[level-membership-for-anesthesiology-category]
17 Coagulation Monitoring
CPB itself induces a “whole-body inflammatory response” because of contact of blood and cellular elements with the extracorporeal circuit. The resultant alterations include leukocyte activation, release of inflammatory mediators, free radical formation, complement activation, kallikrein release, platelet activation, and stimulation of the coagulation and fibrinolytic cascades. This complex interplay of systems induces a coagulopathy characterized by microvascular coagulation, platelet dysfunction, and enhanced fibrinolysis.1,2 The homeostatic perturbations incurred during CPB are a major cause for the postbypass coagulopathy that is seen even after the effects of heparin are reversed with protamine.
The need to monitor anticoagulation during and after surgery is the reason that the cardiac surgical arena has evolved into a major site for the evaluation and use of hemostasis monitors. The rapid and accurate identification of abnormal hemostasis has been the major impetus toward the development of point-of-care tests that can be performed at the bedside or in the operating room. The detection and treatment of specific coagulation disorders in a timely and cost-efficient manner are major goals in hemostasis monitoring for the cardiac surgical patient. This chapter discusses the mechanisms of normal coagulation and how they are affected by CPB. The latter portion discusses the laboratory and point-of-care tests available for monitoring the coagulation system. A comprehensive overview of hemostasis, transfusion medicine, and the management of coagulopathy and bleeding disorders after CPB is provided in Chapters 30 and 31.
Hemostasis

Anticoagulation for Cardiopulmonary Bypass
Heparin acts as an antithrombin III (AT III) agonist and accelerates AT III binding to thrombin.3–5 In the absence of AT III, heparin is clinically ineffective as an anticoagulant; adequate AT III activity is necessary in patients about to undergo heparinization for cardiac surgical procedures.6–9
Monitoring heparin effect
The first clotting time to be used to measure heparin’s effect was the whole-blood clotting time (WBCT) or the Lee–White WBCT. This simply requires whole blood to be placed in a glass tube, maintained at 37°C, and manually tilted until blood fluidity is no longer detected. This test fell out of favor for monitoring the cardiac surgical patient because it was so labor intensive and required the undivided attention of the person performing the test for periods up to 30 minutes. Although the glass surface of the test tube acts as an activator of factor XII, the heparin doses used for cardiac surgery prolong the WBCT to such a profound degree that the test is impractical as a monitor of the effect of heparin during cardiac surgery.10 To speed the clotting time so that the test was appropriate for clinical use, activators were added to the test tubes, and the activated coagulation time (ACT) was introduced into practice.11

Activated Coagulation Time
The ACT was first introduced by Hattersley in 1966 and is still the most widely used monitor of heparin effect during cardiac surgery. Whole blood is added to a test tube containing an activator, diatomaceous earth (celite) or kaolin. The presence of activator augments the contact activation phase of coagulation, which stimulates the intrinsic coagulation pathway. ACT can be performed manually, whereby the operator measures the time interval from when blood is injected into the test tube to when clot is seen along the sides of the tube. More commonly, the ACT is automated as it is in the Hemochron and Hemotec systems. In the automated system, the test tube is placed in a device that warms the sample to 37°C. The Hemochron device (International Technidyne, Edison, NJ) rotates the test tube, which contains celite activator and a small iron cylinder, to which 2 mL whole blood is added. Before clot forms, the cylinder rolls along the bottom of the rotating test tube. When clot forms, the cylinder is pulled away from a magnetic detector, interrupts a magnetic field, and signals the end of the clotting time. Normal ACT values range from 80 to 120 seconds. The Hemochron ACT also can be performed using kaolin as the activator in a similar manner (Figure 17-1).

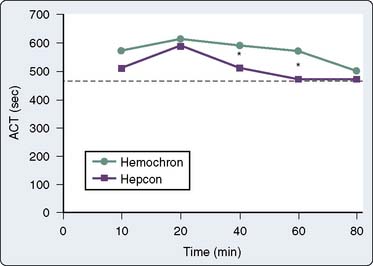
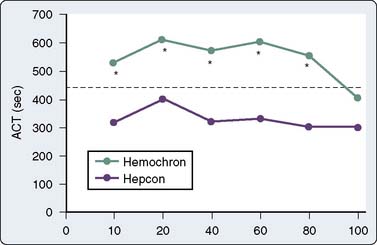
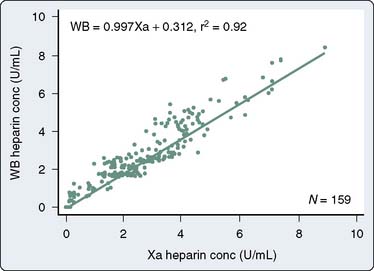
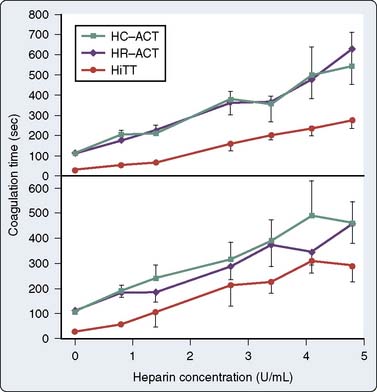
The Hemotec ACT device (Medtronic Hemotec, Parker, CO) is a cartridge with two chambers that contain kaolin activator and is housed in a heat block. Blood (0.4 mL) is placed into each chamber, and a daisy-shaped plunger is raised and passively falls into the chamber. The formation of clot will slow the rate of descent of the plunger, and this decrease in velocity of the plunger is detected by a photo-optical system that signals the end of the ACT test. The Hemochron and Hemotec ACTs have been compared in a number of investigations and have been found to differ significantly at low heparin concentrations.12 However, differences in heparin concentration, activator concentration, and the measurement technique make comparison of these tests difficult and have led to the realization that the Hemochron ACT result and the Hemotec ACT result are not interchangeable. In adult patients given 300 U/kg heparin for CPB, the Hemochron and Hemotec (Hepcon) ACTs were both therapeutic at all time points; however, at two points, the Hemochron ACT was statistically longer13 (Figure 17-2). This difference was even more pronounced in pediatric patients, who have greater heparin consumption rates (Figure 17-3). The apparent “overestimation” of ACT by the Hemochron device during hypothermic CPB may be because of the different volumes of blood that each assay warms to 37°C.
The ACT test can be modified by the addition of heparinase. With this modification, the coagulation status of the patient can be monitored during CPB while the anticoagulant effects of heparin are eliminated. Because this test is a side-by-side comparison of the untreated ACT to the heparinase ACT, it also has the advantage of being a rapid test for the assessment of a circulating heparin-like substance or for residual heparinization after CPB.14
With the introduction of ACT monitoring into the cardiac surgical arena, clinicians have been able to more accurately titrate heparin and protamine dosages.11,15 As a result, many investigators report reductions in blood loss and transfusion requirements, although many of these studies used retrospective analyses.16 The improvements in postoperative hemostasis documented with ACT monitoring are potentially attributable to better intraoperative suppression of microvascular coagulation and improved monitoring of heparin reversal with protamine.17
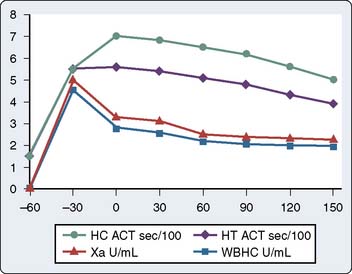
ACT monitoring of heparinization is not without pitfalls, and its use has been criticized because of the extreme variability of the ACT and the absence of a correlation with plasma heparin levels (Figure 17-4). Many factors have been suggested to alter the ACT, and these factors are prevalent during cardiac surgical procedures. When the extracorporeal circuit prime is added to the patient’s blood volume, hemodilution occurs and may theoretically increase ACT. Evidence suggests that this degree of hemodilution alone is not enough to actually alter ACT. Hypothermia increases ACT in a “dose-related” fashion. It has been shown by Culliford et al18 that, although hemodilution and hypothermia significantly increase the ACT of a heparinized blood sample, similar increases do not occur in the absence of added heparin. The effects of platelet alterations are a bit more problematic. At mild-to-moderate degrees of thrombocytopenia, the baseline and heparinized ACT are not affected. It is not until platelet counts are reduced to less than 30,000 to 50,000/μL that ACT may be prolonged.19 Patients treated with platelet inhibitors such as prostacyclin, aspirin, or platelet-membrane-receptor antagonists have a prolonged heparinized ACT compared with patients not treated with platelet inhibitors.20 This ACT prolongation is not related exclusively to decreased levels of platelet factor 4 (PF4) (PF4 is a heparin-neutralizing substance) because it also occurs when blood is anticoagulated with substances that are not neutralized by PF4. Platelet lysis, however, significantly shortens the ACT because of the release of PF4 and other platelet membrane components, which may have heparin-neutralizing activities.21 Gravlee et al22 showed that anesthesia and surgery decrease the ACT and create a hypercoagulable state, possibly by creating a thromboplastic response or through activation of platelets.
During CPB, heparin decay varies substantially and its measurement is problematic because hemodilution and hypothermia alter the metabolism of heparin. In a CPB study, Mabry et al23 found that the consumption of heparin varied from 0.01 to 3.86 U/kg/min and there was no correlation between the initial sensitivity to heparin and the rate of heparin decay.23 In the pediatric population, the consumption of heparin is increased to more than that of adult levels. The heparin administration protocol for pediatric patients undergoing CPB should account for a large volume of distribution, increased consumption, and a shorter elimination half-life. In monitoring the effects of heparin in pediatric patients, the minimum acceptable ACT value should be increased or an additional monitor should be used. The discrepancy between the Hemochron ACT and the Hemotec ACT that is demonstrated in Figure 17-2 is even more pronounced in pediatric patients (see Figure 17-3). Some investigators recommend the maintenance of heparin concentrations in addition to ACT during pediatric congenital heart surgery to ensure that optimal anticoagulation is being achieved.24,25

Cascade POC (Point of Care) System
A completely different technology for measuring the effect of heparin is used by the Cascade POC analyzer (Helena, Beaumont, TX; Formerly Rapid Point Coagulation Analyzer Bayer Diagnostics, Tarrytown, NY). This test system contains disposable cards with celite activator for the measurement of heparin activity. This variation of the ACT is called the “heparin management test” (HMT). This card contains paramagnetic iron oxide particles that move in response to an oscillating magnetic field within the device. When clot formation occurs, movement of the iron oxide particles is decreased and the end of the test is signaled. This system is capable of measuring prothrombin time (PT) and activated partial thromboplastin time (aPTT), which is discussed later. The suitability of this platform for the monitoring of ACT during cardiac surgery has been demonstrated in a variety of clinical studies.26,27 Suitability for monitoring heparinization in the interventional cardiology laboratory also has been reported. HMT correlates well with anti-Xa heparin activity in CPB patients and is less variable than standard ACT measures. In a comparison with ACT, the coefficients of variation were similar between the tests at baseline but were three times greater for the ACT during heparinization. This degree of agreement with plasma anti-Xa measurements has not been demonstrated universally when studying blood from patients undergoing CPB.28

Heparin Resistance
Heparin resistance is documented by an inability to increase the ACT of blood to expected levels despite an adequate dose and plasma concentration of heparin. In many clinical situations, especially when heparin desensitization or a heparin inhibitor is suspected, heparin resistance can be treated by administering increased doses of heparin in a competitive fashion. If an adequately prolonged clotting time is ultimately achieved using greater than expected doses of heparin, a better term than heparin resistance would be heparin tachyphylaxis of “altered heparin responsiveness.” During cardiac surgical procedures, the belief that a safe minimum ACT value of 300 to 400 seconds is required for CPB is based on a few clinical studies and a relative paucity of scientific data. However, inability to attain this degree of anticoagulation in the heparin-resistant patient engenders the fear among cardiac surgical providers that the patient will experience a microvascular consumptive coagulopathy or that clots will form in the extracorporeal circuit. This potential for fibrin formation in the extracorporeal circuit was reported by Young et al,29 who found increased production of fibrin monomer and consumption of fibrinogen and platelets in six of nine rhesus monkeys when the ACT declined to less than 400 seconds. However, Metz and Keats30 reported no adverse effects of thrombosis or excessive bleeding in 51 patients undergoing CPB whose ACT was less than 400 seconds. In a porcine model, the group whose ACT was maintained between 250 and 300 seconds did not have excessive consumption of coagulation factors, increases in fibrin monomer formation, or changes in oxygenator performance compared with the group whose ACT was maintained at greater than 450 seconds.31
Many clinical conditions are associated with heparin resistance.32 Sepsis, liver disease, and pharmacologic agents represent just a few33,34 (Table 17-1). Many investigators have documented decreased levels of AT III secondary to heparin pretreatment,35 whereas others have not found decreased AT III levels.36 Esposito et al37 measured coagulation factor levels in patients receiving preoperative heparin infusions and found that a lower baseline ACT was the only risk factor for predicting heparin resistance compared with patients not receiving preoperative heparin.
TABLE 17-1 Disease States Associated with Heparin Resistance
| Disease State | Comment |
|---|---|
| Newborn | Decreased AT III levels until 6 months of age |
| Venous thromboembolism | May have increased factor VIII level |
| Accelerated clearance of heparin | |
| Pulmonary embolism | Accelerated clearance of heparin |
| Congenital AT III deficiency | 40% to 60% of normal AT III concentration |
| Type I | Reduced synthesis of normal/abnormal AT III |
| Type II | Molecular defect within the AT III molecule |
| Acquired AT III deficiency | < 25% of normal AT III concentration |
| Preeclampsia | Levels unchanged in normal pregnancy |
| Cirrhosis | Decreased protein synthesis |
| Nephrotic syndrome | Increased urinary excretion of AT III |
| DIC | Increased consumption of AT III |
| Heparin pretreatment | 85% of normal AT III concentration because of accelerated clearance |
| Estrogen therapy | |
| Cytotoxic drug therapy (l-asparaginase) | Decreased protein synthesis |
ATIII, antithrombin III; DIC, disseminated intravascular coagulation.
Patients receiving preoperative heparin therapy traditionally require larger heparin doses to achieve a given level of anticoagulation when that anticoagulation is measured by the ACT. Presumably, this “heparin resistance” is due to deficiencies in the level or activity of AT III.34,37–40 Other possible causes include enhanced factor VIII activity and platelet dysfunction causing a decrease in ACT response to heparin. Levy et al40 have shown that the in vitro addition of AT III enhances the ACT response to heparin. Lemmer and Despotis39 demonstrated that this heparin resistance, as measured by the ACT, does not correlate with preoperative AT III levels. It is unclear that these patients have increased heparin requirements during CPB because the ideal ACT and monitoring techniques have yet to be elucidated. Nicholson et al41 demonstrated that the temporal courses of ACT and AT III concentration do not parallel each other, further suggesting that AT III depletion is not the sole cause of heparin resistance during CPB. Lower ACTs (lower than 480s) were tolerated in this study with no adverse outcome. AT III concentrate is available as a heat-treated human product or in recombinant form and represents a reasonable method of treating patients with documented AT III deficiency (Box 17-1). Heparin responsiveness in the form of increased ACT levels is documented both in vitro and in vivo when heparin-resistant patients are treated with AT III.
BOX 17-1. Heparin Resistance
In a multicenter, randomized, placebo-controlled trial using 75 U/kg recombinant AT III, AT III–treated patients received less FFP to augment ACT levels and also had evidence of less hemostatic activation while on CPB.42 There was a trend toward increased blood loss in the AT III group, which is potentially a dose effect that requires further investigation.43,44

Heparin-Induced Thrombocytopenia
The options for treating these patients are few. If the clinician has the luxury of being able to discontinue heparin for a few weeks, often the antibody disappears and allows a brief period of heparinization for CPB without complication.45,46 Changing the tissue source of heparin was an option when bovine heparin was predominantly in use. Some types of low-molecular-weight heparin have been administered to patients with HIT, but reactivity of the particular low-molecular-weight heparin with the patient’s platelets should be confirmed in vitro. Supplementing heparin administration with pharmacologic platelet inhibition using prostacyclin, iloprost, aspirin, or aspirin and dipyridamole have been reported, all with favorable outcomes. The use of tirofiban with unfractionated heparin has been used in this clinical circumstance. Plasmapheresis may be used to reduce antibody levels. The use of heparin could be avoided altogether through anticoagulation with direct thrombin inhibitors such as argatroban, hirudin, or bivalirudin. These thrombin inhibitors have become standard of care in the management of the patient with HIT (Box 17-2). Bivalirudin has been studied in multicenter trials in HIT patients who must undergo CPB.47 Monitoring the direct thrombin inhibitors during CPB is discussed later in this chapter.

Measurement of Heparin Sensitivity
Even in the absence of heparin resistance, patient response to an intravenous bolus of heparin is extremely variable.48 The variability stems from different concentrations of various endogenous heparin-binding proteins such as vitronectin and PF4. This variability exists whether measuring heparin concentration or the ACT; however, variability seems to be greater when measuring the ACT. Because of the large interpatient variation in heparin responsiveness and the potential for heparin resistance, it is critical that a functional monitor of heparin anticoagulation (with or without a measure of heparin concentration) be used in the cardiac surgical patient. Bull et al49 documented a threefold range of ACT response to a 200-U/kg heparin dose and similar discrepancy in heparin decay rates and, thus, recommended the use of individual patient dose–response curves to determine the optimal heparin dose. This is the concept on which point-of-care individual heparin dose–response (HDR) tests are based.
An HDR curve can be generated manually using the baseline ACT and the ACT response to an in vivo or in vitro dose of heparin. Extrapolation to the desired ACT provides the additional heparin dose required for that ACT. Once the actual ACT response to the heparin dose is plotted, further dose–response calculations are made based on the average of the target ACT and the actual ACT (Figure 17-5). This methodology was first described by Bull et al49 and forms the scientific basis for the automated dose–response systems manufactured by Hemochron and Hemotec. The Hemochron RxDx system uses the heparin-response test, which is an ACT with a known quantity of in vitro heparin (3 IU/mL). A dose–response curve is generated that enables calculation of the heparin dose required to attain the target ACT using an algorithm that incorporates the patient’s baseline ACT, estimated blood volume, and heparin-response test. The patient’s heparin sensitivity can be calculated in seconds per international units per milliliter (sec/IU/mL) by dividing the heparin-response test by 3 IU/mL.
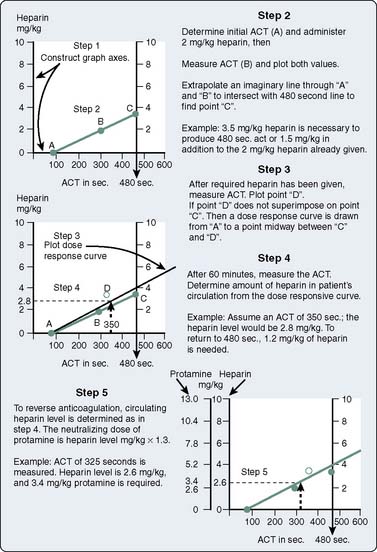
Figure 17-5 Construction of a dose–response curve for heparin. ACT, activated coagulation time.
(From Bull BS, Huse WM, Brauer FS, et al: Heparin therapy during extracorporeal circulation: II. The use of a dose-response curve to individualize heparin and protamine dosage. J Thorac Cardiovasc Surg 69:685–689, 1975.)
The RxDx system also provides an individualized protamine dose using the protamine-response test (PRT). This is an ACT with one of two specific quantities of protamine, depending on the amount of circulating heparin suspected (2 or 3 IU/mL). The protamine dose needed to return the ACT to baseline can be calculated on the basis of a protamine–response curve using the patient’s heparinized ACT, the PRT, and an estimate of the patient’s blood volume. Jobes et al50 reported that the heparin dose directed by the RxDx system resulted in ACT values far greater than the target ACT. In their patients, in vivo heparin sensitivity was higher than in vitro sensitivity. RxDx also resulted in lower protamine doses, lower postoperative mediastinal tube losses, and reduced transfusion requirements compared with a ratio-based system of heparin/protamine administration. In a larger study that standardized the treatment of heparin rebound, the reduced protamine dose was confirmed; however, the reductions in bleeding were not substantiated.51 The use of a protamine dose–response curve has been shown to successfully reduce the protamine dose in vascular surgery compared with standard weight-based protamine dosing.52
The Hepcon HMS system uses the HDR cartridge in the Hepcon instrument (Figure 17-6). Each cartridge houses six chambers. Chambers 1 and 2 contain heparin at a concentration of 2.5 IU/mL, chambers 3 and 4 contain heparin at a concentration of 1.5 IU/mL, and chambers 5 and 6 do not contain heparin. Once information regarding patient weight, height, and CPB prime volume is entered, the information that can be obtained from this test includes the baseline ACT (chambers 5 and 6) and an HDR slope. The dose–response slope, which is the increase in ACT from 1.5 to 2.5 IU/mL heparin, is extrapolated to the desired target ACT or target heparin concentration and the heparin dose is calculated.32,53

