes Arch Clin Exp Ophthalmol. 2005;243:1069–1070.
27 Schwartz SG, Hickey M, Puliafito CA. Bilateral CRAO and CRVO from thrombotic thrombocytopenic purpura: OCT findings and treatment with triamcinolone acetonide and bevacizumab. Ophthal Surg Lasers Imaging. 2006;37:420–422.
28 Schaffer DB. Eye findings in intrauterine infections. Clin Perinatol. 1981;8:415–443.
29 Ortiz JM, Yanoff M, Cameron JD, et al. Disseminated intravascular coagulation in infancy and in the neonate. Ocular findings. Arch Ophthalmol. 1982;100:1413–1415.
30 Brismar G, Schimmelpfennig W. Bilateral exudative retinal detachment in pregnancy. Acta Ophthalmol (Copenh). 1989;67:699–702.
31 Mayo GL, Tolentino MJ. Images in clinical medicine. Central serous chorioretinopathy in pregnancy. N Engl J Med. 2005;353:e6.