[level-membership-for-neurology-category]
Chapter 40 Clinical Neurogenetics
Genetics in Clinical Neurology
Gene Expression, Diversity, and Regulation
Types of Genetic Variation and Mutations
Chromosomal Analysis and Abnormalities
Disorders of Mendelian Inheritance
Mendelian Disease Gene Identification by Linkage Analysis and Chromosome Mapping
Non-Mendelian Patterns of Inheritance
Common Neurological Disorders and Complex Disease Genetics
Whole Genome/Exome Sequencing in Disease Gene Discovery
Future Role of Systems Biology in Neurogenetic Disease
Environmental Contributions to Neurogenetic Disease
Genetics and the Paradox of Disease Definition
Clinical Approach to the Patient with Suspected Neurogenetic Disease
Genetics in Clinical Neurology
Since the discovery of the structure of deoxyribonucleic acid (DNA) and the elucidation of the genetic mechanisms of heredity, clinical neurology has benefited from advances in genetics and neuroscience. This clinically relevant basic research has permitted dissection of the cellular machinery supporting the function of the brain and its connections while establishing causal relationships between such dysfunction, human genetic variation, and various neurological diseases. In the modern practice of neurology, the use of genetics has become widespread, and neurologists are confronted daily with data from an ever-increasing catalogue of genetic studies relating to conditions such as developmental disorders, dementia, ataxia, neuropathy, and epilepsy, to name but a few. The use of genetic information in the clinical evaluation of neurological disease has expanded dramatically over the past decade. More efficient techniques for discovering disease genes has led to a greater availability of genetic testing in the clinic. Approximately one-third of pediatric neurology hospital admissions are related to a genetic diagnosis, and there are many dozens of genetic tests available to the practicing neurologist, including several related to common diseases. This number will surely continue to increase rapidly (Fig. 40.1).
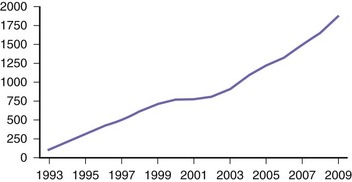
(Data from GeneTests: Medical Genetics Information Resource. Available at http://www.ncbi.nlm.nih.gov/sites/GeneTests/.)
As neuroscience and genetic research have progressed, we have been led to a deeper understanding of the sources and nature of human genetic variation and its relationship to clinical phenotypes. In the past there has been a tendency to consider genetic traits as either present or absent, and correspondingly, patients were either healthy or diseased; this is the traditional view of Mendelian, or single gene, conditions. Although certain relatively rare neurological diseases—Friedreich ataxia or Huntington disease (HD), for example—can be traced to a single causal gene, the common forms of other diseases such as Alzheimer dementia, stroke, epilepsy, or autism usually arise from an interplay of multiple genes, each of which increases disease susceptibility and likely interacts with environmental factors. Subsequently, the realm of the “sporadic” and the “idiopathic” has been challenged by the identification of genetic susceptibility factors, which has sparked a flurry of investigation into a variety of genes and genetic markers that confer a risk of illness yet are not wholly causative. Disease status may lie on the end of a continuum of individual variation and thus can be considered a quantitative rather than purely qualitative trait (Plomin et al., 2009). So, rather than using what might be considered an arbitrary cutoff point, such as a specific number of senile plaques or neuritic tangles that define affected or unaffected patients, one might instead think in terms of a continuum of pathology that relates to different levels of burden or susceptibility.
Gene Expression, Diversity, and Regulation
The basic principles of molecular genetics are outlined in Fig. 40.2 and Table 40.1, and more detailed descriptions can be found elsewhere (Alberts et al., 2008; Griffiths et al., 2002; Lodish et al., 2008; Strachan and Read, 2003). To briefly summarize, deoxyribonucleic acid (DNA), found in the nucleus of all cells, comprises the raw material from which heritable information is transferred among individuals, with the simplest heritable unit being the gene. DNA is composed of a series of individual nucleotides, all of which contain an identical pentose (2′-deoxyribose)-phosphate backbone but differ at an attached base that can be either adenine (A), guanine (G), thymine (T), or cytosine (C). A and G are purine bases and pair with the pyrimidine bases T and C, respectively, to form a double-stranded helical structure which allows for semiconservative bidirectional replication, the means by which DNA is copied in a precise and efficient manner. In total, there are approximately 3.2 billion base pairs in human DNA. By convention, a DNA sequence is described by listing the bases as they are expressed from the 5′ to 3′ direction along the pentose backbone (e.g., 5′-ATGCAT-3′…etc.), as this is the order in which it is typically used by the cellular machinery, also called the sense strand (compare to RNA, later). The opposite paired, or antisense, strand is arranged antiparallel (3′ to 5′) and can also be referred to when discussing sequence; however, by convention this is generally not done unless that strand is also transcribed into RNA.
| Allele | Alternate forms of a locus (gene) |
| Anticipation | Earlier onset and/or worsening severity of disease in successive generations |
| Antisense | Nucleic acid sequence complementary to mRNA |
| Chromosome | Organizational unit of the genome consisting of a linear arrangement of genes |
| Cis-acting | A regulatory nucleotide sequence present on the molecule being regulated |
| Codon | A three-nucleotide sequence representing a single amino acid |
| Complex disease | Disease exhibiting non-Mendelian inheritance involving the interaction of multiple genes and the environment |
| De novo | A mutation newly arising in an individual and not present in either parent |
| Diploid | A genome having paired genetic information, half-normal number is haploid |
| DNA | Deoxyribonucleic acid; used for storage, replication, and inheritance of genetic information |
| Dominant | Allele that determines phenotype when a single copy is present in an individual |
| Endophenotype | Subset of phenotypic characteristics used to group patients manifesting a given trait |
| Exome | Portion of the genome representing only the coding regions of genes |
| Exon | Segment of DNA that is expressed in at least one mature mRNA |
| Expressivity | The range of phenotypes observed with a specific disease-associated genotype |
| Frameshift | DNA mutation that adds or removes nucleotides, affecting which are grouped as codons |
| Gene | Contiguous DNA sequence that codes for a given messenger RNA and its splice variants |
| Genome | A complete set of DNA from a given individual |
| Genotype | The DNA sequence of a gene |
| Haplotype | A group of alleles on the same chromosome close enough to be inherited together |
| Hemizygous | Genes having only a single allele in an individual, such as the X chromosome in males |
| Heteroplasmy | A mixture of multiple mitochondrial genomes in a given individual |
| Heterozygous | Genes having two distinct alleles in an individual at a given locus |
| Homozygous | Genes having two identical alleles in an individual at a given locus |
| Intron | Segment of DNA between exons that is transcribed into RNA but removed by splicing |
| Kilobase | 1000 bases or base-pairs |
| Linkage disequilibrium | The co-occurrence of two alleles more frequently than expected by random chance, suggesting they are in close proximity to one another |
| Locus | Location of a DNA sequence (or a gene) on a chromosome or within the genome |
| Lyonization | The process of random inactivation of one of the pair of X chromosomes in females |
| Marker | Sequence of DNA used to identify a gene or a locus |
| Megabase | 1,000,000 bases or base-pairs |
| Meiosis | Process of cellular division that produces gametes containing a haploid amount of DNA |
| Mendelian | Obeying standard single-gene patterns of inheritance (e.g., recessive or dominant) |
| Microarray | A glass or plastic support (e.g., slide or chip) to which large numbers of DNA molecules can be attached for use in high-throughput genetic analysis |
| Missense | DNA mutation that changes a given codon to represent a different amino acid |
| Mitosis | Process of cellular division during which DNA is replicated |
| Nonsense | DNA mutation that changes a given codon into a translation termination signal |
| Penetrance | The likelihood of a disease-associated genotype to express a specific disease phenotype |
| Phenotype | The clinical manifestations of a given genotype |
| Polymorphism | Sequence variation among individuals, typically not considered to be pathogenic |
| Probe | DNA sequence used for identifying a specific gene or allele |
| Promoter | DNA sequences that regulate transcription of a given gene |
| Protein | Functional cellular macromolecules encoded by a gene |
| Recessive | Allele that determines phenotype only when two copies are present in an individual |
| Relative risk | The ratio of the chance of disease in individuals with a specific genetic susceptibility factor over the chance of disease in those without it |
| Resequencing | A method of identifying clinically relevant genetic variation in a candidate gene of interest by comparing the sequence in individuals with disease to a reference sequence |
| RNA | Ribonucleic acid; expressed form of a gene, called messenger or mRNA if protein coding |
| Sense | Nucleic acid sequence corresponding to mRNA |
| Silent | DNA mutation that changes a given codon but does not alter the corresponding amino acid |
| SNP | Single nucleotide polymorphism |
| Splicing | RNA processing mechanism where introns are removed and exons joined to create mRNA; in alternative splicing, exons are utilized in a regulated manner within a cell or tissue |
| Trans-acting | A regulatory protein that acts on a molecule other than that which expressed it |
| Transcription | Cellular process where DNA sequence is used as template for RNA synthesis |
| Transcriptome | The complete set of RNA transcripts produced by a cell, tissue, or individual |
| Translation | Cellular process where mRNA sequence is converted to protein |
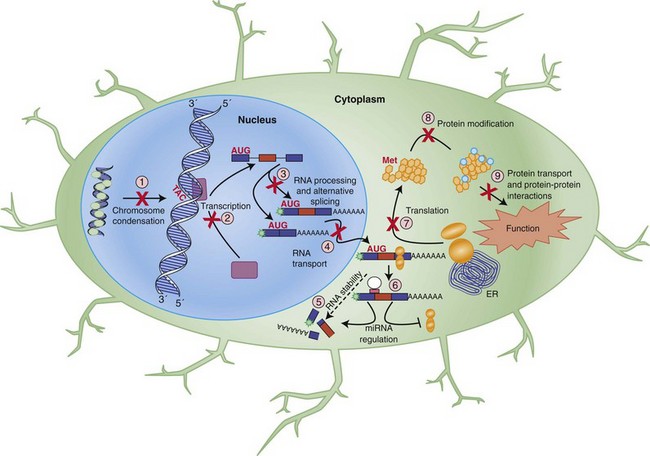
The expression of a gene is tightly and coordinately regulated (see Fig. 40.2), an important consideration for understanding the molecular mechanisms of disease. The typical gene contains one or more promoters, DNA sequences that allow for the binding of a cellular protein complex that includes RNA polymerase and other factors that faithfully copy the DNA in the 5′ to 3′ direction in a process known as transcription. The resulting single-stranded molecule contains a ribose sugar unit in its backbone and, thus the resulting molecule is termed ribonucleic acid, or RNA. RNA also differs from the template DNA by the incorporation of uracil (U) in place of thymine (T), as it also pairs efficiently with adenine, and thymine serves a secondary role in DNA repair that is not necessary in RNA. The sequence of the RNA matches the sense DNA strand and is therefore complementary to (and hence derived from) the antisense strand.
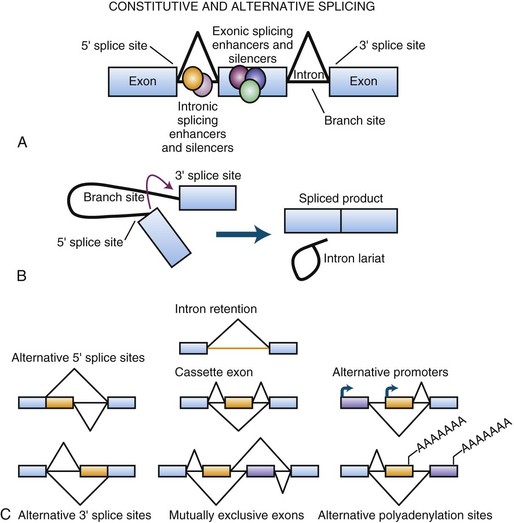
Transcribed coding RNA must be processed to become protein-encoding messenger RNA (mRNA), a term used to differentiate these RNAs from all other types of RNA in the cell. To become mature, RNA is stabilized by modification at the ends with a 7-methylguanosine 5′ cap and a long poly-A 3′ tail. A further critical stage in the maturation of the RNA molecule involves a rearrangement process termed RNA splicing (Fig. 40.3). This is necessary because the expressed coding sequences in DNA, called exons, of virtually every gene are discontinuous and interspersed with long stretches of generally non-conserved intervening sequences referred to as introns. This, along with other mechanisms, likely plays an evolutionary role in the development of new genes by allowing for the shuffling of functional sequences (Babushok et al., 2007). Nascent RNA molecules are recognized by the spliceosome, a protein complex that removes the introns and rejoins the exons. Not every exon is utilized at all times in every RNA derived from a single gene. Exons may be skipped or included in a regulated manner through alternative splicing, which occurs in nearly 95% of all genes to create different isoforms of that mRNA. The dynamic nature of this observation is critical to a complete understanding of cellular gene expression. DNA is essentially a storage molecule, and with few exceptions in the absence of mutagens, its sequence remains static and, aside from a few epigenetic events, is therefore limited to a genetic regulatory role as a transcriptional rheostat. Current estimates place the number of individual human genes at just over 22,000 (Pertea and Salzberg, 2010), so it is difficult to reconcile biological and clinical diversity with simple variations in expression. Alternative splicing provides a means of dramatically elevating this diversity by enabling a single gene to encode multiple proteins with a wide array of functions. Supporting this, recent analysis of RNA complexity in human tissues suggest that there are at least seven alternative splicing events per multi-exon gene, generating over 100,000 alternative splicing events (Pan et al., 2008). Because alternative splicing and other forms of RNA processing can be subject to complex layers of temporal and spatial regulation, particularly in the human brain (Licatalosi and Darnell, 2010; Ward and Cooper, 2010), it is a robust source for both biological diversity and disease-causing mutations (see Polymorphisms and Point Mutations).
DNA to RNA to Protein
The central dogma of genetics has been that DNA is transcribed into RNA that is than translated into protein—the “business” end of the process. So, following its transcription from DNA in the nucleus, mRNA is transported out of the nucleus to the cytoplasm, and possibly to a specific subcellular location depending on the mRNA, where it can be deciphered by the cell. This takes place via interaction with a complex known as the ribosome, which binds the mRNA and converts its genetic information into protein via the process of translation. The ribosome initiates translation at a pre-encoded start site and converts the mRNA sequence into protein until a designated termination site is reached. Sequence information is read in three-nucleotide groups called codons, each of which specifies an individual amino acid. With the four distinct bases, there are mathematically 64 possible codons, but these have an element of redundancy and code for only 20 different amino acids and 3 termination signals (UAG, UGA, and UAA), also called stop codons. The start codon is ATG and codes for methionine. These amino acids are joined by the ribosome to synthesize a protein. This protein, which may undergo further modification, will ultimately carry out a programmed biological function in the cell. Regulation of this process is highly coordinated and important in learning, for example, where activity-dependent translation at the synapse underlies some aspects of synaptic plasticity, which may go awry in certain disorders such as fragile X syndrome and autism (Morrow et al., 2008).
Over the last decade, the discovery of several classes of functional non–protein coding RNAs has added additional complexity to our understanding of how the genetic code is manifest at the level of cellular function. Of these, microRNAs (miRNAs) are increasingly being recognized as vital players in gene regulation and neurological disease (Weinberg and Wood, 2009). Nascent miRNA molecules are processed to form short (approximately 22-nucleotide) RNA duplexes that target endogenous cellular machinery to specific coding RNAs and induce posttranscriptional gene silencing through a diverse repertoire including RNA cleavage, translational blocking, transport to inactive cell sites, or promotion of RNA decay (Filipowicz et al., 2008; Weinberg and Wood, 2009). Depending on the cell and the context, miRNA activity can result in specific gene inactivation, functional repression, or more subtle regulatory effects and may involve multiple RNAs in a given biological pathway (Flynt and Lai, 2008). Estimates suggest that miRNAs may regulate 30% of protein-coding genes, implicating these molecules as important targets for future research into the biology of neurological disease (Filipowicz et al., 2008; Weinberg and Wood, 2009).
It is important to emphasize that most genes are not simply “on” or “off.” In reality, cells maintain strict regulatory control over their genes. Some genes, such as those required for cell structure or maintenance, must be expressed constitutively, but genes with specific precise functions may only be needed in certain cells at certain times under certain conditions. Potential levels of regulation are depicted in Fig. 40.2 and include virtually every stage of gene expression. Initially, genes can be regulated at the level of transcription, ranging from the regulated binding of histone proteins, which leads to chromosome condensation, inactivating genes, to the coordinated activity of protein factors that activate or repress gene transcription in response to cell state, environmental conditions, or other factors. Once expressed, the RNA is subject to processing regulation, particularly through alternative splicing as already discussed. Transport of the mRNA and its translation provide additional steps for cellular regulation. Lastly, the final protein can be subject to control via posttranslational modifications or interactions with other proteins. To operate, all these levels of regulation require trans-acting factors, such as proteins, which stimulate or repress a particular step, as well as cis-acting elements, sequences recognized and bound by the regulatory factors.
These detailed levels of regulation provide a dynamic and expansive capability to precisely control cellular function, essential for growth, development, and survival in an unpredictable environment. However, this also provides many potential points at which disease can arise from disrupted regulation. Consequently, a defective gene could cause disease directly through its own action or indirectly by disrupting regulation of other cellular pathways. For example, the forkhead box P2 (FOXP2) transcription factor regulates the expression of genes thought to be important for the development of spoken language (Konopka et al., 2009). Mutations in this gene cause an autosomal dominant disorder characterized by impairment of speech articulation and language processing (Lai et al., 2001). However, other mutations in this gene are responsible for approximately 1% to 2% of sporadic developmental verbal dyspraxia (MacDermot et al., 2005), likely via downstream effects. Mutation of the methyl-CpG-binding protein 2 (MECP2), which regulates chromatin structure, causes the neurodevelopmental disorder, Rett syndrome, but other mutations in this gene can cause intellectual disability or autism (Gonzales and LaSalle, 2010). Similarly, the FOX1 protein (also called ataxin 2 binding protein 1, or A2BP1), a neuron-specific RNA splicing factor (Underwood et al., 2005) predicted to regulate a large network of genes important to neurodevelopment (Yeo et al., 2009; Zhang et al., 2008), causes autistic spectrum disorder when disrupted (Martin et al., 2007) but has also been implicated as a susceptibility gene associated with both primary biliary cirrhosis (Joshita et al., 2010) and hand osteoarthritis (Zhai et al., 2009), presumably due to downstream effects or specific effects in non-neural tissues. This concept of genes acting on other genes will be further explored later (see Common Neurological Disorders and Complex Disease Genetics).
In addition to the complexity of regulatory mutations that affect gene expression by altering RNA or protein levels or by disrupting RNA splicing, there are certain mutations that do not cause protein dysfunction, but instead have effects restricted to the RNA itself. For example, RNA inclusions are found in several forms of triplet repeat disorders (see Repeat Expansion Disorders) including myotonic dystrophy type 1 and the fragile X–associated tremor/ataxia syndrome (FXTAS) (Garcia-Arocena and Hagerman, 2010; Orr and Zoghbi, 2007). The latter is particularly interesting from a genetic standpoint, because a disorder of late-onset progressive ataxia, tremor, and cognitive impairment occurs in carriers of FMR1 alleles of intermediate sizes, which are not full fragile X–causing mutations (Garcia-Arocena and Hagerman, 2010). FXTAS is a dominant gain-of-function disease that occurs via an entirely different mechanism than the recessive loss-of-function disease, fragile X syndrome (Garcia-Arocena and Hagerman, 2010; Penagarikano et al., 2007). Primary disorders of RNA still represent relatively uncharted territory, and it is likely that more RNA-specific diseases will be identified. This is particularly exciting for many reasons, not the least of which is that certain classes of these disorders may be amendable to therapy (Nakamori and Thornton, 2010; Wheeler et al., 2009).