Heparin Concentration
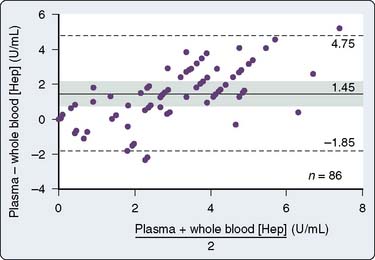
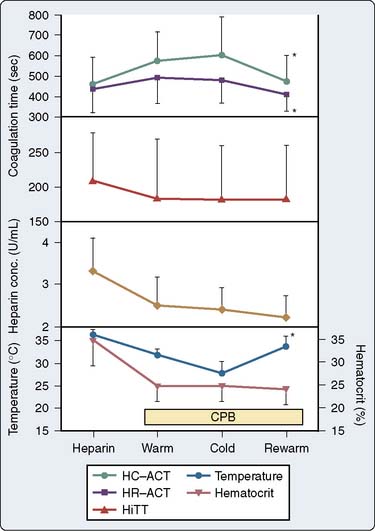
Proponents of ACT measurement to guide anticoagulation for CPB argue that a functional assessment of the anticoagulant effect of heparin is mandatory and that the variability in ACT represents a true variability in the coagulation status of the patient. Opponents argue that during CPB, the sensitivity of the ACT to heparin is altered and ACT does not correlate with heparin concentration or with anti–factor Xa activity measurement. Heparin concentration can be measured using the Hepcon HMS system (Medtronic Hemotec), which uses an automated protamine titration technique. With a cartridge with four or six chambers containing tissue thromboplastin and a series of known protamine concentrations, 0.2 mL whole blood is automatically dispensed into the chambers. The first channel to clot is the channel whose protamine concentration most accurately neutralizes the heparin without a heparin or a protamine excess. Because protamine neutralizes heparin in the ratio of 1 mg protamine per 100 units heparin, the concentration of heparin in the blood sample can be calculated. A cartridge that monitors heparin concentration over a wide range can be used first, followed by another cartridge that can measure heparin concentrations within a more narrow range. The maintenance of a stable heparin concentration rather than a specific ACT level usually results in greater doses of heparin being administered because the hemodilution and hypothermia on CPB increase the sensitivity of the ACT to heparin. The measure of heparin concentration has been shown to correlate more closely with anti–factor Xa activity measurements than the ACT during CPB54 (Figure 17-7), although the precision and bias of the test may not prove to be acceptable for exclusive use clinically (Figure 17-8).
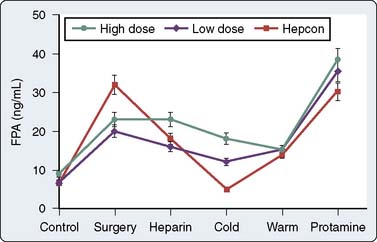
In a small, prospective, randomized study comparing ACT and heparin concentration monitoring, Gravlee et al55 demonstrated increased mediastinal tube drainage after surgery in the heparin concentration group, a finding that was initially attributed to greater total heparin doses. However, heparin rebound was not systematically assessed. In a follow-up study, the authors found a greater incidence of heparin rebound in the heparin concentration group, which, when treated, resulted in no difference in bleeding between the ACT and heparin concentration groups.56 In a prospective, randomized trial, Despotis et al57 demonstrated that by using a transfusion algorithm in association with Hepcon-based heparin management, chest tube drainage was minimally reduced and transfusion of non–red blood cell products could be significantly reduced relative to a group of patients who had ACT-based heparin management. They attributed their results to better preservation of the coagulation system by high heparin doses because the doses of heparin administered in the Hepcon group were nearly twice the doses used in the ACT management group. However, Gravlee et al55 were unable to confirm suppression of ongoing coagulation using Hepcon CPB management. With the exception of during cold CPB, fibrinopeptide A (FPA) levels in patients who had heparin concentration monitoring were virtually indistinguishable from those in patients who had ACT monitoring (Figure 17-9). The Hepcon, however, remains one of the more sensitive tests for detecting residual heparinization after protamine reversal because the heparin concentration can be measured by protamine titration to levels as low as 0.4 IU/mL (see Monitoring for Heparin Rebound section later in this chapter).

High-Dose Thrombin Time
A functional test of heparin-induced anticoagulation that correlates well with heparin levels is the high-dose thrombin time (HiTT; International Technidyne, Edison, NJ). The TT is a clotting time that measures the conversion of fibrinogen to fibrin by thrombin. The TT is prolonged by the presence of heparin and by hypofibrinogenemias or dysfibrinogenemias. Because the TT is sensitive to very low levels of heparin, a high dose of thrombin is necessary in the TT to accurately assay the high doses of heparin used for CPB. The HiTT is performed by adding whole blood to a prewarmed, prehydrated test tube that contains a lyophilized thrombin preparation. After the addition of 1.5 mL blood, the tube is inserted into a Hemochron well and the time to clot formation is measured. In vitro assays indicate that HiTT is equivalent to the ACT in evaluation of the anticoagulant effects of heparin at heparin concentrations in the range of 0 to 4.8 IU/mL (Figure 17-10). Unlike ACT, HiTT is not altered by hemodilution and hypothermia, and has been shown to correlate better with heparin concentration than the ACT during CPB.58 While on CPB, heparin concentration and HiTT decrease, whereas the Hemochron and the Hepcon ACT increase (Figure 17-11). Another potential advantage of HiTT monitoring is for patients receiving aprotinin therapy. In the presence of heparin, aprotinin augments the celite ACT,59 possibly because its kallikrein-inhibiting capacity prolongs activation of the intrinsic coagulation pathway by XIIa. This should not be interpreted to represent enhanced anticoagulation. The kaolin ACT is less affected by aprotinin therapy than the celite ACT, perhaps because kaolin, unlike celite, activates the intrinsic pathway by stimulation of factor XI directly.60 Others have suggested that kaolin binds to aprotinin and reduces the anticoagulant effect of aprotinin in vitro. However, the heparinized kaolin ACT is still somewhat prolonged in the presence of aprotinin. HiTT is not affected by aprotinin therapy and can be used as a measure of heparinization for CPB patients receiving aprotinin therapy.59 The high-dose thromboplastin time is another measure of anticoagulation that is not affected by aprotinin therapy.61 The high-dose thromboplastin time is a WBCT in which celite is replaced by 0.3 mL of rabbit brain thromboplastin to which 1.2 mL blood is added. This test measures the time to coagulation via activation of the extrinsic pathway. This pathway of coagulation is also stimulated during pericardiotomy because of the rich thromboplastin environment of the pericardial cavity.
Heparin neutralization

Protamine Effects on Coagulation Monitoring
Reversal of heparin-induced anticoagulation is most frequently performed with protamine. Biologically, protamine binds to positively charged groups such as phosphate groups and may have important properties in angiogenesis and immune function. Different successful dosing plans have been proposed.62,63 The recommended dose of protamine for heparin reversal is 1 to 1.3 mg protamine per 100 units heparin; however, this dose often results in a protamine excess.
Protamine injection causes adverse hemodynamic effects.64 Protamine reactions have been classified into three types.65 The most common of these reactions is the type I reaction characterized by hypotension. Type II (immunologic) reactions are categorized as IIA (anaphylaxis), IIB (anaphylactoid), and IIC (noncardiogenic pulmonary edema). Type III reactions are heralded by hypotension and catastrophic pulmonary hypertension leading to right-heart failure.33,52,66
In addition to hemodynamic sequelae, protamine has adverse effects on coagulation.67 Large doses prolong the WBCT and the ACT, possibly via thrombin inhibition.68 In animals and in humans, protamine has been associated with thrombocytopenia, likely because of activation of the complement cascade.64 The anticoagulant effect of protamine also may be caused by inhibition of platelet aggregation, alteration in the platelet surface membrane, or depression of the platelet response to various agonists.68–70 These alterations in platelet function result from the presence of the heparin–protamine complex, not protamine alone. Protamine–heparin complexes activate AT III in vitro and result in complement activation. The anticoagulant effects of free protamine occur when protamine is given in doses in excess of those used clinically; however, the risk of free protamine being the cause of a hemostatic defect is small, given the rapid clearance of protamine relative to heparin.

Monitoring for Heparin Rebound
The phenomenon referred to as heparin rebound describes the re-establishment of a heparinized state after heparin has been neutralized with protamine. Various explanations for heparin rebound have been proposed.9,56,71,72 The most commonly postulated is that rapid distribution and clearance of protamine occur shortly after protamine administration, leaving unbound heparin remaining after protamine clearance. Furthermore, endogenous heparin antagonists have an even shorter life span than protamine and are eliminated rapidly, resulting in free heparin concentrations. Also possible is the release of heparin from tissues considered heparin storage sites (endothelium, connective tissues). Endothelial cells bind and depolymerize heparin via PF4. Uptake into the cells of the reticuloendothelial system, vascular smooth muscle, and extracellular fluid may account for the storage of heparin that contributes to reactivation of heparin anticoagulation, referred to as heparin rebound.9
Residual low levels of heparin can be detected by sensitive heparin concentration monitoring in the first hour after protamine reversal and can be present for up to 6 hours after surgery. Gravlee et al’s56 study suggests that without careful monitoring for heparin rebound in the postoperative period, increased bleeding as a result of heparin rebound may occur, specifically when greater doses of heparin have been administered. Monitoring for heparin rebound can be accomplished using tests that are sensitive to low levels of circulating heparin.52,57,73,74 These tests are also useful monitors for confirmation of heparin neutralization at the conclusion of CPB (see later).

Heparin Neutralization Monitors
To administer the appropriate dose of protamine at the conclusion of CPB, it would be ideal to measure the concentration of heparin present and give the dose of protamine necessary to neutralize only the circulating heparin. As a result of heparin metabolism and elimination, which vary considerably among individuals, the dose of protamine required to reverse a given dose of heparin decreases over time. Furthermore, protamine antagonizes the anti-IIa effects of heparin more effectively than the anti-Xa effects and, thus, varies in its potency depending on the source of heparin and its anti-IIa properties. Administration of a large fixed dose of protamine or a dose based on the total heparin dose given is no longer the standard of care and may result in an increased incidence of protamine-related adverse effects. An optimal dose of protamine is desired because unneutralized heparin results in clinical bleeding and an excess of protamine may produce an undesired coagulopathy. The use of individualized protamine dose–response curves uniformly results in a reduced protamine dose and has been shown to reduce postoperative bleeding.49,73 One such dose–response test, the Hemochron PRT test, is an ACT performed on a heparinized blood sample that contains a known quantity of protamine. With knowledge of the ACT, PRT, and the estimated blood volume of the patient, the protamine dose needed to neutralize the existing heparin level can be extrapolated. The Hepcon instrument also has a PRT, which is the protamine titration assay. The chamber that clots first contains the dose of protamine that most closely approximates the circulating dose of heparin. The protamine dose required for its neutralization is calculated on the basis of a specified heparin/protamine dose ratio by measuring the circulating heparin level.
At the levels of heparinization needed for cardiac surgery, tests that are sensitive to heparin become unclottable. ACT is relatively insensitive to heparin and is ideal for monitoring anticoagulation at high heparin levels but is too insensitive to accurately diagnose incomplete heparin neutralization. Reiner et al75 showed that ACT had a high predictive value for adequate anticoagulation (confirmed by laboratory aPTT) when longer than 225 seconds but was poorly predictive for inadequate anticoagulation when shorter than 225 seconds. The low levels of heparin present when heparin is incompletely neutralized are best measured by other more sensitive tests of heparin-induced anticoagulation, such as heparin concentration, aPTT, and TT. Thus, after CPB, confirmation of return to the unanticoagulated state should be performed with a sensitive test for heparin anticoagulation76–79 (Box 17-3).
Thrombin Time
TT is the time it takes for the conversion of fibrinogen to fibrin clot when blood or plasma is exposed to thrombin. Fibrin strands form in seconds. Detection of fibrin formation using standard laboratory equipment involves incubation of the blood or plasma sample within the chamber in which an optical or electrical probe sits. A detector senses either movement of the probe or the creation of an electrical field (electrical detection) because of fibrin formation and hence signals the end of the test. Hemochron manufactures a point-of-care TT test that uses a lyophilized preparation of thrombin in a Hemochron test tube to which 1 mL blood is added. Identification of fibrin formation in a Hemochron machine uses the standard Hemochron technology described previously with the ACT. The manufacturer suggests that the normal TT is 39 to 53 seconds for whole blood and 43 to 68 seconds for citrated blood. Because the TT specifically measures the activity of thrombin, it is very sensitive to heparin-induced enhancement of AT III activity. It is a useful test in the post-CPB period for differentiating the cause of bleeding when both PT and aPTT are prolonged because it excludes the intrinsic and extrinsic coagulation pathway limbs and evaluates the conversion of factor I to Ia. The TT is increased in the presence of heparin, hypofibrinogenemia, dysfibrinogenemia, amyloidosis, or antibodies to thrombin.80 The TT is also increased in the presence of fibrin degradation products if the systemic fibrinogen concentration is low.
Bedside Tests of Heparin Neutralization
Hepcon measures heparin concentration via a protamine titration assay. Cartridges with varying ranges of protamine concentration are available for use. The cartridge with the lower concentration of protamine in the titration is useful for the detection of residual circulating heparin and is sensitive to levels of heparin as low as 0.2 IU/mL. Whole-blood PT and aPTT assays are sensitive to deficiencies in coagulation factors and overly sensitive to low levels of heparin (aPTT); they lack specificity in assessing residual heparinization. The heparin-neutralized thrombin time (HNTT) is a TT assay with a small dose of protamine sufficient to neutralize 1.5 IU/mL heparin. Because the TT is increased in the presence of heparin, hypofibrinogenemia, or dysfibrinogenemia, HNTT and TT should be performed together to discriminate among these three causes. A normal HNTT in the presence of an increased TT virtually confirms residual heparin effect and would indicate the need for protamine administration. If HNTT is prolonged as well as the TT, the cause of bleeding may be attributed either to a fibrinogen problem or to a concentration of heparin greater than that which could be neutralized by the HNTT. In one study comparing bedside monitors of anticoagulation, the TT-HNTT difference bore a significant correlation with the aPTT increase. Using the line of best fit, aPTT elevation of 1.5 times the control corresponded to a 31-second difference in the TT and HNTT, indicating a convenient threshold value of TT-HNTT for the administration of protamine.80

Platelet Factor 4
A component of the alpha granule of platelets, PF4 binds to and inactivates heparin. The physiologic role of PF4 is that at the site of vascular injury, PF4 is released from platelets, binds heparin (or heparin-like compounds), and promotes thrombin and clot formation. PF4 adequately neutralizes heparin inhibition of factors Xa and IIa and may be superior to protamine in neutralizing anti-Xa effects. Animal data suggest that PF4 is devoid of the adverse hemodynamic effects seen with protamine and that it may be able to be infused more rapidly.81,82 Heparin reversal has been documented by WBCT, ACT, and heparin concentration at a PF4 concentration of 40 U/mL, approximately twice the reversal dose of protamine.74 Levy et al83 subsequently found the reversal dose of PF4 to be approximately 60 U/mL and documented similar ACT and viscoelastic measurements of clot formation compared with protamine.

Heparinase
Heparinase (Neutralase I) is an enzyme that specifically degrades heparin by catalyzing cleavage of the saccharide bonds found in the heparin molecule. As demonstrated by the ACT, heparinase in a dose of 5 mg/kg has been shown to successfully neutralize heparin effects in healthy volunteers and in patients who have undergone CPB. A dose of 7 mg/kg has been demonstrated to be even more efficacious in returning ACT to baseline values. Doses sufficient to neutralize a dose of 300 IU/kg of heparin had no significant hemodynamic effects in a canine model.84 Investigators have not found any platelet-depressive effects of heparinase in contrast with the well-documented platelet dysfunction associated with protamine therapy.69 Return to the unanticoagulated state after the use of heparinase has been confirmed using ACT monitoring or heparin concentration monitoring.
Tests of coagulation

Bedside Tests of Coagulation
Many investigators have studied the former Ciba Corning Biotrack system for monitoring anticoagulation in different clinical scenarios. For patients receiving oral anticoagulant therapy, the Biotrack 512 monitor has been found to be suitable for monitoring PT and INR. Reiner et al75 compared the bedside Biotrack aPTT with the laboratory aPTT and heparin level in patients receiving therapeutic heparinization after interventional cardiac catheterization. The authors found a strong correlation (r = 0.89) between the Biotrack aPTT and the aPTT from the hospital laboratory. The correlation between Biotrack aPTT and heparin level was not strong, probably because of the many other factors such as heparin neutralization and clearance that affect the heparin concentration in vivo.85 Another study in patients receiving heparin compared the Ciba Corning Biotrack aPTT assay with standard laboratory aPTT and documented that Biotrack was less sensitive to heparin than the laboratory aPTT; however, the correlation coefficient of these two tests was r = 0.82.86 In patients on warfarin therapy, the Biotrack aPTT was more sensitive than the laboratory aPTT and yielded consistently greater results for aPTT value. In another study in patients being anticoagulated for nonsurgical applications, the bedside aPTT was similar to the standard aPTT in its prediction of treatment in simple therapeutic algorithms. However, in more complex clinical situations, there was less agreement between the bedside aPTT and laboratory aPTT.87
In a comparison of bedside coagulation monitors after cardiac surgical procedures, Reich et al80 documented acceptable accuracy and precision levels for Hemochron and Ciba Corning Biotrack PT in comparison with standard laboratory plasma PT, making them potentially valuable for use in the perioperative period. Neither Hemochron nor Ciba Corning aPTT reached this level of clinical competence compared with standard laboratory tests. Others have documented that this monitor seems to be more precise for PT than for aPTT.88 Because of rapid turnaround times, these point-of-care coagulation monitors may be useful in predicting patients who will bleed after cardiac surgery89 and have also been used successfully in transfusion algorithms to decrease the number of allogeneic blood products given to cardiac surgical patients.90,91

Measures of Fibrin Formation
One such experimental situation is the use of heparin-bonded extracorporeal circuits during CPB with the expectation that thrombin activation and fibrin formation will be minimized. Coating of the extracorporeal circuit with the heparin ligand makes the circuit more biocompatible such that the inflammatory response elicited is diminished or nonexistent. Heparin-bonded circuits have been extensively studied and considered advantageous because of their ability to reduce the inflammatory response to CPB. Human studies reveal decreases in enzymes that mark leukocyte activation, thus showing a reduction in the whole-body inflammatory response similar to that seen with leukocyte-depletion techniques.92–94
Further enhancements in biocompatibility include less leukocyte activation and preserved platelet function. Reductions in thrombin generation have been difficult to document.95 The increases in the fibrinogen fragment F1.2 and in D-dimer levels when heparin-coated circuits are used are similar to those seen when uncoated circuits are used.96,97 Human studies have documented less bleeding and reduced transfusion requirements with the use of a heparin-coated extracorporeal circulation when these circuits are used in conjunction with a reduced systemic heparin dose.98,99 In this circumstance, markers of thrombin generation are increased even greater than those in patients in whom full-dose heparin and uncoated circuits are used. In fact, increases are seen in fibrinopeptide A, prothrombin fragment F1.2, thrombin–antithrombin III complexes, D-dimers, and plasminogen activators during CPB and after protamine administration regardless of whether heparin-coated circuits are used and regardless of heparin dose.100 Despite this, the use of reduced heparin doses and coated circuitry has resulted in diminished transfusion requirements and reduced chest tube drainage volumes without evidence of complications.97,101 Because microvascular coagulation is not fully inhibited, the use of a reduced dose of heparin cannot be systematically advocated and should be implemented with caution.

Monitoring fibrinolysis
When fibrinolysis is a result of enhanced activation of the coagulation system, secondary fibrinolysis ensues. A well-known extreme form of secondary fibrinolysis is seen during disseminated intravascular coagulation, when both systemic coagulation and fibrinolysis are occurring in excess. During CPB, fibrinolysis is most likely secondary to the microvascular coagulation that is occurring despite attempts at suppression using high doses of heparin.102,103


End Products of Fibrin Degradation
Other methods for quantifying fibrinolysis include measurement of the end products of fibrin degradation. Fibrin degradation products are the result of the cleavage of fibrin monomers and polymers and can be measured using a latex agglutination assay. When plasmin cleaves cross-linked fibrin, dimeric units are formed that comprise one D-domain from each of two adjacent fibrin units. These “D-dimers” are frequently measured by researchers in clinical and laboratory investigations. They are measured by either enzyme-linked immunosorbent assays or latex agglutination techniques and, thus, are not available for on-site use. Controversy still exists regarding whether D-dimer level or fibrin degradation products are the most sensitive test for detecting fibrinolysis, but most agree that the presence of D-dimers is the most specific for cross-linked fibrin degradation.104
Monitoring the thrombin inhibitors
A new class of drugs, the selective thrombin inhibitors, is a viable alternative to heparin anticoagulation for CPB. These agents include hirudin, argatroban, and other experimental agents. A major advantage of these agents over heparin is that they are able to effectively inhibit clot-bound thrombin in an AT III–independent fashion.105 The platelet thrombin receptor is believed to be the focus of thrombin’s procoagulant effects in states of thrombosis such as after coronary artery angioplasty. Because surface-bound thrombin is more effectively suppressed, thrombin generation can be reduced at lower levels of systemic anticoagulation than are achieved during anticoagulation by the heparin–AT III complex. This translates into less bleeding despite the lack of a clinically useful antidote for the thrombin antagonists.106,107 Thrombin antagonists are also not susceptible to neutralization by PF4 and, thus, are not neutralized at endothelial sites where activated platelets reside. They are also useful in patients with HIT in whom the administration of heparin and subsequent antibody-induced platelet aggregation would be dangerous.108 The lack of a potent antidote (such as protamine) and a prolonged duration of action are the major reasons that hirudin and other thrombin inhibitors have not found widespread clinical acceptance for use in CPB procedures (see Chapter 31).

Hirudin
Hirudin, a coagulation inhibitor isolated from the salivary glands of the medicinal leech (Hirudo medicinalis), is a potent inhibitor of thrombin that, unlike heparin, acts independently of AT III and inhibits clot-bound thrombin, as well as fluid-phase thrombin. Hirudin does not require a cofactor and is not susceptible to neutralization by PF4. This would seem to be beneficial in patients in whom platelet activation and thrombosis are potential problems. Recombinant hirudin was administered as a 0.25-mg/kg bolus and an infusion to maintain the hirudin concentration at 2.5 μg/mL, as determined by the ecarin clotting time in studies by Koster et al.109–117 The ecarin clotting time, modified for use in the Cascade POC analyzer, has been used in large series of patients with HIT.109–114 Compared with standard treatment with heparin or low-molecular-weight heparins, recombinant hirudin–treated patients maintained platelet counts and hemoglobin levels, and had few bleeding complications, if renal function was normal.115,116 Hirudin is a small molecule (molecular weight, 7 kDa) that is eliminated by the kidney and is easily hemofiltered at the end of CPB.117 In patients with abnormal renal function, bivalirudin is preferable to hirudin. An alternative treatment in this setting would be administration of unfractionated heparin with a platelet antagonist, such as tirofiban, to prevent the hyperaggregability of platelets that occurs in patients with HIT.116

Bivalirudin
Bivalirudin is a small 20-amino acid molecule with a plasma half-life of 24 minutes. It is a synthetic derivative of hirudin and thus acts as a direct thrombin inhibitor. Bivalirudin binds to both the catalytic binding site and the anion-binding exosite on fluid-phase and clot-bound thrombin. The part of the molecule that binds to thrombin is actually cleaved by thrombin itself, so the elimination of bivalirudin activity is independent of specific organ metabolism. Bivalirudin has been used successfully as an anticoagulant in interventional cardiology procedures as a replacement for heparin therapy. In fact, in interventional cardiology, bivalirudin has been associated with less bleeding and equivalent ischemic outcomes compared with heparin plus a platelet inhibitor.118 This may be the result of bivalirudin being both an antithrombin anticoagulant and an antithrombin at the level of the platelet. Merry et al119 showed equivalence with regard to bleeding outcomes and an improvement in graft flow after off-pump cardiac surgery when bivalirudin was used (0.75-mg/kg bolus, 1.75-mg/kg/hr infusion). Case reports confirm the safety of bivalirudin use during CPB.120–122 Multicenter clinical trials comparing bivalirudin with heparin anticoagulation in off-pump surgery123 and in CPB124 demonstrated “noninferiority” of bivalirudin. Efficacy of anticoagulation and markers of blood loss were similar in the two groups, suggesting that bivalirudin can be a safe and effective anticoagulant in CPB. These multicenter trials used the ACT as the monitor of anticoagulant activity during surgery, but ideal monitoring is performed using the ecarin clotting time, as seen with hirudin.125 The ecarin clotting time has a closer correlation with anti-IIa activity and plasma drug levels than does the ACT. For this reason, standard ACT monitoring during antithrombin therapy is not preferred if ecarin clotting time can be measured. A plasma-modified ACT can be used to more accurately assay the anticoagulant effects of the thrombin inhibitor drugs than ACT. This test requires the addition of exogenous plasma and, thus, is not readily available as a point-of-care assay.126
The anticoagulant effects of the thrombin antagonists can be monitored using the ACT, aPTT, or the TT. The bleeding time also may be prolonged. In a canine CPB model, dogs receiving a synthetic thrombin inhibitor had less postoperative blood loss and greater platelet counts than those receiving heparin; however, those who received a large dose of the thrombin inhibitor still had ACT increases at 2 hours after CPB.127 There were no differences in hemodynamics noted in the groups.
Bivalirudin has been compared favorably with heparin in patients undergoing coronary angioplasty for unstable angina.128 The half-life of aPTT prolongation is approximately 40 minutes, and reductions in formation of fibrinopeptide A are evidence of thrombin inhibition and fibrinogen preservation. Careful monitoring should be used because there may be a rebound prothrombotic state after cessation of therapy, which could lead to recurrence of anginal symptoms (Box 17-4).
Monitoring platelet function

Platelet Count
Numerous events occur during cardiac surgical procedures that predispose patients to platelet-related hemostasis defects. The two major categories are thrombocytopenia and qualitative platelet defects. Thrombocytopenia commonly occurs during cardiac surgery as a result of hemodilution, sequestration, and destruction by nonendothelial surfaces. Platelet counts commonly decline to 100,000/μL or slightly less; however, the final platelet count is greatly dependent on the starting value and the duration of platelet destructive interventions (i.e., CPB).129 Between 10,000/μL and 100,000/μL, bleeding time decreases directly; however, at platelet counts greater than 50,000/μL, neither the bleeding time nor platelet count has any correlation with postoperative bleeding in cardiac surgical patients. In contrast, platelet size or mean platelet volume does have some correlation with hemostatic function. Larger, younger platelets are more hemostatically active than smaller ones.130,131 Mean platelet volume multiplied by the platelet count gives an estimation of overall platelet mass and is referred to as the “plateletcrit.” It is important to appreciate the inverse relation between platelet volume and platelet count when using a measure such as the plateletcrit to assess the viability of the existing platelet population. Because the mean platelet volume is dependent on the method of specimen collection, the anticoagulant used, and temperature of the storage conditions, its reproducibility is dependent on standardized laboratory procedures.
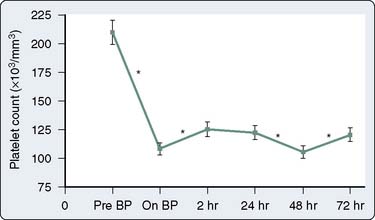
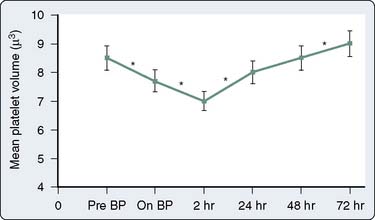
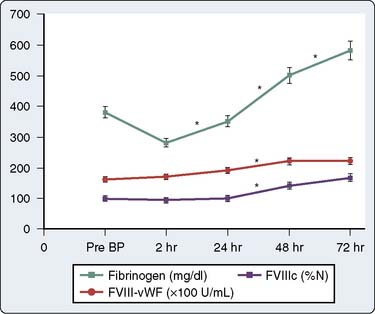
Qualitative platelet defects occur more commonly than thrombocytopenia during CPB procedures. The range of possible causes of platelet dysfunction includes traumatic extracorporeal techniques, pharmacologic therapy, hypothermia, and fibrinolysis; the hemostatic insult increases with the duration of time spent on CPB.132 The use of bubble oxygenators, noncoated extracorporeal circulation, and cardiotomy suctioning may cause platelets to become activated, initiate the release reaction, and partly deplete platelets of the contents of their alpha granules. Many of these changes are only transiently associated with CPB. Khuri et al133 characterized the hematologic changes associated with CPB in a group of 85 patients. Whereas the platelet count declines and reaches a plateau at 2 hours after CPB, mean platelet volume reaches its nadir at 2 hours after CPB and then begins to increase during the ensuing 72 hours131,133 (Figures 17-12 and 17-13). The relative thrombocytopenia seen up to 72 hours after cardiac surgery is not consistently associated with a bleeding diathesis. Similarly, the clotting proteins fibrinogen, factor VIII–von Willebrand factor, and factor VIII-C also increase to levels greater than baseline in the 2 to 72 hours after CPB (Figure 17-14).
Large doses of heparin have been shown to reduce the ability of the platelets to aggregate and to reduce clot strength.68 This effect is not reversed when protamine is administered; however, it may be mitigated by the prophylactic administration of aprotinin.134,135 The adverse effects of heparin on platelet function may be because of its ability to inhibit the formation of thrombin, the most potent in vivo platelet activator.136 However, heparin also activates the fibrinolytic system, a system that, through plasmin and other activators, has the ability to depress platelet function through other mechanisms. In an extracorporeal baboon model, intravenous heparin administration resulted in increases in plasmin activity, in the quantity of immunoreactive plasmin light chain, and in immunoreactive fibrinogen fragment E.1,137 In addition, various degrees of fibrinolysis occur after CPB. Circulating plasmin causes dissolution of the GP Ib platelet receptor and decreases the adhesiveness of platelets. Because fibrinolysis is partly responsible for the platelet dysfunction seen after heparin administration and CPB, the efficacy of antifibrinolytic agents as hemostatic drugs can be better appreciated. In addition to reducing platelet adhesiveness to von Willebrand factor, the fibrin degradation products formed depress platelet responsiveness to agonists.138,139
Protamine–heparin complexes and protamine alone also contribute to platelet depression after CPB. Mild-to-moderate degrees of hypothermia are associated with reversible degrees of platelet activation and platelet dysfunction,140 which may be partly mitigated by the use of aprotinin therapy.141 Overall, the potential coagulation benefits of normothermic CPB compared with hypothermic CPB require further study in well-conducted randomized trials (Box 17-5).

Bleeding Time
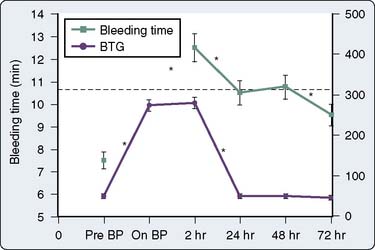
Numerous prospective blinded investigations have confirmed that bleeding time has little or no value in predicting excessive hemorrhage after cardiac surgery.142,143 Even in patients receiving therapeutic doses of aspirin, an increase in bleeding time does not necessarily translate to an increase in mediastinal tube drainage or transfusions if reinfusion and blood conservation techniques are used aggressively. There is substantial evidence that platelet-directed therapy in the form of platelet transfusions or desmopressin acetate shortens a prolonged bleeding time in patients with clinical hemorrhage.144,145 In a study of 85 patients undergoing CPB, Khuri et al133 demonstrated that bleeding time becomes abnormally increased during CPB and does not return to baseline even by 72 hours after surgery, whereas markers of platelet activation return to baseline by 24 hours after surgery (Figure 17-15). Because the bleeding time does not follow the temporal course of postoperative coagulopathy, the bleeding time may be a nonspecific and impractical test for detecting an existing platelet defect but may be suitable for following patient response to platelet-directed therapies.

Aggregometry
Activated platelets undergo aggregation, which is initially a reversible process. Activation also induces the release of substances from alpha and dense platelet granules and platelet lysosomes. Because platelet granules contain many platelet agonists, the release of granular contents further stimulates platelet activation and is responsible for the secondary phase of platelet aggregation. This secondary phase of platelet aggregation is dependent on the release of thromboxane and other substances from the platelet granules, is an energy-consuming process, and is irreversible (Table 17-2).
| Ligand | Receptor | Properties |
|---|---|---|
| Collagen | GP Ia/IIa, GP IIb/IIIa, GP IV | Adhesion, aggregation, secretion |
| Thrombospondin | GP IV, αvβ3 | Adhesion, antiadhesion |
| vWF | GP Ib/IX, GP IIb/IIIa | Adhesion |
| Fibrinogen | GP IIb/IIIa | Aggregation |
| Laminin | GP Ic/IIa | Attachment |
| Vitronectin | αvβ3, GP IIb/IIIa | αvβ3 = vitronectin receptor |
| Fibronectin | GP Ic/IIa, GP IIb/IIIa | Attachment, spreading |
GP, glycoprotein; vWF, von Willebrand factor.
Defects in platelet aggregation can be seen in patients with storage pool deficiency, Bernard–Soulier syndrome, or Glanzmann’s thrombasthenia and in patients taking salicylates. Impaired platelet aggregation has been demonstrated to occur after extracorporeal circulation, but investigators have had difficulty showing a correlation between impaired aggregation and clinical bleeding.146,147 One ex vivo study demonstrated a significant correlation between platelet aggregation and 3-hour postoperative bleeding; however, correlations were greatest when preoperative aggregometry was performed using whole-blood samples and when postoperative measurements were performed using platelet-rich plasma.147 The assessment of the platelet defect induced by aspirin consumption is more sensitive when whole-blood aggregometry is performed. The extreme sensitivity of this assay to minor defects in platelet function has resulted in a high negative predictive value but a rather low positive predictive value for bleeding. Its inability to be performed easily in the clinical arena has designated platelet aggregometry as strictly a research tool with occasional clinical applications.

Platelet-Mediated Force Transduction
An instrument that measures the force developed by platelets during clot retraction has been shown to be directly related to platelet concentration and function148 (Hemodyne). The apparatus consists of a cup and a parallel upper plate. The cup is filled with blood or the platelet-containing solution, and the upper plate is lowered onto the clotting solution. Clot forms and adheres to the outer edges of the cup and to the plate above. A thin layer of oil is deposited onto the surfaces that are exposed to air. The upper plate is coupled to a displacement transducer that translates displacement caused by platelet retraction into a force. Normal values for platelet force development have been suggested by the investigators.70 The antiplatelet effects of heparin have been evaluated using this force retractometer. Using this instrument, investigators have shown that high heparin concentrations completely abolish platelet force generation.68 Furthermore, the concentration of protamine required to reverse the anticoagulant effects of heparin is not sufficient to reverse these antiplatelet effects. The antiplatelet effects of protamine alone also have been evaluated using this monitor.

Fluorescence Flow Cytometry
Flow cytometry is ideal for the detection of low concentrations of specific proteins within a large population of cells. These proteins either may be static portions of the platelet surface or dynamic products of platelet activation. The platelet release reaction enables specific integrin proteins, which are a part of the platelet alpha granule membrane, to incorporate themselves into the platelet surface membrane through a mechanism analogous to exocytosis. A portion of the GP IIb/IIIa receptor is also a protein of the alpha granule membrane that becomes exposed on the surface membrane of the platelet in response to platelet activation. Flow cytometry allows for the detection and quantification of many of these surface membrane constituents as a result of immunofluorescent innovations.149
Flow cytometry techniques have been enhanced by the development of specific monoclonal antibodies, which recognize antigens on the platelet (or white blood cell) surface. Antibodies developed are so specific that different ligand binding sites can be measured on the same GP IIb/IIIa molecule that characterizes different phases of receptor activation. Some of the epitopes for which monoclonal antibodies have been developed include PADGEM and GMP-140 (markers of platelet activation), the activated GP IIb/IIIa complex, and the GP Ib receptor.150–152 A large number of monoclonal antibodies are available for identification of specific platelet ligand-binding sites (Table 17-3). Antibodies that bind specifically to activated platelets but minimally to unstimulated platelets are referred to as “activation dependent.” In utilizing activation-dependent monoclonal antibodies, flow cytometry measures the platelet reactivity or response to the addition of platelet agonists. The technique of flow cytometry can be performed using whole blood or platelet-rich plasma. The fluorescent-labeled monoclonal antibody directed against a specific platelet membrane protein is quantified by the flow cytometer, which is an instrument equipped with a laser or a light source of a specific excitation wavelength. Light scatter data are collected that help to differentiate platelets from other cellular particles. Fluorescent antibody detection is expressed as percentage of the total number of particles or as fluorescence intensity.
| Antibody Binding Site (Other Name) | Antibodies Available | Requirement for Binding/Functional Activity |
|---|---|---|
| GP Ib (CD42b) | AP-1; 6D1 | von Willebrand receptor; platelet adhesion |
| GP IX | FMC25 | Platelet adhesion to endothelium |
| GP IIb/IIIa complex (αIIBβ3, CD41) | 7E3; 10E5; 4F10; A2A9 | Fibrinogen receptor; platelet aggregation |
| GP IIb/IIIa (αIIBβ3, CD41a) | PAC1 | Active conformation of GP IIb/IIIa only |
| Fibrinogen | 2G5; 9F9 | Receptor-induced changes because of bound fibrinogen |
| GP IIb heavy chain of GP IIb/IIIa | P2; PMI-1 | Ligand-induced changes in GP IIb |
| IIIa portion of GP IIb/IIIa (CD61) | AP6; Ab15; Y2/51 | Receptor bound by fibrinogen |
| GMP140 (CD62P, P-selectin) | S12; KC4; VH10 | alpha granule membrane protein, mediates platelet–leukocyte interactions |
| LAMP-1 (CD63) | CLB-gran/12; H5G11 | Lysosome membrane protein, expressed after platelet secretion |
| 40-kDa protein | D495 | Dense granule membrane protein |
| Thrombospondin | P8 | Bound thrombospondin |
| Factor VIIIa light chain | 1B3 | Present on a procoagulant surface |
| Factor Va light chain | V237 | Present on a procoagulant surface |
GMP, granule membrane protein; GP, glycoprotein; LAMP, lysosme membrane protein.
The ability to specifically identify platelet defects by fluorescence flow cytometry has greatly aided in the characterization of hematologic disease states such as the Bernard–Soulier syndrome and Glanzmann’s thrombasthenia.153,154 In the cardiac surgical arena, flow cytometry has aided in diagnosing the disorders of platelet function induced by CPB and protamine administration.69,140,155,156 Kestin et al136 used flow cytometric techniques to study the effects of CPB on the in vivo time-dependent upregulation of P-selectin in blood emerging from a bleeding wound. They showed that P-selectin expression is depressed after heparinization and during CPB, and recovers at approximately 2 hours after the conclusion of CPB. In contrast, in vitro activation of CPB blood with the platelet agonist phorbol myristate acetate did not reveal this depression of P-selectin expression at any time point. Using another platelet activator (thrombin-receptor agonist peptide) and flow cytometry, others did not demonstrate depression of P-selectin expression early in CPB but did so after 90 minutes of CPB and after protamine administration.157
Much uncertainty still exists regarding GP Ib receptor modulation in response to CPB. Using flow cytometry, George et al149 found a modest reduction in platelet surface GP Ib during CPB. However, a subsequent study by van Oeveren et al,2 confirming a reduction in GP Ib, subjected the platelets to centrifugation and processing techniques that might have induced in vitro artifactual platelet activation. Kestin et al136 used monoclonal antibodies to many epitopes expressed on GP Ib and concluded that expression of this receptor is not reduced during CPB.
As a result of the many monoclonal antibodies directed at specific epitopes on the GP IIb/IIIa receptor, investigations into the dynamics of this receptor during CPB have yielded variable results. Some studies have confirmed modest reductions in the expression of GP IIb/IIIa, although not all have used whole-blood techniques. Rinder et al,155 using a whole-blood technique, demonstrated a small decrease in GP IIb/IIIa expression. Monoclonal antibodies are available that bind to the GP IIb/IIIa fibrinogen binding site, and others are available that recognize receptor-bound fibrinogen. Flow cytometric techniques also have helped to characterize the mechanisms of action of several pharmacologic agents that have shown hemostatic potential in the perioperative period.69
Bedside platelet function testing