Types of Genetic Variation and Mutations
Rare versus Common Variation
As dictated by the principles of natural selection, most genetic variation is not deleterious, and the induced phenotypic variability can be beneficial as a source on which evolution may act. From a clinical standpoint, it is helpful to dichotomize genetic variation into common and rare variation, while accepting that genetic variation is likely a continuum, and the choice of cutoff could be considered arbitrary. Rare genetic variants are of low frequency in the population (<1% frequency), either because they are deleterious and selected against, or because they are new and most often benign. Common genetic variation (>1% to 5% population frequency), on the other hand, is either adaptive, neutral, or not deleterious enough to be subject to strong negative selection; such variants are referred to as polymorphisms. The preeminent genetic model has been that common disease susceptibility is related to common genetic variation, and more rare forms of disease are caused by rare genetic variants, so-called mutations, which act in a Mendelian fashion. In contrast, common variants or polymorphisms may increase susceptibility for disease, but alone are not sufficient to cause disease (see Common Neurological Disorders and Complex Disease Genetics).
Polymorphisms and Point Mutations
The most prevalent form of genetic polymorphism is the single nucleotide polymorphism (SNP), which occurs on average every 300 to 1000 base pairs in the human genome. Most of these SNPs are relatively benign on their own and do not directly cause disease, so for the purposes of this initial discussion, we will concern ourselves primarily with mutations: rare genetic variants sufficient to cause disease. Pathogenic mutations can occur in numerous ways and vary from single nucleotide changes to gross rearrangements of chromosomes (Fig. 40.4). Owing to the large volume of DNA in the human genome, heritable mutations can arise spontaneously in the germline over time through errors in DNA replication or from DNA damage by metabolic or environmental sources despite the constant surveillance of extensive cellular preventive proofreading and repair mechanisms. Thus, mutations can be inherited from the parent or occur de novo in the germline. An example of a common de novo variant is trisomy 21, which causes Down syndrome (discussed further in Chromosomal Analysis and Abnormalities). The smallest pathogenic alterations, termed point mutations, involve a change in a single nucleotide within a DNA sequence. A point mutation can result in one of three possible effects with respect to protein: (1) a change to a different amino acid, called a missense mutation, (2) a change to a termination codon, called a nonsense mutation, or (3) creation of a new sequence that is silent with regard to protein sequence but alters some aspect of gene regulation, such as RNA splicing or transcriptional expression levels. Nonsense mutations can cause premature truncation of a protein, whereas a missense mutation can affect a protein in different ways depending on the chemical properties of the new amino acid and whether the change is located in a region of functional importance.
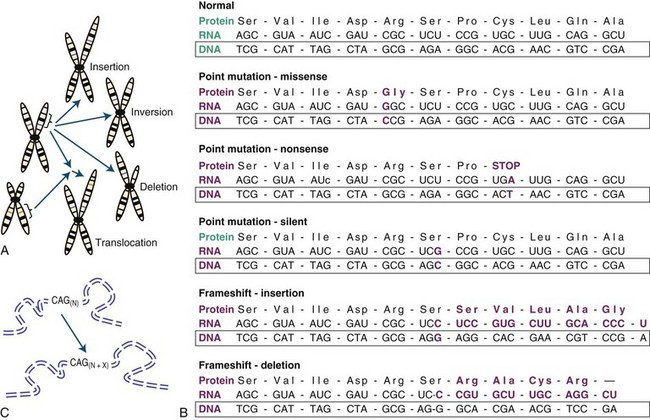
It should be emphasized that not all point mutations are disease-causing variants, although until recently, many considered that a premature stop codon was a “smoking gun.” Whole genome sequencing demonstrates that more than 100 such nonsense mutations may exist per genome, and the vast majority are expected to be relatively benign (Lupski et al., 2010; see Whole Genome/Exome Sequencing in Disease Gene Discovery). So in many cases, the pathogenicity of rare variants is not immediately discernable, and without strong statistical or functional evidence, labeling such genetic variation a mutation is premature and may be misleading. It is likely that most of these, including some variants thought previously to cause rare Mendelian diseases, may simply be benign genetic variation. This is because even a complete knockout of one allele caused by a premature stop codon (haploinsufficiency) may have no discernable effect on gene function for a majority of genes in the human genome (Lupski et al., 2010; Ng et al., 2009; Yngvadottir et al., 2009).
Occasionally, silent coding mutations or point mutations in noncoding regions may be significant for disease if they damage sequences important for gene expression (e.g., transcriptional and/or RNA processing regulatory elements). It has been estimated that up to half of all disease-causing mutations impact RNA splicing, which can have dire consequences given the importance of splicing to regulated gene expression. Such is the case for frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), where in some populations, the most common mutations disrupt splicing, causing a pathogenic imbalance in tau isoforms (D’Souza and Schellenberg, 2005). As for noncoding mutations, given the large volume of such sequences in the human genome—perhaps up to 96%—and our still imprecise ability to predict sequences required for regulation or to interpret identified sequence changes without direct experimentation, the majority of these mutations likely go unrecognized. It is hoped that the next generation of sequencing and bioinformatic technologies will allow for a better understanding of the role of these types of mutation in human disease.
Structural Chromosomal Abnormalities and Copy Number Variation
Small deletions and insertions can occur through slippage and strand mispairing at regions of short, tandem DNA repeats during replication. If the deletion or insertion is not a multiple of three, a frameshift will result, which leads to the translation of an altered protein sequence from the site of the mutation. On a larger scale, errors of chromosomal replication or recombination can result in inversions, translocations, deletions, duplications, or insertions (Stankiewicz and Lupski, 2010). When the region of deletion or duplication is greater than 1 kb, this is referred to as a copy number variation (CNV). Copy number variation is far more common than previously suspected, and it is estimated that at least 4% of the human genome varies in copy number (Conrad et al., 2010; Redon et al., 2006), much of which is commonly observed in the population and benign (Conrad et al., 2010). However, some rare CNVs such as the recurrent chromosome 17p12 duplication underlying most cases of Charcot-Marie-Tooth type 1A (Shchelochkov et al., 2010) or the alpha-synuclein triplication that can cause Parkinson disease (PD)(Singleton et al., 2003) are pathogenic and act in a Mendelian fashion. Even though such changes may be extensive, they may not be pathogenic if they do not disrupt expression of any key genes. This is particularly true for balanced translocations where genetic material is rearranged between chromosomes, yet no significant portion is actually lost. Although an individual with such a condition may be normal, if the germline is affected, their offspring may receive unbalanced chromosomal material and consequently develop a clinical phenotype (Kovaleva and Shaffer, 2003). CNVs will be discussed in greater detail when we consider common and complex disease genetics (see Copy Number Variation and Comparative Genomic Hybridization).
Repeat Expansion Disorders
Most mutations thus far discussed pass from parent to offspring unaltered, and in large affected families, the identical mutation can potentially be traced back generations. In contrast, there is a specific class of mutation, the repeat expansion (Orr and Zoghbi, 2007) (Table 40.2), which is unstable and can present with earlier onset and increasing severity in successive generations, a process known as anticipation. There are several examples of diseases caused by expanded repeats in coding sequence (e.g., most spinocerebellar ataxias, HD), as well as examples in noncoding sequence (e.g., fragile X syndrome, myotonic dystrophy) and within an intron (e.g., Friedreich ataxia). Interestingly, virtually all these disorders show neurological symptoms that can include such features as ataxia, intellectual disability, dementia, myotonia, or epilepsy, depending on the disease. The most common repeated sequence seen in these diseases is the CAG triplet, which codes for glutamine and expansion of which is seen in a variety of the spinocerebellar ataxias (SCAs) including SCA types 1, 2, 3, 6, 7, 17, and dentatorubropallidoluysian atrophy (DRPLA). In addition to protein-specific effects, these disorders likely share a common pathogenesis due to the presence of the polyglutamine repeat regions. In some disorders, the phenotype can be quite different depending on the number of repeats, such as in the FMR1 gene, where more than 200 CCG repeats causes fragile X syndrome, but repeats in the premutation range of 60 to 200, from which fully expanded alleles arise, can result in FXTAS or premature ovarian failure (Oostra and Willemsen, 2009). Although in general, the underlying mutation is similar, each specific repeat expansion has distinct effects on its corresponding gene, and thus in addition to varying phenotypes, they may also show very different inheritance patterns as illustrated later (see Disorders of Mendelian Inheritance).
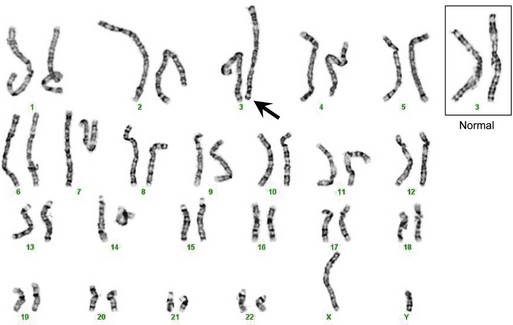
Chromosomal Analysis and Abnormalities
The DNA coding for an individual gene is generally too small to be visualized microscopically, but it is possible to observe the chromosomes as they condense during mitosis as part of cell division (Griffiths et al., 2002; Strachan and Read, 2003). Traditionally, various staining techniques (e.g., Giemsa) are applied, producing a detailed pattern of banding along the chromosomes that are then photographed and aligned for comparative analysis. This arrangement and analysis of the chromosomes is known as a karyotype (Fig. 40.5). Through these methods, it is possible to visually identify large chromosomal deletions, duplications, or rearrangements. If high-resolution banding techniques are employed, structural alterations on the order of as small as 3 Mb (3 million base pairs) can be detected. More sophisticated techniques can also be employed, such as fluorescent in situ hybridization (FISH). In this method, a short DNA sequence, or probe, that corresponds to a chromosomal region of interest is hybridized with the patient’s DNA and detected visually via excitation of a fluorescent label. FISH can improve on visual resolution by 10- to 100-fold and is in common use for detection of a large number of well-defined genetic syndromes (Speicher and Carter, 2005) such as 15q duplication syndrome, DiGeorge syndrome (22q11 deletion), and Smith-Magenis syndrome (17p11 deletion).
More recent technological developments involving microarray technology (Geschwind, 2003) permit screening of the entire genome at high resolution (from kilobase to single nucleotide level) and are rapidly replacing techniques based on microscopic analysis. This technology is responsible for the emerging appreciation for the structural chromosomal variation in humans mentioned earlier, most of which is submicroscopic. For this section, we will focus on chromosomal alterations that can be detected microscopically, since the clinical implications of many small or rare structural variants identified are not yet clear (see Copy Number Variation and Comparative Genomic Hybridization).
The most common chromosomal abnormalities encountered clinically involve sporadic aneuploidy, either a deletion leaving one chromosome, or a monosomy, or a duplication leaving three chromosomes, or a trisomy (Strachan and Read, 2003). This occurs most frequently via nondisjunction, whereby chromosomes fail to separate during meiosis in the production of the gametes. The majority of aneuploidies are lethal, although there are a few that are viable and will be briefly discussed. Monosomy X (45,XO), also called Turner syndrome, is seen in approximately 1 of every 5000 births and results in sterile females of small stature with a variety of mild physical deformities including webbing of the neck, multiple nevi, and hand and elbow variations, with a very specific cognitive profile in patients with the full deletion (Strachan and Read, 2003). Individuals with additional copies of the X chromosome are also seen. While both females (47,XXX) and males (47,XXY) may have varying degrees of learning disabilities, especially involving language and attention (Geschwind et al., 2000), the males are referred to as having Klinefelter syndrome (KS) due to a phenotype also involving gynecomastia and infertility. XYY males have cognitive profiles similar to XXY males but several studies have suggested more severe social and behavioral problems in some individuals, especially increased aggression, which is rare in KS. Trisomy 21 (47, +21), or Down syndrome, includes profound intellectual impairment, flat faces with prominent epicanthal folds, and a predisposition to cardiac disease. At 1 in approximately 700 births, this is the most common genetic cause of intellectual disability and is associated with advanced maternal age at the time of conception. The other aneuploidies which can survive to term (trisomy 13 [47, +13], Edwards syndrome; trisomy 18 [47, +18], Patau syndrome) have much more severe phenotypes with drastically decreased viability, and death generally occurs within weeks to months after birth.
Disorders of Mendelian Inheritance
In this section we will consider genetic disorders caused by mutation of a single gene. Associating a clinical disease phenotype to the mutation of a specific gene has long been the goal of clinically based, or translational, neuroscience. It is expected that gene identification will eventually lead to an understanding of the disease etiology as well as more accurate diagnosis and better treatments. The ability to determine the genetic nature of most single-gene disease is ultimately based upon the laws of inheritance devised by Mendel in the late 1800s (Griffiths et al., 2002). To summarize these findings in a clinical context, the assumption is made that a phenotypic trait (or in this example, a disease) is caused by the alteration of a single gene. It is important to emphasize that this assumption does not always hold true, particularly for the more complex genetic diseases, as we will discuss later, but it is still true for many diseases seen by neurologists, and more than 3000 Mendelian conditions have been identified to date (OMIM, 2010). Now, if we accept the premise that a given disease is caused by a single gene, we know that for any individual, the gene exists as a pair of alleles with one copy from each parent. However, the alleles may not be equal, and one member of the pair may control the phenotype despite the presence of the other copy. In this case, we say that allele is dominant over the other, the latter of which is labeled as recessive. Depending on the gene and the mutation, as discussed later, a disease allele may be either dominant or recessive. Next, during the development of the gametes, these alleles segregate randomly in a process independent from all other genes. Therefore, the chance of a child receiving a particular allele is entirely random. If these laws all hold true, the observed inheritance of the clinical disease in families will follow a specific pattern that can be used to identify the nature of the causative gene. Although diseases showing Mendelian inheritance are either rare conditions or rare forms of common conditions (e.g., early-onset Alzheimer dementia or PD), identification of such genes are seminal biological advances that can have enormous impact on our understanding of these neurological conditions.
Autosomal Dominant Disorders
Autosomal dominant inheritance is characterized by direct transmission of the disorder from parent to child (Fig. 40.6). Affected individuals are seen in all generations, and a vertical line can be drawn on the pedigree to illustrate the passage of the disorder. Since only one deleterious copy of the disease gene is necessary, risk of transmission from an affected parent is 50%. Since the disorder is autosomal, there is no sex preference, and both males and females can present with the disease. One caveat involves the concept of penetrance, or the percent likelihood that a trait will manifest in a person with a specific genotype. A dominant gene is considered to have complete penetrance if all individuals with a given mutation develop disease. In practice, however, many autosomal dominant genes show varying degrees of penetrance or expressivity, most likely due to the influence of other genes and environmental factors.
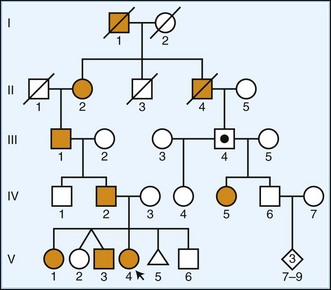
There are over 400 examples of diseases with neurological phenotypes that show autosomal dominant inheritance (OMIM, 2010). These conditions include hyperkalemic periodic paralysis (voltage-gated sodium channel NaV1.4 on chromosome 17, often caused by missense mutations), HD (Huntington on chromosome 4, caused by CAG repeat expansion), SCA type 3 (ataxin-3 on chromosome 14, caused by CAG repeat expansion), Charcot-Marie-Tooth type 1B (myelin protein zero on chromosome 1, often caused by missense mutations), early-onset familial Alzheimer disease (AD)(presenilin-1, often caused by missense mutations), frontotemporal dementia with parkinsonism (microtubule-associated protein tau on chromosome 17, often caused by missense or splicing mutations), tuberous sclerosis type 1 (hamartin on chromosome 9, often caused by nonsense mutations and frameshifts), neurofibromatosis type 1 (neurofibromin on chromosome 17, caused by point mutations, frameshifts, and splicing mutations), and familial amyotrophic lateral sclerosis (ALS) (superoxide dismutase-1 on chromosome 21, caused by missense mutations), to name a few. Even rare Mendelian forms of more common syndromes such as epilepsy or sleep disorders (e.g., familial advanced sleep-phase syndrome) have been identified. More detailed lists can be found using the recommended online resources (Table 40.3).
| Disease-specific and gene-specific resources | GeneReviews at GeneTests University of Washington, Seattle, WA USA US National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/sites/GeneTests/ |
| Locus Specific Mutation Databases Human Genome Variation Society, Australia http://www.hgvs.org/dblist/glsdb.html |
|
| Neuromuscular Disease Center Washington University, St. Louis, MO USA http://neuromuscular.wustl.edu/ |
|
| Online Mendelian Inheritance in Man Johns Hopkins University, Baltimore, MD USA US National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/omim/ |
|
| Clinical genetic testing and clinical trials | ClinicalTrials.gov US National Institutes of Health http://clinicaltrials.gov/ |
| GeneTests University of Washington, Seattle, WA USA US National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/sites/GeneTests/ |
|
| Genomic variation and other genome resources | Alternative Splicing and Transcript Diversity Database European Molecular Biology Laboratory—European Bioinformatics Institute http://www.ebi.ac.uk/astd/ |
| Catalog of Published Genome-Wide Association Studies US National Human Genome Research Institute http://www.genome.gov/gwastudies/ |
|
| Ensembl Databases European Molecular Biology Laboratory—European Bioinformatics Institute Wellcome Trust Sanger Institute, UK http://www.ensembl.org/ |
|
| Database of Genomic Variants The Centre for Applied Genomics, Canada http://projects.tcag.ca/variation/ |
|
| International HapMap Project http://hapmap.ncbi.nlm.nih.gov/index.html |
|
| Single Nucleotide Polymorphism Database US National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/projects/SNP/ |
|
| 1000 Genomes Project http://www.1000genomes.org/ |
Autosomal Recessive Disorders
Autosomal recessive inheritance is characterized by lack of intergenerational transmission, in contrast to dominantly inherited disorders (Fig. 40.7). Affected individuals are seen in single generations, often separated by one or more unaffected generations. Because two deleterious copies of the disease gene are necessary, transmission requires both parents to be either affected or carriers. In the most common scenario when both parents are carriers, the risk of an affected child is 25% (50% from each parent). As with all autosomal disorders, there is no sex preference, and both males and females can present with the disease. In families showing this mode of inheritance, it is important to ask about consanguinity. In rare cases of families with considerable inbreeding, recessive alleles may be so common as to cause disease in successive generations, creating a pseudodominant pattern of inheritance.