Thromboelastography
The coaguloviscometers that were developed in the 1920s formed the basis of viscoelastic coagulation testing that is now known as thromboelastography. Thromboelastography in its current form was developed by Hartert in 1948 and has been used in many different clinical scenarios to diagnose coagulation abnormalities.158–160 Although not yet truly portable, the thromboelastograph (TEG; Haemoscope, Niles, IL [now Haemonetics, Braintree, MA]) can be performed “onsite” either in the operating room or in a laboratory and provides a rapid whole- blood analysis that yields information about clot formation and clot dissolution (Table 17-4). Within minutes, information is obtained regarding the integrity of the coagulation cascade, platelet function, platelet–fibrin interactions, and fibrinolysis. The principle is as follows: Whole blood (0.36 mL) is placed into a plastic cuvette into which a plastic pin is suspended; this plastic pin is attached to a torsion wire that is coupled to an amplifier and recorded; a thin layer of oil is added to the surface of the blood to prevent drying of the specimen; and the cuvette oscillates through an arc of 4 degrees, 45 minutes at 37°C. When the blood is liquid, movement of the cuvette does not affect the pin. However, as clot begins to form, the pin becomes coupled to the motion of the cuvette and the torsion wire generates a signal that is recorded. The recorded tracing can be stored by computer, and the parameters of interest are calculated using a simple software package. Alternatively, the tracing can be generated online with a recording speed of 2 mm/min. The tracing generated has a characteristic conformation that is the signature of the TEG (Figure 17-16).
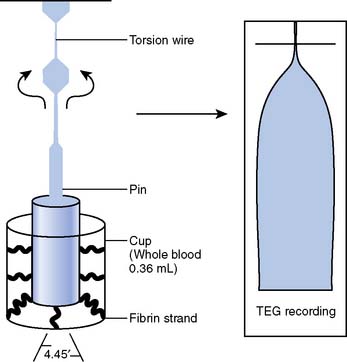
Figure 17-16 Schematic diagram of the thromboelastograph (TEG) instrumentation (left) and a sample tracing (right).
(From Mallett SV, Cox DJA: Thromboelastography. Br J Anaesth 69:307–313, 1992.)
Characteristic TEG tracings can be recognized to be indicative of particular coagulation defects. A prolonged R value indicates a deficiency in coagulation factor activity or level and is seen typically in patients with liver disease and in patients on anticoagulants such as Coumadin or heparin. MA and a angle are reduced in states associated with platelet dysfunction or thrombocytopenia and are reduced even further in the presence of a fibrinogen defect. LY30, or the lysis index at 30 minutes after MA, is increased in conjunction with fibrinolysis. These particular signature tracings are depicted in Figure 17-17.
Figure 17-17 Signature thromboelastograph tracings.
(From Mallett SV, Cox DJA: Thromboelastography. Br J Anaesth 69:307–313, 1992.)
TEG is a useful tool for diagnosing and treating perioperative coagulopathy in patients undergoing cardiac surgical procedures because of a variety of potential coagulation defects that may exist.159 Within 15 to 30 minutes, on-site information is available regarding the integrity of the coagulation system, the platelet function, fibrinogen function, and fibrinolysis.160,161 With the addition of heparinase, TEG can be performed during CPB and can provide valuable and timely information regarding coagulation status.162 Because TEG is a viscoelastic test and evaluates whole-blood hemostasis interactions, it is suggested that TEG is a more accurate predictor of postoperative hemorrhage than routine coagulation tests that analyze individual components of the hemostasis system. A number of clinical trials have confirmed that, in cardiac surgical patients, TEG has a greater predictive value and greater specificity than routine coagulation tests for diagnosing patients known as “bleeders.”163 Tuman et al160 studied 42 patients, of whom 9 were classified as bleeders. A routine coagulation screen consisting of ACT, PT, aPTT, and platelet count had only a 33% accuracy for predicting bleeding, whereas TEG and Sonoclot (Sienco, Morrison, CO; another viscoelastic test) had 88% and 74% accuracy, respectively. Mongan and Hosking164 also found that TEG abnormalities predict postoperative bleeding, and using TEG parameters, they also were able to identify a population of patients who respond to therapy with desmopressin acetate. In a prospective study of 16-hour postoperative blood loss, Gravlee et al165 reported on 897 cardiac surgical patients in whom routine coagulation tests were measured immediately on heparin reversal in the operating room. The weak correlations and poor predictive values of these tests confirm that these tests perform poorly as predictors of bleeding. TEG was not studied in this trial.
In a large, retrospective evaluation in more than 1000 patients, Spiess et al166 found that the institution of a transfusion algorithm using TEG resulted in a significant reduction in the incidence of mediastinal exploration and in the rate of transfusion of allogeneic blood products. Because of its ease of use and application at the bedside, TEG has been used in many research settings to assess drug effects on platelet function and clot strength.167,168 Information from the TEG also has been used to guide clinical decision making during orthotopic liver transplantation, obstetric procedures, and vascular surgical procedures.168–171

Thromboelastography Modifications
Thromboelastography was originally performed using recalcified citrated whole blood or celite activator. The addition of recombinant human tissue factor as an activator can be used to accelerate the rate of thrombin formation and, thus, the formation of fibrin.172 This serves to shorten the time required for development of the MA. Because the MA primarily is reflective of clot strength and platelet function, this information can be obtained more quickly with tissue factor enhancement. The recombinant tissue factor is a thromboplastin agent and is available from a number of manufacturers.
An application of thromboelastography in the clinical arena is its use in monitoring GP IIb/IIIa-receptor blockade and ADP-receptor blockade in patients treated with specific antiplatelet agents. TEG with tissue factor acceleration speeds the appearance of MA and is accurate for monitoring the platelet inhibition by large concentrations of GP IIb/IIIa-receptor blockers. Using this technique with platelet-rich plasma, researchers have used the reduction of the MA as an index of platelet inhibition by GP IIb/IIIa-receptor blockers in the catheterization laboratory.171 Comparison with the baseline MA yields a relative measure of the degree of platelet inhibition. Thrombin-receptor agonist peptide (TRAP)–induced aggregation correlates strongly with the TEG values measured in this fashion.171
Because the MA is a function of the platelet–fibrinogen interaction, a reduction in the MA can be accomplished by the addition of potent GP IIb/IIIa-receptor blockade to the assay. The resultant MA, in the presence of excessively high GP IIb/IIIa-receptor blockade, primarily is due to the fibrinogen concentration and the strength of fibrin alone. This value (called Maf) correlates strongly with plasma fibrinogen concentration.173,174
The thienopyridine ADP-receptor blockers, clopidogrel and ticlopidine, are widely used in cardiovascular medicine. The ability to measure the platelet defect induced by these drugs is difficult unless sophisticated laboratory techniques such as ADP-aggregometry are used. Aggregometry yields accurate results; however, it is not readily available in the perioperative period as a point-of-care test. Native TEG analysis does not measure the thienopyridine-induced platelet defect because the formation of thrombin in the assay has an overwhelming effect on the development of the TEG MA. A modification of the TEG removes thrombin from the assay and studies a nonthrombin clot, strengthened by the addition of ADP. Figure 17-18 depicts the different signature TEG tracings that are used to calculate the platelet contribution to MA when a platelet inhibitor is present. This assay was specifically created to measure the platelet inhibition by ADP antagonists such as clopidogrel and is referred to as the “platelet mapping assay.” The MAkh is the maximal activation of platelets and fibrin, and is the largest amplitude that can be achieved. The MAf is the maximal amplitude that is obtained when a thrombin-depleted fibrin clot is formed without a platelet contribution. The MApi is the MAf contribution plus the platelet contribution. MApi is created by adding an activator such as ADP to the MAf assay (for clopidogrel testing). Only platelets that can be activated by ADP contribute to the MApi. The following formula calculates the percentage reduction in platelet activity using this assay:

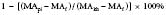
Clopidogrel, ticlopidine, and even aspirin inhibition now can be studied at the point-of-care using this modification.175,176

Sonoclot
Another test of viscoelastic properties of blood is the Sonoclot. In 1975, von Kaulla introduced the Sonoclot (Sienco, Wheat Ridge, CO) which is a coagulation analyzer that measures the changing impedance on an ultrasonic probe that is immersed in a coagulating blood sample.177 The machine consists of a plastic disposable probe mounted on an ultrasonic transducer that vibrates vertically at 200 Hz. The probe is immersed to a standard depth into 0.4 mL whole blood or plasma. The fluid exerts a force, or resistance, on the probe, which does not allow it to vibrate freely, and when the sample begins to clot, fibrin strands form on the tip of the probe and further increase the resistance. The electronic mechanism that drives the probe’s vibration also acts as a transducer, measures the impedance to vibration, and, subsequently, converts it to a signal on paper. This signature of the Sonoclot reflects coagulation in real time, from the start of fibrin formation, to fibrin cross-linkage, platelet-mediated clot strengthening, and, eventually, to clot retraction and fibrinolysis.
Immersion of the probe into the blood sample causes an initial increase in the signal because of the increased impedance to vibration by fluid relative to air. The onset time is the time for initial fibrin formation and is defined as the time taken to reach an amplitude of 1 mm. This time also has been referred to as the “SonACT” (Figure 17-19). Further increase in the signal occurs because of an increased rate of fibrinogen conversion to fibrin. The rate of rise of this first peak (R1) is expressed as the percentage of the peak amplitude per unit time (normal values, 18% to 45%). After R1, there is a shoulder or a dip before the rise in amplitude that characterizes R2. This shoulder is a result of the action of platelets and fibrin in producing clot retraction. As the clot retracts from the walls of the cuvette, the impedance to vibration briefly decreases. As fibrinogen converts to fibrin and fibrin polymerizes, the speed of clot formation and the platelet–fibrin interactions are reflected by the slope R2, the second wave. In the presence of greater concentrations of fibrinogen, a larger clot mass is represented by a greater amplitude of the R2 wave because of a greater impedance to vibration. The amplitude of the peak of R2 is therefore related to the concentration of normal functional fibrinogen. The subsequent downward slope, R3, occurs as a result of platelet-mediated clot retraction that causes plasma expulsion and clot size diminution, and thus a lower impedance. The magnitude of the R3 drop is reflective of platelet number and function. Figure 17-19 demonstrates the normal Sonoclot tracing. Figure 17-20 depicts a tracing in a patient with dysfunctional or deficient fibrinogen levels. When fibrinolysis occurs, the signal decreases even further to baseline values. The time for fibrinolysis to occur varies with each sample and usually is seen on Sonoclot analysis only if a patient has accelerated fibrinolysis.
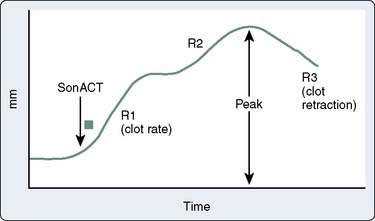
Figure 17-19 Normal Sonoclot tracing.
(From Hett DA, Walker D, Pilkington SN, Smith DC: Sonoclot analysis. Br J Anaesth 75:771–776, 1995, by permission of BMJ Publishing Group.)
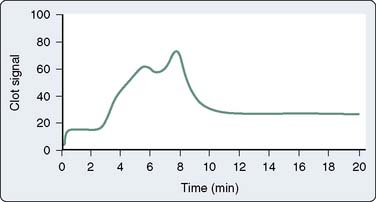
Figure 17-20 Sonoclot tracing in a patient with fibrinogen deficiency.
(From Hett DA, Walker D, Pilkington SN, Smith DC: Sonoclot analysis. Br J Anaesth 75:771–776, 1995, by permission of BMJ Publishing Group.)
Sonoclot and TEG have been studied in the surgical arena because of their ability to measure viscoelastic properties of coagulation and their on-site applications. Tuman et al160 found an accuracy of 74% using Sonoclot and an accuracy of 88% using TEG to predict bleeding after cardiac surgery. The reason that viscoelastic tests have been able to predict bleeding so successfully probably relates to their ability to measure platelet function, a major determinant of postoperative hemostasis.178 Significant correlations have been documented between specific Sonoclot parameters and platelet count and coagulation factor assays. This reproducibility has allowed the use of Sonoclot to also predict and successfully treat coagulation abnormalities in patients undergoing liver transplantation.179 Like the TEG, Sonoclot is also useful in the diagnosis of hypercoagulable states.180
A direct comparison of TEG with Sonoclot is not likely to reveal a significant advantage of one test over the other because both tests measure the dynamic process of hemostasis by transducing the impedance changes that occur as blood clots. Both tests are easy to perform, can be performed on whole blood, and can be conveniently located in the operating room. Tuman et al160 did not document a statistical difference between these two tests in clinical accuracy; however, they did show the viscoelastic tests to be superior to routine plasma coagulation testing. Sonoclot may provide coagulation information earlier because the initial deflection occurs when the first fibrin strands are forming. However, both TEG and Sonoclot accurately reflect the platelet–fibrin interactions required for clot formation.

ROTEM (Rotational Thrombelastometry)
The ROTEM gives a viscoelastic measurement of clot strength in whole blood (Pentapharm, Munich, Germany). A small amount of blood and coagulation activators is added to a disposable cuvette that is then placed in a heated cuvette holder. A disposable pin (sensor) that is fixed on the tip of a rotating shaft is lowered into the whole-blood sample. The loss of elasticity on clotting of the sample leads to changes in the rotation of the shaft that is detected by the reflection of light on a small mirror attached to the shaft. A detector records the axis rotation over time, and this rotation is translated into a graph or thromboelastogram.160,161
The main descriptive parameters associated with ROTEM are the following:
ROTEM is approved for use in coagulation monitoring in Europe, and its use and familiarity are highest there. Spalding et al’s161 recent study has shown that implementation of ROTEM-guided coagulation management is useful in the choice of an appropriate therapeutic option in the bleeding patient. This reduces costs by avoiding administration of costly component therapy such as fresh-frozen plasma, cryoprecipitate, platelet concentrates, or antifibrinolytic agents. Its use in cardiac surgery and in transfusion algorithms is likely to be similar to that of TEG. ROTEM is currently seeking approval as a coagulation monitoring device from the Food and Drug Administration in the United States.

Tests of Platelet Response to Agonists
HemoSTATUS
Despite the introduction of numerous point-of-care coagulation analyzers that allow for rapid determination of a patient’s coagulation status, the qualitative measure of platelet function, at the bedside, remains an elusive challenge. HemoSTATUS (Medtronic, Parker, CO) is a point-of-care platelet function assay that used the Hepcon HMS monitoring system to measure platelet reactivity. A six-channel cartridge measures the heparinized kaolin-activated ACT without platelet activator (channels 1 and 2) and with incrementally increasing doses of platelet-activating factor [PAF] (channels 3 to 6). The ACT of the PAF-activated channels will be shortened because of the ability of activated platelets to speed coagulation. The respective doses of PAF in channels 3 through 6 are 1.25, 6.25, 12.5, and 150 nmol/L. For each of channels 3 through 6, the degree of shortening of the ACT as a ratio to the ACT without PAF is the “clot ratio” and is calculated as 1 − (ACTactivated/ACTcontrol). The “maximal” clot ratio is the clot ratio for channel 6 that was derived using blood from healthy volunteers (Figure 17-21). A comparison of the patient clot ratio to the maximal clot ratio (derived from normal volunteers) yields a comparative measure of platelet function, termed the “percentage of maximal platelet function.”
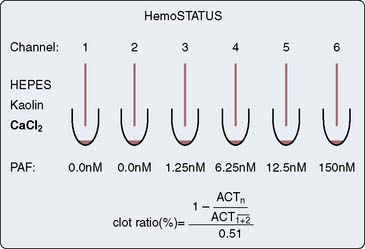
The potential ability to measure the qualitative function of platelets using a point-of-care assay provides innumerable advantages for clinicians caring for cardiac surgical patients. Platelet dysfunction is one of the more common hemostasis defects incurred during CPB, yet it is difficult to specifically measure platelet function rapidly and at the bedside. Viscoelastic tests conveniently measure platelet function, but their use in transfusion algorithms is limited by a lack of specificity to the measure of platelet dysfunction. Transfusion algorithms like the one published by Despotis et al,91 which have been suggested to result in reduced transfusions in cardiac surgical patients, have incorporated only the measure of platelet number because the on-site ability to measure platelet function has been so elusive. Inclusion of a measure of platelet function into a transfusion algorithm potentially would reduce allogeneic transfusions even further.
An initial investigation of HemoSTATUS in cardiac surgical patients was performed by Despotis et al.144 The authors studied 150 patients and conducted multivariate analyses to evaluate the relation between postoperative blood loss and multiple demographic, operative, and hemostatic measurements. They demonstrated a significant correlation between HemoSTATUS measurements on arrival in the intensive care unit and 4-hour postoperative mediastinal tube drainage (r = −0.85, channel 5; r = −0.82, channel 6). The accuracy of a number of hemostasis assays was measured using receiver operating characteristic curves for the detection of excessive mediastinal tube drainage. The highest predictability for bleeding was found in both the channel 5 clot ratio and the bleeding time. The PT, aPTT, and platelet count had much lower predictive value. HemoSTATUS-derived clot ratios also had the capability to detect enhanced platelet function after the administration of pharmacologic platelet therapy (desmopressin acetate) and after the transfusion of platelet concentrates. Subsequent investigations in cardiac surgical patients have confirmed a significant yet weak correlation of HemoSTATUS with postoperative bleeding, but have not found this test to be superior to TEG or routine coagulation tests in its predictive value.163
Validation studies have been performed to compare the assessment of platelet reactivity using HemoSTATUS with that measured by fluorescence flow cytometry. The percentage reduction in platelet function at multiple time points during cardiac surgery was compared using the two methodologies using each patient’s baseline platelet function as the control. Both tests reflected a similar degree of platelet dysfunction at the time points 90 minutes into CPB, after protamine, and at intensive care unit arrival. Differences in the type of assay and the type of platelet activators used (thrombin peptide vs. PAF) may have accounted for the inability of the tests to correlate with each other at all of the time points studied.157 This POC platelet function assay is no longer supported commercially, nor available.
Other Agonist-Activated Platelet Function Tests
VerifyNow (formerly marketed as Ultegra; Accumetrics, San Diego, CA) is a point-of-care monitor designed specifically to measure the platelet response to a TRAP. This technology was approved by the U.S. Food and Drug Administration for use as a platelet function assay. In whole blood, it measures TRAP activation-induced platelet agglutination of fibrinogen-coated beads using an optical detection system. After anticoagulated whole blood is added to the mixing chamber, the platelets become activated if they are responsive to the agonist. The activated GP IIB/IIIa receptors on the platelets bind to adjacent platelets via the fibrinogen on the beads and cause agglutination of the blood and the beads. Light transmittance through the chamber is measured and increases as agglutination increases, much like standard aggregometry. Antithrombotic drug effects cause a diminished agglutination (measured by light transmittance); thus, the degree of platelet inhibition can be quantified. Direct pharmacologic blockade of GP IIB/IIIa receptors with a GP IIb/IIIa antagonist is detected with high accuracy using this device and TRAP agonist. VerifyNow has been especially useful in accurately measuring receptor inhibition in the invasive cardiology patients receiving GP IIb/IIIa–inhibiting drugs.181–183 More recent cartridges using arachidonic acid as the agonist have been developed that can accurately assess aspirin-induced platelet dysfunction. Through inhibition of arachidonic acid, indirect prevention of GP IIb/IIIa expression is accomplished. The antiplatelet effects of the drug clopidogrel also can be measured using a VerifyNow cartridge that incorporates ADP as the agonist.184 Each of these drug effects can be measured using the appropriate cartridge of the VerifyNow device.185
The Hemostatometer has been renamed the Clot Signature Analyzer (CSA; Xylum, Scarsdale, NY). Whole blood is maintained under a constant driving pressure of 60 mm Hg as it is forced out into a synthetic vessel. The pressure distally in the vessel is monitored. The tubing of this synthetic vessel is perforated, and the distal pressure decline is measured. The time to restoration of this distal pressure will be a function of development of a platelet plug. Thus, the time for initial closure is a measure of platelet function. A subsequent time is measured, and that is the time to complete pressure loss caused by complete vessel occlusion. This clot is the result of coagulation and clot formation, and the time to the second pressure decline is reflective of coagulation function. Another chamber of this device contains a collagen-coated fibril on which platelets adhere and form a plug. A similar pressure measurement technique indicates the formation of a platelet thrombus. This point-of-care assay has been used to measure platelet reactivity in high-risk patients with atherosclerotic coronary artery disease.186–188 However, in cardiac surgical patients, preoperative platelet reactivity did not have predictive accuracy for bleeding.189 Data evaluating the Clot Signature Analyzer in the postoperative period to predict bleeding are lacking.
The Platelet Function Analyzer (PFA-100; Dade Behring, Miami, FL) is a monitor of platelet adhesive capacity that is currently approved by the U.S. Food and Drug Administration and is valuable in its diagnostic abilities to identify drug-induced platelet abnormalities, platelet dysfunction of von Willebrand disease, and other acquired and congenital platelet defects.190,191 The test is conducted as a modified in vitro bleeding time. Whole blood is drawn through a chamber by vacuum and is perfused across an aperture in a collagen membrane coated with an agonist (epinephrine or ADP). Platelet adhesion and formation of aggregates seal the aperture, thus indicating the “closure time” measured by the PFA-100.192,193 In cardiac surgical patients, the preoperative PFA-100 closure time significantly correlated with postoperative blood loss (r = 0.41; P = 0.022).194 However, preliminary evidence with post-CPB sampling and with in vitro addition of GP IIb/IIIa–inhibiting drugs suggests that these closure times may exceed those measurable using standard testing with the PFA-100.195
“Platelet Works” (Helena Laboratories, Beaumont, TX) is a test that uses the principle of the platelet count ratio to assess platelet reactivity. The instrument is a Coulter counter that measures the platelet count in a standard EDTA-containing tube. Platelet count also is measured in tubes containing the platelet agonist ristocetin, ADP, epinephrine, collagen, or thrombin. Addition of blood to these agonist tubes causes platelets to activate, adhere to the tube, and effectively be eliminated from the platelet count. The ratio of the activated platelet count to the nonactivated platelet count is a function of the reactivity of the platelets. Early investigation in cardiac surgical patients indicated that this assay is useful in providing a platelet count, and that it is capable of measuring the platelet dysfunction that accompanies CPB196 (Box 17-6). Even more essential is the need to measure the antiplatelet effects of the oral antithrombotic agents used to treat cardiovascular patients with intracoronary stents. The point-of-care tests that are being promulgated currently are those that can monitor the effects of drugs like clopidogrel, prasugrel, and aspirin.197–199
Impact Cone and Plate(let) Analyzer (CPA; DiaMed Cressier, Switzerland)
In the Impact cone and plate(let) analyzer, whole blood is exposed to uniform shear by the spinning of a cone in a standardized cup. This allows for platelet function testing under conditions that mimic physiologic blood flow, thus achieving the most accurate pattern of platelet function. After automated staining, platelet adhesion to the cup is evaluated by image analysis software. The test yields two parameters: average size and surface coverage, which determine platelet function in terms of adhesion and aggregation. These values constitute a general platelet function parameter. This device identifies both congenital and acquired platelet defects, as well as the effects of antiplatelet drugs including GP IIb/IIIa antagonists, aspirin, and clopidogrel.200–203 Recent studies suggest the Impact cone and plate(let) analyzer appears to be a useful tool for testing perioperative platelet function and may help in predicting postoperative blood loss.204 Widespread experience with this instrument is limited because it has only recently become commercially available.
Multiplate Analyzer (Dynabyte Medical, Munich, Germany)
Multiplate analyzer is a test of platelet function in whole blood using impedance aggregometry.205,206 First introduced in 2005, it is one of the most widely applied platelet aggregometers in Europe today. The analysis is performed in a single-use test cell, which incorporates a magnetic stirrer, as well as two independent impedance sensors. Activated platelets adhere and aggregate on the electrodes and, thus, enhance the electrical resistance between them. Typically, 300 μL buffered citrated blood is added to 300 μL isotonic saline and then analyzed. The device provides five channels for parallel determinations, as well as automatic analysis, calibration, and documentation using an integrated computer system. Several specific test reagents are available for stimulation of different receptors or activation of signal transduction pathways of platelets to detect changes induced by drugs, as well as by acquired or hereditary platelet disorders. After an incubation time of 3 minutes, the selected agonist solution is added and the increase in electrical impedance is recorded continuously for 6 minutes. The resistance change is transformed to arbitrary aggregation units (AUs) and plotted against time. The area under the aggregation curve is used to quantify the aggregation response and is expressed in units (1 unit corresponds with 10 AU/min). The mean values of the two independent determinations are expressed as the area under the curve of the aggregation tracing (Figure 17-22). The system has a high sensitivity for antiplatelet drugs (aspirin, clopidogrel, prasugrel, IIbIIIa-antagonists).207 Studies also have shown that Multiplate analysis is predictive for transfusion requirements in cardiac surgery205,206,208–210 and can be predictive of thromboembolism in stent patients who are nonresponsive to platelet inhibitors.211 This device is not currently available in the United States.
1 Khuri S.F., Valeri C.R., Loscalzo J., et al. Heparin causes platelet dysfunction and induces fibrinolysis before cardiopulmonary bypass. Ann Thorac Surg. 1995;60:1008.
2 van Oeveren W., Harder M.P., Roozendaal K.J., et al. Aprotinin protects platelets against the initial effect of cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1990;99:788. discussion 796
3 Marciniak E. Physiology of antithrombin III. Ric Clin Lab. 1984;14:475.
4 Marciniak E., Gora-Maslak G. Enhancement by heparin of thrombin-induced antithrombin III proteolysis: Its relation to the molecular weight and anticoagulant activity of heparin. Thromb Res. 1982;28:411.
5 Marciniak E., Romond E.H. Catabolism and distribution of functionally heterogeneous human antithrombin III. J Lab Clin Med. 1987;109:89.
6 Marciniak E. Thrombin-induced proteolysis of human antithrombin III: An outstanding contribution of heparin. Br J Haematol. 1981;48:325.
7 Hashimoto K., Yamagishi M., Sasaki T., et al. Heparin and antithrombin III levels during cardiopulmonary bypass: Correlation with subclinical plasma coagulation. Ann Thorac Surg. 1994;58:799. discussion 804
8 Heller E.L., Paul L. Anticoagulation management in a patient with an acquired antithrombin III deficiency. J Extra Corpor Technol. 2001;33:245.
9 Teoh K.H., Young E., Bradley C.A., Hirsh J. Heparin binding proteins. Contribution to heparin rebound after cardiopulmonary bypass. Circulation. 1993;88:II-420.
10 Jaberi M., Bell W.R., Benson D.W. Control of heparin therapy in open-heart surgery. J Thorac Cardiovasc Surg. 1974;67:133.
11 Roth J.A., Cukingnan R.A., Scott C.R. Use of activated coagulation time to monitor heparin during cardiac surgery. Ann Thorac Surg. 1979;28:69.
12 Reich D.L., Zahl K., Perucho M.H., Thys D.M. An evaluation of two activated clotting time monitors during cardiac surgery. J Clin Monit. 1992;8:33.
13 Horkay F., Martin P., Rajah S.M., Walker D.R. Response to heparinization in adults and children undergoing cardiac operations. Ann Thorac Surg. 1992;53:822.
14 Despotis G.J., Summerfield A.L., Joist J.H., et al. In vitro reversal of heparin effect with heparinase: Evaluation with whole blood prothrombin time and activated partial thromboplastin time in cardiac surgical patients. Anesth Analg. 1994;79:670.
15 Jobes D.R., Schwartz A.J., Ellison N., et al. Monitoring heparin anticoagulation and its neutralization. Ann Thorac Surg. 1981;31:161.
16 Niinikoski J., Laato M., Laaksonen V., et al. Use of activated clotting time to monitor anticoagulation during cardiac surgery. Scand J Thorac Cardiovasc Surg. 1984;18:57.
17 Babka R., Colby C., El-Etr A., Pifarre R. Monitoring of intraoperative heparinization and blood loss following cardiopulmonary bypass surgery. J Thorac Cardiovasc Surg. 1977;73:780.
18 Culliford A.T., Gitel S.N., Starr N., et al. Lack of correlation between activated clotting time and plasma heparin during cardiopulmonary bypass. Ann Surg. 1981;193:105.
19 Ammar T., Fisher C.F., Sarier K., Coller B.S. The effects of thrombocytopenia on the activated coagulation time. Anesth Analg. 1996;83:1185.
20 Ammar T., Scudder L.E., Coller B.S. In vitro effects of the platelet glycoprotein IIb/IIIa receptor antagonist c7E3 Fab on the activated clotting time. Circulation. 1997;95:614.
21 Bode A.P., Lust R.M. Masking of heparin activity in the activated coagulation time (ACT) by platelet procoagulant activity. Thromb Res. 1994;73:285.
22 Gravlee G.P., Whitaker C.L., Mark L.J., et al. Baseline activated coagulation time should be measured after surgical incision. Anesth Analg. 1990;71:549.
23 Mabry C.D., Read R.C., Thompson B.W., et al. Identification of heparin resistance during cardiac and vascular surgery. Arch Surg. 1979;114:129.
24 Andrew M., Ofosu F., Schmidt B., et al. Heparin clearance and ex vivo recovery in newborn piglets and adult pigs. Thromb Res. 1988;52:517.
25 Andrew M., MacIntyre B., MacMillan J., et al. Heparin therapy during cardiopulmonary bypass in children requires ongoing quality control. Thromb Haemost. 1993;70:937.
26 Papaconstantinou C., Radegran K. Use of the activated coagulation time in cardiac surgery. Effects on heparin-protamine dosages and bleeding. Scand J Thorac Cardiovasc Surg. 1981;15:213-215.
27 Roth J.A., Cukingnan R.A., Scott C.R. Use of activated coagulation time to monitor heparin during cardiac surgery. Ann Thorac Surg. 1979;28:69-72.
28 Flom-Halvorsen H.I., Ovrum E., Abdelnoor M., et al. Assessment of heparin anticoagulation: Comparison of two commercially available methods. Ann Thorac Surg. 1999;67:1012-1016. discussion 6–7
29 Young J.A., Kisker C.T., Doty D.B. Adequate anticoagulation during cardiopulmonary bypass determined by activated clotting time and the appearance of fibrin monomer. Ann Thorac Surg. 1978;26:231.
30 Metz S., Keats A.S. Low activated coagulation time during cardiopulmonary bypass does not increase postoperative bleeding. Ann Thorac Surg. 1990;49:440.
31 Cardoso P.F., Yamazaki F., Keshavjee S., et al. A reevaluation of heparin requirements for cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1991;101:153.
32 Gravlee G.P., Brauer S.D., Roy R.C., et al. Predicting the pharmacodynamics of heparin: A clinical evaluation of the Hepcon System 4. J Cardiothorac Anesth. 1987;1:379.
33 Utley J.R. Pathophysiology of cardiopulmonary bypass: current issues. J Card Surg. 1990;5:177.
34 Young E., Prins M., Levine M.N., Hirsh J. Heparin binding to plasma proteins, an important mechanism for heparin resistance. Thromb Haemost. 1992;67:639.
35 Marciniak E., Gockerman J.P. Heparin-induced decrease in circulating antithrombin-III. Lancet. 1977;2:581.
36 Linden M.D., Schneider M., Baker S., et al. Decreased concentration of antithrombin after preoperative therapeutic heparin does not cause heparin resistance during cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 2004;18:131.
37 Esposito R.A., Culliford A.T., Colvin S.B., et al. Heparin resistance during cardiopulmonary bypass. The role of heparin pretreatment. J Thorac Cardiovasc Surg. 1983;85:346.
38 Kanbak M. The treatment of heparin resistance with antithrombin III in cardiac surgery. Can J Anaesth. 1999;46:581.
39 Lemmer J.H.Jr, Despotis G.J. Antithrombin III concentrate to treat heparin resistance in patients undergoing cardiac surgery. J Thorac Cardiovasc Surg. 2002;123:213.
40 Levy J.H., Montes F., Szlam F., Hillyer C.D. The in vitro effects of antithrombin III on the activated coagulation time in patients on heparin therapy. Anesth Analg. 2000;90:1076.
41 Nicholson S.C., Keeling D.M., Sinclair M.E., Evans R.D. Heparin pretreatment does not alter heparin requirements during cardiopulmonary bypass. Br J Anaesth. 2001;87:844.
42 Avidan M.S., Levy J.H., van Aken H., et al. Recombinant human antithrombin III restores heparin responsiveness and decreases activation of coagulation in heparin-resistant patients during cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2005;130:107-113.
43 Avidan M.S., Levy J.H., Scholz J., et al. A phase III, double-blind, placebo-controlled, multicenter study on the efficacy of recombinant human antithrombin in heparin-resistant patients scheduled to undergo cardiac surgery necessitating cardiopulmonary bypass. Anesthesiology. 2005;102:276-284.
44 Lobato R.L., Despotis G.J., Levy J.H., et al. Anticoagulation management during cardiopulmonary bypass: A survey of 54 North American institutions. J Thorac Cardiovasc Surg. 2010;139:1665-1666.
45 Warkentin T.E., Kelton J.G. Temporal aspects of heparin-induced thrombocytopenia. N Engl J Med. 2001;344:1286-1292.
46 Warkentin T.E., Greinacher A. Heparin-induced thrombocytopenia and cardiac surgery. Ann Thorac Surg. 2003;76:638.
47 Koster A., Dyke C.M., Aldea G., et al. Bivalirudin during cardiopulmonary bypass in patients with previous or acute heparin-induced thrombocytopenia and heparin antibodies: Results of the CHOOSE-ON trial. Ann Thorac Surg. 2007;83:572-577.
48 Mabry C.D., Thompson B.W., Read R.C. Activated clotting time (ACT) monitoring of intraoperative heparinization in peripheral vascular surgery. Am J Surg. 1979;138:894.
49 Bull B.S., Huse W.M., Brauer F.S., Korpman R.A. Heparin therapy during extracorporeal circulation. II. The use of a dose-response curve to individualize heparin and protamine dosage. J Thorac Cardiovasc Surg. 1975;69:685.
50 Jobes D.R., Aitken G.L., Shaffer G.W. Increased accuracy and precision of heparin and protamine dosing reduces blood loss and transfusion in patients undergoing primary cardiac operations. J Thorac Cardiovasc Surg. 1995;110:36.
51 Shore-Lesserson L., Reich D.L., DePerio M. Heparin and protamine titration do not improve haemostasis in cardiac surgical patients. Can J Anaesth. 1998;45:10.
52 Szalados J.E., Ouriel K., Shapiro J.R. Use of the activated coagulation time and heparin dose-response curve for the determination of protamine dosage in vascular surgery. J Cardiothorac Vasc Anesth. 1994;8:515.
53 Despotis G.J., Levine V., Joiner-Maier D., Joist J.H. A comparison between continuous infusion versus standard bolus administration of heparin based on monitoring in cardiac surgery. Blood Coagul Fibrinolysis. 1997;8:419.
54 Despotis G.J., Summerfield A.L., Joist J.H., et al. Comparison of activated coagulation time and whole blood heparin measurements with laboratory plasma anti-Xa heparin concentration in patients having cardiac operations. J Thorac Cardiovasc Surg. 1994;108:1076.
55 Gravlee G.P., Haddon W.S., Rothberger H.K., et al. Heparin dosing and monitoring for cardiopulmonary bypass. A comparison of techniques with measurement of subclinical plasma coagulation. J Thorac Cardiovasc Surg. 1990;99:518.
56 Gravlee G.P., Rogers A.T., Dudas L.M., et al. Heparin management protocol for cardiopulmonary bypass influences postoperative heparin rebound but not bleeding. Anesthesiology. 1992;76:393.