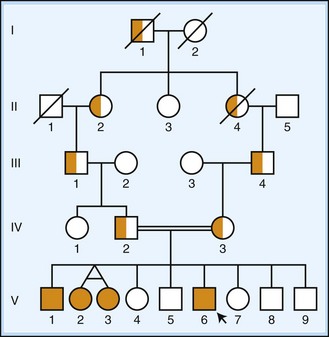
Fig. 40.7 Autosomal recessive inheritance. A pedigree diagram is shown, using standard nomenclature as described in Fig. 40.6. Carriers of disease are indicated by half-filled icons. Individuals V-2 and V-3 illustrate the diagramming of monozygotic twins. Consanguineous mating is indicated by a doubled line. An autosomal recessive pedigree demonstrates indirect transmission of disease without a sex preference, often in a single generation (occasionally described as horizontal). On average, 25% of offspring of two carriers are affected.
As mentioned for the autosomal dominant disorders, diseases that share this mode of inheritance may have very distinct types of underlying mutations. Upwards of 600 disorders with autosomal recessive inheritance show neurological symptoms (OMIM, 2010). Examples include Friedreich ataxia (frataxin on chromosome 9, caused by intronic GAA repeat expansion), spinal muscular atrophy type 1 (survival of motor neuron 1 on chromosome 5, caused by deletion of exon 7), Wilson disease (ATPase, Cu++ transporting, beta-polypeptide on chromosome 13, often caused by missense mutations), Tay-Sachs disease (hexosaminidase A on chromosome 15, commonly caused by frameshift, splicing, or nonsense mutations), glycogen storage type II or Pompe disease (acid alpha-glucosidase gene on chromosome 17, often caused by point mutations, splicing mutations, and exon deletions), phenylketonuria (phenylalanine hydroxylase on chromosome 12, often caused by missense mutations), and ataxia-telangiectasia (ataxia-telangiectasia mutated on chromosome 11, often caused by point mutations and splicing mutations). More detailed lists can be found using the recommended online resources (see Table 40.3).
Sex-Linked (X-Linked) Disorders
Recessive X-linked transmission is characterized by the presence of disease in males only (Fig. 40.8). Affected males cannot pass the disease on to their sons, but all their daughters must inherent the abnormal X chromosome and are, therefore, obligate carriers. A carrier female has a 50% chance of passing the disease allele to a child, but all males receiving it will be affected. Dominant X-linked transmission (see Fig. 40.8) is similar, except carrier females are affected and transmit the disease to 50% of their children irrespective of their sex. Affected males usually show a more severe phenotype, or may even exhibit lethality, and transmit the disease to all of their daughters and none of their sons.
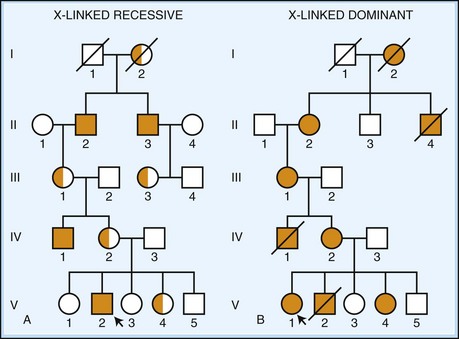
Fig. 40.8 X-linked inheritance. A, X-linked recessive disease. A pedigree diagram is shown using standard nomenclature as described in Fig. 40.6. Carriers of disease are indicated by half-filled icons. Disease manifests only in hemizygous males. Fathers cannot pass the disease to their sons, but all daughters of an affected male are obligate carriers of disease. Carrier females have a 50% chance to pass on the disease gene and can have affected sons. In some cases, a female carrier can be mildly symptomatic, usually due to non-random lyonization. B, X-linked dominant disease. A pedigree diagram is shown using standard nomenclature as described in Fig. 40.6. Disease manifests in heterozygous females (although severity may be affected by lyonization). The mutant gene is either lethal in males (as shown here) or has a much more severe phenotype. Affected females pass on the disease 50% of the time.
Over 100 X-linked disorders with neurological phenotypes are known (OMIM, 2010). The majority of these X-linked disorders are recessive, and as seen for the autosomal diseases, mutation type varies widely among the different disorders. Some examples include X-linked adrenoleukodystrophy (ATP-binding cassette subfamily D member 1, commonly caused by missense and frameshift mutations), Duchenne muscular dystrophy (dystrophin, commonly caused by deletions), Emery-Dreifuss muscular dystrophy-1 (emerin, often caused by nonsense mutations), Menkes disease (ATPase, Cu++-transporting, alpha-polypeptide, commonly caused by frameshifts, nonsense mutations, and splicing mutations), Fabry disease (alpha-galactosidase A, commonly caused by point mutations, gene rearrangements, and splicing mutations), and Pelizaeus-Merzbacher disease (proteolipid protein-1, often caused by duplications and missense mutations). X-linked dominant disorders include Rett syndrome (methyl-CpG-binding protein-2, often due to missense and nonsense mutations), incontinentia pigmenti (inhibitor of kappa light polypeptide gene enhancer in B cells, kinase gamma [IKBKG], often due to deletions), and Aicardi syndrome (gene unknown). More detailed lists can be found using the recommended online resources (see Table 40.3).
Mendelian Disease Gene Identification by Linkage Analysis and Chromosome Mapping
As mentioned previously, patterns of inheritance can be utilized to locate genes responsible for disease. Traditionally, genes showing Mendelian patterns of inheritance can be physically mapped and identified through linkage analysis (Altshuler et al., 2008; Pulst, 2003) (Fig. 40.9). In this technique, one attempts to find a known region of DNA, termed a marker, which is co-inherited (segregates) with the disease being studied and subsequently uses the location of that marker to find the disease gene. Although in principle, two points on the same chromosome theoretically segregate independently from one another, the recombination process that mediates this (termed crossing-over because maternal and paternal chromosomes swap segments during gamete formation) is statistically more likely to separate points that are far apart from one another than those that are close. Segments of DNA that segregate together are described as being linked. If the degree of linkage exceeds that expected by chance, the regions are said to be in disequilibrium and are therefore in close proximity. By using naturally occurring DNA polymorphisms as locational markers, the physical mapping of an unknown disease gene is possible, although the mapped region will likely contain other genes as well. Depending on the size of the family, the generational distance of affected individuals sampled, and the density of the markers being used, the region containing the disease gene is narrowed down to a size more amenable to further detailed analysis. Subsequent analysis, usually DNA sequencing of likely candidate genes, is then performed to locate a mutation that segregates with the affected members of the original family. Many genes important to neurological disease have been identified in this way, including the genes for HD, Duchenne muscular dystrophy, Wilson disease, neurofibromatosis type 1, Von Hippel-Lindau syndrome, torsion dystonia 1, Friedreich ataxia, myotonic dystrophy type 1, hyperkalemic periodic paralysis, familial advanced sleep-phase syndrome, and many others. Although still useful clinically for large families, utilization of this technique is not possible for many diseases because of small family sizes and/or lack of power due to insufficient generational separation between affected individuals in the pedigree.
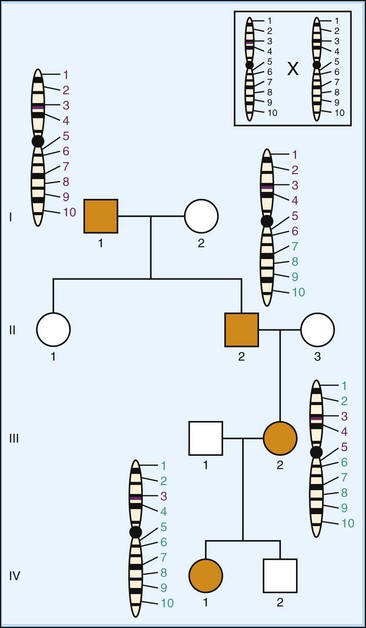
Fig. 40.9 Linkage analysis. A pedigree is depicted as in Fig. 40.6, showing autosomal dominant inheritance of disease (filled icons). Transmission of the chromosome containing the mutant gene (purple line) is illustrated for all affected individuals. Numbers represent the location of specific chromosomal markers (e.g., single nucleotide polymorphisms or other sequences). Purple numbers represent markers originally from the mutant chromosome in individual I-1. With each mating, there is potential crossing over between regions of homologous chromosomes (insert), likely resulting in the separation of markers spaced far apart along the chromosome. In this example, examination of all affected individuals shows the disease segregates with marker 3, and the two are therefore in linkage disequilibrium, suggesting they are near one another. Once identified, the marker location can be used to select candidate genes for sequencing to identify the causative gene and mutation in the family.
Non-Mendelian Patterns of Inheritance
Mitochondrial Disorders
Mitochondria are double-membraned organelles responsible for energy production within the cell via the process of oxidative phosphorylation, which relies on the transfer of electrons through a chain of protein complexes within the inner mitochondrial membrane. Disruption of mitochondrial function can lead to a variety of diseases with multisystem involvement, including prominent neurological symptoms (DiMauro and Hirano, 2009; Zeviani and Carelli, 2007). Mitochondria possess their own genome with 37 genes. Because mitochondria are cytoplasmic and the majority of cytoplasm within the zygote is derived from the egg and not the sperm, disorders involving mitochondrial DNA are inherited through the maternal line (Fig. 40.10). A single cell contains many mitochondria which all replicate independently of the nuclear DNA, so it is possible that a mutation in the mitochondrial genome may be present in some of the mitochondria but not others, a condition termed heteroplasmy. This proportion can affect whether a disease is expressed and, if so, what tissues are affected if a minimum threshold of abnormal mitochondria is reached. Heteroplasmy may also change over time as cells divide and the mitochondria are redistributed. Some examples of such disorders include MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes, caused by point mutations within the gene encoding mitochondrial tRNALEU), MERRF (myoclonic epilepsy with ragged red fibers, caused by point mutations within the gene encoding mitochondrial tRNALYS), and LHON (Leber hereditary optic neuropathy, most often caused by point mutations in either of two mitochondrial genes encoding complex I subunits, ND4 or ND6).
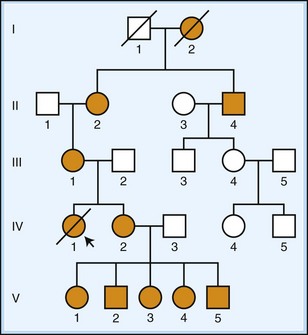
Fig. 40.10 Mitochondrial (maternal) inheritance. A pedigree diagram is shown using standard nomenclature as described in Fig. 40.6. As the mutant gene is carried in the mitochondrial genome, disease is passed on to all the offspring of affected females (see text). Males can be affected but cannot pass on disease. Severity and onset of the disease may be affected by heteroplasmy, the proportion of abnormal mitochondria per cell, as illustrated by a severe phenotype seen in patient IV-1.
Because the mitochondria themselves contain only a few genes, the majority of mitochondrial proteins, including the machinery responsible for the replication and repair of the mitochondrial genome, are all encoded by nuclear genes. Since these genes are located within the nuclear genome, despite the fact that their mutation gives rise to dysfunctional mitochondria, the disease will show a Mendelian pattern of inheritance. Some examples include infantile-onset SCA (twinkle on chromosome 10, autosomal recessive, caused by missense mutations), progressive external ophthalmoplegia A2 (adenine nucleotide translocator 1 on chromosome 4, autosomal dominant, caused by missense mutations), and Charcot-Marie-Tooth type 2A2 (mitofusin-2 on chromosome 1, autosomal dominant, often caused by missense mutations). Interestingly, various mutations, commonly missense, of the nuclear gene DNA polymerase gamma (POLG) on chromosome 15, which encodes the polymerase responsible for both replication and repair of the mitochondrial genome, cause a wide variety of diverse phenotypes with different modes of inheritance (Hudson and Chinnery, 2006). These include the autosomal recessive Alpers syndrome of encephalopathy, seizures, and liver failure, an autosomal dominant form of chronic progressive external ophthalmoplegia, and autosomal recessive phenotypes of cerebellar ataxia and peripheral neuropathy, among others.
Imprinting
For most genes, expression is controlled by distinct cellular processes that operate irrespective of the gene’s parental origin. However, for some genes, expression in the offspring differs depending on whether the allele was maternally or paternally inherited, and such genes are described as being imprinted (Spencer, 2009). Imprinting arises from epigenetic modifications such as DNA or histone methylation, which are parent-specific alterations that do not change the actual DNA sequence (Fig. 40.11). One example of this is sex-specific DNA methylation that occurs for some genes during the formation of gametes. In the offspring, the methylated gene is bound by histone proteins forming transcriptionally inactive heterochromatin. This allows all gene expression to be driven by the allele derived from the other parent. This can be dynamic depending on the gene, and the magnitude of differential expression between the alleles can vary based on stage of development, tissue type, and possibly other factors. Deletion of an imprinted region or defective imprinting in gametogenesis can lead to disease as illustrated by observations involving chromosome 15q (Lalande and Calciano, 2007). In this example, differential methylation affects the expression of multiple genes, and loss of maternal patterning can lead to Angelman syndrome, characterized by intellectual impairment, epilepsy, ataxia, and inappropriate laughter, while loss of the paternal pattern causes Prader-Willi syndrome, associated with intellectual impairment, obesity, and behavioral problems. The most common mechanism involves de novo deletion of the imprinted region from one parent, although in some cases, defective imprinting can also occur during gametogenesis. In the majority of cases, defective imprinting occurs spontaneously and is therefore unlikely to recur in families; however, imprinting defects can rarely be due to small deletions involving sequences important for regulating parent-specific methylation.
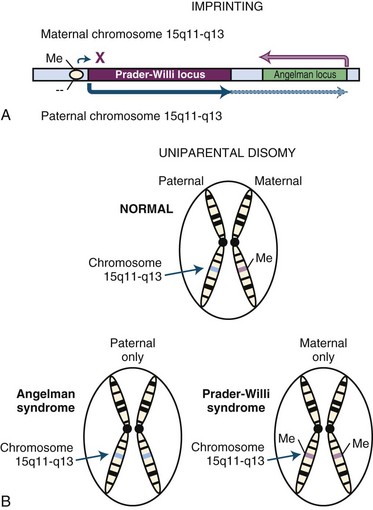
Uniparental Disomy
Uniparental disomy arises when pairs of chromosomes are inherited from the same parent, either in their entirety or in large segments due to segregation errors or chromosomal rearrangement (Kotzot, 2008) (see Fig. 40.11). The uniparentally inherited chromosomes can be identical (isodisomic) or different (heterodisomic). In families where the parents lack underlying chromosomal abnormalities, these events usually occur spontaneously and are unlikely to recur. Disease can result from effects related to loss of chromosomal imprinting, pairing of an autosomal recessive mutation, pairing of an X-linked recessive mutation in a female child, or from the generation of a mosaic trisomy. The disorders most commonly associated with this mechanism are the Prader-Willi and Angelman syndromes, discussed previously for imprinting disorders, which can arise from maternal and paternal uniparental disomy, respectively, due to a loss of the imprinting pattern from the missing parental allele. Down syndrome can also rarely result from a mosaic trisomy. There are several examples in the literature of single cases where an autosomal recessive disease arose in a child from uniparental disomy pairing an abnormal allele from a carrier parent, including disorders such as abetalipoproteinemia, Bloom syndrome, autosomal recessive deafness-1A, spinal muscular atrophy, cystic fibrosis, and others (Zlotogora, 2004).
Common Neurological Disorders and Complex Disease Genetics
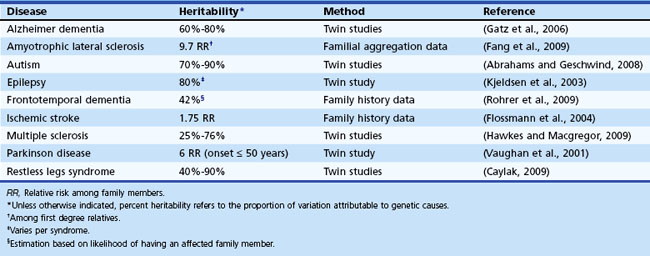
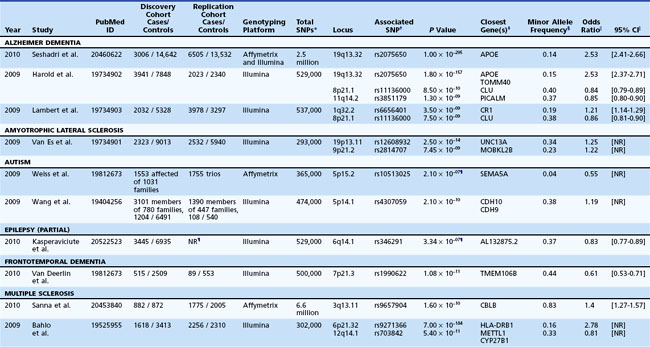
To this point, we have focused on Mendelian neurological disease, in which mutations of a single gene are sufficient to cause disease. Neurological diseases with Mendelian inheritance are rare in most populations, and account for less than 5% of those with common conditions such as Alzheimer dementia. Yet, many of the common neurological diseases seen worldwide have significant genetic contributions (Table 40.4). For example, twin studies have shown high heritability (≥60%) for Alzheimer dementia (Gatz et al., 2006) and autism (Abrahams and Geschwind, 2008; Freitag, 2007), increased relative risk is seen in first-degree relatives of probands with ALS (approximately 10-fold) (Fang et al., 2009) and epilepsy (about 2.5-fold) (Helbig et al., 2008), and a variety of studies support a degree of heritability in PD (Belin and Westerlund, 2008) and cerebrovascular disease (Matarin et al., 2010). But even when family history is present, the mode of inheritance is not clear, and no major disease-causing Mendelian mutations are usually identified in the majority of cases. So in contrast to the single-gene Mendelian disorders previously discussed, these common complex genetic conditions appear to be genetically heterogeneous and multifactorial, likely involving interplay between multiple genes, each with small effect size, and environmental factors, none of which are sufficient to be causal, but each of which increases susceptibility to the disorder. This is the basis of the “common disease–common variant” (CDCV) model, which has driven most research into common genetic diseases (Schork et al., 2009). The alternative model is that rather than common SNPs, multiple inherited rare variants of small to intermediate effect size or de novo mutations with large effect size underlie genetic risk for common disorders. The difficulty with assessing this latter proposition is that until the very recent advent of efficient whole genome or whole exome sequencing, genome-wide identification of such rare variants was not feasible. In contrast, efficient genome-wide assessment of common variation has been possible for several years and has been applied to numerous neurological disorders (Table 40.5). Still, the true nature of the type of genetic variation underlying most complex disease is not known, but major advances are being made. Here we discuss the strategies currently being used, starting with genome-wide screening for common variation.