57 Despotis G.J., Joist J.H., Hogue C.W.Jr, et al. The impact of heparin concentration and activated clotting time monitoring on blood conservation. A prospective, randomized evaluation in patients undergoing cardiac operation. J Thorac Cardiovasc Surg. 1995;110:46.
58 Wang J.S., Lin C.Y., Karp R.B. Comparison of high-dose thrombin time with activated clotting time for monitoring of anticoagulant effects of heparin in cardiac surgical patients. Anesth Analg. 1994;79:9.
59 Wang J.S., Lin C.Y., Hung W.T., Karp R.B. Monitoring of heparin-induced anticoagulation with kaolin-activated clotting time in cardiac surgical patients treated with aprotinin. Anesthesiology. 1992;77:1080.
60 Dietrich W., Dilthey G., Spannagl M., et al. Influence of high-dose aprotinin on anticoagulation, heparin requirement, and celite- and kaolin-activated clotting time in heparin-pretreated patients undergoing open-heart surgery. A double-blind, placebo-controlled study. Anesthesiology. 1995;83:679. discussion 29A
61 Tabuchi N., Njo T.L., Tigchelaar I., et al. Monitoring of anticoagulation in aprotinin-treated patients during heart operation. Ann Thorac Surg. 1994;58:774.
62 Dercksen S.J., Linssen G.H. Monitoring of blood coagulation in open heart surgery. II. Use of individualized dosages of heparin and protamine controlled by activated coagulation times. Acta Anaesthesiol Belg. 1980;31:121.
63 Dercksen S.J., Linssen G.H. Monitoring of blood coagulation in open heart surgery. I. Effects of conventional dosages of heparin and protamine. Acta Anaesthesiol Belg. 1980;31:113.
64 Ellison N., Jobes D.R., Schwartz A.J. Implications of anticoagulant therapy. Int Anesthesiol Clin. 1982;20:121.
65 Jobes D.R. Safety issues in heparin and protamine administration for extracorporeal circulation. J Cardiothorac Vasc Anesth. 1998;12:17.
66 Habazettl H., Conzen P.F., Vollmar B., et al. Effect of leukopenia on pulmonary hypertension after heparin-protamine in pigs. J Appl Physiol. 1992;73:44.
67 Warkentin T.E., Crowther M.A. Reversing anticoagulants both old and new. Can J Anaesth. 2002;49:S11.
68 Carr M.E.Jr, Carr S.L. At high heparin concentrations, protamine concentrations which reverse heparin anticoagulant effects are insufficient to reverse heparin anti-platelet effects. Thromb Res. 1994;75:617.
69 Ammar T., Fisher C.F. The effects of heparinase 1 and protamine on platelet reactivity. Anesthesiology. 1997;86:1382.
70 Carr M.E.Jr. Measurement of platelet force: The Hemodyne hemostasis analyzer. Clin Lab Manage Rev. 1995;9:312.
71 Martin P., Horkay F., Gupta N.K., et al. Heparin rebound phenomenon—much ado about nothing? Blood Coagul Fibrinolysis. 1992;3:187.
72 Jobes D.R., Schwartz A.J., Ellison N. Heparin rebound (letter). J Thorac Cardiovasc Surg. 1981;82:940.
73 LaDuca F.M., Zucker M.L., Walker C.E. Assessing heparin neutralization following cardiac surgery: Sensitivity of thrombin time-based assays versus protamine titration methods. Perfusion. 1999;14:181.
74 Ohata T., Sawa Y., Ohtake S., et al. Clinical role of blood heparin level monitoring during open heart surgery. Jpn J Thorac Cardiovasc Surg. 1999;47:600.
75 Reiner J.S., Coyne K.S., Lundergan C.F., Ross A.M. Bedside monitoring of heparin therapy: Comparison of activated clotting time to activated partial thromboplastin time. Cathet Cardiovasc Diagn. 1994;32:49.
76 D’Ambra M. Restoration of the normal coagulation process: Advances in therapies to antagonize heparin. J Cardiovasc Pharmacol. 1996;27:S58.
77 Martindale S.J., Shayevitz J.R., D’Errico C. The activated coagulation time: Suitability for monitoring heparin effect and neutralization during pediatric cardiac surgery. J Cardiothorac Vasc Anesth. 1996;10:458.
78 Moriau M., Masure R., Hurlet A., et al. Haemostasis disorders in open heart surgery with extracorporeal circulation. Importance of the platelet function and the heparin neutralization. Vox Sang. 1977;32:41.
79 Shigeta O., Kojima H., Hiramatsu Y., et al. Low-dose protamine based on heparin-protamine titration method reduces platelet dysfunction after cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1999;118:354.
80 Reich D.L., Yanakakis M.J., Vela-Cantos F.P., et al. Comparison of bedside coagulation monitoring tests with standard laboratory tests in patients after cardiac surgery. Anesth Analg. 1993;77:673.
81 Williams R.D., D’Ambra M.N., Maione T.E., et al. Recombinant platelet factor 4 reversal of heparin in human cardiopulmonary bypass blood. J Thorac Cardiovasc Surg. 1994;108:975.
82 Cook J.J., Niewiarowski S., Yan Z., et al. Platelet factor 4 efficiently reverses heparin anticoagulation in the rat without adverse effects of heparin-protamine complexes. Circulation. 1992;85:1102.
83 Levy J.H., Cormack J.G., Morales A. Heparin neutralization by recombinant platelet factor 4 and protamine. Anesth Analg. 1995;81:35.
84 Michelsen L.G., Kikura M., Levy J.H., et al. Heparinase I (Neutralase) reversal of systemic anticoagulation. Anesthesiology. 1996;85:339.
85 Bain B., Forster T., Sleigh B. Heparin and the activated partial thromboplastin time—a difference between the in-vitro and in-vivo effects and implications for the therapeutic range. Am J Clin Pathol. 1980;74:668.
86 Ray M.J., Carroll P.A., Just S.J., et al. The effect of oral anticoagulant therapy on APTT results from a bedside coagulation monitor. J Clin Monit. 1994;10:97.
87 Werner M., Gallagher J.V., Ballo M.S., Karcher D.S. Effect of analytic uncertainty of conventional and point-of-care assays of activated partial thromboplastin time on clinical decisions in heparin therapy. Am J Clin Pathol. 1994;102:237.
88 Samama C.M., Quezada R., Riou B., et al. Intraoperative measurement of activated partial thromboplastin time and prothrombin time with a new compact monitor. Acta Anaesthesiol Scand. 1994;38:232.
89 Nuttall G.A., Oliver W.C., Beynen F.M., et al. Determination of normal versus abnormal activated partial thromboplastin time and prothrombin time after cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 1995;9:355.
90 Despotis G.J., Santoro S.A., Spitznagel E., et al. Prospective evaluation and clinical utility of on-site monitoring of coagulation in patients undergoing cardiac operation. J Thorac Cardiovasc Surg. 1994;107:271.
91 Despotis G.J., Grishaber J.E., Goodnough L.T. The effect of an intraoperative treatment algorithm on physicians’ transfusion practice in cardiac surgery. Transfusion. 1994;34:290.
92 Borowiec J., Bagge L., Saldeen T., Thelin S. Biocompatibility reflected by haemostasis variables during cardiopulmonary bypass using heparin-coated circuits. Thorac Cardiovasc Surg. 1997;45:163.
93 Jansen P.G., te Velthuis H., Huybregts R.A., et al. Reduced complement activation and improved postoperative performance after cardiopulmonary bypass with heparin-coated circuits. J Thorac Cardiovasc Surg. 1995;110:829.
94 Gu Y.J., Boonstra P.W., Rijnsburger A.A., et al. Cardiopulmonary bypass circuit treated with surface-modifying additives: A clinical evaluation of blood compatibility. Ann Thorac Surg. 1998;65:1342.
95 Gorman R.C., Ziats N., Rao A.K., et al. Surface-bound heparin fails to reduce thrombin formation during clinical cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1996;111:1. discussion 11,
96 Baufreton C., Jansen P.G., Le Besnerais P., et al. Heparin coating with aprotinin reduces blood activation during coronary artery operations. Ann Thorac Surg. 1997;63:50.
97 Ovrum E., Holen E.A., Tangen G., et al. Completely heparinized cardiopulmonary bypass and reduced systemic heparin: Clinical and hemostatic effects. Ann Thorac Surg. 1995;60:365.
98 Aldea G.S., Doursounian M., O’Gara P., et al. Heparin-bonded circuits with a reduced anticoagulation protocol in primary CABG: A prospective, randomized study. Ann Thorac Surg. 1996;62:410.
99 Weiss B.M., von Segesser L.K., Turina M.I., et al. Perioperative course and recovery after heparin-coated cardiopulmonary bypass: Low-dose versus high-dose heparin management. J Cardiothorac Vasc Anesth. 1996;10:464.
100 Kuitunen A.H., Heikkila L.J., Salmenpera M.T. Cardiopulmonary bypass with heparin-coated circuits and reduced systemic anticoagulation. Ann Thorac Surg. 1997;63:438.
101 Ovrum E., Brosstad F., Am Holen E., et al. Effects on coagulation and fibrinolysis with reduced versus full systemic heparinization and heparin-coated cardiopulmonary bypass. Circulation. 1995;92:2579.
102 Muller N., Popov-Cenic S., Buttner W., et al. Studies of fibrinolytic and coagulation factors during open heart surgery. II. Postoperative bleeding tendency and changes in the coagulation system. Thromb Res. 1975;7:589.
103 Umlas J. Fibrinolysis and disseminated intravascular coagulation in open heart surgery. Transfusion. 1976;16:460.
104 Whitten C.W., Greilich P.E., Ivy R., et al. D-dimer formation during cardiac and noncardiac thoracic surgery. Anesth Analg. 1999;88:1226.
105 Weitz J., Klement P. Bivalirudin as an alternative anticoagulant to heparin in cardiopulmonary bypass: Data from a porcine model (abstract). Anesthesiology. 2003;99:A.
106 Lincoff A.M., Bittl J.A., Kleiman N.S., et al. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE] trial). Am J Cardiol. 2004;93:1092.
107 Merry A.F. Bivalirudin, blood loss, and graft patency in coronary artery bypass surgery. Semin Thromb Hemost. 2004;30:337.
108 Greinacher A. The use of direct thrombin inhibitors in cardiovascular surgery in patients with heparin-induced thrombocytopenia. Semin Thromb Hemost. 2004;30:315.
109 Koster A., Kuppe H., Hetzer R., et al. Emergent cardiopulmonary bypass in five patients with heparin-induced thrombocytopenia type II employing recombinant hirudin. Anesthesiology. 1998;89:777.
110 Koster A., Crystal G.J., Kuppe H., Mertzlufft F. Acute heparin-induced thrombocytopenia type II during cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 2000;14:300.
111 Koster A., Hansen R., Kuppe H., et al. Recombinant hirudin as an alternative for anticoagulation during cardiopulmonary bypass in patients with heparin-induced thrombocytopenia type II: A 1-year experience in 57 patients. J Cardiothorac Vasc Anesth. 2000;14:243.
112 Koster A., Loebe M., Hansen R., et al. A quick assay for monitoring recombinant hirudin during cardiopulmonary bypass in patients with heparin-induced thrombocytopenia type II: Adaptation of the ecarin clotting time to the act II device. J Thorac Cardiovasc Surg. 2000;119:1278.
113 Koster A., Kuppe H., Crystal G.J., Mertzlufft F. Cardiovascular surgery without cardiopulmonary bypass in patients with heparin-induced thrombocytopenia type II using anticoagulation with recombinant hirudin. Anesth Analg. 2000;90:292.
114 Koster A., Hansen R., Grauhan O., et al. Hirudin monitoring using the TAS ecarin clotting time in patients with heparin-induced thrombocytopenia type II. J Cardiothorac Vasc Anesth. 2000;14:249.
115 Koster A., Pasic M., Bauer M., et al. Hirudin as anticoagulant for cardiopulmonary bypass: Importance of preoperative renal function. Ann Thorac Surg. 2000;69:37.
116 Koster A., Loebe M., Mertzlufft F., et al. Cardiopulmonary bypass in a patient with heparin induced thrombocytopenia II and impaired renal function using heparin and the platelet GP IIb/IIIa inhibitor tirofiban as anticoagulant. Ann Thorac Surg. 2000;70:2160.
117 Koster A., Merkle F., Hansen R., et al. Elimination of recombinant hirudin by modified ultrafiltration during simulated cardiopulmonary bypass: Assessment of different filter systems. Anesth Analg. 2000;91:265.
118 Maroo A., Lincoff A.M. Bivalirudin in PCI: An overview of the REPLACE Trial. Semin Thromb Hemost. 2004;30:329.
119 Merry A.F., Raudkivi P.J., Middleton N.G., et al. Bivalirudin versus heparin and protamine in off-pump coronary artery bypass surgery. Ann Thorac Surg. 2004;77:925.
120 Davis Z., Anderson R., Short D., et al. Favorable outcome with bivalirudin anticoagulation during cardiopulmonary bypass. Ann Thorac Surg. 2003;75:264.
121 Gordon G., Rastegar H., Schumann R., et al. Successful use of bivalirudin for cardiopulmonary bypass in a patient with heparin-induced thrombocytopenia. J Cardiothorac Vasc Anesth. 2003;17:632.
122 Vasquez J.C., Vichiendilokkul A., Mahmood S., Baciewicz F.A.Jr. Anticoagulation with bivalirudin during cardiopulmonary bypass in cardiac surgery. Ann Thorac Surg. 2002;74:2177.
123 Smedira N.G., Dyke C.M., Koster A., et al. Anticoagulation with bivalirudin for off-pump coronary artery bypass grafting: The results of the EVOLUTION-OFF study. J Thorac Cardiovasc Surg. 2006;131:686-692.
124 Dyke C.M., Smedira N.G., Koster A., et al. A comparison of bivalirudin to heparin with protamine reversal in patients undergoing cardiac surgery with cardiopulmonary bypass: The EVOLUTION-ON study. J Thorac Cardiovasc Surg. 2006;131:533-539.
125 Koster A., Spiess B., Chew D.P., et al. Effectiveness of bivalirudin as a replacement for heparin during cardiopulmonary bypass in patients undergoing coronary artery bypass grafting. Am J Cardiol. 2004;93:356.
126 McDonald S.B., Kattapurum B.M., Saleem R., et al. Monitoring hirudin anticoagulation in two patients undergoing cardiac surgery with a plasma-modified act method. Anesthesiology. 2002;97:509-512.
127 Chomiak P.N., Walenga J.M., Koza M.J., et al. Investigation of a thrombin inhibitor peptide as an alternative to heparin in cardiopulmonary bypass surgery. Circulation. 1993;88:II-407.
128 Bittl J.A., Strony J., Brinker J.A., et al. Treatment with bivalirudin (Hirulog) as compared with heparin during coronary angioplasty for unstable or postinfarction angina. Hirulog Angioplasty Study Investigators. N Engl J Med. 1995;333:764.
129 Zilla P., Fasol R., Deutsch M., et al. Whole blood aggregometry and platelet adenine nucleotides during cardiac surgery. Scand J Thorac Cardiovasc Surg. 1988;22:165.
130 Halbmayer W.M., Haushofer A., Radek J., et al. Platelet size, fibrinogen and lipoprotein(a) in coronary heart disease. Coron Artery Dis. 1995;6:397.
131 Boldt J., Zickmann B., Benson M., et al. Does platelet size correlate with function in patients undergoing cardiac surgery? Intensive Care Med. 1993;19:44.
132 Despotis G.J., Filos K.S., Zoys T.N., et al. Factors associated with excessive postoperative blood loss and hemostatic transfusion requirements: A multivariate analysis in cardiac surgical patients. Anesth Analg. 1996;82:13.
133 Khuri S.F., Wolfe J.A., Josa M., et al. Hematologic changes during and after cardiopulmonary bypass and their relationship to the bleeding time and nonsurgical blood loss. J Thorac Cardiovasc Surg. 1992;104:94.
134 Bertolino G., Locatelli A., Noris P., et al. Platelet composition and function in patients undergoing cardiopulmonary bypass for heart surgery. Haematologica. 1996;81:116.
135 Boldt J., Schindler E., Knothe C., et al. Does aprotinin influence endothelial-associated coagulation in cardiac surgery? J Cardiothorac Vasc Anesth. 1994;8:527.
136 Kestin A.S., Valeri C.R., Khuri S.F., et al. The platelet function defect of cardiopulmonary bypass. Blood. 1993;82:107.
137 Upchurch G.R., Valeri C.R., Khuri S.F., et al. Effect of heparin on fibrinolytic activity and platelet function in vivo. Am J Physiol. 1996;271:H528.
138 Adelman B., Michelson A.D., Loscalzo J., et al. Plasmin effect on platelet glycoprotein Ib- von Willebrand factor interactions. Blood. 1985;65:32.
139 Adelman B., Michelson A.D., Greenberg J., Handin R.I. Proteolysis of platelet glycoprotein Ib by plasmin is facilitated by plasmin lysine-binding regions. Blood. 1986;68:1280.
140 Michelson A.D., MacGregor H., Barnard M.R., et al. Reversible inhibition of human platelet activation by hypothermia in vivo and in vitro. Thromb Haemost. 1994;71:633.
141 Boldt J., Zickmann B., Czeke A., et al. Blood conservation techniques and platelet function in cardiac surgery. Anesthesiology. 1991;75:426.
142 Burns E.R., Billett H.H., Frater R.W., Sisto D.A. The preoperative bleeding time as a predictor of postoperative hemorrhage after cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1986;92:310.
143 Burns E.R., Lawrence C. Bleeding time. A guide to its diagnostic and clinical utility. Arch Pathol Lab Med. 1989;113:1219.
144 Despotis G.J., Levine V., Filos K.S., et al. Evaluation of a new point-of-care test that measures PAF-mediated acceleration of coagulation in cardiac surgical patients. Anesthesiology. 1996;85:1311.
145 Despotis G.J., Levine V., Saleem R., et al. Use of point-of-care test in identification of patients who can benefit from desmopressin during cardiac surgery: A randomized controlled trial. Lancet. 1999;354:106.
146 Mohr R., Martinowitz U., Lavee J., et al. The hemostatic effect of transfusing fresh whole blood versus platelet concentrates after cardiac operations. J Thorac Cardiovasc Surg. 1988;96:530.
147 Ray M.J., Hawson G.A., Just S.J., et al. Relationship of platelet aggregation to bleeding after cardiopulmonary bypass. Ann Thorac Surg. 1994;57:981.
148 Carr M.E.Jr. In vitro assessment of platelet function. Transfus Med Rev. 1997;11:106.
149 George J.N., Pickett E.B., Saucerman S., et al. Platelet surface glycoproteins. Studies on resting and activated platelets and platelet membrane microparticles in normal subjects, and observations in patients during adult respiratory distress syndrome and cardiac surgery. Clin Invest. 1986;78:340.
150 Adelman B., Michelson A.D., Handin R.I., Ault K.A. Evaluation of platelet glycoprotein Ib by fluorescence flow cytometry. Blood. 1985;66:423.
151 Michelson A.D., Barnard M.R. Thrombin-induced changes in platelet membrane glycoproteins Ib, IX, and IIb-IIIa complex. Blood. 1987;70:1673.
152 Nieuwenhuis H.K., van Oosterhout J.J., Rozemuller E., et al. Studies with a monoclonal antibody against activated platelets: Evidence that a secreted 53,000-molecular weight lysosome-like granule protein is exposed on the surface of activated platelets in the circulation. Blood. 1987;70:838.
153 Michelson A.D. Flow cytometric analysis of platelet surface glycoproteins: Phenotypically distinct subpopulations of platelets in children with chronic myeloid leukemia. J Lab Clin Med. 1987;110:346.
154 LaRosa C.A., Rohrer M.J., Benoit S.E., et al. Neutrophil cathepsin G modulates the platelet surface expression of the glycoprotein (GP) Ib-IX complex by proteolysis of the von Willebrand factor binding site on GPIb alpha and by a cytoskeletal-mediated redistribution of the remainder of the complex. Blood. 1994;84:158.
155 Rinder C.S., Mathew J.P., Rinder H.M., et al. Modulation of platelet surface adhesion receptors during cardiopulmonary bypass. Anesthesiology. 1991;75:563.
156 Sloand J.A., Sloand E.M. Studies on platelet membrane glycoproteins and platelet function during hemodialysis. J Am Soc Nephrol. 1997;8:799.
157 Shore-Lesserson L., Ammar T., DePerio M., et al. Platelet-activated clotting time does not measure platelet reactivity during cardiac surgery. Anesthesiology. 1999;91:362.
158 Miller B.E., Mochizuki T., Levy J.H., et al. Predicting and treating coagulopathies after cardiopulmonary bypass in children. Anesth Analg. 1997;85:1196.
159 Spiess B.D. Thromboelastography and cardiopulmonary bypass. Semin Thromb Hemost. 1995;21:27.
160 Tuman K.J., Spiess B.D., McCarthy R.J., Ivankovich A.D. Comparison of viscoelastic measures of coagulation after cardiopulmonary bypass. Anesth Analg. 1989;69:69.
161 Spalding G.J., Hartrumpf M., Sierig T., et al. Cost reduction of perioperative coagulation management in cardiac surgery: Value of “bedside” thrombelastography (ROTEM). Eur J Cardiothorac Surg. 2007;31:1052-1057.
162 Tuman K.J., McCarthy R.J., Djuric M., et al. Evaluation of coagulation during cardiopulmonary bypass with a heparinase-modified thromboelastographic assay. J Cardiothorac Vasc Anesth. 1994;8:144.
163 Ereth M.H., Nuttall G.A., Klindworth J.T., et al. Does the platelet-activated clotting test (HemoSTATUS) predict blood loss and platelet dysfunction associated with cardiopulmonary bypass? Anesth Analg. 1997;85:259.
164 Mongan P.D., Hosking M.P. The role of desmopressin acetate in patients undergoing coronary artery bypass surgery. A controlled clinical trial with thromboelastographic risk stratification. Anesthesiology. 1992;77:38.
165 Gravlee G.P., Arora S., Lavender S.W., et al. Predictive value of blood clotting tests in cardiac surgical patients. Ann Thorac Surg. 1994;58:216.
166 Spiess B.D., Gillies B.S., Chandler W., Verrier E. Changes in transfusion therapy and reexploration rate after institution of a blood management program in cardiac surgical patients. J Cardiothorac Vasc Anesth. 1995;9:168.
167 Whitten C.W., Allison P.M., Latson T.W., et al. Evaluation of laboratory coagulation and lytic parameters resulting from autologous whole blood transfusion during primary aortocoronary artery bypass grafting. J Clin Anesth. 1996;8:229.
168 Greilich P.E., Alving B.M., O’Neill K.L., et al. A modified thromboelastographic method for monitoring c7E3 Fab in heparinized patients. Anesth Analg. 1997;84:31.
169 Kang Y.G., Martin D.J., Marquez J., et al. Intraoperative changes in blood coagulation and thromboelastographic monitoring in liver transplantation. Anesth Analg. 1985;64:888.
170 Martin L.K., Kang Y., De Wolf A.M. Coagulation changes immediately following liver graft reperfusion. Transplant Proc. 1991;23:1946.
171 Khurana S., Mattson J.C., Westley S., et al. Monitoring platelet glycoprotein IIb/IIIa-fibrin interaction with tissue factor-activated thromboelastography. J Lab Clin Med. 1997;130:401.
172 Shore-Lesserson L., Manspeizer H.E., DePerio M., et al. Thromboelastography-guided transfusion algorithm reduces transfusions in complex cardiac surgery. Anesth Analg. 1999;88:312.
173 Gottumukkala V.N., Sharma S.K., Philip J. Assessing platelet and fibrinogen contribution to clot strength using modified thromboelastography in pregnant women. Anesth Analg. 1999;89:1453.
174 Greilich P.E., Alving B.M., Longnecker D., et al. Near-site monitoring of the antiplatelet drug abciximab using the Hemodyne analyzer and modified thromboelastograph. J Cardiothorac Vasc Anesth. 1999;13:58.
175 Craft R.M., Chavez J.J., Bresee S.J., et al. A novel modification of the Thromboelastograph assay, isolating platelet function, correlates with optical platelet aggregation. J Lab Clin Med. 2004;143:301.
176 Shore-Lesserson L., Fischer G., Sanders J., et al. Clopidogrel induces a platelet aggregation defect that is partially mitigated by ex-vivo addition of aprotinin. Anesthesiology. 2004;101:A.
177 Hett D.A., Walker D., Pilkington S.N., Smith D.C. Sonoclot analysis. Br J Anaesth. 1995;75:771.
178 Stern M.P., DeVos-Doyle K., Viguera M.G., Lajos T.Z. Evaluation of post-cardiopulmonary bypass Sonoclot signatures in patients taking nonsteroidal anti-inflammatory drugs. J Cardiothorac Anesth. 1989;3:730.
179 Chapin J.W., Becker G.L., Hulbert B.J., et al. Comparison of Thromboelastograph and Sonoclot coagulation analyzers for assessing coagulation status during orthotopic liver transplantation. Transplant Proc. 1989;21:3539.
180 Peck S.D. Evaluation of the in vitro detection of the hypercoagulable state using the thrombin generation test and plasma clot impedance test. Thromb Haemost. 1979;42:764.
181 Smith J.W., Steinhubl S.R., Lincoff A.M., et al. Rapid platelet-function assay: An automated and quantitative cartridge-based method. Circulation. 1999;99:620.
182 Coller B.S., Lang D., Scudder L.E. Rapid and simple platelet function assay to assess glycoprotein IIb/IIIa receptor blockade. Circulation. 1997;95:860.
183 Coller B.S., Folts J.D., Scudder L.E., Smith S.R. Antithrombotic effect of a monoclonal antibody to the platelet glycoprotein IIb/IIIa receptor in an experimental animal model. Blood. 1986;68:783.
184 Paniccia R., Antonucci E., Gori A.M., et al. Different methodologies for evaluating the effect of clopidogrel on platelet function in high-risk coronary artery disease patients. J Thromb Haemost. 2007;5:1835-1838.
185 van Werkum J.W., Harmsze A.M., Elsenberg E.H., et al. The use of the VerifyNow system to monitor antiplatelet therapy: A review of the current evidence. Platelets. 2008;19:479-488.
186 Ikarugi H., Taka T., Nakajima S., et al. Norepinephrine, but not epinephrine, enhances platelet reactivity and coagulation after exercise in humans. J Appl Physiol. 1999;86:133.
187 John L.C., Rees G.M., Kovacs I.B. Inhibition of platelet function by heparin. An etiologic factor in postbypass hemorrhage. J Thorac Cardiovasc Surg. 1993;105:816.
188 Kovacs I.B., Mayou S.C., Kirby J.D. Infusion of a stable prostacyclin analogue, iloprost, to patients with peripheral vascular disease: Lack of antiplatelet effect but risk of thromboembolism. Am J Med. 1991;90:41.
189 Ratnatunga C.P., Rees G.M., Kovacs I.B. Preoperative hemostatic activity and excessive bleeding after cardiopulmonary bypass. Ann Thorac Surg. 1991;52:250.
190 Bock M., De Haan J., Beck K.H., et al. Standardization of the PFA-100 (R) platelet function test in 105 mmol/L buffered citrate: Effect of gender, smoking, and oral contraceptives. Br J Haematol. 1999;106:898.
191 Escolar G., Cases A., Vinas M., et al. Evaluation of acquired platelet dysfunctions in uremic and cirrhotic patients using the platelet function analyzer (PFA-100): Influence of hematocrit elevation. Haematologica. 1999;84:614.
192 Kundu S.K., Heilmann E.J., Sio R., et al. Description of an in vitro platelet function analyzer—PFA-100. Semin Thromb Hemost. 1995;21:106.
193 Mammen E.F., Comp P.C., Gosselin R., et al. PFA-100 system: A new method for assessment of platelet dysfunction. Semin Thromb Hemost. 1998;24:195.
194 Wahba A., Sander S., Birnbaum D.E. Are in-vitro platelet function tests useful in predicting blood loss following open heart surgery? Thorac Cardiovasc Surg. 1998;46:228.
195 Nuttall G., Oliver W.C.Jr, Ereth M.H., et al. The PFA 100, correlation with platelet aggregometry with GP IIB/IIIA receptor antagonists (abstract). Anesthesiology. 1999;91:A518.
196 Carville D.G., Schleckser P.A., Guyer K.E., et al. Whole blood platelet function assay on the ICHOR point-of-care hematology analyzer. J Extra Corpor Technol. 1998;30:171.
197 Slaughter T.F., Sreeram G., Sharma A.D., et al. Reversible shear-mediated platelet dysfunction during cardiac surgery as assessed by the PFA-100 platelet function analyzer. Blood Coagul Fibrinolysis. 2001;12:85.
198 Steinhubl S.R., Talley J.D., Braden G.A., et al. Point-of-care measured platelet inhibition correlates with a reduced risk of an adverse cardiac event after percutaneous coronary intervention: Results of the GOLD (AU-Assessing Ultegra) multicenter study. Circulation. 2001;103:2572.
199 Faraday N., Guallar E., Sera V.A., et al. Utility of whole blood hemostatometry using the clot signature analyzer for assessment of hemostasis in cardiac surgery. Anesthesiology. 2002;96:1115.
200 Kenet G., Lubetsky A., Shenkman B., et al. Cone and platelet analyser (CPA): A new test for the prediction of bleeding among thrombocytopenic patients. Br J Haematol. 1998;101:255-259.
201 Varon D., Lashevski I., Brenner B., et al. Cone and plate(let) analyzer: Monitoring glycoprotein IIb/IIIa anatgonists and von Willebrand disease replacement therapy by testing platelet disposition under flow conditions. Am Heart J. 1998;135(5 Pt 2 Su):S187-S193.
202 Shenkman B., Schneiderman J., Tamarin I., et al. Testing the effect of GPIIb-IIIa antagonist in patients undergoing carotid stenting: Correlation between standard aggregometry, flow cytometry and the cone and plate(let) analyzer (CPA) methods. Thromb Res. 2001;102:311-317.
203 Savion N., Varon D. Impact-the cona and plate(let) analyzer: Testing platelet function and anti-platelet drug response. Pathophysiol Haemost Thromb. 2006;35:83-88.
204 Gerrah R., Brill A., Tshori S., et al. Using cone and plate(let) analyzer to predict bleeding in cardiac surgery. Asian Cardiovasc Thorac Ann. 2006;14:310-315.
205 Rahe-Meyer N., Winterhalter M., Boden A., et al. Platelet concentrates transfusion in cardiac surgery and platelet function assessment by multiple electrode aggregometry. Acta Anaesthesiol Scand. 2009;53:168-175.
206 Rahe-Meyer N., Winterhalter M., Hartmann J., et al. An evaluation of cyclooxygenase-1 inhibition before coronary artery surgery: Aggregometry versus patient self-reporting. Anesth Analg. 2008;107:1791-1797.
207 Velik-Salchner C., Maier S., Innerhofer P., et al. Point-of-care whole blood impedance aggregometry versus classical light transmission aggregometry for detecting aspirin and clopidogrel: The results of a pilot study. Anesth Analg. 2008;107:1798-1806.
208 Velik-Salchner C., Maier S., Innerhofer P., et al. An assessment of cardiopulmonary bypass-induced changes in platelet function using whole blood and classical light transmission aggregometry: The results of a pilot study. Anesth Analg. 2009;108:1747-1754.
209 Mengistu A.M., Röhm K.D., Boldt J., et al. The influence of aprotinin and tranexamic acid on platelet function and postoperative blood loss in cardiac surgery. Anesth Analg. 2008;107:391-397.
210 Mengistu A.M., Wolf M.W., Boldt J., et al. Evaluation of a new platelet function analyzer in cardiac surgery: A comparison of modified thromboelastography and whole-blood aggregometry. J Cardiothorac Vasc Anesth. 2008;22:40-46.
211 Sibbing D., Braun S., Morath T., et al. Platelet reactivity after clopidogrel treatment assessed with point-of-care analysis and early drug-eluting stent thrombosis. J Am Coll Cardiol. 2009;53:849-856.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
17 Coagulation Monitoring
CPB itself induces a “whole-body inflammatory response” because of contact of blood and cellular elements with the extracorporeal circuit. The resultant alterations include leukocyte activation, release of inflammatory mediators, free radical formation, complement activation, kallikrein release, platelet activation, and stimulation of the coagulation and fibrinolytic cascades. This complex interplay of systems induces a coagulopathy characterized by microvascular coagulation, platelet dysfunction, and enhanced fibrinolysis.1,2 The homeostatic perturbations incurred during CPB are a major cause for the postbypass coagulopathy that is seen even after the effects of heparin are reversed with protamine.
The need to monitor anticoagulation during and after surgery is the reason that the cardiac surgical arena has evolved into a major site for the evaluation and use of hemostasis monitors. The rapid and accurate identification of abnormal hemostasis has been the major impetus toward the development of point-of-care tests that can be performed at the bedside or in the operating room. The detection and treatment of specific coagulation disorders in a timely and cost-efficient manner are major goals in hemostasis monitoring for the cardiac surgical patient. This chapter discusses the mechanisms of normal coagulation and how they are affected by CPB. The latter portion discusses the laboratory and point-of-care tests available for monitoring the coagulation system. A comprehensive overview of hemostasis, transfusion medicine, and the management of coagulopathy and bleeding disorders after CPB is provided in Chapters 30 and 31.
Hemostasis
Anticoagulation for Cardiopulmonary Bypass
Heparin acts as an antithrombin III (AT III) agonist and accelerates AT III binding to thrombin.3–5 In the absence of AT III, heparin is clinically ineffective as an anticoagulant; adequate AT III activity is necessary in patients about to undergo heparinization for cardiac surgical procedures.6–9
Monitoring heparin effect
The first clotting time to be used to measure heparin’s effect was the whole-blood clotting time (WBCT) or the Lee–White WBCT. This simply requires whole blood to be placed in a glass tube, maintained at 37°C, and manually tilted until blood fluidity is no longer detected. This test fell out of favor for monitoring the cardiac surgical patient because it was so labor intensive and required the undivided attention of the person performing the test for periods up to 30 minutes. Although the glass surface of the test tube acts as an activator of factor XII, the heparin doses used for cardiac surgery prolong the WBCT to such a profound degree that the test is impractical as a monitor of the effect of heparin during cardiac surgery.10 To speed the clotting time so that the test was appropriate for clinical use, activators were added to the test tubes, and the activated coagulation time (ACT) was introduced into practice.11
Activated Coagulation Time
The ACT was first introduced by Hattersley in 1966 and is still the most widely used monitor of heparin effect during cardiac surgery. Whole blood is added to a test tube containing an activator, diatomaceous earth (celite) or kaolin. The presence of activator augments the contact activation phase of coagulation, which stimulates the intrinsic coagulation pathway. ACT can be performed manually, whereby the operator measures the time interval from when blood is injected into the test tube to when clot is seen along the sides of the tube. More commonly, the ACT is automated as it is in the Hemochron and Hemotec systems. In the automated system, the test tube is placed in a device that warms the sample to 37°C. The Hemochron device (International Technidyne, Edison, NJ) rotates the test tube, which contains celite activator and a small iron cylinder, to which 2 mL whole blood is added. Before clot forms, the cylinder rolls along the bottom of the rotating test tube. When clot forms, the cylinder is pulled away from a magnetic detector, interrupts a magnetic field, and signals the end of the clotting time. Normal ACT values range from 80 to 120 seconds. The Hemochron ACT also can be performed using kaolin as the activator in a similar manner (Figure 17-1).
The Hemotec ACT device (Medtronic Hemotec, Parker, CO) is a cartridge with two chambers that contain kaolin activator and is housed in a heat block. Blood (0.4 mL) is placed into each chamber, and a daisy-shaped plunger is raised and passively falls into the chamber. The formation of clot will slow the rate of descent of the plunger, and this decrease in velocity of the plunger is detected by a photo-optical system that signals the end of the ACT test. The Hemochron and Hemotec ACTs have been compared in a number of investigations and have been found to differ significantly at low heparin concentrations.12 However, differences in heparin concentration, activator concentration, and the measurement technique make comparison of these tests difficult and have led to the realization that the Hemochron ACT result and the Hemotec ACT result are not interchangeable. In adult patients given 300 U/kg heparin for CPB, the Hemochron and Hemotec (Hepcon) ACTs were both therapeutic at all time points; however, at two points, the Hemochron ACT was statistically longer13 (Figure 17-2). This difference was even more pronounced in pediatric patients, who have greater heparin consumption rates (Figure 17-3). The apparent “overestimation” of ACT by the Hemochron device during hypothermic CPB may be because of the different volumes of blood that each assay warms to 37°C.
The ACT test can be modified by the addition of heparinase. With this modification, the coagulation status of the patient can be monitored during CPB while the anticoagulant effects of heparin are eliminated. Because this test is a side-by-side comparison of the untreated ACT to the heparinase ACT, it also has the advantage of being a rapid test for the assessment of a circulating heparin-like substance or for residual heparinization after CPB.14
With the introduction of ACT monitoring into the cardiac surgical arena, clinicians have been able to more accurately titrate heparin and protamine dosages.11,15 As a result, many investigators report reductions in blood loss and transfusion requirements, although many of these studies used retrospective analyses.16 The improvements in postoperative hemostasis documented with ACT monitoring are potentially attributable to better intraoperative suppression of microvascular coagulation and improved monitoring of heparin reversal with protamine.17
ACT monitoring of heparinization is not without pitfalls, and its use has been criticized because of the extreme variability of the ACT and the absence of a correlation with plasma heparin levels (Figure 17-4). Many factors have been suggested to alter the ACT, and these factors are prevalent during cardiac surgical procedures. When the extracorporeal circuit prime is added to the patient’s blood volume, hemodilution occurs and may theoretically increase ACT. Evidence suggests that this degree of hemodilution alone is not enough to actually alter ACT. Hypothermia increases ACT in a “dose-related” fashion. It has been shown by Culliford et al18 that, although hemodilution and hypothermia significantly increase the ACT of a heparinized blood sample, similar increases do not occur in the absence of added heparin. The effects of platelet alterations are a bit more problematic. At mild-to-moderate degrees of thrombocytopenia, the baseline and heparinized ACT are not affected. It is not until platelet counts are reduced to less than 30,000 to 50,000/μL that ACT may be prolonged.19 Patients treated with platelet inhibitors such as prostacyclin, aspirin, or platelet-membrane-receptor antagonists have a prolonged heparinized ACT compared with patients not treated with platelet inhibitors.20 This ACT prolongation is not related exclusively to decreased levels of platelet factor 4 (PF4) (PF4 is a heparin-neutralizing substance) because it also occurs when blood is anticoagulated with substances that are not neutralized by PF4. Platelet lysis, however, significantly shortens the ACT because of the release of PF4 and other platelet membrane components, which may have heparin-neutralizing activities.21 Gravlee et al22 showed that anesthesia and surgery decrease the ACT and create a hypercoagulable state, possibly by creating a thromboplastic response or through activation of platelets.
During CPB, heparin decay varies substantially and its measurement is problematic because hemodilution and hypothermia alter the metabolism of heparin. In a CPB study, Mabry et al23 found that the consumption of heparin varied from 0.01 to 3.86 U/kg/min and there was no correlation between the initial sensitivity to heparin and the rate of heparin decay.23 In the pediatric population, the consumption of heparin is increased to more than that of adult levels. The heparin administration protocol for pediatric patients undergoing CPB should account for a large volume of distribution, increased consumption, and a shorter elimination half-life. In monitoring the effects of heparin in pediatric patients, the minimum acceptable ACT value should be increased or an additional monitor should be used. The discrepancy between the Hemochron ACT and the Hemotec ACT that is demonstrated in Figure 17-2 is even more pronounced in pediatric patients (see Figure 17-3). Some investigators recommend the maintenance of heparin concentrations in addition to ACT during pediatric congenital heart surgery to ensure that optimal anticoagulation is being achieved.24,25
Cascade POC (Point of Care) System
A completely different technology for measuring the effect of heparin is used by the Cascade POC analyzer (Helena, Beaumont, TX; Formerly Rapid Point Coagulation Analyzer Bayer Diagnostics, Tarrytown, NY). This test system contains disposable cards with celite activator for the measurement of heparin activity. This variation of the ACT is called the “heparin management test” (HMT). This card contains paramagnetic iron oxide particles that move in response to an oscillating magnetic field within the device. When clot formation occurs, movement of the iron oxide particles is decreased and the end of the test is signaled. This system is capable of measuring prothrombin time (PT) and activated partial thromboplastin time (aPTT), which is discussed later. The suitability of this platform for the monitoring of ACT during cardiac surgery has been demonstrated in a variety of clinical studies.26,27 Suitability for monitoring heparinization in the interventional cardiology laboratory also has been reported. HMT correlates well with anti-Xa heparin activity in CPB patients and is less variable than standard ACT measures. In a comparison with ACT, the coefficients of variation were similar between the tests at baseline but were three times greater for the ACT during heparinization. This degree of agreement with plasma anti-Xa measurements has not been demonstrated universally when studying blood from patients undergoing CPB.28
Heparin Resistance
Heparin resistance is documented by an inability to increase the ACT of blood to expected levels despite an adequate dose and plasma concentration of heparin. In many clinical situations, especially when heparin desensitization or a heparin inhibitor is suspected, heparin resistance can be treated by administering increased doses of heparin in a competitive fashion. If an adequately prolonged clotting time is ultimately achieved using greater than expected doses of heparin, a better term than heparin resistance would be heparin tachyphylaxis of “altered heparin responsiveness.” During cardiac surgical procedures, the belief that a safe minimum ACT value of 300 to 400 seconds is required for CPB is based on a few clinical studies and a relative paucity of scientific data. However, inability to attain this degree of anticoagulation in the heparin-resistant patient engenders the fear among cardiac surgical providers that the patient will experience a microvascular consumptive coagulopathy or that clots will form in the extracorporeal circuit. This potential for fibrin formation in the extracorporeal circuit was reported by Young et al,29 who found increased production of fibrin monomer and consumption of fibrinogen and platelets in six of nine rhesus monkeys when the ACT declined to less than 400 seconds. However, Metz and Keats30 reported no adverse effects of thrombosis or excessive bleeding in 51 patients undergoing CPB whose ACT was less than 400 seconds. In a porcine model, the group whose ACT was maintained between 250 and 300 seconds did not have excessive consumption of coagulation factors, increases in fibrin monomer formation, or changes in oxygenator performance compared with the group whose ACT was maintained at greater than 450 seconds.31
Many clinical conditions are associated with heparin resistance.32 Sepsis, liver disease, and pharmacologic agents represent just a few33,34 (Table 17-1). Many investigators have documented decreased levels of AT III secondary to heparin pretreatment,35 whereas others have not found decreased AT III levels.36 Esposito et al37 measured coagulation factor levels in patients receiving preoperative heparin infusions and found that a lower baseline ACT was the only risk factor for predicting heparin resistance compared with patients not receiving preoperative heparin.
TABLE 17-1 Disease States Associated with Heparin Resistance
| Disease State | Comment |
|---|---|
| Newborn | Decreased AT III levels until 6 months of age |
| Venous thromboembolism | May have increased factor VIII level |
| Accelerated clearance of heparin | |
| Pulmonary embolism | Accelerated clearance of heparin |
| Congenital AT III deficiency | 40% to 60% of normal AT III concentration |
| Type I | Reduced synthesis of normal/abnormal AT III |
| Type II | Molecular defect within the AT III molecule |
| Acquired AT III deficiency | < 25% of normal AT III concentration |
| Preeclampsia | Levels unchanged in normal pregnancy |
| Cirrhosis | Decreased protein synthesis |
| Nephrotic syndrome | Increased urinary excretion of AT III |
| DIC | Increased consumption of AT III |
| Heparin pretreatment | 85% of normal AT III concentration because of accelerated clearance |
| Estrogen therapy | |
| Cytotoxic drug therapy (l-asparaginase) | Decreased protein synthesis |
ATIII, antithrombin III; DIC, disseminated intravascular coagulation.
Patients receiving preoperative heparin therapy traditionally require larger heparin doses to achieve a given level of anticoagulation when that anticoagulation is measured by the ACT. Presumably, this “heparin resistance” is due to deficiencies in the level or activity of AT III.34,37–40 Other possible causes include enhanced factor VIII activity and platelet dysfunction causing a decrease in ACT response to heparin. Levy et al40 have shown that the in vitro addition of AT III enhances the ACT response to heparin. Lemmer and Despotis39 demonstrated that this heparin resistance, as measured by the ACT, does not correlate with preoperative AT III levels. It is unclear that these patients have increased heparin requirements during CPB because the ideal ACT and monitoring techniques have yet to be elucidated. Nicholson et al41 demonstrated that the temporal courses of ACT and AT III concentration do not parallel each other, further suggesting that AT III depletion is not the sole cause of heparin resistance during CPB. Lower ACTs (lower than 480s) were tolerated in this study with no adverse outcome. AT III concentrate is available as a heat-treated human product or in recombinant form and represents a reasonable method of treating patients with documented AT III deficiency (Box 17-1). Heparin responsiveness in the form of increased ACT levels is documented both in vitro and in vivo when heparin-resistant patients are treated with AT III.
BOX 17-1. Heparin Resistance
In a multicenter, randomized, placebo-controlled trial using 75 U/kg recombinant AT III, AT III–treated patients received less FFP to augment ACT levels and also had evidence of less hemostatic activation while on CPB.42 There was a trend toward increased blood loss in the AT III group, which is potentially a dose effect that requires further investigation.43,44
Heparin-Induced Thrombocytopenia