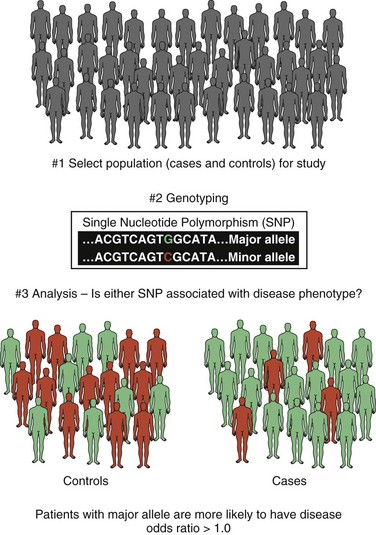
Common Variants and Genome-Wide Association Studies
As already discussed, genetic linkage provides a means of localizing a disease gene to a specific region of a chromosome by using a DNA marker that tracks with affected individuals within families. Linkage analysis, while not without value in genetically complex disease, is less powered than genetic association studies for identification of common variation in complex genetic disease. Genetic association studies assess whether one or more of a defined set of genetic variants are increased or decreased in a disease versus a control population. If a genetic variant is observed in individuals with disease significantly more often or less than expected by chance, that variant is said to be associated with the disease. When one or a few genes are studied, this is a candidate gene association study. When common variants from across the entire genome are studied in this manner, the result is a genome-wide association study, or GWAS (Mullen et al., 2009; Simon-Sanchez and Singleton, 2008) (Fig. 40.12). Original genetic association studies were conducted with a small number of candidate genes, but advances in technology have permitted GWAS in thousands of subjects in a wide variety of human diseases, including dozens of neurological conditions (see Table 40.5). Although the SNPs themselves may directly influence the disease under study, most often this is not the case, and SNPs are best thought of as markers for the location of a gene(s) or region relevant to the disease. In fact, most alleles of the second major type of common genetic variation, CNVs, are mostly captured by SNPs (Conrad et al., 2010) and can be identified by the common SNP genotyping platforms, allowing GWASs to evaluate the contribution of common inherited CNVs as well as SNPs.
The model that underlies the value of GWAS is based on the concept that common disease is predicted to arise from the interplay of effects caused by common polymorphisms in multiple genes, as well as environmental and other factors. The aim of a GWAS is to identify these common variants that correlate to risk for the disease in question but do not alone cause the disease. Because the effect size, or increase in odds for a disease, is expected to be small (negative selection would have removed strongly deleterious variants from the population), and many independent genetic markers are tested, large sample sizes are needed to have power to detect genome-wide association. This is further compounded by two of the many major factors challenging GWASs of common neurological diseases, phenotypic and genetic heterogeneity. Phenotypic heterogeneity describes the wide and variable clinical spectrum patients with a particular neurological disorder or syndrome (e.g., frontotemporal dementia, epilepsy, multiple sclerosis, autism) manifest. Genetic heterogeneity refers to the notion that even in those with a relatively homogeneous phenotype, many different genetic factors may be contributing in different individuals to lead to the same phenotype. Both of these forms of heterogeneity require large samples to have adequate power to detect genetic risk factors of even moderate size. The smaller the effect of any given genetic variant, the larger the sample size needed to detect that variant. One strategy that may increase power is to study intermediate phenotypes, or endophenotypes, that may be more related to individual genetic risk factors than the broad clinical diagnosis of a disorder, such as specific measures of language or social behavior in autism (Abrahams and Geschwind, 2008; Alarcon et al., 2008; Vernes et al., 2008). Alternatively, such phenotypes can be used to identify more homogeneous subgroups of patients, such as those with specific forms of pathology, as in TAR DNA-binding protein (TDP-43) inclusion-positive frontotemporal dementia (FTD), which may have improved power in a recent FTD GWAS by reducing heterogeneity (Van Deerlin et al., 2010).
Efficiently generating the extensive genotype data necessary for a GWAS has been made possible using microarray technology (Coppola and Geschwind, 2006; Geschwind, 2003). In this type of experiment, specific fragments of DNA corresponding to the sequences of the target SNPs are immobilized in a grid pattern across a glass slide, termed the array. Genomic DNA from individual cases and controls is fluorescently labeled, hybridized to the slide, and the signals from laser-induced dye excitation are collected. The readout will be a map of the SNP pattern for each patient. Data cleaning, quality control, and statistical analysis are performed to determine whether any SNPs are associated with patients more than controls. Given the large number of independent tests performed in a GWAS, statistical significance is commonly set at 5 × 10−8 (McCarthy et al., 2008; Wellcome Trust Case Control Consortium, 2007) to correct for multiple comparisons. It is now also considered standard to demonstrate that any statistically significant association identified is present in more than one study population, providing an independent replication of the initial finding. Study power and replication may also both be aided by the availability of shared GWAS data (Box 40.1).
Box 40.1 Genetic Data Repositories and Data Sharing
Sharing genetic data is very important, and this is emphasized clearly in relation to genome-wide association study (GWAS) data which, because it is produced on common platforms typing essentially the same genetic variation in multiple populations, has great value beyond its original intended purpose. By sharing this data, other researchers have the opportunity to virtually perform GWAS analysis on populations they would not necessarily be able to evaluate. Since large sample sizes increase the power of GWAS, and few single groups can recruit enough patients for a well powered GWAS, this permits pooling and reanalysis of data collected in many laboratories on a single neuropsychiatric disease such as schizophrenia (e.g., Purcell et al., 2009; The International Schizophrenia Consortium at http://pngu.mgh.harvard.edu/isc/). It also permits study across diseases that may share common etiologies, such as amyotrophic lateral sclerosis and frontotemporal dementia (van Es et al., 2009) or autism and schizophrenia (e.g., Cantor and Geschwind, 2008; Psychiatric GWAS Consortium at https://pgc.unc.edu/index.php).
Additionally, the population could be resorted based on other known variables, SNPs could be excluded or grouped during analysis, different methods of analysis could be applied to the raw data, or data from individual members could be extracted for use in other studies (Purcell et al., 2009). Because of the benefits of this versatility, many funding organizations, including the National Institutes of Health, and major scientific journals, such as Nature and Science, have policies in place for investigators to make GWAS data available to other researchers. In some cases, disease-specific repositories have been established for the purpose of sharing both the biomaterials and genetic information, such as the Autism Genetic Resource Exchange (AGRE at http://www.agre.org/) and the NIMH Human Genetics Initiative repository (https://www.nimhgenetics.org/nimh_human_genetics_initiative/).
Recently published genome-wide association studies of interest involving neurological disease are shown in Table 40.5. One example illustrating the use of a GWAS in complex disease is from a 2009 study by Ikram and colleagues, who performed a GWAS using a population of 19,602 white persons, of whom 1544 had strokes (Ikram et al., 2009). They identified two intergenic SNPs on chromosome 12p13 with significant genome-wide association for total stroke implicating the NINJ2 gene, which is a cell-adhesion molecule found in radial glia (Ikram et al., 2009). Replication in an independent cohort confirmed the association of one SNP with a combined hazard ratio of 1.29 for ischemic stroke in white persons (Ikram et al., 2009). The mechanism of how NINJ2 increases risk for ischemic stroke is unclear at this time, but the results of this GWAS open up a new avenue of research by highlighting it as a candidate for future molecular and cellular studies into stroke etiology.
Similarly, for the most common neurodegenerative dementia, Alzheimer dementia, recent GWASs have benefited from large numbers of available cases and expanded the loci known to be associated with disease beyond the apolipoprotein E locus to include other neuronal molecules such as BIN1 and PICALM, which are involved in clathrin-mediated endocytosis and intracellular trafficking, and the apolipoprotein, CLU (Harold et al., 2009; Lambert et al., 2009; Seshadri et al., 2010). In Parkinson disease, recent GWASs in large cohorts of European and Japanese patients identified alpha-synuclein (SNCA) and LRRK2 as susceptibility loci (Edwards et al., 2010; Satake et al., 2009; Simon-Sanchez et al., 2009), which is notable because both genes also give rise to autosomal dominant forms of parkinsonism. The tau protein (MAPT), another gene responsible for autosomal dominant forms of parkinsonism, was also found to be associated with disease in European populations (Edwards et al., 2010; Simon-Sanchez et al., 2009). Together these results suggest a commonality between Mendelian and sporadic forms of this disorder.
It is important that physicians have a clear understanding of the meaning of GWAS results so as to be able to differentiate common variants associated with disease from disease-causing mutations. A potential error to be avoided in the clinical interpretation of GWAS data is directly equating the findings to the future development of the disease. It must be reiterated that the finding of an association with a common variant does not equal the finding of a disease gene. By definition, these common variants must have low penetrance, otherwise they would not be so common in normal individuals, and they would likely act in a more Mendelian way. Furthermore, such variants might be associated with disease modifiers—for example, genes acting either upstream or downstream in pathways where disruption or dysregulation can lead to the disease, or perhaps genes involved in the production or regulation of factors involved in such pathways. Instead of directly causing disease, such modifier genes confer a risk of disease, the magnitude of which is sometimes not directly quantifiable because it involves interaction with other genes and the environment. Therefore, for most conditions, reported GWAS information cannot be directly translated into a clinical setting, because the presence of the variant does not necessarily lead to the disease in most cases, particularly for the more rare disorders. As an example, one of the strongest and best-known identified associations, the apolipoprotein E ε4 allele detected in sporadic AD, with an odds ratio of 4 (Coon et al., 2007), has such an inconsistent predictive value that it is not recommended for routine use in disease prediction nor as a typical part of most clinical dementia evaluations (Knopman et al., 2001). Despite this, some commercial organizations have begun to create direct-to-consumer tests for genetic variation associated with disease. As the public has become more aware of the impact of genetics on health and disease, there has been a growing desire for preemptive screening, particularly for individuals with family members afflicted with common disease. In response to this need, genetic variation screening tests are often marketed as a means of assessing the potential for future development of disease. Given the caveats discussed, there is no definitive means at present to accurately define an individual’s risk of disease based on the presence of one or more associated common variants. It is important for the physician to be aware of this insofar as patients may contact them regarding such testing, and it should be emphasized that any positive results would have unclear predictive value.
There are examples of clinically important allelic variants identified by other methods, so such expectations for GWAS in neurological disease are not unfounded. One such illustration is the variation seen in the cytochrome P450 isoenzyme, CYP2C9, which is responsible for the metabolism of a number of clinically relevant pharmaceutical agents, in particular the anticoagulant, warfarin (Sanderson et al., 2005). The major allele, CYP2C9*1, is seen in more than 95% of Asian and African populations, but multiple variants commonly exist in European and Caucasian populations, including CYP2C9*2 and CYP2C9*3, both of which reduce warfarin metabolism (Sanderson et al., 2005). In one study, 20% of patients carried either CYP2C9*2 or CYP2C9*3 and required a mean reduction of their warfarin dosage by 27% to maintain an optimal therapeutic range, reflected by an increased relative risk of bleeding of about 2.3 (Sanderson et al., 2005). Although the relative risk in this example is still greater than typically seen in most GWASs, it demonstrates how common variant risk information can potentially affect the care of an individual patient. As we discover more regarding the nature of complex genetic disease, new ways of utilizing this information clinically will likely be determined. In the meantime, the value of GWAS data, especially from a pharmacogenomic research perspective, is significant; it can help identify new genes, pathways, and biological networks related to disease that may have therapeutic benefit (Box 40.2).
Box 40.2 Pharmacogenetics and Personalized Medicine
In addition to contributing to disease susceptibility, genetic variation can have other medically applicable roles. One of the most highly anticipated benefits for genetic research is the capability of tailoring medical or pharmacological therapies to target a patient’s disease based on their individual genotype, the so-called concept of personalized medicine. The initial application of this concept is in the optimization of drug effects and minimization of toxic side effects based on genotype, termed pharmacogenetics (Holmes et al., 2009). Although this field has not yet advanced to the point of routine clinic use, there are examples of the potential utility and the benefit to patients we may hope to see in the near future. In the management of stroke, genetic variation has been found to impact patient response to antiplatelet agents and anticoagulants (Meschia, 2009; see main text) and influence statin-associated myopathy (Link et al., 2008; Meschia, 2009). In a recent GWAS analysis, 85 patients with myopathy were identified from an initial group of over 12,000 patients taking simvastatin, and association was demonstrated in both this cohort and a large replication cohort with a SNP in the SLCO1B1 gene (Link et al., 2008). SLCO1B1 encodes a membrane protein that mediates liver uptake of various drugs including statins, and in the presence of the associated SNP, the odds ratio for myopathy was 4.3 when heterozygous and 17.4 when homozygous (Link et al., 2008), clearly reflecting a need to modify statin treatment in such patients.
A number of practical issues will have to be solved before such testing can achieve widespread use in the clinic, particularly determinations of the clinical benefit and cost-effectiveness in specific diseases and populations (Holmes et al., 2009; Meschia, 2009; Swen et al., 2007); however, recent rapid advancements in technology, such as next-generation DNA sequencing, may prove beneficial in this arena.
Rare Variants and Candidate Gene Resequencing
So far, common variation is only able to explain a small percentage of genetic risk for common neurological disease. The other major model that attempts to explain what is currently referred to as the missing heritability (Manolio et al., 2009) in complex genetic disease implicates rare variants with medium to high penetrance instead of more common ones with low penetrance (Schork et al., 2009) (Fig. 40.13). Rare variants are defined as DNA alterations that are found in less than 1% of most populations or, in some cases, are “private” and only seen in specific affected families. In this model, one or more rare variants, alone or in combination with common variants, produce the disease in question. A GWAS is not well suited to detect these variants, because they are rare and most likely to be relatively recent mutations that do not segregate on common haplotypes measured in these studies. Even when they do, they do not occur in high enough frequency in the general population to provide statistical power for their detection using current sample sizes. Detection generally requires resequencing of potentially involved candidate genes in a defined population of patients and controls. One major difficulty of such investigations is that the baseline level of rare variation among normal humans is not clearly established. Studies such as the 1000 Genomes Project (http://www.1000genomes.org/) are attempting to catalog normal human variation within the 0.1% to 1% range, so researchers will be able to better define this class of rare variants and develop more effective strategies for their detection.
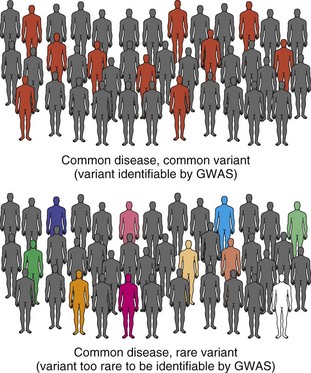
An example of this approach involves the developmental disorder, autism, where sequencing of the gene, contactin-associated protein-like 2 (CNTNAP2), in 635 patients with autism spectrum disorder (ASD) and 942 controls found 13 rare variants unique to patients, including one which was seen in 4 patients from 3 unrelated families (Bakkaloglu et al., 2008). Recessively inherited mutations in CNTNAP2 in an Amish family with a syndromic form of autism with epilepsy provided the most convincing evidence for the causal role of mutations in this gene (Strauss et al., 2006). Interestingly, this same gene illustrates that the common disease–rare variant and common disease–common variant hypotheses are not mutually exclusive, since common variants in this gene modulate language function in ASD and other conditions (Alarcon et al., 2008; Vernes et al., 2008). Exciting advances in DNA sequencing (see Whole Genome/Exome Sequencing in Disease Gene Discovery) will allow us to finally analyze many whole genomes and understand to what extent common and/or rare variants contribute to many common neurological diseases.
Copy Number Variation and Comparative Genomic Hybridization
The majority of variation and disease-causing mutations discussed to this point have centered around single base pair changes in DNA sequence. However, as previously described in Structural Chromosomal Abnormalities and Copy Number Variation, the CNV (Beckmann et al., 2007; Stankiewicz and Lupski, 2010; Wain et al., 2009; Zhang et al., 2009) (Fig. 40.14) actually represents more total real estate in our genome. Advances in methods such as the advent of the microarray indicate that such changes occur quite commonly (at 10−4 to 10−6 per locus per generation) compared to single nucleotide changes (10−8 per base pair per generation on average) (Lupski, 2007). Overall, CNVs are estimated to represent at least 4% (Conrad et al., 2010) and potentially up to 13% of the total human genome (Redon et al., 2006; Stankiewicz and Lupski, 2010). The high frequency of these events may reflect an evolutionary advantage of CNVs as a mechanism for producing genetic diversity (Zhang et al., 2009) but also implies that clinically relevant CNVs are quite likely to occur de novo more frequently than point mutations (Table 40.6). CNVs can potentially cause disease in numerous ways, including disruption of a gene’s coding region (which could cause a dominant effect or release a recessive effect on the homologous allele) or by altering regulated gene expression via positive or negative dosage effects. If the CNV itself results in the disease phenotype, it could be transmitted as a Mendelian disorder, as is the case for Charcot-Marie-Tooth type 1A. Such CNVs may be examples of rare variants in the common disease model. Alternatively, their contribution may be more subtle and insidious, with low penetrance and variable expressivity contributing to the risk of a complex genetic disease, such as in autism (Bucan et al., 2009).
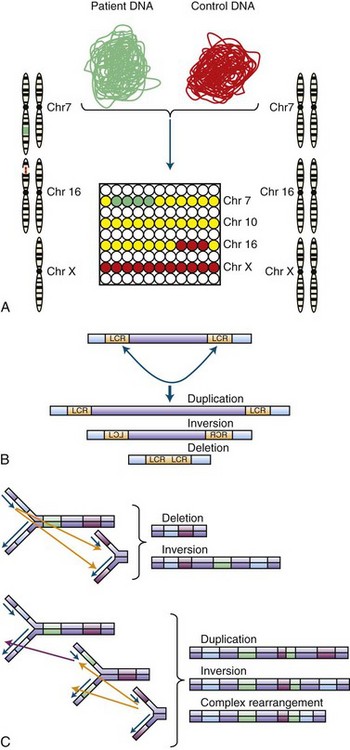
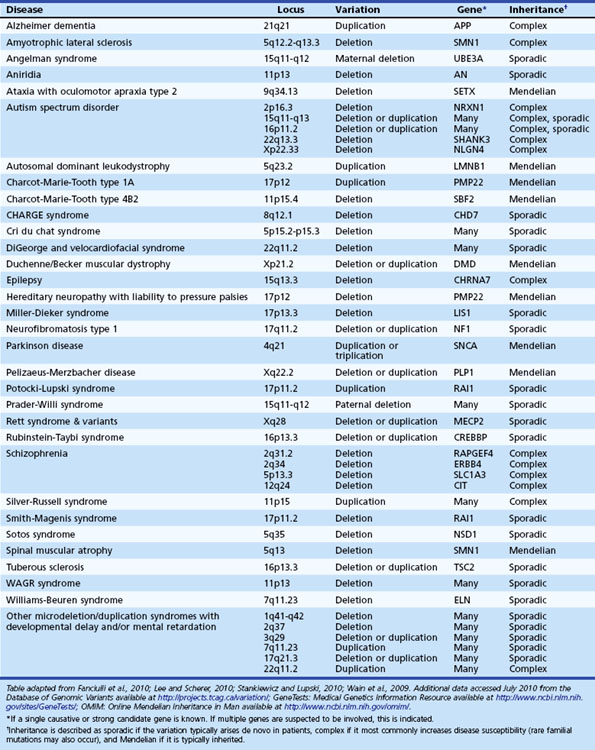
CNVs can be detected via essentially the same microarray technology used to detect SNPs, with only a few minor adjustments. In this case, DNA probes corresponding to specific chromosomal regions are placed on an array and hybridized with differentially fluorescent-labeled genomic DNA from the individual being studied and from a reference genomic DNA sample, a technique termed array comparative genomic hybridization (CGH) (also called chromosomal microarray analysis) (see Fig. 40.14, A). The average ratio of fluorescence is normalized across the array and then evaluated for each probe. If both samples hybridize to a given probe equally, the corresponding DNA region is present equally in both samples. However, if the DNA sample being studied hybridizes more or less intensely than the reference sample, it must contain either more or less of the chromosomal region in question, thus indicating a copy number variation at that location. The minimum size of a CNV that can be detected by this method is limited to the genomic distance between the minimum number of probes needed to observe a statistically significant signal change, but is usually on the order of kilobases for the highest resolution arrays. The same microarrays used to genotype SNPs in GWASs may also be used to detect CNVs, incorporating both intensity and inheritance data. Array CGH essentially produces a molecular karyotype capable of detecting genomic structural changes with much finer detail than routine microscopic methods. In most major diagnostic labs, this method has replaced microscopic karyotyping and FISH, the latter of which is now used for confirmation.
CNVs are also particularly important for neurodevelopmental disorders, with de novo CNVs present in more than 5% of patients with intellectual disability (ID) (Koolen et al., 2009) or ASD (Bucan et al., 2009; Marshall et al., 2008; Pinto et al., 2010; Sebat et al., 2007). Based on these findings, array CGH is now clinically indicated in children with a wide range of neurodevelopmental disabilities including ID and ASD (Miller DT et al., 2010). These studies also revealed several potential new autism candidate genes as well as novel biological pathways for future study of disease pathogenesis (Bucan et al., 2009; Pinto et al., 2010). Remarkably, de novo CNVs are also associated with schizophrenia (Stefansson et al., 2008; Walsh et al., 2008), especially childhood-onset forms, and some of the same CNVs observed in ASD are also observed in schizophrenia (Cantor and Geschwind, 2008), suggesting some shared liability between what were previously considered clinically distinct conditions.
Whole Genome/Exome Sequencing in Disease Gene Discovery
The identification of disease genes and their mutations hinges on the capability to sequence DNA to assess for detrimental alterations. The standard method of DNA sequencing technology currently in use is called Sanger sequencing. Although effective and accurate, the high throughput of this method is severely limited by reaction time and length of read, which is less than 1 kilobase. Recently, another new technology has been developed, termed next-generation sequencing (NextGen) (Metzker, 2010), that can rapidly generate large amounts of high-quality DNA sequence information in a relatively inexpensive and efficient manner (Table 40.7). The sequence of the human genome was derived using Sanger sequencing over a 13-year period, and subsequent Sanger sequencing of human genomes took roughly a year, but next-generation sequencing can currently accomplish the same feat in weeks. Therefore, it is now possible to rapidly interrogate an individual patient’s DNA on a genome-wide level for unknown disease-causing mutations. Several different technologies exist under the next-generation sequencing umbrella and cannot be fully described here (for details, see Metzker, 2010). The same technology can also be applied to mRNA to study gene expression and/or alternative splicing on a genome-wide basis. This technology has dramatically reduced the cost of sequencing an entire genome to less than 1% of the cost of Sanger technology (Metzker, 2010), and this is expected to reach a level comparable to current clinical testing, such as Sanger sequencing–based genetic panels, in the near future. Questions regarding data storage, analysis, and quality control, as well as translation to a clinical setting, still remain but will hopefully be answered soon, allowing the integration of the technology into the clinician’s repertoire (Geschwind and Konopka, 2009). The clinical utility of this approach was demonstrated recently by Lupski and colleagues, who sequenced the whole genome of the proband in a family with a previously undiagnosed form of Charcot-Marie-Tooth (CMT) disease type 1 (Lupski et al., 2010). By comparing the proband’s genome sequence to the human genome reference sequence, over 3.4 million SNPs and 234 CNVs were detected and subsequently paired down using a more detailed analysis until compound heterozygous mutations were identified in the SH3TC2 gene on chromosome 5, a gene previously shown to cause a different form of CMT, CMT type 4C (Lupski et al., 2010). The new mutations identified within this single family revealed an unexpected level of complexity in this Mendelian disorder, suggesting that such comprehensive sequencing methods may be clinically necessary to identify novel disease-causing mutations in known disease genes if they lead to phenotypic variation.
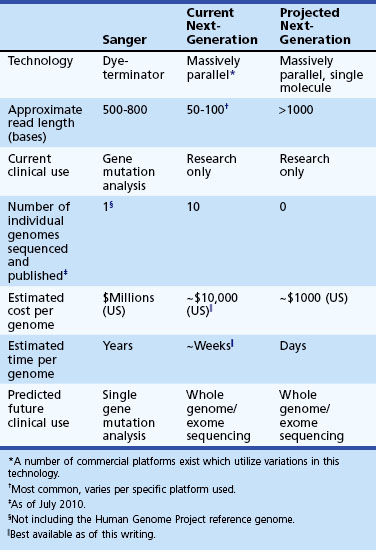
Although extremely powerful, the challenges of data interpretation and analysis may slow the arrival of whole genome sequencing to the clinic. As an initial step in the transition of this technology to the clinical arena, a significant reduction in cost, data volume, and degree of analysis can be achieved by selecting only genomic regions containing protein-coding information for sequencing, a process called exome sequencing (Choi et al., 2009; Hedges et al., 2009; Ng et al., 2009). These coding sequences are initially enriched from a pool of total genomic DNA and then subjected to next-generation sequencing. Although this would be unable to detect relevant noncoding or structural events such as copy number variation, it should prove useful as a means of evaluating Mendelian disorders caused by coding mutations. This has been illustrated by recent reports using this technology to detect novel mutations causing distal arthrogryposis type 2A (Freeman-Sheldon syndrome) (Ng et al., 2009), to confirm an unanticipated diagnosis of congenital chloride diarrhea (Choi et al., 2009), and to elucidate the gene underlying postaxial acrofacial dysostosis (Miller syndrome) (Ng et al., 2010).
In addition to identification of Mendelian mutations, this technology also allows for a more detailed exploration of complex genetic variation. In studies of common disease, it may prove a more effective means of assessing the contributions of rare variants than other methods such as a GWAS (Cirulli and Goldstein, 2010). Additionally, it may also identify novel types of variation such as double- and triple-nucleotide polymorphisms, which generate amino acid changes more than 90% and 99% of the time, respectively, and occur at 1% the density of SNPs (Rosenfeld et al., 2010). Future studies will have to further assess the contribution of such novel DNA changes to human disease, but the current findings confirm that next-generation sequencing technology will be able to uncover new types of functional genomic variation.
Lastly, whole genome sequencing may also provide new information regarding environmental contributions to disease. Recently, whole genome sequencing was reported from a pair of monozygotic twins who were discordant for multiple sclerosis (Baranzini et al., 2010). No significant genomic, transcriptional, or epigenetic changes were found to explain disease disconcordance among these twins (Baranzini et al., 2010), suggesting there may be other critical genetic or epigenetic factors not examined by this study, or that key differences may lie in other cell types, or that as-yet-undetermined environmental factors are contributing to disease—conclusions which would not be possible to establish without next-generation sequencing technology.
Future Role of Systems Biology in Neurogenetic Disease
The complex relationship between genetic risk variants, even when they are inherited in a Mendelian fashion, and clinical features, or the relationship of these mutations to disease pathophysiology, presents significant challenges to the use of genetics for diagnosis and therapeutics. Furthermore, the majority of studies investigating genetic disorders have focused on the discovery and molecular analysis of the disease genes themselves, as these would intuitively appear to be the most immediately useful in diagnosis and potential treatment. There are some examples, such as metabolic disease and enzyme replacement therapy (Beck, 2010), which support this practice. However, for many more diseases, including virtually all neurodegenerative disorders, knowledge of the specific causative gene has not immediately yielded new curative therapies but has instead raised many new questions regarding the underlying molecular etiology of the disease. The hope is that research into these underlying mechanisms will uncover new therapeutic targets; toward that goal, the technologies discussed have made greater amounts of information available for scientific analysis than ever before. For example, microarrays can be used to study not only genome-wide genetic variation via SNPs as described earlier but also variations in gene expression (Fig. 40.15). For this method, the array platform contains probes that are complementary to genome-wide mRNA sequences, and the study is performed by hybridizing the array with fluorescently labeled mRNA collected from either patients or controls. The intensity of the fluorescent signal can be used to determine and compare the relative levels of expression for each gene across the samples. Similar techniques can also be used to evaluate RNA splicing with probes that correspond to all the exons in a given gene and then assessing samples for their alternative usage in cases and controls. With the availability of this genome-wide data, encompassing both genetic variation and gene expression in clinically-evaluated patients and controls, it becomes possible to incorporate and synthesize the totality of this information together in ways which assess phenotype, genetic variation, and gene expression simultaneously in a more comprehensive way. This field of study, known as systems biology, strives to use these sets of information to develop detailed genetic pathways to identify related genes and genetic programs relevant to disease (Geschwind and Konopka, 2009) (see Fig. 40.15). Such integrative analysis has begun to accelerate our understanding of disease pathogenesis and generate new insights into more effective treatment strategies, which will only improve as we learn more and the techniques improve.
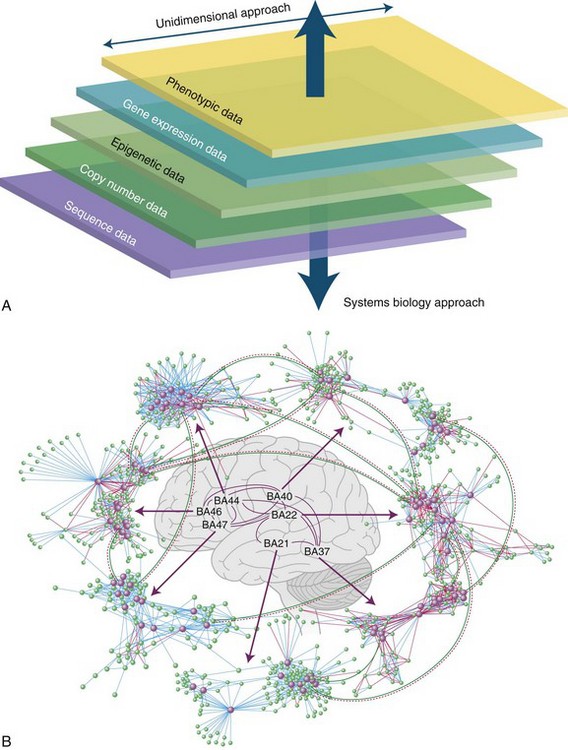
One example of this type of systems biology approach involves using gene expression data, such as from microarray studies, to group individual genes according to their degree of coexpression, forming functionally related gene expression modules. These modules are then graphed according to the interconnectivity of their members which produces a network of correlations centered around one or more key genes, termed hubs, which functionally drive the association either directly or indirectly. Further assessment of these hub genes and their connections can identify potentially important genes and biological pathways affected in disease. Such techniques have already been applied to dementia (Miller JA et al., 2008; Miller JA et al., 2010), schizophrenia (Torkamani et al., 2010), and ALS (Saris et al., 2009). These early systems biology studies illustrate the versatility of such an approach and the potential impact these studies can have on research into complex disease pathogenesis.
Environmental Contributions to Neurogenetic Disease
Although this chapter has principally dealt with the molecular aspect of neurogenetic disease, the contributions of the environment cannot be overlooked, particularly for complex genetic disease. Aside from perhaps the few Mendelian disorders with complete penetrance, all genetic disorders are likely either influenced directly by environmental factors or indirectly by the influence of the environment on other aspects of the patient’s genetic background. Despite this, we still know very little regarding the precise role of the environment in the development of neurogenetic disease, and this is therefore an important area requiring further study (Reis and Roman, 2007). Monozygotic twin studies and animal studies have both indicated that environmental influences can affect the development/severity of Mendelian genetic disease, as well as more complex disorders, but precisely how this occurs in a genetically susceptible individual remains a mystery. Many suggestions have been postulated for various disorders, including exposures to diverse physical, chemical, or biological insults, but an overall comprehensive picture has yet to develop. For example, multiple sclerosis (MS) is a complex neurological disease that likely results from a combination of genetic susceptibility and environmental contributions (Handel et al., 2010) and is one of the most well-studied neurological disorders for environmental influence. Several environmental factors have been postulated to play a role in the development of multiple sclerosis. These include vitamin D levels, which may explain epidemiological findings that MS risk is associated with geographical location in childhood and month of birth; exposure to Epstein-Barr virus, which is associated with increased MS risk if it occurs after the age of 15 years; and smoking, which appears to increase MS risk and can worsen established disease course (Handel et al., 2010). If such environmental influences could be linked to specific molecular and/or cellular events that may trigger disease in genetically susceptible individuals, it would have a dramatic impact on our understanding of disease pathogenesis, our treatment of established patients, and our recommended preventive strategies to reduce disease. The influx of new genetic information identifying risk factors for complex disease is expected to stimulate research into the impact of the environment on these variants (Traynor, 2009), ideally translating into improvements in our understanding of the environmental effects on neurogenetic disease.
Genetics and the Paradox of Disease Definition
In Mendelian disorders, X-linked adrenoleukodystrophy is a prime example of this paradox. In a single family, all with the same mutation, neurological phenotypes may range from an inflammatory cerebral demyelination to a noninflammatory distal axonopathy to a behavioral phenotype similar to attention deficit hyperactive disorder or autism spectrum disorder (Moser et al., 2005), despite all family members carrying the identical genetic diagnosis. With regard to complex disease, frontotemporal dementia spectrum disorders provide another salient example, as families with the same mutations can have vastly different clinical features ranging from purely psychiatric to motor neuron disease, parkinsonism, cortical basal degeneration, progressive supranuclear palsy, or dementia, either singly or in combination (van Swieten and Heutink, 2008). A similar scenario can be observed in epilepsy, where broad seizure phenotypes are seen in some familial forms of epilepsy (Helbig et al., 2008). Conversely, identification of Mendelian mutations can lead to a broadening of disease definition, as has been the case in Friedreich ataxia, where adults with a distinct late-onset phenotype are now frequently identified (Bhidayasiri et al., 2005), or in adult polyglucosan body disease, a progressive myeloneuropathy discovered to be the adult form of glycogen storage disease type IV, which can lead to fatal liver complications in children (Lossos et al., 2009). What is further remarkable is that genetic findings in certain Mendelian forms of PD question the notion of pathology as the gold standard. Here, certain families with mutations in the LRRK2 gene lack Lewy body pathology, yet have clear dopamine-responsive PD (Zimprich et al., 2004). This raises the question as to what is the gold standard, as the absence of Lewy bodies would not be consistent with a pathological PD diagnosis. Seen from this perspective, it is clear that neither pathology, genetic findings, nor clinical phenotypes can be interpreted in isolation, and it is the combination of these characteristics that define a disease. As we gather more genetic information about neurological disorders in the coming years, our definitions of these diseases will certainly expand and change. Identifying disease-causing mutations and/or establishing a genetic risk profile will provide further knowledge regarding disease etiology, with implications for counseling, further diagnostic workup, and eventually for treatment—described in greater detail next.
Clinical Approach to the Patient with Suspected Neurogenetic Disease
Evaluation and Diagnosis
Evaluation and diagnosis benefit from the arsenal of genetic testing available for single gene disorders and for genomic variation. Many commercial laboratories offer testing for Mendelian disease genes, and in some settings, genetic testing has become as routine as other common blood tests. However, because genetic testing carries additional implications for a patient and their family, particularly with regard to heritability of disease, it is important that it be used appropriately and that patients be fully educated prior to such testing. Important points to consider for genetic testing are summarized in Table 40.8. Although how the testing is incorporated into a clinical evaluation strategy will vary by disease, a general principle is that most genetic disease is diagnosed clinically via a thorough history (including family history) and physical examination. A complete evaluation for nongenetic causes should be performed as appropriate prior to any genetic testing so that possible treatments can be initiated in a timely manner. Genetic testing should only be used to confirm a clinical suspicion, not for screening purposes, because currently this is low yield and not cost-effective in the majority of cases. Specialist referral to a tertiary center is appropriate for all cases where a diagnostically useful clinical phenotype cannot be established. Genetic counseling (see later) should be provided, either by a physician or a licensed genetic counselor, prior to testing to ensure that patients understand the nature of the test and the possible results. When testing is ordered, it should be based on phenotype and supported by mode of inheritance if this can be determined. Testing of an asymptomatic minor is never indicated for a genetic disease where there is no treatment or cure. Knowledge of the disease status without chance for treatment may have many negative consequences.
Table 40.8 The Neurogenetic Evaluation and the Clinical Utilization of Genetic Testing
| Establish the phenotype | All patients in whom a genetic diagnosis is suspected require a thorough physical examination and clinical history including a detailed family history. Differential diagnosis is established based on phenotype. Genetic etiologies should be considered in all cases where there is a positive family history of disease. |
| Rule out non-genetic etiologies | With the exception of suspected genetic diseases with known disease-modifying treatments, patients should be fully evaluated for non-genetic causes of disease prior to the initiation of genetic testing, as these are generally more amenable to treatment. |
| Order genetic testing based on phenotype | Genetic testing should not be used as a screening tool. Physicians suspecting a hereditary disorder but unable to arrive at a diagnostically useful clinical phenotype should refer these patients for further evaluation at a tertiary center specializing in such cases. |
| Use disease biomarkers when available | Cost management should be maintained through the use of biomarker testing whenever possible, with genetic testing as the confirmatory step in diagnosis to obtain the genotype for clinical trials, research studies, and genotype-phenotype clinical correlations. |
| Avoid genetic panels | Disease- or inheritance-based multigene panels should be discouraged in routine clinical practice, as these are not a cost-effective use of patient resources. There may, however, be a role for small focused panels in specific disorders with heterogeneous phenotypes. |
| Provide genetic counseling | Genetic counseling (by a physician, geneticist, or genetic counselor) should be provided to all patients for whom genetic testing is recommended. Follow-up counseling should be provided to all patients with a positive gene test and offered to family members who may be at risk or disease carriers. Any and all ethical concerns should be fully addressed. |
| Utilize new technology in challenging cases | Whole genome and/or exome sequencing, when clinically available, could potentially be an appropriate consideration for patients with suspected genetic disease and complete negative genetic and non-genetic evaluations. |
The types of single-gene testing available vary per laboratory and gene (Table 40.9). The most comprehensive (and expensive) testing type is full gene sequencing, where all coding regions, as well as approximately 50 bases in each intron/exon junction, are sequenced for the presence of mutation. This will detect all coding point mutations and splice-site mutations as well as small insertions and deletions but will miss more detailed structural variation. Importantly, novel coding mutations can be detected in this way. Targeted sequence analysis (also called select exon testing) consists of specific sequencing reactions designed to only detect one or a few previously identified mutations. This will not detect any sequence variations outside of the limited region of the gene being searched. For repeat disorders, there are specific tests to identify the relevant expansions using either polymerase chain reaction (PCR) or Southern blotting, a hybridization-based DNA sizing technique. Larger deletions or duplications (e.g., copy number variations) can be detected by quantitative PCR methods or by comparative genomic hybridization. It is important to be aware of the type of testing being ordered; in some cases, such as select exon testing, a negative result does not exclude mutations elsewhere in the gene being tested. Interpretation of these genetic results may be straightforward, for example, if no mutations are present or if known pathogenic changes are found. In contrast, interpretation may be complicated if novel sequence variants of unknown pathological significance are identified. Inconclusive results may require interpretation by a specialist and/or further testing to determine the likelihood of pathogenicity.