The options for treating these patients are few. If the clinician has the luxury of being able to discontinue heparin for a few weeks, often the antibody disappears and allows a brief period of heparinization for CPB without complication.45,46 Changing the tissue source of heparin was an option when bovine heparin was predominantly in use. Some types of low-molecular-weight heparin have been administered to patients with HIT, but reactivity of the particular low-molecular-weight heparin with the patient’s platelets should be confirmed in vitro. Supplementing heparin administration with pharmacologic platelet inhibition using prostacyclin, iloprost, aspirin, or aspirin and dipyridamole have been reported, all with favorable outcomes. The use of tirofiban with unfractionated heparin has been used in this clinical circumstance. Plasmapheresis may be used to reduce antibody levels. The use of heparin could be avoided altogether through anticoagulation with direct thrombin inhibitors such as argatroban, hirudin, or bivalirudin. These thrombin inhibitors have become standard of care in the management of the patient with HIT (Box 17-2). Bivalirudin has been studied in multicenter trials in HIT patients who must undergo CPB.47 Monitoring the direct thrombin inhibitors during CPB is discussed later in this chapter.
Measurement of Heparin Sensitivity
Even in the absence of heparin resistance, patient response to an intravenous bolus of heparin is extremely variable.48 The variability stems from different concentrations of various endogenous heparin-binding proteins such as vitronectin and PF4. This variability exists whether measuring heparin concentration or the ACT; however, variability seems to be greater when measuring the ACT. Because of the large interpatient variation in heparin responsiveness and the potential for heparin resistance, it is critical that a functional monitor of heparin anticoagulation (with or without a measure of heparin concentration) be used in the cardiac surgical patient. Bull et al49 documented a threefold range of ACT response to a 200-U/kg heparin dose and similar discrepancy in heparin decay rates and, thus, recommended the use of individual patient dose–response curves to determine the optimal heparin dose. This is the concept on which point-of-care individual heparin dose–response (HDR) tests are based.
An HDR curve can be generated manually using the baseline ACT and the ACT response to an in vivo or in vitro dose of heparin. Extrapolation to the desired ACT provides the additional heparin dose required for that ACT. Once the actual ACT response to the heparin dose is plotted, further dose–response calculations are made based on the average of the target ACT and the actual ACT (Figure 17-5). This methodology was first described by Bull et al49 and forms the scientific basis for the automated dose–response systems manufactured by Hemochron and Hemotec. The Hemochron RxDx system uses the heparin-response test, which is an ACT with a known quantity of in vitro heparin (3 IU/mL). A dose–response curve is generated that enables calculation of the heparin dose required to attain the target ACT using an algorithm that incorporates the patient’s baseline ACT, estimated blood volume, and heparin-response test. The patient’s heparin sensitivity can be calculated in seconds per international units per milliliter (sec/IU/mL) by dividing the heparin-response test by 3 IU/mL.
Figure 17-5 Construction of a dose–response curve for heparin. ACT, activated coagulation time.
(From Bull BS, Huse WM, Brauer FS, et al: Heparin therapy during extracorporeal circulation: II. The use of a dose-response curve to individualize heparin and protamine dosage. J Thorac Cardiovasc Surg 69:685–689, 1975.)
The RxDx system also provides an individualized protamine dose using the protamine-response test (PRT). This is an ACT with one of two specific quantities of protamine, depending on the amount of circulating heparin suspected (2 or 3 IU/mL). The protamine dose needed to return the ACT to baseline can be calculated on the basis of a protamine–response curve using the patient’s heparinized ACT, the PRT, and an estimate of the patient’s blood volume. Jobes et al50 reported that the heparin dose directed by the RxDx system resulted in ACT values far greater than the target ACT. In their patients, in vivo heparin sensitivity was higher than in vitro sensitivity. RxDx also resulted in lower protamine doses, lower postoperative mediastinal tube losses, and reduced transfusion requirements compared with a ratio-based system of heparin/protamine administration. In a larger study that standardized the treatment of heparin rebound, the reduced protamine dose was confirmed; however, the reductions in bleeding were not substantiated.51 The use of a protamine dose–response curve has been shown to successfully reduce the protamine dose in vascular surgery compared with standard weight-based protamine dosing.52
The Hepcon HMS system uses the HDR cartridge in the Hepcon instrument (Figure 17-6). Each cartridge houses six chambers. Chambers 1 and 2 contain heparin at a concentration of 2.5 IU/mL, chambers 3 and 4 contain heparin at a concentration of 1.5 IU/mL, and chambers 5 and 6 do not contain heparin. Once information regarding patient weight, height, and CPB prime volume is entered, the information that can be obtained from this test includes the baseline ACT (chambers 5 and 6) and an HDR slope. The dose–response slope, which is the increase in ACT from 1.5 to 2.5 IU/mL heparin, is extrapolated to the desired target ACT or target heparin concentration and the heparin dose is calculated.32,53
Heparin Concentration
Proponents of ACT measurement to guide anticoagulation for CPB argue that a functional assessment of the anticoagulant effect of heparin is mandatory and that the variability in ACT represents a true variability in the coagulation status of the patient. Opponents argue that during CPB, the sensitivity of the ACT to heparin is altered and ACT does not correlate with heparin concentration or with anti–factor Xa activity measurement. Heparin concentration can be measured using the Hepcon HMS system (Medtronic Hemotec), which uses an automated protamine titration technique. With a cartridge with four or six chambers containing tissue thromboplastin and a series of known protamine concentrations, 0.2 mL whole blood is automatically dispensed into the chambers. The first channel to clot is the channel whose protamine concentration most accurately neutralizes the heparin without a heparin or a protamine excess. Because protamine neutralizes heparin in the ratio of 1 mg protamine per 100 units heparin, the concentration of heparin in the blood sample can be calculated. A cartridge that monitors heparin concentration over a wide range can be used first, followed by another cartridge that can measure heparin concentrations within a more narrow range. The maintenance of a stable heparin concentration rather than a specific ACT level usually results in greater doses of heparin being administered because the hemodilution and hypothermia on CPB increase the sensitivity of the ACT to heparin. The measure of heparin concentration has been shown to correlate more closely with anti–factor Xa activity measurements than the ACT during CPB54 (Figure 17-7), although the precision and bias of the test may not prove to be acceptable for exclusive use clinically (Figure 17-8).
In a small, prospective, randomized study comparing ACT and heparin concentration monitoring, Gravlee et al55 demonstrated increased mediastinal tube drainage after surgery in the heparin concentration group, a finding that was initially attributed to greater total heparin doses. However, heparin rebound was not systematically assessed. In a follow-up study, the authors found a greater incidence of heparin rebound in the heparin concentration group, which, when treated, resulted in no difference in bleeding between the ACT and heparin concentration groups.56 In a prospective, randomized trial, Despotis et al57 demonstrated that by using a transfusion algorithm in association with Hepcon-based heparin management, chest tube drainage was minimally reduced and transfusion of non–red blood cell products could be significantly reduced relative to a group of patients who had ACT-based heparin management. They attributed their results to better preservation of the coagulation system by high heparin doses because the doses of heparin administered in the Hepcon group were nearly twice the doses used in the ACT management group. However, Gravlee et al55 were unable to confirm suppression of ongoing coagulation using Hepcon CPB management. With the exception of during cold CPB, fibrinopeptide A (FPA) levels in patients who had heparin concentration monitoring were virtually indistinguishable from those in patients who had ACT monitoring (Figure 17-9). The Hepcon, however, remains one of the more sensitive tests for detecting residual heparinization after protamine reversal because the heparin concentration can be measured by protamine titration to levels as low as 0.4 IU/mL (see Monitoring for Heparin Rebound section later in this chapter).
High-Dose Thrombin Time
A functional test of heparin-induced anticoagulation that correlates well with heparin levels is the high-dose thrombin time (HiTT; International Technidyne, Edison, NJ). The TT is a clotting time that measures the conversion of fibrinogen to fibrin by thrombin. The TT is prolonged by the presence of heparin and by hypofibrinogenemias or dysfibrinogenemias. Because the TT is sensitive to very low levels of heparin, a high dose of thrombin is necessary in the TT to accurately assay the high doses of heparin used for CPB. The HiTT is performed by adding whole blood to a prewarmed, prehydrated test tube that contains a lyophilized thrombin preparation. After the addition of 1.5 mL blood, the tube is inserted into a Hemochron well and the time to clot formation is measured. In vitro assays indicate that HiTT is equivalent to the ACT in evaluation of the anticoagulant effects of heparin at heparin concentrations in the range of 0 to 4.8 IU/mL (Figure 17-10). Unlike ACT, HiTT is not altered by hemodilution and hypothermia, and has been shown to correlate better with heparin concentration than the ACT during CPB.58 While on CPB, heparin concentration and HiTT decrease, whereas the Hemochron and the Hepcon ACT increase (Figure 17-11). Another potential advantage of HiTT monitoring is for patients receiving aprotinin therapy. In the presence of heparin, aprotinin augments the celite ACT,59 possibly because its kallikrein-inhibiting capacity prolongs activation of the intrinsic coagulation pathway by XIIa. This should not be interpreted to represent enhanced anticoagulation. The kaolin ACT is less affected by aprotinin therapy than the celite ACT, perhaps because kaolin, unlike celite, activates the intrinsic pathway by stimulation of factor XI directly.60 Others have suggested that kaolin binds to aprotinin and reduces the anticoagulant effect of aprotinin in vitro. However, the heparinized kaolin ACT is still somewhat prolonged in the presence of aprotinin. HiTT is not affected by aprotinin therapy and can be used as a measure of heparinization for CPB patients receiving aprotinin therapy.59 The high-dose thromboplastin time is another measure of anticoagulation that is not affected by aprotinin therapy.61 The high-dose thromboplastin time is a WBCT in which celite is replaced by 0.3 mL of rabbit brain thromboplastin to which 1.2 mL blood is added. This test measures the time to coagulation via activation of the extrinsic pathway. This pathway of coagulation is also stimulated during pericardiotomy because of the rich thromboplastin environment of the pericardial cavity.
Heparin neutralization
Protamine Effects on Coagulation Monitoring
Reversal of heparin-induced anticoagulation is most frequently performed with protamine. Biologically, protamine binds to positively charged groups such as phosphate groups and may have important properties in angiogenesis and immune function. Different successful dosing plans have been proposed.62,63 The recommended dose of protamine for heparin reversal is 1 to 1.3 mg protamine per 100 units heparin; however, this dose often results in a protamine excess.
Protamine injection causes adverse hemodynamic effects.64 Protamine reactions have been classified into three types.65 The most common of these reactions is the type I reaction characterized by hypotension. Type II (immunologic) reactions are categorized as IIA (anaphylaxis), IIB (anaphylactoid), and IIC (noncardiogenic pulmonary edema). Type III reactions are heralded by hypotension and catastrophic pulmonary hypertension leading to right-heart failure.33,52,66
In addition to hemodynamic sequelae, protamine has adverse effects on coagulation.67 Large doses prolong the WBCT and the ACT, possibly via thrombin inhibition.68 In animals and in humans, protamine has been associated with thrombocytopenia, likely because of activation of the complement cascade.64 The anticoagulant effect of protamine also may be caused by inhibition of platelet aggregation, alteration in the platelet surface membrane, or depression of the platelet response to various agonists.68–70 These alterations in platelet function result from the presence of the heparin–protamine complex, not protamine alone. Protamine–heparin complexes activate AT III in vitro and result in complement activation. The anticoagulant effects of free protamine occur when protamine is given in doses in excess of those used clinically; however, the risk of free protamine being the cause of a hemostatic defect is small, given the rapid clearance of protamine relative to heparin.
Monitoring for Heparin Rebound
The phenomenon referred to as heparin rebound describes the re-establishment of a heparinized state after heparin has been neutralized with protamine. Various explanations for heparin rebound have been proposed.9,56,71,72 The most commonly postulated is that rapid distribution and clearance of protamine occur shortly after protamine administration, leaving unbound heparin remaining after protamine clearance. Furthermore, endogenous heparin antagonists have an even shorter life span than protamine and are eliminated rapidly, resulting in free heparin concentrations. Also possible is the release of heparin from tissues considered heparin storage sites (endothelium, connective tissues). Endothelial cells bind and depolymerize heparin via PF4. Uptake into the cells of the reticuloendothelial system, vascular smooth muscle, and extracellular fluid may account for the storage of heparin that contributes to reactivation of heparin anticoagulation, referred to as heparin rebound.9
Residual low levels of heparin can be detected by sensitive heparin concentration monitoring in the first hour after protamine reversal and can be present for up to 6 hours after surgery. Gravlee et al’s56 study suggests that without careful monitoring for heparin rebound in the postoperative period, increased bleeding as a result of heparin rebound may occur, specifically when greater doses of heparin have been administered. Monitoring for heparin rebound can be accomplished using tests that are sensitive to low levels of circulating heparin.52,57,73,74 These tests are also useful monitors for confirmation of heparin neutralization at the conclusion of CPB (see later).
Heparin Neutralization Monitors
To administer the appropriate dose of protamine at the conclusion of CPB, it would be ideal to measure the concentration of heparin present and give the dose of protamine necessary to neutralize only the circulating heparin. As a result of heparin metabolism and elimination, which vary considerably among individuals, the dose of protamine required to reverse a given dose of heparin decreases over time. Furthermore, protamine antagonizes the anti-IIa effects of heparin more effectively than the anti-Xa effects and, thus, varies in its potency depending on the source of heparin and its anti-IIa properties. Administration of a large fixed dose of protamine or a dose based on the total heparin dose given is no longer the standard of care and may result in an increased incidence of protamine-related adverse effects. An optimal dose of protamine is desired because unneutralized heparin results in clinical bleeding and an excess of protamine may produce an undesired coagulopathy. The use of individualized protamine dose–response curves uniformly results in a reduced protamine dose and has been shown to reduce postoperative bleeding.49,73 One such dose–response test, the Hemochron PRT test, is an ACT performed on a heparinized blood sample that contains a known quantity of protamine. With knowledge of the ACT, PRT, and the estimated blood volume of the patient, the protamine dose needed to neutralize the existing heparin level can be extrapolated. The Hepcon instrument also has a PRT, which is the protamine titration assay. The chamber that clots first contains the dose of protamine that most closely approximates the circulating dose of heparin. The protamine dose required for its neutralization is calculated on the basis of a specified heparin/protamine dose ratio by measuring the circulating heparin level.
At the levels of heparinization needed for cardiac surgery, tests that are sensitive to heparin become unclottable. ACT is relatively insensitive to heparin and is ideal for monitoring anticoagulation at high heparin levels but is too insensitive to accurately diagnose incomplete heparin neutralization. Reiner et al75 showed that ACT had a high predictive value for adequate anticoagulation (confirmed by laboratory aPTT) when longer than 225 seconds but was poorly predictive for inadequate anticoagulation when shorter than 225 seconds. The low levels of heparin present when heparin is incompletely neutralized are best measured by other more sensitive tests of heparin-induced anticoagulation, such as heparin concentration, aPTT, and TT. Thus, after CPB, confirmation of return to the unanticoagulated state should be performed with a sensitive test for heparin anticoagulation76–79 (Box 17-3).
Thrombin Time
TT is the time it takes for the conversion of fibrinogen to fibrin clot when blood or plasma is exposed to thrombin. Fibrin strands form in seconds. Detection of fibrin formation using standard laboratory equipment involves incubation of the blood or plasma sample within the chamber in which an optical or electrical probe sits. A detector senses either movement of the probe or the creation of an electrical field (electrical detection) because of fibrin formation and hence signals the end of the test. Hemochron manufactures a point-of-care TT test that uses a lyophilized preparation of thrombin in a Hemochron test tube to which 1 mL blood is added. Identification of fibrin formation in a Hemochron machine uses the standard Hemochron technology described previously with the ACT. The manufacturer suggests that the normal TT is 39 to 53 seconds for whole blood and 43 to 68 seconds for citrated blood. Because the TT specifically measures the activity of thrombin, it is very sensitive to heparin-induced enhancement of AT III activity. It is a useful test in the post-CPB period for differentiating the cause of bleeding when both PT and aPTT are prolonged because it excludes the intrinsic and extrinsic coagulation pathway limbs and evaluates the conversion of factor I to Ia. The TT is increased in the presence of heparin, hypofibrinogenemia, dysfibrinogenemia, amyloidosis, or antibodies to thrombin.80 The TT is also increased in the presence of fibrin degradation products if the systemic fibrinogen concentration is low.
Bedside Tests of Heparin Neutralization
Hepcon measures heparin concentration via a protamine titration assay. Cartridges with varying ranges of protamine concentration are available for use. The cartridge with the lower concentration of protamine in the titration is useful for the detection of residual circulating heparin and is sensitive to levels of heparin as low as 0.2 IU/mL. Whole-blood PT and aPTT assays are sensitive to deficiencies in coagulation factors and overly sensitive to low levels of heparin (aPTT); they lack specificity in assessing residual heparinization. The heparin-neutralized thrombin time (HNTT) is a TT assay with a small dose of protamine sufficient to neutralize 1.5 IU/mL heparin. Because the TT is increased in the presence of heparin, hypofibrinogenemia, or dysfibrinogenemia, HNTT and TT should be performed together to discriminate among these three causes. A normal HNTT in the presence of an increased TT virtually confirms residual heparin effect and would indicate the need for protamine administration. If HNTT is prolonged as well as the TT, the cause of bleeding may be attributed either to a fibrinogen problem or to a concentration of heparin greater than that which could be neutralized by the HNTT. In one study comparing bedside monitors of anticoagulation, the TT-HNTT difference bore a significant correlation with the aPTT increase. Using the line of best fit, aPTT elevation of 1.5 times the control corresponded to a 31-second difference in the TT and HNTT, indicating a convenient threshold value of TT-HNTT for the administration of protamine.80
Platelet Factor 4
A component of the alpha granule of platelets, PF4 binds to and inactivates heparin. The physiologic role of PF4 is that at the site of vascular injury, PF4 is released from platelets, binds heparin (or heparin-like compounds), and promotes thrombin and clot formation. PF4 adequately neutralizes heparin inhibition of factors Xa and IIa and may be superior to protamine in neutralizing anti-Xa effects. Animal data suggest that PF4 is devoid of the adverse hemodynamic effects seen with protamine and that it may be able to be infused more rapidly.81,82 Heparin reversal has been documented by WBCT, ACT, and heparin concentration at a PF4 concentration of 40 U/mL, approximately twice the reversal dose of protamine.74 Levy et al83 subsequently found the reversal dose of PF4 to be approximately 60 U/mL and documented similar ACT and viscoelastic measurements of clot formation compared with protamine.
Heparinase
Heparinase (Neutralase I) is an enzyme that specifically degrades heparin by catalyzing cleavage of the saccharide bonds found in the heparin molecule. As demonstrated by the ACT, heparinase in a dose of 5 mg/kg has been shown to successfully neutralize heparin effects in healthy volunteers and in patients who have undergone CPB. A dose of 7 mg/kg has been demonstrated to be even more efficacious in returning ACT to baseline values. Doses sufficient to neutralize a dose of 300 IU/kg of heparin had no significant hemodynamic effects in a canine model.84 Investigators have not found any platelet-depressive effects of heparinase in contrast with the well-documented platelet dysfunction associated with protamine therapy.69 Return to the unanticoagulated state after the use of heparinase has been confirmed using ACT monitoring or heparin concentration monitoring.