Table 40.9 Types of Genetic Testing
| Type of Test | Sequence Variant(s) Identified | Sequence Variant(s) Missed or Not Accurately Determined |
|---|---|---|
| Gene sequencing | Point mutations* Frameshifts Splicing mutations† Polymorphisms |
Noncoding variants‡ Copy number variations§ Repeat expansions¶ |
| Select exon sequencing (Targeted mutation analysis) | Known predefined variants Target region only¶,# Point mutations* Frameshifts Splicing mutations† Polymorphisms |
Variants outside target region# Point mutations* Frameshifts Splicing mutations† Polymorphisms Noncoding variants‡ Copy number variations§ Repeat expansions¶ |
| Repeat expansion testing‖ (Targeted mutation analysis) | Repeat expansion in the specific gene tested | Point mutations* Frameshifts Splicing mutations† Polymorphisms Noncoding variants‡ Copy number variations§ |
| Gene copy number variation (Deletion/duplication testing) | Copy number variation§ of gene tested | Point mutations* Frameshifts Splicing mutations† Polymorphisms Noncoding variants‡ Repeat expansions¶ |
| Chromosomal microarray analysis** (Comparative genomic hybridization) | Genome-wide copy number variations†† | Point mutations* Frameshifts Splicing mutations† Polymorphisms Noncoding variants‡ Repeat expansions¶ |
| Whole exome sequencing**,‡‡ | Point mutations* Frameshifts Splicing mutations† Polymorphisms |
Noncoding variants‡ Copy number variations§ Repeat expansions¶ |
| Whole genome sequencing**,‡‡ | Point mutations* Frameshifts Splicing mutations† Polymorphisms Noncoding variants‡ Copy number variations§ |
Repeat expansions¶ |
* Includes missense, nonsense, and silent mutations.
† Includes only those involving splice sites and exonic splicing regulatory sequences.
‡ Includes promoter mutations and noncoding splicing regulatory elements.
§ Arbitrarily defined here as any deletion/duplication/insertion larger than detectable by Sanger sequencing.
‖ Targeted mutation analysis using either polymerase chain reaction (PCR) and/or Southern blot is preferred, as sequencing may be inaccurate due to the large size of many repeat regions.
¶ Only detectable if sequencing is performed as opposed to individual mutation-specific detection methods.
# Size and number of region(s) targeted varies per individual test.
** Genome-wide testing method.
†† Minimum size of CNVs detected and density of genomic coverage varies per test.
‡‡ Not yet available clinically.
Common diseases must be approached in a different manner, because detailed phenotype alone cannot always predict the mutation to test for, particularly when assessing genomic variation. Still, the goal remains to develop strategies incorporating known genetic information into a systematic protocol designed to maximize diagnostic capability while minimizing cost and unnecessary testing (Lintas and Persico, 2009). Tests such as chromosomal microarray analysis are clinically available to search genome-wide for disease-causing CNVs and are recommended for sporadic causes in disorders such as intellectual disability or autism where CNVs have been found responsible for a reasonable percentage of disease (Geschwind and Spence, 2008; Miller DT et al., 2010). Use of such testing in sporadic adult-onset disease is less clear, so the physician is advised to refer to current published guidelines for the disease in question before ordering. For more specific phenotypes, other available tests include those assessing for CNVs (often called simply deletions/duplications) involving individual genes or specific chromosomal regions. Overall, interpretation of CNV results can be challenging, particularly if the CNV was previously unreported. Here, the parents will often need to be evaluated to determine whether the CNV in question is inherited or de novo. As already discussed for DNA sequence changes, such findings may require interpretation by a specialist and/or further testing to determine the likelihood of pathogenicity.
Genetic Counseling
Establishing a precise genetic diagnosis will definitively establish the means of inheritance of a disorder and is extremely useful in genetic counseling and family planning, particularly for disorders that show incomplete penetrance. However, unlike other tests typically ordered by physicians, a positive diagnosis carries implications not only for individual patients but for the entire family. Genetic counseling, therefore, should be provided in all cases where genetic testing is recommended, either by an experienced neurologist, a geneticist, or a licensed genetic counselor. Follow-up counseling should also be provided to all patients with a positive test result and, in many cases, offered to other family members who may be at risk for disease or as carriers. Physicians must be aware of the various ethical implications involved in such testing (Ensenauer et al., 2005). One area of particular importance in this regard involves considerations of genetic testing in asymptomatic individuals, especially minors. This stems in part from concerns that have been raised regarding risks of depression and suicide in asymptomatic individuals diagnosed with fatal genetic disease, although this is not well established, and further study will be important for determining best practices. For minors, standard practice dictates that unless there is disease-modifying therapy available for them, they should not be tested if asymptomatic until they reach an age to consent to such testing and are properly counseled as to the implications. Counseling regarding prenatal testing and assisted reproduction are other topics of relevance to patients of reproductive age. Current reproductive medicine techniques such as in vitro fertilization and preimplantation genetic testing, by assuring that offspring will not harbor the mutation in question, can aid couples concerned about the risk for passing on inherited conditions. Other ethical considerations may also apply, depending on the disease and specific family/patient circumstances.
Prognosis and Treatment
Although the majority of genetic diseases are not curable, therapies do exist for many of them. Defining the genetic etiology of a patient’s disease allows for utilization of the published literature on symptomatic treatments and pharmacotherapy that may benefit a specific condition. Phenylketonuria is an excellent example of this, since dietary restriction of phenylalanine initiated soon after birth will prevent cognitive impairment and enable virtually normal development (Burgard et al., 1999). More importantly, new clinical trials are being developed frequently and can be offered to patients with an established diagnosis. Many disease-based patient registries exist to facilitate this.
The ultimate goal of translational neuroscience is to utilize advances in our understanding of disease at the molecular level to aid in the treatment of patients in the clinic. Recent new treatments, which take advantage of the molecular aspects of these disorders, show promise in the clinic and the laboratory. Such treatments include enzyme replacement therapy for metabolic disorders such as the severe fatal glycogen storage disorder Pompe disease, where use of recombinant acid α-glucosidase in 18 infants prior to 6 months of age enabled all to live to the age of 18 months, a 99% reduction in death, as well as reduced their risk of death or invasive ventilation by 92% compared to historical controls (Kishnani et al., 2007). Work in animal models has suggested potential new pharmacological treatments, such as a recent research study which demonstrated that the use of histone-deacetylase inhibitors can unsilence expanded frataxin alleles in a Friedreich ataxia mouse model, restoring wild-type gene expression levels and reversing cellular transcription changes associated with frataxin deficiency (Rai et al., 2008). Targeted molecules have been designed to correct specific disease-causing biological defects, as shown by recent work where antisense oligonucleotides were used to block mutations that promote splicing defects in the ataxia-telangiectasia mutated (ATM) gene in cell lines from patients with ataxia-telangiectasia, leading to restoration of functional protein (Du et al., 2007). Such newer techniques may markedly exceed the therapeutic benefit of current options, such as in Duchenne muscular dystrophy where patients can expect only moderate short-term benefit (up to 2 years) from the gold standard, glucocorticosteroid treatment (Manzur et al., 2008; Wood et al., 2010). Newer molecular strategies such as dystrophin splice-modulation, which promotes exon skipping via antisense oligonucleotides to bypass point mutations or frameshifts, may potentially resolve the primary defect and has shown promising results in early clinical trials (Wood et al., 2010). Novel treatments aimed at genetic modification of disease are also in development, as was seen in a recent study where investigators used RNA interference techniques to specifically degrade and thus silence the disease allele in a rat model of SCA type 3, resulting in a reduction in neuropathological changes in the brain (Alves et al., 2008). New technologies such as next-generation sequencing and the use of systems biology approaches to disease are expected to lead to additional new innovations. With these advances, the future of clinical neurogenetics is full of promise and stands poised to answer the challenge stated most eloquently by Bernard Baruch (1870-1965): “There are no such things as incurables; there are only things for which [medicine] has not found a cure.”
Abrahams B.S., Geschwind D.H. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341-355.
Alarcon M., Abrahams B.S., Stone J.L., et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82(1):150-159.
Alberts B., Johnson A., Lewis J., et al. Molecular Biology of the Cell, fifth ed. New York: Garland Science; 2008.
Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322(5903):881-888.
Alves S., Nascimento-Ferreira I., Auregan G., et al. Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS One. 2008;3(10):e3341.
Babushok D.V., Ostertag E.M., Kazazian H.H.Jr. Current topics in genome evolution: molecular mechanisms of new gene formation. Cell Mol Life Sci. 2007;64(5):542-554.
Bakkaloglu B., O’Roak B.J., Louvi A., et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. 2008;82(1):165-173.
Baranzini S.E., Mudge J., van Velkinburgh J.C., et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464(7293):1351-1356.
Beck M. Therapy for lysosomal storage disorders. IUBMB Life. 2010;62(1):33-40.
Beckmann J.S., Estivill X., Antonarakis S.E. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8(8):639-646.
Belin A.C., Westerlund M. Parkinson’s disease: a genetic perspective. FEBS J. 2008;275(7):1377-1383.
Bhidayasiri R., Perlman S.L., Pulst S.M., et al. Late-onset Friedreich ataxia: phenotypic analysis, magnetic resonance imaging findings, and review of the literature. Arch Neurol. 2005;62(12):1865-1869.
Bucan M., Abrahams B.S., Wang K., et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5(6):e1000536.
Burgard P., Bremer H.J., Buhrdel P., et al. Rationale for the German recommendations for phenylalanine level control in phenylketonuria, 1997. Eur J Pediatr. 1999;158(1):46-54.
Cantor R.M., Geschwind D.H. Schizophrenia: genome, interrupted. Neuron. 2008;58(2):165-167.
Caylak E. The genetics of sleep disorders in humans: narcolepsy, restless legs syndrome, and obstructive sleep apnea syndrome. Am J Med Genet A. 2009;149A(11):2612-2626.
Choi M., Scholl U.I., Ji W., et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096-19101.
Cirulli E.T., Goldstein D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415-425.
Conrad D.F., Pinto D., Redon R., et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704-712.
Coon K.D., Myers A.J., Craig D.W., et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry. 2007;68(4):613-618.
Coppola G., Geschwind D.H. Technology insight: querying the genome with microarrays–progress and hope for neurological disease. Nat Clin Pract Neurol. 2006;2(3):147-158.
D’Souza I., Schellenberg G.D. Regulation of tau isoform expression and dementia. Biochim Biophys Acta. 2005;1739(2-3):104-115.
DiMauro S., Hirano M. Pathogenesis and treatment of mitochondrial disorders. Adv Exp Med Biol. 2009;652:139-170.
Du L., Pollard J.M., Gatti R.A. Correction of prototypic ATM splicing mutations and aberrant ATM function with antisense morpholino oligonucleotides. Proc Natl Acad Sci U S A. 2007;104(14):6007-6012.
Edwards T.L., Scott W.K., Almonte C., et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74(2):97-109.
Ensenauer R.E., Michels V.V., Reinke S.S. Genetic testing: practical, ethical, and counseling considerations. Mayo Clin Proc. 2005;80(1):63-73.
Fanciulli M., Petretto E., Aitman T.J. Gene copy number variation and common human disease. Clin Genet. 2010;77(3):201-213.
Fang F., Kamel F., Lichtenstein P., et al. Familial aggregation of amyotrophic lateral sclerosis. Ann Neurol. 2009;66(1):94-99.
Filipowicz W., Bhattacharyya S.N., Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9(2):102-114.
Flossmann E., Schulz U.G., Rothwell P.M. Systematic review of methods and results of studies of the genetic epidemiology of ischemic stroke. Stroke. 2004;35(1):212-227.
Flynt A.S., Lai E.C. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. 2008;9(11):831-842.
Freitag C.M. The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007;12(1):2-22.
Garcia-Arocena D., Hagerman P.J. Advances in understanding the molecular basis of FXTAS. Hum Mol Genet. 2010;19(R1):R83-R89.
Gatz M., Reynolds C.A., Fratiglioni L., et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168-174.
Geschwind D., Spence S. Genetics of autism. Continuum. 2008;14(2):49-64.
Geschwind D.H. DNA microarrays: translation of the genome from laboratory to clinic. Lancet Neurol. 2003;2(5):275-282.
Geschwind D.H., Boone K.B., Miller B.L., et al. Neurobehavioral phenotype of Klinefelter syndrome. Ment Retard Dev Disabil Res Rev. 2000;6(2):107-116.
Geschwind D.H., Konopka G. Neuroscience in the era of functional genomics and systems biology. Nature. 2009;461(7266):908-915.
Gonzales M.L., LaSalle J.M. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr Psychiatry Rep. 2010;12(2):127-134.
Griffiths A.J.F., Gelbart W.M., Lewontin R.C., et al. Modern Genetic Analysis: Integrating Genes and Genomes, second ed. New York: W. H. Freeman & Co.; 2002.
Handel A.E., Giovannoni G., Ebers G.C., et al. Environmental factors and their timing in adult-onset multiple sclerosis. Nat Rev Neurol. 2010;6(3):156-166.
Hawkes C.H., Macgregor A.J. Twin studies and the heritability of MS., a conclusion. Mult Scler. 2009;15(6):661-667.
Harold D., Abraham R., Hollingworth P., et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088-1093.
Hedges D.J., Burges D., Powell E., et al. Exome sequencing of a multigenerational human pedigree. PLoS One. 2009;4(12):e8232.
Helbig I., Scheffer I.E., Mulley J.C., et al. Navigating the channels and beyond: unravelling the genetics of the epilepsies. Lancet Neurol. 2008;7(3):231-245.
Holmes M.V., Shah T., Vickery C., et al. Fulfilling the promise of personalized medicine? Systematic review and field synopsis of pharmacogenetic studies. PLoS One. 2009;4(12):e7960.
Hudson G., Chinnery P.F. Mitochondrial DNA polymerase-gamma and human disease. Hum Mol Genet. 2006;15:R244-R252. Spec No 2
Ikram M.A., Seshadri S., Bis J.C., et al. Genomewide association studies of stroke. N Engl J Med. 2009;360(17):1718-1728.
Joshita S., Umemura T., Yoshizawa K., et al. A2BP1 as a novel susceptible gene for primary biliary cirrhosis in Japanese patients. Hum Immunol. 2010;71(5):520-524.
Kishnani P.S., Corzo D., Nicolino M., et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68(2):99-109.
Kjeldsen M.J., Corey L.A., Christensen K., et al. Epileptic seizures and syndromes in twins: the importance of genetic factors. Epilepsy Res. 2003;55(1-2):137-146.
Knopman D.S., DeKosky S.T., Cummings J.L., et al. Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56(9):1143-1153.
Konopka G., Bomar J.M., Winden K., et al. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature. 2009;462(7270):213-217.
Koolen D.A., Pfundt R., de Leeuw N., et al. Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Hum Mutat. 2009;30(3):283-292.
Kotzot D. Complex and segmental uniparental disomy updated. J Med Genet. 2008;45(9):545-556.
Kovaleva N.V., Shaffer L.G. Under-ascertainment of mosaic carriers of balanced homologous acrocentric translocations and isochromosomes. Am J Med Genet A. 2003;121A(2):180-187.
Lai C.S., Fisher S.E., Hurst J.A., et al. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413(6855):519-523.
Lalande M., Calciano M.A. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64(7-8):947-960.
Lambert J.C., Heath S., Even G., et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094-1099.
Lee C., Scherer S.W. The clinical context of copy number variation in the human genome. Expert Rev Mol Med. 2010;12:e8.
Licatalosi D.D., Darnell R.B. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010;11(1):75-87.
Link E., Parish S., Armitage J., et al. SLCO1B1 variants and statin-induced myopathy–a genomewide study. N Engl J Med. 2008;359(8):789-799.
Lintas C., Persico A.M. Autistic phenotypes and genetic testing: state-of-the-art for the clinical geneticist. J Med Genet. 2009;46(1):1-8.
Lodish H., Berk A., Kaiser C.A., et al. Molecular Cell Biology, sixth ed. New York: W. H. Freeman; 2008.
Lossos A., Klein C.J., McEvoy K.M., et al. A 63-year-old woman with urinary incontinence and progressive gait disorder. Neurology. 2009;72(18):1607-1613.
Lupski J.R. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39(Suppl. 7):S43-S47.
Lupski J.R., Reid J.G., Gonzaga-Jauregui C., et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362(13):1181-1191.
MacDermot K.D., Bonora E., Sykes N., et al. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet. 2005;76(6):1074-1080.
Manolio T.A., Collins F.S., Cox N.J., et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753.
Manzur A.Y., Kuntzer T., Pike M., et al. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;1:CD003725.
Marshall C.R., Noor A., Vincent J.B., et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477-488.
Martin C.L., Duvall J.A., Ilkin Y., et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet B Neuropsychiatr Genet. 2007;144(7):869-876.
Matarin M., Singleton A., Hardy J., et al. The genetics of ischaemic stroke. J Intern Med. 2010;267(2):139-155.
McCarthy M.I., Abecasis G.R., Cardon L.R., et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356-369.
Meschia J.F. Pharmacogenetics and stroke. Stroke. 2009;40(11):3641-3645.
Metzker M.L. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11(1):31-46.
Miller D.T., Adam M.P., Aradhya S., et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749-764.
Miller J.A., Horvath S., Geschwind D.H. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107(28):12698-12703.
Miller J.A., Oldham M.C., Geschwind D.H. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J Neurosci. 2008;28(6):1410-1420.
Morrow E.M., Yoo S.Y., Flavell S.W., et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321(5886):218-223.
Moser H.W., Raymond G.V., Dubey P. Adrenoleukodystrophy: new approaches to a neurodegenerative disease. JAMA. 2005;294(24):3131-3134.
Mullen S.A., Crompton D.E., Carney P.W., et al. A neurologist’s guide to genome-wide association studies. Neurology. 2009;72(6):558-565.
Nakamori M., Thornton C. Epigenetic changes and non-coding expanded repeats. Neurobiol Dis. 2010;39(1):21-27.
Ng S.B., Buckingham K.J., Lee C., et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30-35.
Ng S.B., Turner E.H., Robertson P.D., et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272-276.
Online Mendelian Inheritance in Man, OMIM (TM). McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD). Available at http://www.ncbi.nlm.nih.gov/omim/, 2010. Accessed July
Oostra B.A., Willemsen R. FMR1: a gene with three faces. Biochim Biophys Acta. 2009;1790(6):467-477.
Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575-621.
Pan Q., Shai O., Lee L.J., et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413-1415.
Penagarikano O., Mulle J.G., Warren S.T. The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet. 2007;8:109-129.
Pertea M., Salzberg S.L. Between a chicken and a grape: estimating the number of human genes. Genome Biol. 2010;11(5):206.
Pinto D., Pagnamenta A.T., Klei L., et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368-372.
Plomin R., Haworth C.M., Davis O.S. Common disorders are quantitative traits. Nat Rev Genet. 2009;10(12):872-878.
Pulst S.M. Neurogenetics: single gene disorders. J Neurol Neurosurg Psychiatry. 2003;74(12):1608-1614.
Purcell S.M., Wray N.R., Stone J.L., et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748-752.
Rai M., Soragni E., Jenssen K., et al. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One. 2008;3(4):e1958.
Redon R., Ishikawa S., Fitch K.R., et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444-454.
Reis J., Roman G.C. Environmental neurology: a promising new field of practice and research. J Neurol Sci. 2007;262(1-2):3-6.
Rohrer J.D., Guerreiro R., Vandrovcova J., et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73(18):1451-1456.
Rosenfeld J.A., Malhotra A.K., Lencz T. Novel multi-nucleotide polymorphisms in the human genome characterized by whole genome and exome sequencing. Nucleic Acids Res. 2010.
Sanderson S., Emery J., Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med. 2005;7(2):97-104.
Saris C.G., Horvath S., van Vught P.W., et al. Weighted gene co-expression network analysis of the peripheral blood from amyotrophic lateral sclerosis patients. BMC Genomics. 2009;10:405.
Satake W., Nakabayashi Y., Mizuta I., et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303-1307.
Schork N.J., Murray S.S., Frazer K.A., et al. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19(3):212-219.
Sebat J., Lakshmi B., Malhotra D., et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445-449.
Seshadri S., Fitzpatrick A.L., Ikram M.A., et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303(18):1832-1840.
Shchelochkov O.A., Cheung S.W., Lupski J.R. Genomic and clinical characteristics of microduplications in chromosome 17. Am J Med Genet A. 2010;152A(5):1101-1110.
Simon-Sanchez J., Schulte C., Bras J.M., et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308-1312.
Simon-Sanchez J., Singleton A. Genome-wide association studies in neurological disorders. Lancet Neurol. 2008;7(11):1067-1072.
Singleton A.B., Farrer M., Johnson J., et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841.
Speicher M.R., Carter N.P. The new cytogenetics: blurring the boundaries with molecular biology. Nat Rev Genet. 2005;6(10):782-792.
Spencer H.G. Effects of genomic imprinting on quantitative traits. Genetica. 2009;136(2):285-293.
Stankiewicz P., Lupski J.R. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437-455.
Stefansson H., Rujescu D., Cichon S., et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455(7210):232-236.
Strachan T., Read A.P. Human Molecular Genetics, third ed. London: Garland Science; 2003.
Strauss K.A., Puffenberger E.G., Huentelman M.J., et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354(13):1370-1377.
Swen J.J., Huizinga T.W., Gelderblom H., et al. Translating pharmacogenomics: challenges on the road to the clinic. PLoS Med. 2007;4(8):e209.
Torkamani A., Dean B., Schork N.J., et al. Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Res. 2010;20(4):403-412.
Traynor B.J. The era of genomic epidemiology. Neuroepidemiology. 2009;33(3):276-279.
Underwood J.G., Boutz P.L., Dougherty J.D., et al. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol. 2005;25(22):10005-10016.
Van Deerlin V.M., Sleiman P.M., Martinez-Lage M., et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42(3):234-239.
van Es M.A., Veldink J.H., Saris C.G., et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41(10):1083-1087.
van Swieten J.C., Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 2008;7(10):965-974.
Vaughan J.R., Davis M.B., Wood N.W. Genetics of parkinsonism: a review. Ann Hum Genet. 2001;65(Pt 2):111-126.
Vernes S.C., Newbury D.F., Abrahams B.S., et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008;359(22):2337-2345.
Wain L.V., Armour J.A., Tobin M.D. Genomic copy number variation, human health, and disease. Lancet. 2009;374(9686):340-350.
Walsh T., McClellan J.M., McCarthy S.E., et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539-543.
Ward A.J., Cooper T.A. The pathobiology of splicing. J Pathol. 2010;220(2):152-163.
Weinberg M.S., Wood M.J. Short non-coding RNA biology and neurodegenerative disorders: novel disease targets and therapeutics. Hum Mol Genet. 2009;18(R1):R27-R39.
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661-678.
Wheeler T.M., Sobczak K., Lueck J.D., et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325(5938):336-339.
Wood M.J., Gait M.J., Yin H. RNA-targeted splice-correction therapy for neuromuscular disease. Brain. 2010;133(Pt 4):957-972.
Yeo G.W., Coufal N.G., Liang T.Y., et al. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol. 2009;16(2):130-137.
Yngvadottir B., Xue Y., Searle S., et al. A genome-wide survey of the prevalence and evolutionary forces acting on human nonsense SNPs. Am J Hum Genet. 2009;84(2):224-234.
Zeviani M., Carelli V. Mitochondrial disorders. Curr Opin Neurol. 2007;20(5):564-571.
Zhai G., van Meurs J.B., Livshits G., et al. A genome-wide association study suggests that a locus within the ataxin 2 binding protein 1 gene is associated with hand osteoarthritis: the Treat-OA consortium. J Med Genet. 2009;46(9):614-616.
Zhang C., Zhang Z., Castle J., et al. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008;22(18):2550-2563.
Zhang F., Gu W., Hurles M.E., et al. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451-481.
Zimprich A., Biskup S., Leitner P., et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601-607.
Zlotogora J. Parents of children with autosomal recessive diseases are not always carriers of the respective mutant alleles. Hum Genet. 2004;114(6):521-526.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 40 Clinical Neurogenetics
Genetics in Clinical Neurology
Gene Expression, Diversity, and Regulation
Types of Genetic Variation and Mutations
Chromosomal Analysis and Abnormalities
Disorders of Mendelian Inheritance
Mendelian Disease Gene Identification by Linkage Analysis and Chromosome Mapping
Non-Mendelian Patterns of Inheritance
Common Neurological Disorders and Complex Disease Genetics
Whole Genome/Exome Sequencing in Disease Gene Discovery
Future Role of Systems Biology in Neurogenetic Disease
Environmental Contributions to Neurogenetic Disease
Genetics and the Paradox of Disease Definition
Clinical Approach to the Patient with Suspected Neurogenetic Disease
Genetics in Clinical Neurology
Since the discovery of the structure of deoxyribonucleic acid (DNA) and the elucidation of the genetic mechanisms of heredity, clinical neurology has benefited from advances in genetics and neuroscience. This clinically relevant basic research has permitted dissection of the cellular machinery supporting the function of the brain and its connections while establishing causal relationships between such dysfunction, human genetic variation, and various neurological diseases. In the modern practice of neurology, the use of genetics has become widespread, and neurologists are confronted daily with data from an ever-increasing catalogue of genetic studies relating to conditions such as developmental disorders, dementia, ataxia, neuropathy, and epilepsy, to name but a few. The use of genetic information in the clinical evaluation of neurological disease has expanded dramatically over the past decade. More efficient techniques for discovering disease genes has led to a greater availability of genetic testing in the clinic. Approximately one-third of pediatric neurology hospital admissions are related to a genetic diagnosis, and there are many dozens of genetic tests available to the practicing neurologist, including several related to common diseases. This number will surely continue to increase rapidly (Fig. 40.1).
(Data from GeneTests: Medical Genetics Information Resource. Available at http://www.ncbi.nlm.nih.gov/sites/GeneTests/.)
As neuroscience and genetic research have progressed, we have been led to a deeper understanding of the sources and nature of human genetic variation and its relationship to clinical phenotypes. In the past there has been a tendency to consider genetic traits as either present or absent, and correspondingly, patients were either healthy or diseased; this is the traditional view of Mendelian, or single gene, conditions. Although certain relatively rare neurological diseases—Friedreich ataxia or Huntington disease (HD), for example—can be traced to a single causal gene, the common forms of other diseases such as Alzheimer dementia, stroke, epilepsy, or autism usually arise from an interplay of multiple genes, each of which increases disease susceptibility and likely interacts with environmental factors. Subsequently, the realm of the “sporadic” and the “idiopathic” has been challenged by the identification of genetic susceptibility factors, which has sparked a flurry of investigation into a variety of genes and genetic markers that confer a risk of illness yet are not wholly causative. Disease status may lie on the end of a continuum of individual variation and thus can be considered a quantitative rather than purely qualitative trait (Plomin et al., 2009). So, rather than using what might be considered an arbitrary cutoff point, such as a specific number of senile plaques or neuritic tangles that define affected or unaffected patients, one might instead think in terms of a continuum of pathology that relates to different levels of burden or susceptibility.
Gene Expression, Diversity, and Regulation
The basic principles of molecular genetics are outlined in Fig. 40.2 and Table 40.1, and more detailed descriptions can be found elsewhere (Alberts et al., 2008; Griffiths et al., 2002; Lodish et al., 2008; Strachan and Read, 2003). To briefly summarize, deoxyribonucleic acid (DNA), found in the nucleus of all cells, comprises the raw material from which heritable information is transferred among individuals, with the simplest heritable unit being the gene. DNA is composed of a series of individual nucleotides, all of which contain an identical pentose (2′-deoxyribose)-phosphate backbone but differ at an attached base that can be either adenine (A), guanine (G), thymine (T), or cytosine (C). A and G are purine bases and pair with the pyrimidine bases T and C, respectively, to form a double-stranded helical structure which allows for semiconservative bidirectional replication, the means by which DNA is copied in a precise and efficient manner. In total, there are approximately 3.2 billion base pairs in human DNA. By convention, a DNA sequence is described by listing the bases as they are expressed from the 5′ to 3′ direction along the pentose backbone (e.g., 5′-ATGCAT-3′…etc.), as this is the order in which it is typically used by the cellular machinery, also called the sense strand (compare to RNA, later). The opposite paired, or antisense, strand is arranged antiparallel (3′ to 5′) and can also be referred to when discussing sequence; however, by convention this is generally not done unless that strand is also transcribed into RNA.
| Allele | Alternate forms of a locus (gene) |
| Anticipation | Earlier onset and/or worsening severity of disease in successive generations |
| Antisense | Nucleic acid sequence complementary to mRNA |
| Chromosome | Organizational unit of the genome consisting of a linear arrangement of genes |
| Cis-acting | A regulatory nucleotide sequence present on the molecule being regulated |
| Codon | A three-nucleotide sequence representing a single amino acid |
| Complex disease | Disease exhibiting non-Mendelian inheritance involving the interaction of multiple genes and the environment |
| De novo | A mutation newly arising in an individual and not present in either parent |
| Diploid | A genome having paired genetic information, half-normal number is haploid |
| DNA | Deoxyribonucleic acid; used for storage, replication, and inheritance of genetic information |
| Dominant | Allele that determines phenotype when a single copy is present in an individual |
| Endophenotype | Subset of phenotypic characteristics used to group patients manifesting a given trait |
| Exome | Portion of the genome representing only the coding regions of genes |
| Exon | Segment of DNA that is expressed in at least one mature mRNA |
| Expressivity | The range of phenotypes observed with a specific disease-associated genotype |
| Frameshift | DNA mutation that adds or removes nucleotides, affecting which are grouped as codons |
| Gene | Contiguous DNA sequence that codes for a given messenger RNA and its splice variants |
| Genome | A complete set of DNA from a given individual |
| Genotype | The DNA sequence of a gene |
| Haplotype | A group of alleles on the same chromosome close enough to be inherited together |
| Hemizygous | Genes having only a single allele in an individual, such as the X chromosome in males |
| Heteroplasmy | A mixture of multiple mitochondrial genomes in a given individual |
| Heterozygous | Genes having two distinct alleles in an individual at a given locus |
| Homozygous | Genes having two identical alleles in an individual at a given locus |
| Intron | Segment of DNA between exons that is transcribed into RNA but removed by splicing |
| Kilobase | 1000 bases or base-pairs |
| Linkage disequilibrium | The co-occurrence of two alleles more frequently than expected by random chance, suggesting they are in close proximity to one another |
| Locus | Location of a DNA sequence (or a gene) on a chromosome or within the genome |
| Lyonization | The process of random inactivation of one of the pair of X chromosomes in females |
| Marker | Sequence of DNA used to identify a gene or a locus |
| Megabase | 1,000,000 bases or base-pairs |
| Meiosis | Process of cellular division that produces gametes containing a haploid amount of DNA |
| Mendelian | Obeying standard single-gene patterns of inheritance (e.g., recessive or dominant) |
| Microarray | A glass or plastic support (e.g., slide or chip) to which large numbers of DNA molecules can be attached for use in high-throughput genetic analysis |
| Missense | DNA mutation that changes a given codon to represent a different amino acid |
| Mitosis | Process of cellular division during which DNA is replicated |
| Nonsense | DNA mutation that changes a given codon into a translation termination signal |
| Penetrance | The likelihood of a disease-associated genotype to express a specific disease phenotype |
| Phenotype | The clinical manifestations of a given genotype |
| Polymorphism | Sequence variation among individuals, typically not considered to be pathogenic |
| Probe | DNA sequence used for identifying a specific gene or allele |
| Promoter | DNA sequences that regulate transcription of a given gene |
| Protein | Functional cellular macromolecules encoded by a gene |
| Recessive | Allele that determines phenotype only when two copies are present in an individual |
| Relative risk | The ratio of the chance of disease in individuals with a specific genetic susceptibility factor over the chance of disease in those without it |
| Resequencing | A method of identifying clinically relevant genetic variation in a candidate gene of interest by comparing the sequence in individuals with disease to a reference sequence |
| RNA | Ribonucleic acid; expressed form of a gene, called messenger or mRNA if protein coding |
| Sense | Nucleic acid sequence corresponding to mRNA |
| Silent | DNA mutation that changes a given codon but does not alter the corresponding amino acid |
| SNP | Single nucleotide polymorphism |
| Splicing | RNA processing mechanism where introns are removed and exons joined to create mRNA; in alternative splicing, exons are utilized in a regulated manner within a cell or tissue |
| Trans-acting | A regulatory protein that acts on a molecule other than that which expressed it |
| Transcription | Cellular process where DNA sequence is used as template for RNA synthesis |
| Transcriptome | The complete set of RNA transcripts produced by a cell, tissue, or individual |
| Translation | Cellular process where mRNA sequence is converted to protein |
The expression of a gene is tightly and coordinately regulated (see Fig. 40.2), an important consideration for understanding the molecular mechanisms of disease. The typical gene contains one or more promoters, DNA sequences that allow for the binding of a cellular protein complex that includes RNA polymerase and other factors that faithfully copy the DNA in the 5′ to 3′ direction in a process known as transcription. The resulting single-stranded molecule contains a ribose sugar unit in its backbone and, thus the resulting molecule is termed ribonucleic acid, or RNA. RNA also differs from the template DNA by the incorporation of uracil (U) in place of thymine (T), as it also pairs efficiently with adenine, and thymine serves a secondary role in DNA repair that is not necessary in RNA. The sequence of the RNA matches the sense DNA strand and is therefore complementary to (and hence derived from) the antisense strand.
Transcribed coding RNA must be processed to become protein-encoding messenger RNA (mRNA), a term used to differentiate these RNAs from all other types of RNA in the cell. To become mature, RNA is stabilized by modification at the ends with a 7-methylguanosine 5′ cap and a long poly-A 3′ tail. A further critical stage in the maturation of the RNA molecule involves a rearrangement process termed RNA splicing (Fig. 40.3). This is necessary because the expressed coding sequences in DNA, called exons, of virtually every gene are discontinuous and interspersed with long stretches of generally non-conserved intervening sequences referred to as introns. This, along with other mechanisms, likely plays an evolutionary role in the development of new genes by allowing for the shuffling of functional sequences (Babushok et al., 2007). Nascent RNA molecules are recognized by the spliceosome, a protein complex that removes the introns and rejoins the exons. Not every exon is utilized at all times in every RNA derived from a single gene. Exons may be skipped or included in a regulated manner through alternative splicing, which occurs in nearly 95% of all genes to create different isoforms of that mRNA. The dynamic nature of this observation is critical to a complete understanding of cellular gene expression. DNA is essentially a storage molecule, and with few exceptions in the absence of mutagens, its sequence remains static and, aside from a few epigenetic events, is therefore limited to a genetic regulatory role as a transcriptional rheostat. Current estimates place the number of individual human genes at just over 22,000 (Pertea and Salzberg, 2010), so it is difficult to reconcile biological and clinical diversity with simple variations in expression. Alternative splicing provides a means of dramatically elevating this diversity by enabling a single gene to encode multiple proteins with a wide array of functions. Supporting this, recent analysis of RNA complexity in human tissues suggest that there are at least seven alternative splicing events per multi-exon gene, generating over 100,000 alternative splicing events (Pan et al., 2008). Because alternative splicing and other forms of RNA processing can be subject to complex layers of temporal and spatial regulation, particularly in the human brain (Licatalosi and Darnell, 2010; Ward and Cooper, 2010), it is a robust source for both biological diversity and disease-causing mutations (see Polymorphisms and Point Mutations).
DNA to RNA to Protein
The central dogma of genetics has been that DNA is transcribed into RNA that is than translated into protein—the “business” end of the process. So, following its transcription from DNA in the nucleus, mRNA is transported out of the nucleus to the cytoplasm, and possibly to a specific subcellular location depending on the mRNA, where it can be deciphered by the cell. This takes place via interaction with a complex known as the ribosome, which binds the mRNA and converts its genetic information into protein via the process of translation. The ribosome initiates translation at a pre-encoded start site and converts the mRNA sequence into protein until a designated termination site is reached. Sequence information is read in three-nucleotide groups called codons, each of which specifies an individual amino acid. With the four distinct bases, there are mathematically 64 possible codons, but these have an element of redundancy and code for only 20 different amino acids and 3 termination signals (UAG, UGA, and UAA), also called stop codons. The start codon is ATG and codes for methionine. These amino acids are joined by the ribosome to synthesize a protein. This protein, which may undergo further modification, will ultimately carry out a programmed biological function in the cell. Regulation of this process is highly coordinated and important in learning, for example, where activity-dependent translation at the synapse underlies some aspects of synaptic plasticity, which may go awry in certain disorders such as fragile X syndrome and autism (Morrow et al., 2008).
Over the last decade, the discovery of several classes of functional non–protein coding RNAs has added additional complexity to our understanding of how the genetic code is manifest at the level of cellular function. Of these, microRNAs (miRNAs) are increasingly being recognized as vital players in gene regulation and neurological disease (Weinberg and Wood, 2009). Nascent miRNA molecules are processed to form short (approximately 22-nucleotide) RNA duplexes that target endogenous cellular machinery to specific coding RNAs and induce posttranscriptional gene silencing through a diverse repertoire including RNA cleavage, translational blocking, transport to inactive cell sites, or promotion of RNA decay (Filipowicz et al., 2008; Weinberg and Wood, 2009). Depending on the cell and the context, miRNA activity can result in specific gene inactivation, functional repression, or more subtle regulatory effects and may involve multiple RNAs in a given biological pathway (Flynt and Lai, 2008). Estimates suggest that miRNAs may regulate 30% of protein-coding genes, implicating these molecules as important targets for future research into the biology of neurological disease (Filipowicz et al., 2008; Weinberg and Wood, 2009).
It is important to emphasize that most genes are not simply “on” or “off.” In reality, cells maintain strict regulatory control over their genes. Some genes, such as those required for cell structure or maintenance, must be expressed constitutively, but genes with specific precise functions may only be needed in certain cells at certain times under certain conditions. Potential levels of regulation are depicted in Fig. 40.2 and include virtually every stage of gene expression. Initially, genes can be regulated at the level of transcription, ranging from the regulated binding of histone proteins, which leads to chromosome condensation, inactivating genes, to the coordinated activity of protein factors that activate or repress gene transcription in response to cell state, environmental conditions, or other factors. Once expressed, the RNA is subject to processing regulation, particularly through alternative splicing as already discussed. Transport of the mRNA and its translation provide additional steps for cellular regulation. Lastly, the final protein can be subject to control via posttranslational modifications or interactions with other proteins. To operate, all these levels of regulation require trans-acting factors, such as proteins, which stimulate or repress a particular step, as well as cis-acting elements, sequences recognized and bound by the regulatory factors.
These detailed levels of regulation provide a dynamic and expansive capability to precisely control cellular function, essential for growth, development, and survival in an unpredictable environment. However, this also provides many potential points at which disease can arise from disrupted regulation. Consequently, a defective gene could cause disease directly through its own action or indirectly by disrupting regulation of other cellular pathways. For example, the forkhead box P2 (FOXP2) transcription factor regulates the expression of genes thought to be important for the development of spoken language (Konopka et al., 2009). Mutations in this gene cause an autosomal dominant disorder characterized by impairment of speech articulation and language processing (Lai et al., 2001). However, other mutations in this gene are responsible for approximately 1% to 2% of sporadic developmental verbal dyspraxia (MacDermot et al., 2005), likely via downstream effects. Mutation of the methyl-CpG-binding protein 2 (MECP2), which regulates chromatin structure, causes the neurodevelopmental disorder, Rett syndrome, but other mutations in this gene can cause intellectual disability or autism (Gonzales and LaSalle, 2010). Similarly, the FOX1 protein (also called ataxin 2 binding protein 1, or A2BP1), a neuron-specific RNA splicing factor (Underwood et al., 2005) predicted to regulate a large network of genes important to neurodevelopment (Yeo et al., 2009; Zhang et al., 2008), causes autistic spectrum disorder when disrupted (Martin et al., 2007) but has also been implicated as a susceptibility gene associated with both primary biliary cirrhosis (Joshita et al., 2010) and hand osteoarthritis (Zhai et al., 2009), presumably due to downstream effects or specific effects in non-neural tissues. This concept of genes acting on other genes will be further explored later (see Common Neurological Disorders and Complex Disease Genetics).
In addition to the complexity of regulatory mutations that affect gene expression by altering RNA or protein levels or by disrupting RNA splicing, there are certain mutations that do not cause protein dysfunction, but instead have effects restricted to the RNA itself. For example, RNA inclusions are found in several forms of triplet repeat disorders (see Repeat Expansion Disorders) including myotonic dystrophy type 1 and the fragile X–associated tremor/ataxia syndrome (FXTAS) (Garcia-Arocena and Hagerman, 2010; Orr and Zoghbi, 2007). The latter is particularly interesting from a genetic standpoint, because a disorder of late-onset progressive ataxia, tremor, and cognitive impairment occurs in carriers of FMR1 alleles of intermediate sizes, which are not full fragile X–causing mutations (Garcia-Arocena and Hagerman, 2010). FXTAS is a dominant gain-of-function disease that occurs via an entirely different mechanism than the recessive loss-of-function disease, fragile X syndrome (Garcia-Arocena and Hagerman, 2010; Penagarikano et al., 2007). Primary disorders of RNA still represent relatively uncharted territory, and it is likely that more RNA-specific diseases will be identified. This is particularly exciting for many reasons, not the least of which is that certain classes of these disorders may be amendable to therapy (Nakamori and Thornton, 2010; Wheeler et al., 2009).
Types of Genetic Variation and Mutations
Rare versus Common Variation
As dictated by the principles of natural selection, most genetic variation is not deleterious, and the induced phenotypic variability can be beneficial as a source on which evolution may act. From a clinical standpoint, it is helpful to dichotomize genetic variation into common and rare variation, while accepting that genetic variation is likely a continuum, and the choice of cutoff could be considered arbitrary. Rare genetic variants are of low frequency in the population (<1% frequency), either because they are deleterious and selected against, or because they are new and most often benign. Common genetic variation (>1% to 5% population frequency), on the other hand, is either adaptive, neutral, or not deleterious enough to be subject to strong negative selection; such variants are referred to as polymorphisms. The preeminent genetic model has been that common disease susceptibility is related to common genetic variation, and more rare forms of disease are caused by rare genetic variants, so-called mutations, which act in a Mendelian fashion. In contrast, common variants or polymorphisms may increase susceptibility for disease, but alone are not sufficient to cause disease (see Common Neurological Disorders and Complex Disease Genetics).
Polymorphisms and Point Mutations
The most prevalent form of genetic polymorphism is the single nucleotide polymorphism (SNP), which occurs on average every 300 to 1000 base pairs in the human genome. Most of these SNPs are relatively benign on their own and do not directly cause disease, so for the purposes of this initial discussion, we will concern ourselves primarily with mutations: rare genetic variants sufficient to cause disease. Pathogenic mutations can occur in numerous ways and vary from single nucleotide changes to gross rearrangements of chromosomes (Fig. 40.4). Owing to the large volume of DNA in the human genome, heritable mutations can arise spontaneously in the germline over time through errors in DNA replication or from DNA damage by metabolic or environmental sources despite the constant surveillance of extensive cellular preventive proofreading and repair mechanisms. Thus, mutations can be inherited from the parent or occur de novo in the germline. An example of a common de novo variant is trisomy 21, which causes Down syndrome (discussed further in Chromosomal Analysis and Abnormalities). The smallest pathogenic alterations, termed point mutations, involve a change in a single nucleotide within a DNA sequence. A point mutation can result in one of three possible effects with respect to protein: (1) a change to a different amino acid, called a missense mutation, (2) a change to a termination codon, called a nonsense mutation, or (3) creation of a new sequence that is silent with regard to protein sequence but alters some aspect of gene regulation, such as RNA splicing or transcriptional expression levels. Nonsense mutations can cause premature truncation of a protein, whereas a missense mutation can affect a protein in different ways depending on the chemical properties of the new amino acid and whether the change is located in a region of functional importance.
It should be emphasized that not all point mutations are disease-causing variants, although until recently, many considered that a premature stop codon was a “smoking gun.” Whole genome sequencing demonstrates that more than 100 such nonsense mutations may exist per genome, and the vast majority are expected to be relatively benign (Lupski et al., 2010; see Whole Genome/Exome Sequencing in Disease Gene Discovery). So in many cases, the pathogenicity of rare variants is not immediately discernable, and without strong statistical or functional evidence, labeling such genetic variation a mutation is premature and may be misleading. It is likely that most of these, including some variants thought previously to cause rare Mendelian diseases, may simply be benign genetic variation. This is because even a complete knockout of one allele caused by a premature stop codon (haploinsufficiency) may have no discernable effect on gene function for a majority of genes in the human genome (Lupski et al., 2010; Ng et al., 2009; Yngvadottir et al., 2009).
Occasionally, silent coding mutations or point mutations in noncoding regions may be significant for disease if they damage sequences important for gene expression (e.g., transcriptional and/or RNA processing regulatory elements). It has been estimated that up to half of all disease-causing mutations impact RNA splicing, which can have dire consequences given the importance of splicing to regulated gene expression. Such is the case for frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), where in some populations, the most common mutations disrupt splicing, causing a pathogenic imbalance in tau isoforms (D’Souza and Schellenberg, 2005). As for noncoding mutations, given the large volume of such sequences in the human genome—perhaps up to 96%—and our still imprecise ability to predict sequences required for regulation or to interpret identified sequence changes without direct experimentation, the majority of these mutations likely go unrecognized. It is hoped that the next generation of sequencing and bioinformatic technologies will allow for a better understanding of the role of these types of mutation in human disease.
Structural Chromosomal Abnormalities and Copy Number Variation
Small deletions and insertions can occur through slippage and strand mispairing at regions of short, tandem DNA repeats during replication. If the deletion or insertion is not a multiple of three, a frameshift will result, which leads to the translation of an altered protein sequence from the site of the mutation. On a larger scale, errors of chromosomal replication or recombination can result in inversions, translocations, deletions, duplications, or insertions (Stankiewicz and Lupski, 2010). When the region of deletion or duplication is greater than 1 kb, this is referred to as a copy number variation (CNV). Copy number variation is far more common than previously suspected, and it is estimated that at least 4% of the human genome varies in copy number (Conrad et al., 2010; Redon et al., 2006), much of which is commonly observed in the population and benign (Conrad et al., 2010). However, some rare CNVs such as the recurrent chromosome 17p12 duplication underlying most cases of Charcot-Marie-Tooth type 1A (Shchelochkov et al., 2010) or the alpha-synuclein triplication that can cause Parkinson disease (PD)(Singleton et al., 2003) are pathogenic and act in a Mendelian fashion. Even though such changes may be extensive, they may not be pathogenic if they do not disrupt expression of any key genes. This is particularly true for balanced translocations where genetic material is rearranged between chromosomes, yet no significant portion is actually lost. Although an individual with such a condition may be normal, if the germline is affected, their offspring may receive unbalanced chromosomal material and consequently develop a clinical phenotype (Kovaleva and Shaffer, 2003). CNVs will be discussed in greater detail when we consider common and complex disease genetics (see Copy Number Variation and Comparative Genomic Hybridization).
Repeat Expansion Disorders
Most mutations thus far discussed pass from parent to offspring unaltered, and in large affected families, the identical mutation can potentially be traced back generations. In contrast, there is a specific class of mutation, the repeat expansion (Orr and Zoghbi, 2007) (Table 40.2), which is unstable and can present with earlier onset and increasing severity in successive generations, a process known as anticipation. There are several examples of diseases caused by expanded repeats in coding sequence (e.g., most spinocerebellar ataxias, HD), as well as examples in noncoding sequence (e.g., fragile X syndrome, myotonic dystrophy) and within an intron (e.g., Friedreich ataxia). Interestingly, virtually all these disorders show neurological symptoms that can include such features as ataxia, intellectual disability, dementia, myotonia, or epilepsy, depending on the disease. The most common repeated sequence seen in these diseases is the CAG triplet, which codes for glutamine and expansion of which is seen in a variety of the spinocerebellar ataxias (SCAs) including SCA types 1, 2, 3, 6, 7, 17, and dentatorubropallidoluysian atrophy (DRPLA). In addition to protein-specific effects, these disorders likely share a common pathogenesis due to the presence of the polyglutamine repeat regions. In some disorders, the phenotype can be quite different depending on the number of repeats, such as in the FMR1 gene, where more than 200 CCG repeats causes fragile X syndrome, but repeats in the premutation range of 60 to 200, from which fully expanded alleles arise, can result in FXTAS or premature ovarian failure (Oostra and Willemsen, 2009). Although in general, the underlying mutation is similar, each specific repeat expansion has distinct effects on its corresponding gene, and thus in addition to varying phenotypes, they may also show very different inheritance patterns as illustrated later (see Disorders of Mendelian Inheritance).
Chromosomal Analysis and Abnormalities
The DNA coding for an individual gene is generally too small to be visualized microscopically, but it is possible to observe the chromosomes as they condense during mitosis as part of cell division (Griffiths et al., 2002; Strachan and Read, 2003). Traditionally, various staining techniques (e.g., Giemsa) are applied, producing a detailed pattern of banding along the chromosomes that are then photographed and aligned for comparative analysis. This arrangement and analysis of the chromosomes is known as a karyotype (Fig. 40.5). Through these methods, it is possible to visually identify large chromosomal deletions, duplications, or rearrangements. If high-resolution banding techniques are employed, structural alterations on the order of as small as 3 Mb (3 million base pairs) can be detected. More sophisticated techniques can also be employed, such as fluorescent in situ hybridization (FISH). In this method, a short DNA sequence, or probe, that corresponds to a chromosomal region of interest is hybridized with the patient’s DNA and detected visually via excitation of a fluorescent label. FISH can improve on visual resolution by 10- to 100-fold and is in common use for detection of a large number of well-defined genetic syndromes (Speicher and Carter, 2005) such as 15q duplication syndrome, DiGeorge syndrome (22q11 deletion), and Smith-Magenis syndrome (17p11 deletion).
More recent technological developments involving microarray technology (Geschwind, 2003) permit screening of the entire genome at high resolution (from kilobase to single nucleotide level) and are rapidly replacing techniques based on microscopic analysis. This technology is responsible for the emerging appreciation for the structural chromosomal variation in humans mentioned earlier, most of which is submicroscopic. For this section, we will focus on chromosomal alterations that can be detected microscopically, since the clinical implications of many small or rare structural variants identified are not yet clear (see Copy Number Variation and Comparative Genomic Hybridization).
The most common chromosomal abnormalities encountered clinically involve sporadic aneuploidy, either a deletion leaving one chromosome, or a monosomy, or a duplication leaving three chromosomes, or a trisomy (Strachan and Read, 2003). This occurs most frequently via nondisjunction, whereby chromosomes fail to separate during meiosis in the production of the gametes. The majority of aneuploidies are lethal, although there are a few that are viable and will be briefly discussed. Monosomy X (45,XO), also called Turner syndrome, is seen in approximately 1 of every 5000 births and results in sterile females of small stature with a variety of mild physical deformities including webbing of the neck, multiple nevi, and hand and elbow variations, with a very specific cognitive profile in patients with the full deletion (Strachan and Read, 2003). Individuals with additional copies of the X chromosome are also seen. While both females (47,XXX) and males (47,XXY) may have varying degrees of learning disabilities, especially involving language and attention (Geschwind et al., 2000), the males are referred to as having Klinefelter syndrome (KS) due to a phenotype also involving gynecomastia and infertility. XYY males have cognitive profiles similar to XXY males but several studies have suggested more severe social and behavioral problems in some individuals, especially increased aggression, which is rare in KS. Trisomy 21 (47, +21), or Down syndrome, includes profound intellectual impairment, flat faces with prominent epicanthal folds, and a predisposition to cardiac disease. At 1 in approximately 700 births, this is the most common genetic cause of intellectual disability and is associated with advanced maternal age at the time of conception. The other aneuploidies which can survive to term (trisomy 13 [47, +13], Edwards syndrome; trisomy 18 [47, +18], Patau syndrome) have much more severe phenotypes with drastically decreased viability, and death generally occurs within weeks to months after birth.
Disorders of Mendelian Inheritance
In this section we will consider genetic disorders caused by mutation of a single gene. Associating a clinical disease phenotype to the mutation of a specific gene has long been the goal of clinically based, or translational, neuroscience. It is expected that gene identification will eventually lead to an understanding of the disease etiology as well as more accurate diagnosis and better treatments. The ability to determine the genetic nature of most single-gene disease is ultimately based upon the laws of inheritance devised by Mendel in the late 1800s (Griffiths et al., 2002). To summarize these findings in a clinical context, the assumption is made that a phenotypic trait (or in this example, a disease) is caused by the alteration of a single gene. It is important to emphasize that this assumption does not always hold true, particularly for the more complex genetic diseases, as we will discuss later, but it is still true for many diseases seen by neurologists, and more than 3000 Mendelian conditions have been identified to date (OMIM, 2010). Now, if we accept the premise that a given disease is caused by a single gene, we know that for any individual, the gene exists as a pair of alleles with one copy from each parent. However, the alleles may not be equal, and one member of the pair may control the phenotype despite the presence of the other copy. In this case, we say that allele is dominant over the other, the latter of which is labeled as recessive. Depending on the gene and the mutation, as discussed later, a disease allele may be either dominant or recessive. Next, during the development of the gametes, these alleles segregate randomly in a process independent from all other genes. Therefore, the chance of a child receiving a particular allele is entirely random. If these laws all hold true, the observed inheritance of the clinical disease in families will follow a specific pattern that can be used to identify the nature of the causative gene. Although diseases showing Mendelian inheritance are either rare conditions or rare forms of common conditions (e.g., early-onset Alzheimer dementia or PD), identification of such genes are seminal biological advances that can have enormous impact on our understanding of these neurological conditions.
Autosomal Dominant Disorders
Autosomal dominant inheritance is characterized by direct transmission of the disorder from parent to child (Fig. 40.6). Affected individuals are seen in all generations, and a vertical line can be drawn on the pedigree to illustrate the passage of the disorder. Since only one deleterious copy of the disease gene is necessary, risk of transmission from an affected parent is 50%. Since the disorder is autosomal, there is no sex preference, and both males and females can present with the disease. One caveat involves the concept of penetrance, or the percent likelihood that a trait will manifest in a person with a specific genotype. A dominant gene is considered to have complete penetrance if all individuals with a given mutation develop disease. In practice, however, many autosomal dominant genes show varying degrees of penetrance or expressivity, most likely due to the influence of other genes and environmental factors.
There are over 400 examples of diseases with neurological phenotypes that show autosomal dominant inheritance (OMIM, 2010). These conditions include hyperkalemic periodic paralysis (voltage-gated sodium channel NaV1.4 on chromosome 17, often caused by missense mutations), HD (Huntington on chromosome 4, caused by CAG repeat expansion), SCA type 3 (ataxin-3 on chromosome 14, caused by CAG repeat expansion), Charcot-Marie-Tooth type 1B (myelin protein zero on chromosome 1, often caused by missense mutations), early-onset familial Alzheimer disease (AD)(presenilin-1, often caused by missense mutations), frontotemporal dementia with parkinsonism (microtubule-associated protein tau on chromosome 17, often caused by missense or splicing mutations), tuberous sclerosis type 1 (hamartin on chromosome 9, often caused by nonsense mutations and frameshifts), neurofibromatosis type 1 (neurofibromin on chromosome 17, caused by point mutations, frameshifts, and splicing mutations), and familial amyotrophic lateral sclerosis (ALS) (superoxide dismutase-1 on chromosome 21, caused by missense mutations), to name a few. Even rare Mendelian forms of more common syndromes such as epilepsy or sleep disorders (e.g., familial advanced sleep-phase syndrome) have been identified. More detailed lists can be found using the recommended online resources (Table 40.3).
| Disease-specific and gene-specific resources | GeneReviews at GeneTests University of Washington, Seattle, WA USA US National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/sites/GeneTests/ |
| Locus Specific Mutation Databases Human Genome Variation Society, Australia http://www.hgvs.org/dblist/glsdb.html |
|
| Neuromuscular Disease Center Washington University, St. Louis, MO USA http://neuromuscular.wustl.edu/ |
|
| Online Mendelian Inheritance in Man Johns Hopkins University, Baltimore, MD USA US National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/omim/ |
|
| Clinical genetic testing and clinical trials | ClinicalTrials.gov US National Institutes of Health http://clinicaltrials.gov/ |
| GeneTests University of Washington, Seattle, WA USA US National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/sites/GeneTests/ |
|
| Genomic variation and other genome resources | Alternative Splicing and Transcript Diversity Database European Molecular Biology Laboratory—European Bioinformatics Institute http://www.ebi.ac.uk/astd/ |
| Catalog of Published Genome-Wide Association Studies US National Human Genome Research Institute http://www.genome.gov/gwastudies/ |
|
| Ensembl Databases European Molecular Biology Laboratory—European Bioinformatics Institute Wellcome Trust Sanger Institute, UK http://www.ensembl.org/ |
|
| Database of Genomic Variants The Centre for Applied Genomics, Canada http://projects.tcag.ca/variation/ |
|
| International HapMap Project http://hapmap.ncbi.nlm.nih.gov/index.html |
|
| Single Nucleotide Polymorphism Database US National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/projects/SNP/ |
|
| 1000 Genomes Project http://www.1000genomes.org/ |
Autosomal Recessive Disorders
Autosomal recessive inheritance is characterized by lack of intergenerational transmission, in contrast to dominantly inherited disorders (Fig. 40.7
[/not-level-membership-for-neurology-category]
